
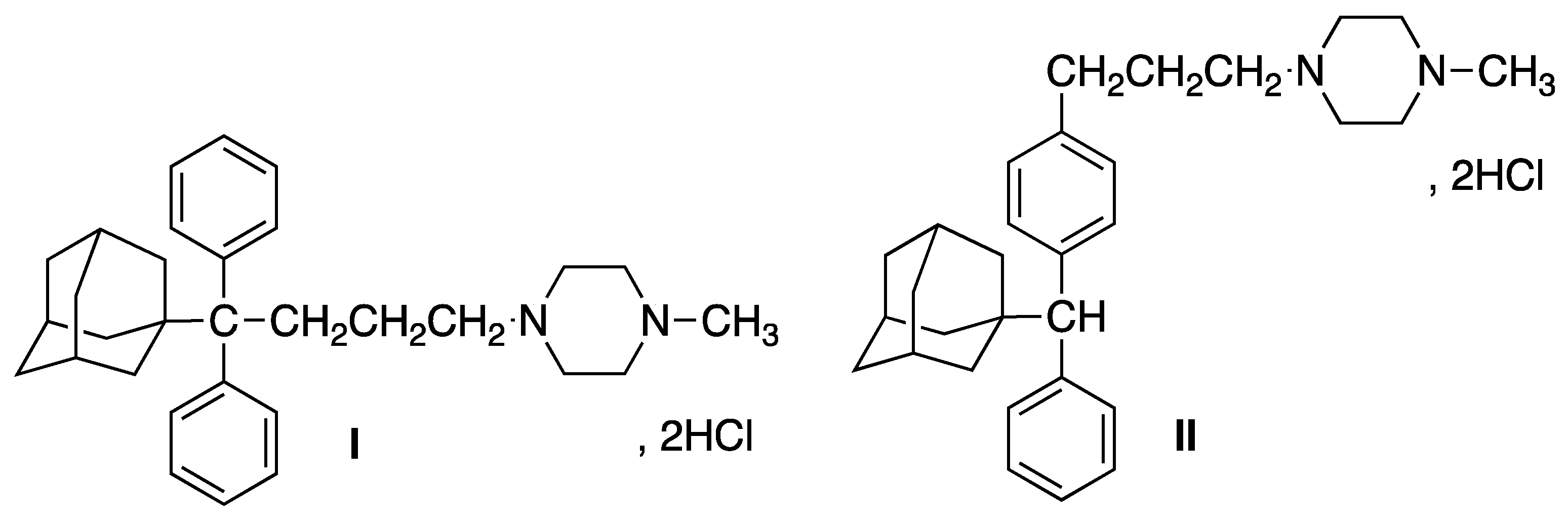
Biophysical Evaluation and In Vitro Controlled Release of Two Isomeric Adamantane Phenylalkylamines with Antiproliferative/Anticancer and Analgesic Activity

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Post Compression Parameters
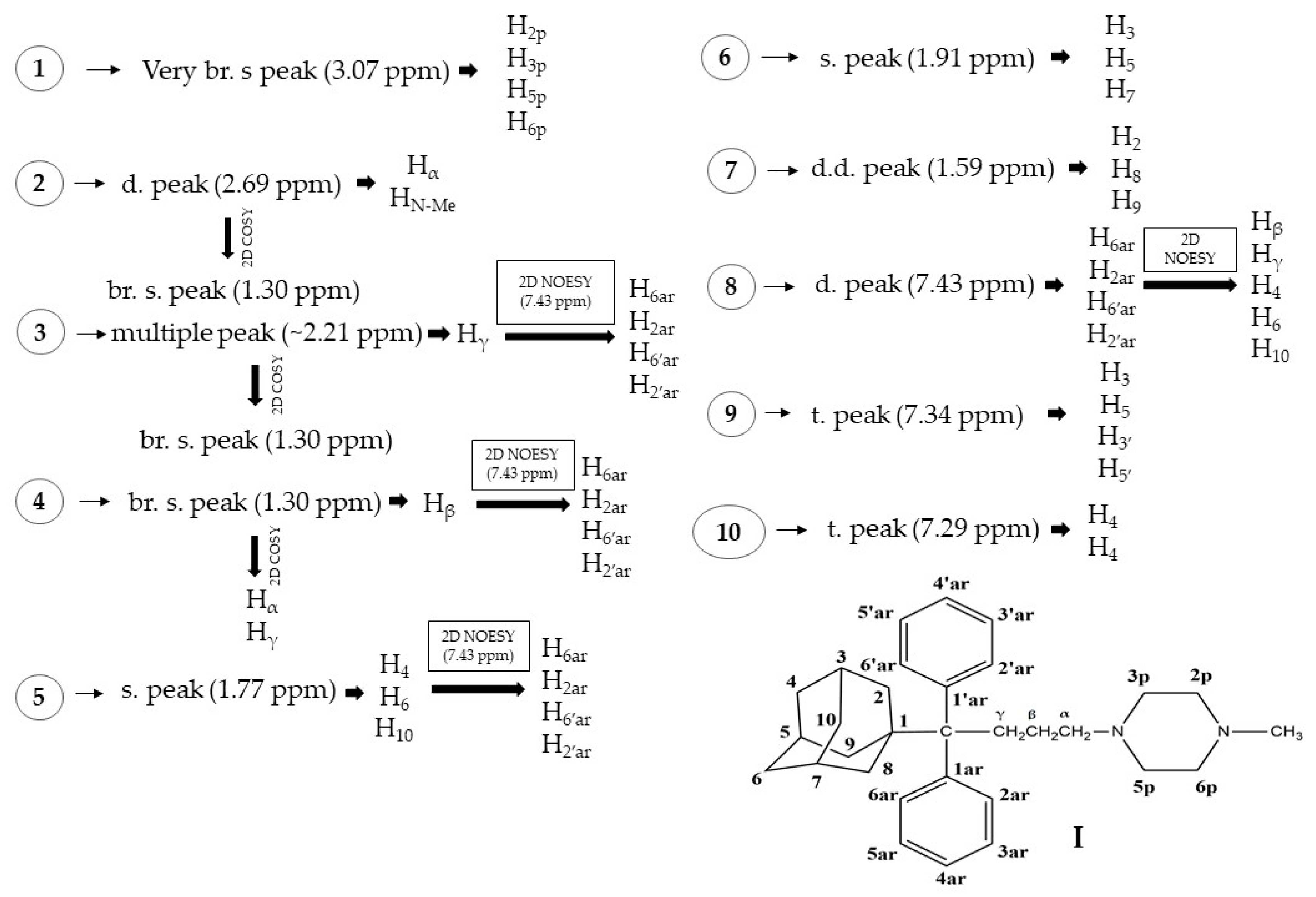
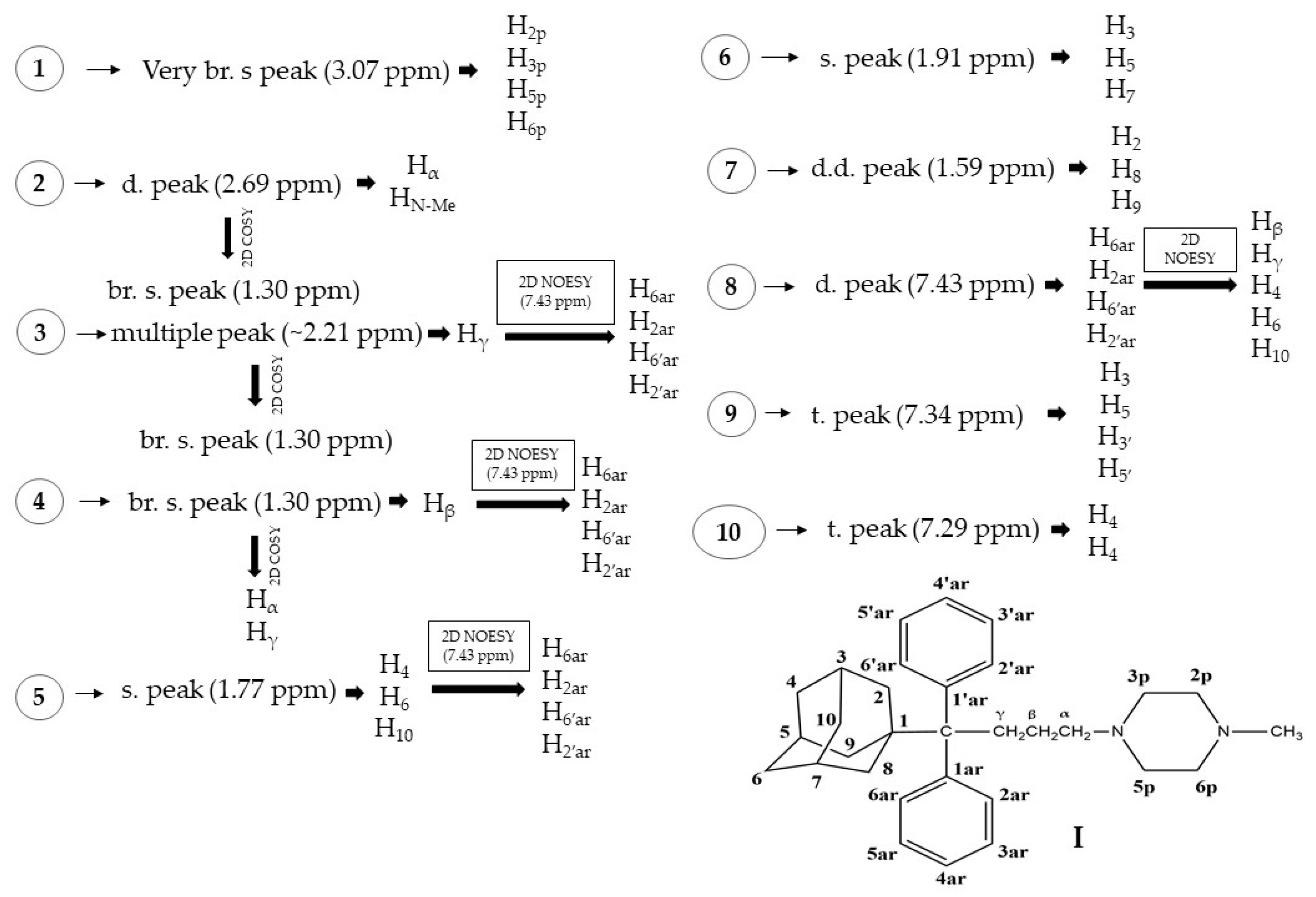
2.2. NMR
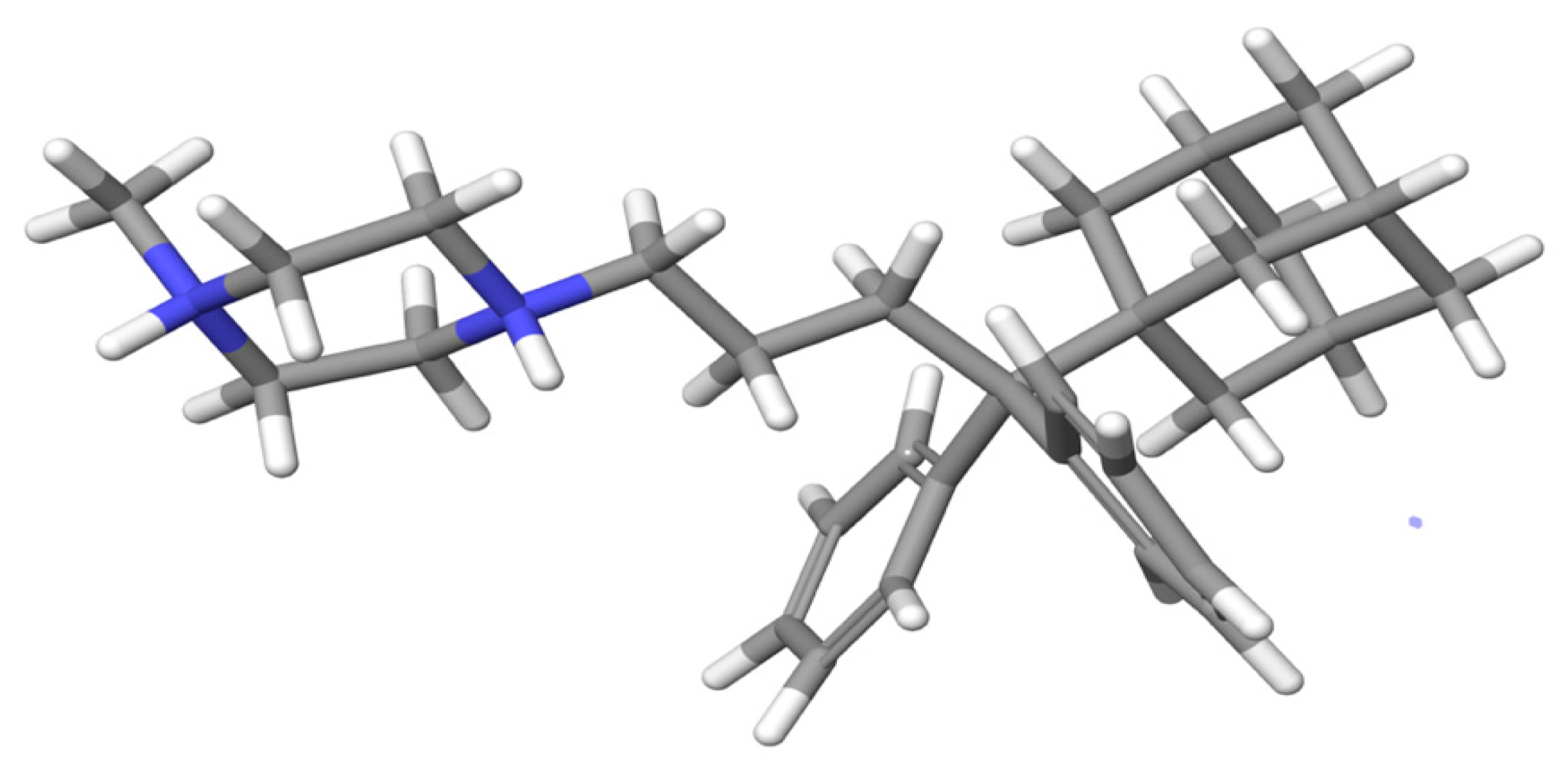
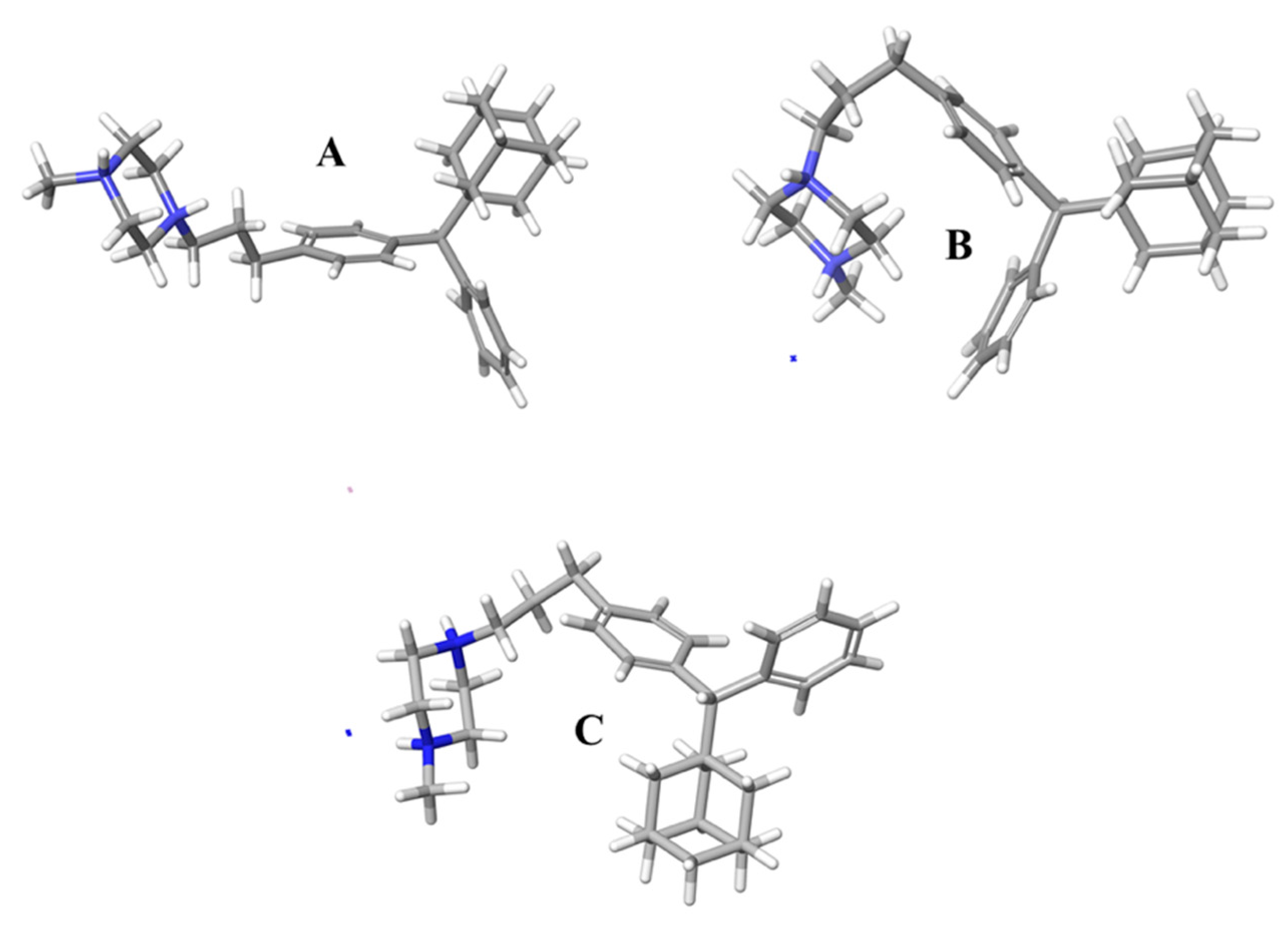
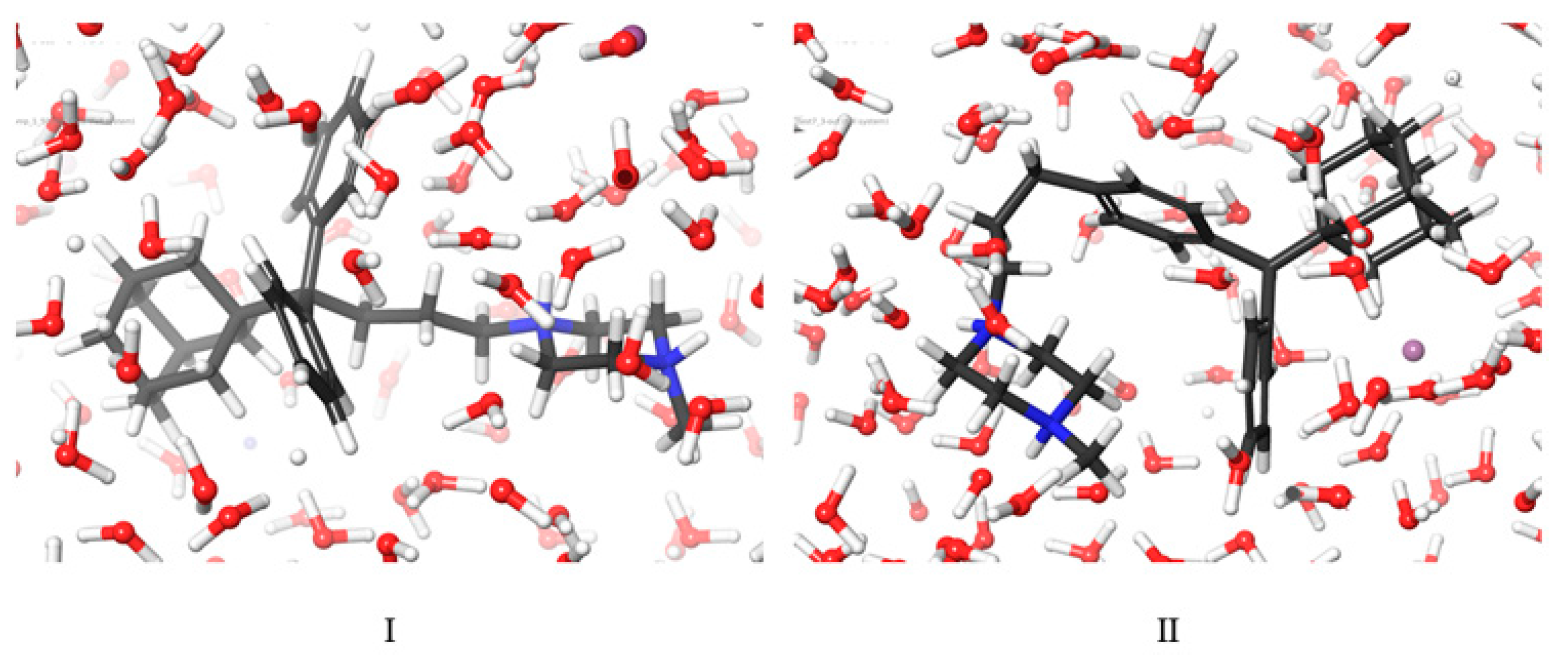
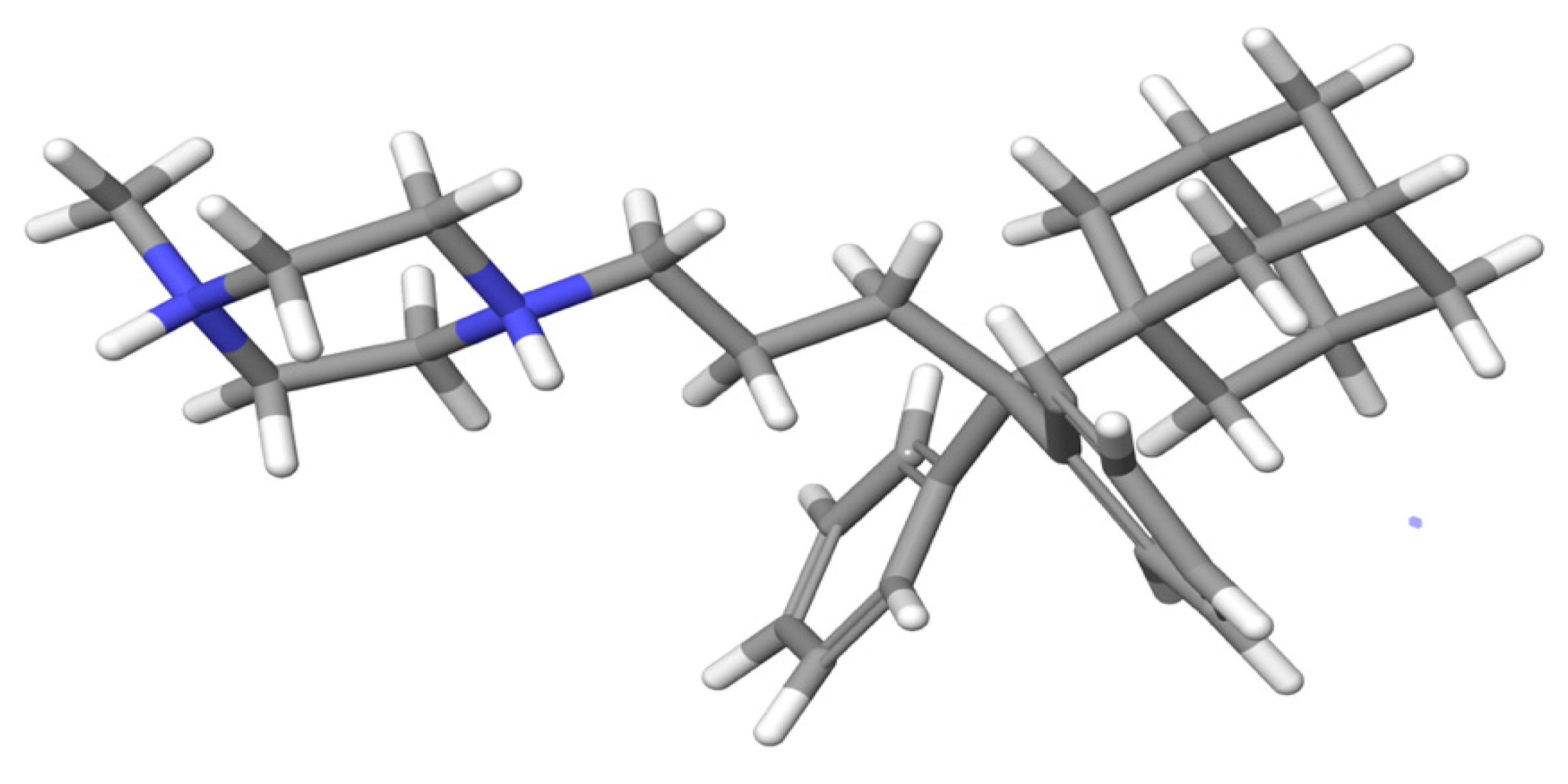
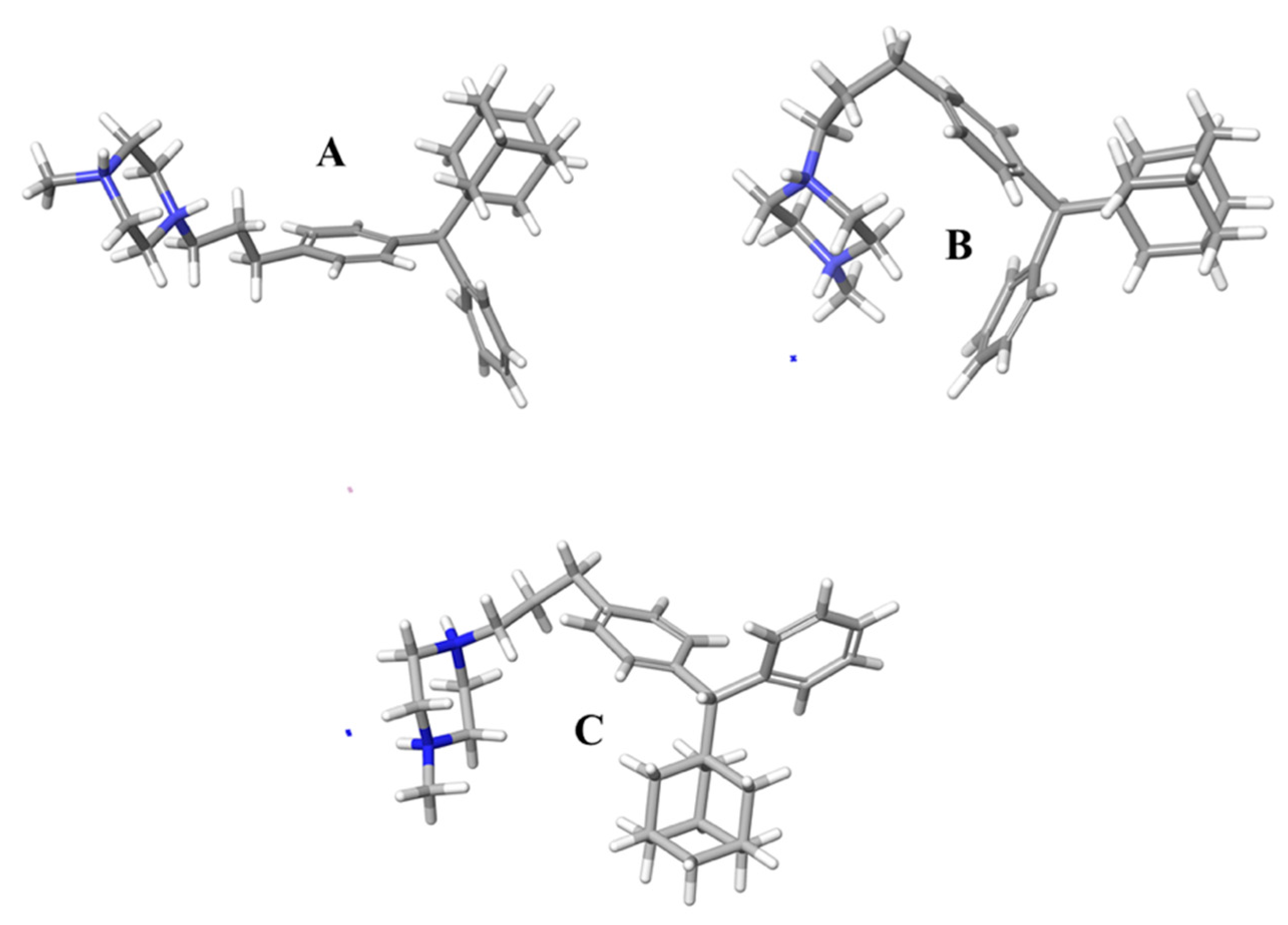
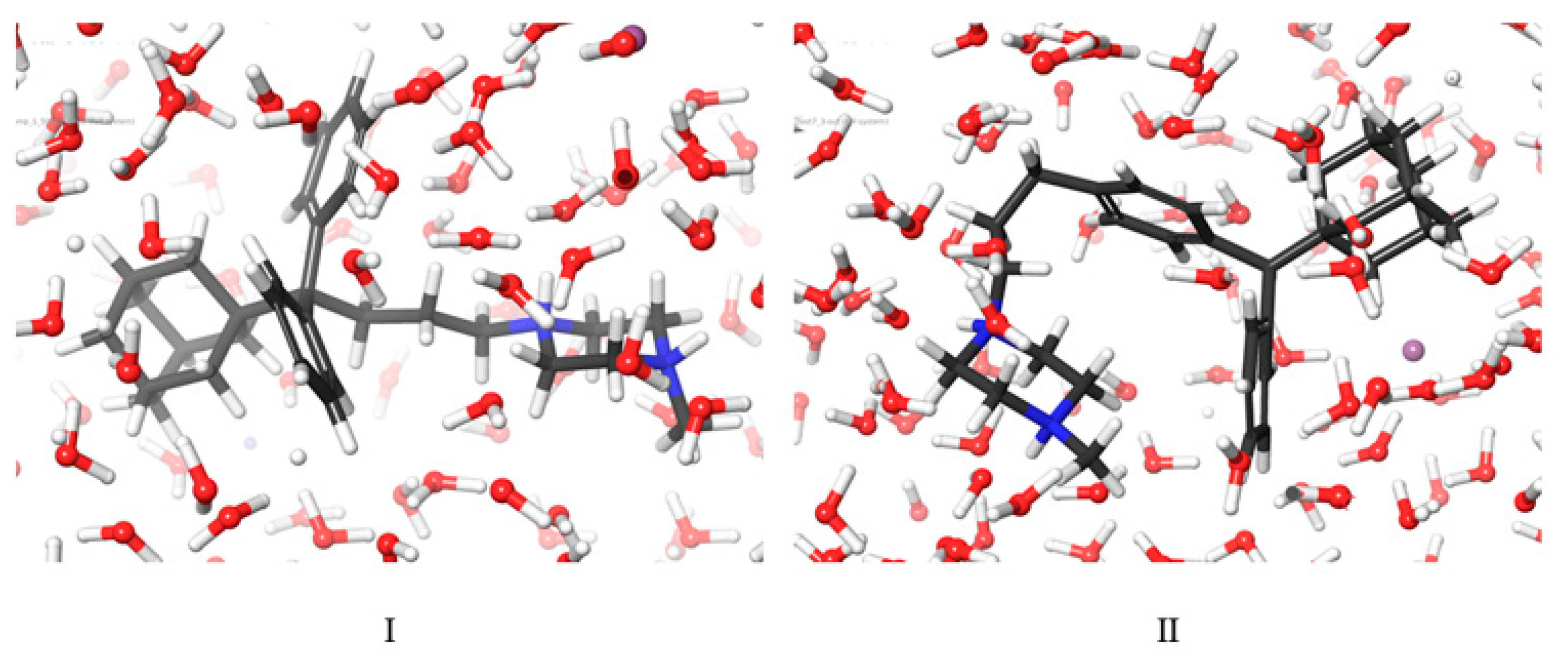
2.3. Molecular Dynamic Results
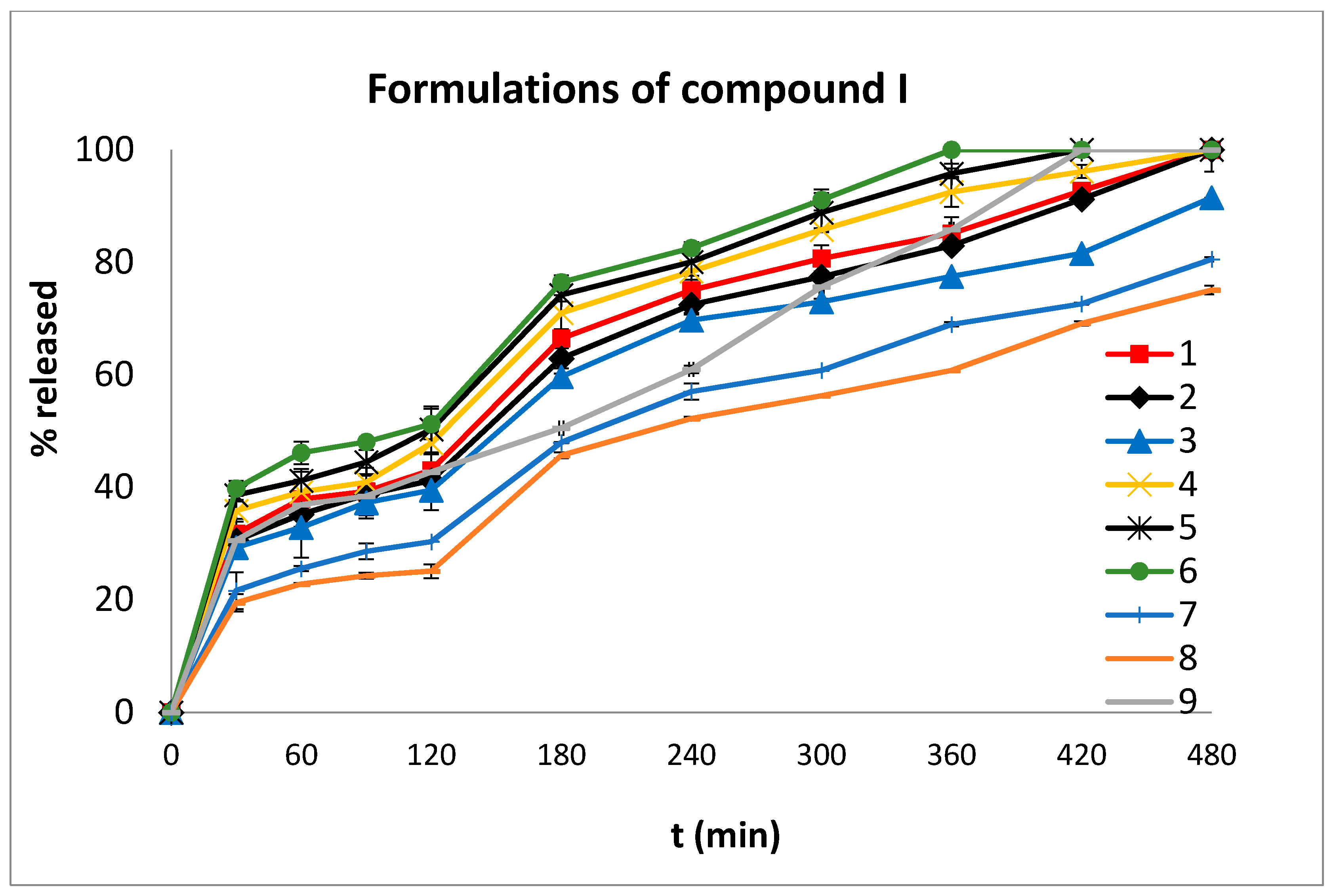
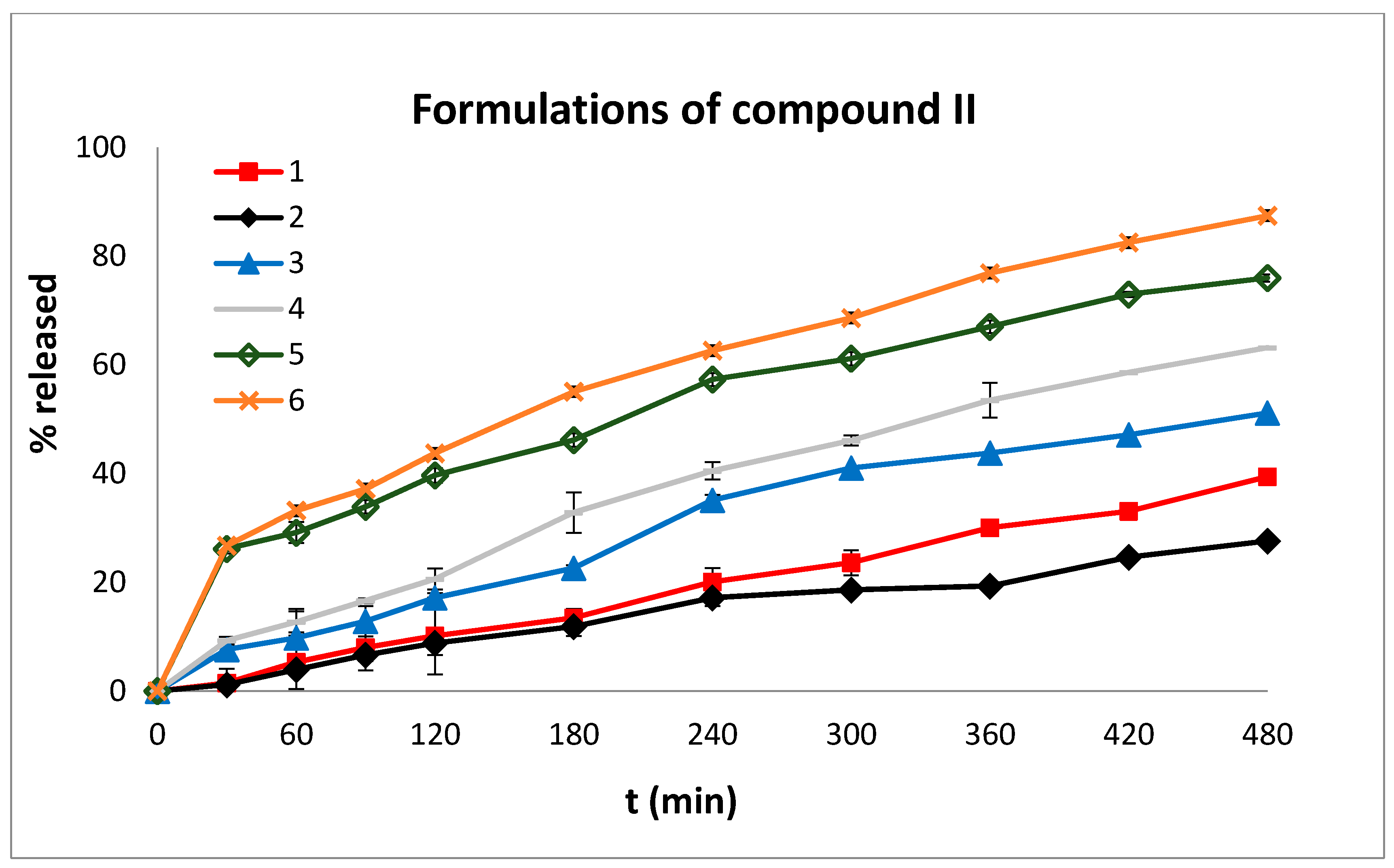
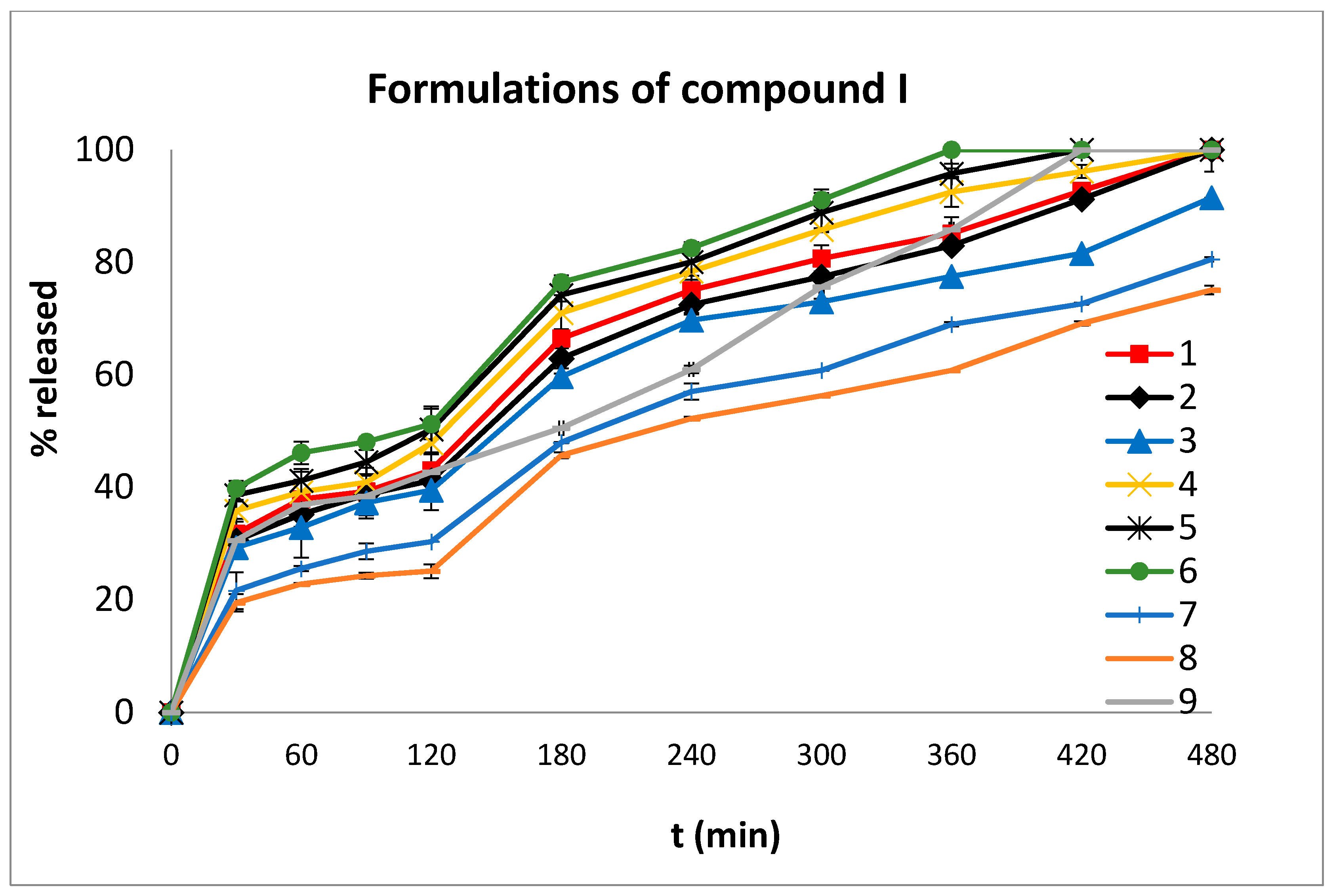
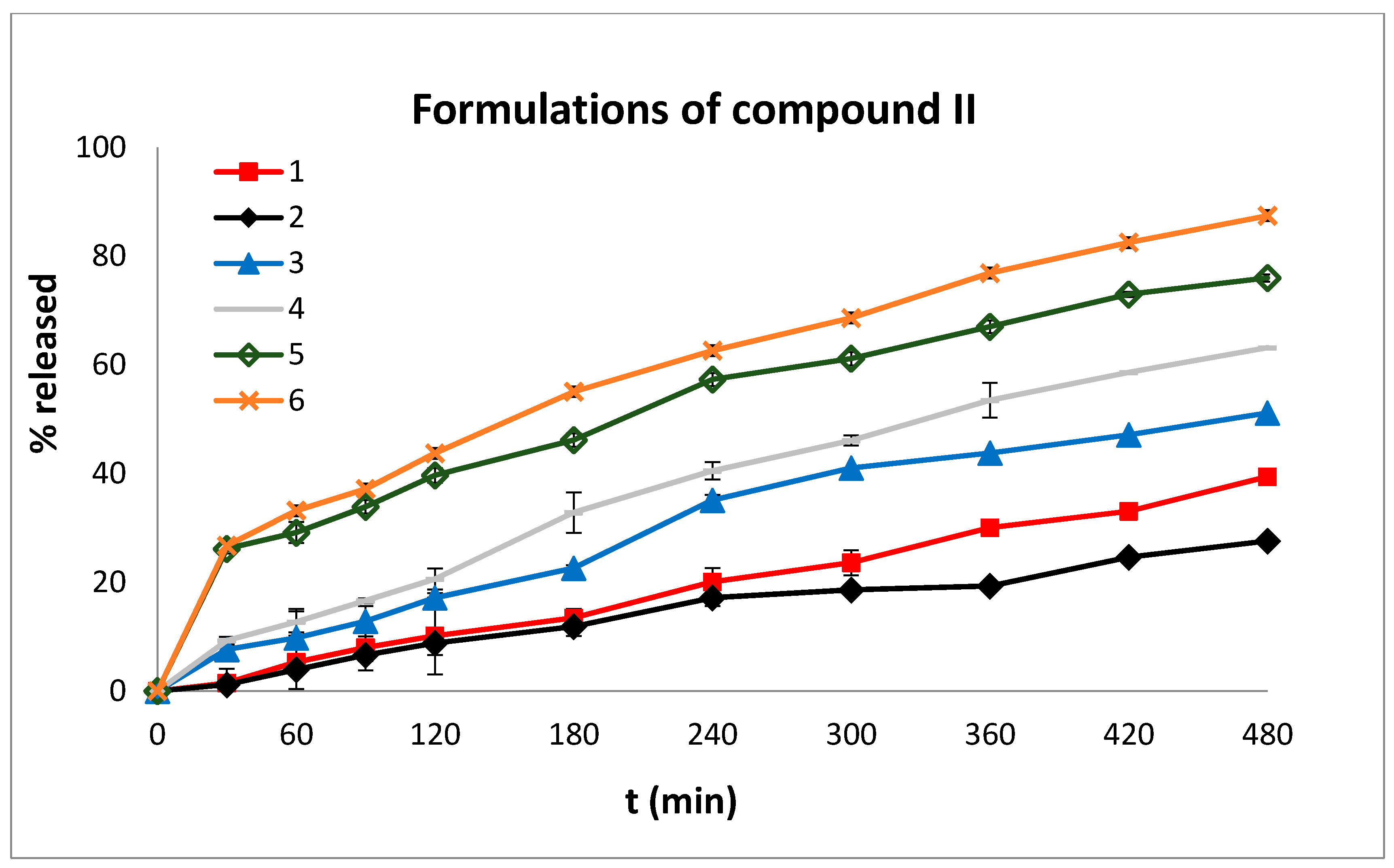
2.4. In Vitro Dissolution Studies
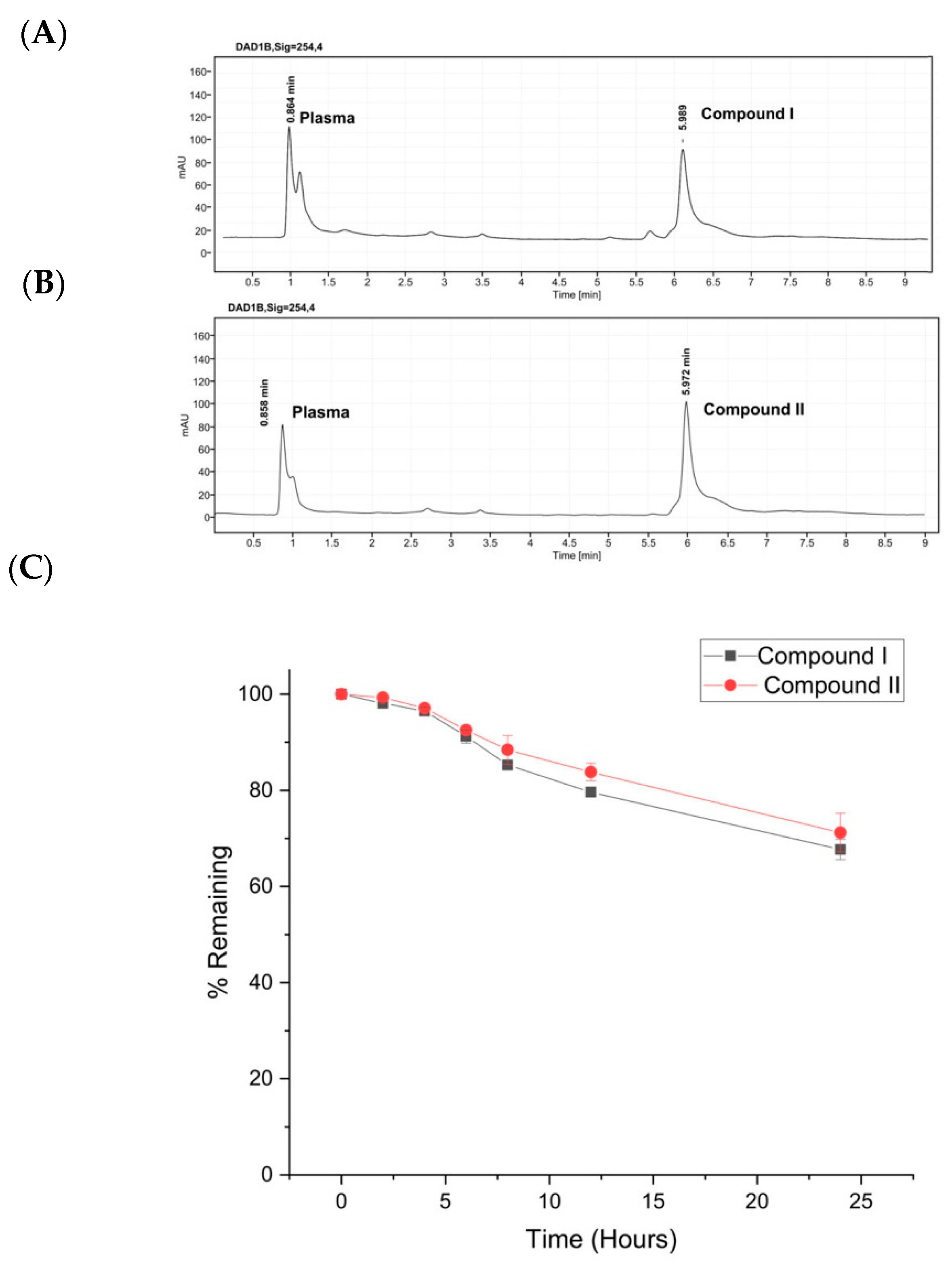
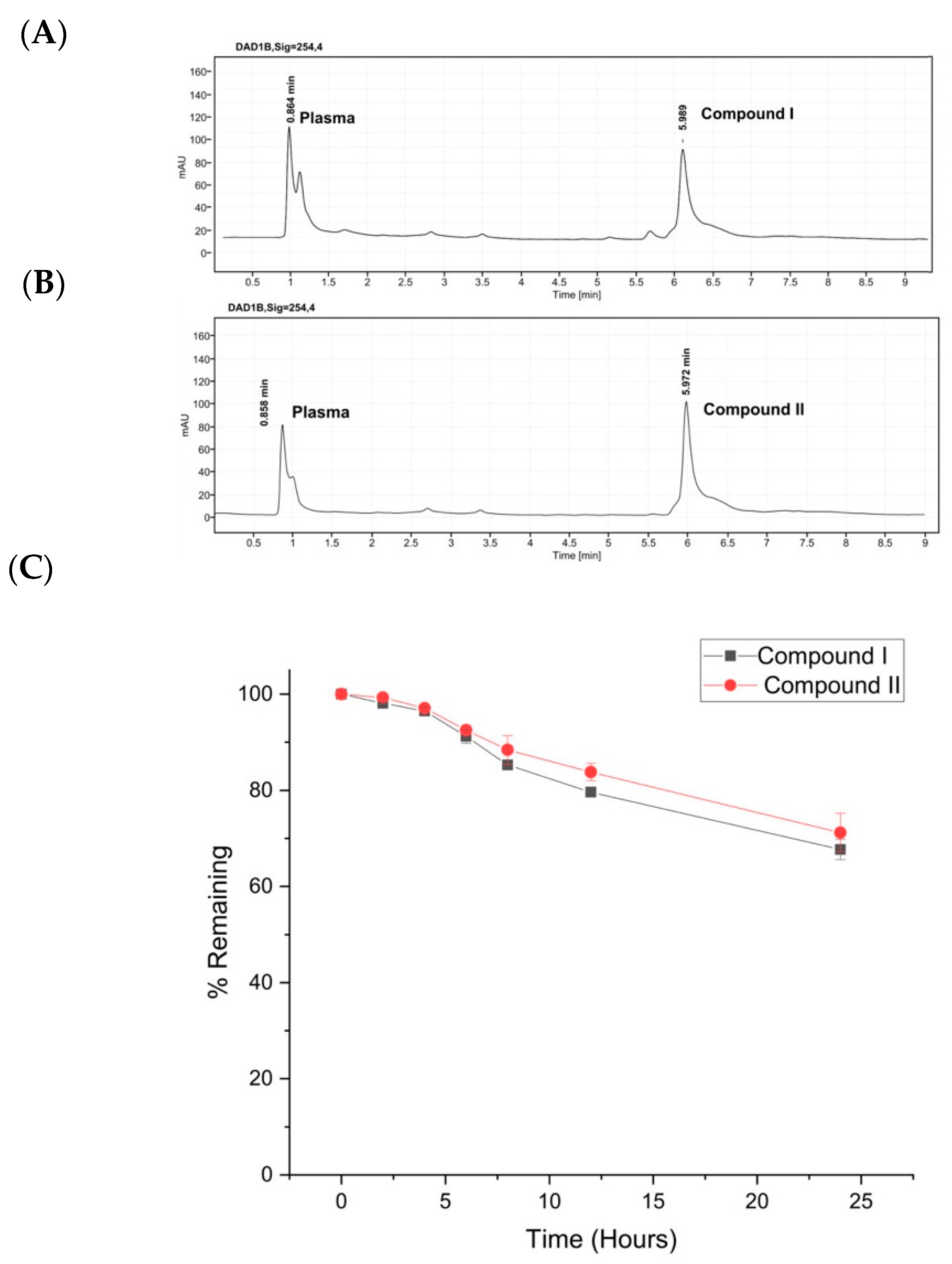
2.5. Plasma Stability
2.6. Results of the Pharmacokinetics and Toxicity Properties of the Two Compounds
3. Materials and Methods
3.1. Formulation of Compounds I and II into Modified Release Tablets
3.2. Post Compression Parameters
3.3. In Vitro Dissolution Studies
3.4. Stability of Compounds I and II in Human Plasma
3.5. NMR Experiments
3.6. Molecular Dynamics Simulations Method
3.7. Methods to Predict Pharmacokinetic and Toxicity of These Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tyagi, P.; Pechenov, S.; Anand Subramony, J. Oral peptide delivery: Translational challenges due to physiological effects. J. Control. Release 2018, 287, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. An overview of clinical and commercial impact of drug delivery systems. J. Control. Release 2014, 190, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghi, S.; Lee, W.K.; Kong, S.N.; Shetty, A.; Drum, C.L. Oral administration of protein nanoparticles: An emerging route to disease treatment. Pharmacol. Res. 2020, 158, 104685. [Google Scholar] [CrossRef] [PubMed]
- Landis, M.S.; Bhattachar, S.; Yazdanian, M.; Morrison, J. Commentary: Why Pharmaceutical Scientists in Early Drug Discovery Are Critical for Influencing the Design and Selection of Optimal Drug Candidates. AAPS PharmSciTech 2018, 19, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riganas, S.; Papanastasiou, I.; Foscolos, G.B.; Tsotinis, A.; Dimas, K.; Kourafalos, V.N.; Eleutheriades, A.; Moutsos, V.I.; Khan, H.; Margarita, P.; et al. New adamantane derivatives with sigma affinity and antiproliferative activity. Med. Chem. 2012, 8, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Riganas, S.; Papanastasiou, I.; Foscolos, G.B.; Tsotinis, A.; Serin, G.; Mirjolet, J.-F.; Dimas, K.; Kourafalos, V.N.; Eleutheriades, A.; Moutsos, V.I.; et al. New adamantane phenylalkylamines with σ-receptor binding affinity and anticancer activity, associated with putative antagonism of neuropathic pain. J. Med. Chem. 2012, 55, 10241–10261. [Google Scholar] [CrossRef] [PubMed]
- Riganas, S.; Papanastasiou, I.; Foscolos, G.B.; Tsotinis, A.; Bourguignon, J.J.; Serin, G.; Mirjolet, J.F.; Dimas, K.; Kourafalos, V.N.; Eleutheriades, A.; et al. Synthesis, σ1, σ2-receptors binding affinity and antiproliferative action of new C1-substituted adamantanes. Bioorganic Med. Chem. 2012, 20, 3323–3331. [Google Scholar] [CrossRef]
- Papanastasiou, I.; Tsotinis, A.; Kolocouris, N.; Nikas, S.P.; Vamvakides, A. New aminoadamantane derivatives with antiproliferative activity. Med. Chem. Res. 2014, 23. [Google Scholar] [CrossRef]
- Papanastasiou, I.; Riganas, S.; Foscolos, G.B.; Tsotinis, A.; Akhtar, S.A.; Khan, M.A.; Rahman, K.M.; Thurston, D.E. Synthesis and cytotoxicity of 4-(2-Adamantyl)phenylalkylamines. Lett. Org. Chem. 2015, 12, 319–323. [Google Scholar] [CrossRef]
- Koperniku, A.; Foscolos, A.-S.; Papanastasiou, I.; Foscolos, G.B.; Tsotinis, A.; Schols, D. 4-(1-Adamantyl)phenylalkylamines with potential antiproliferative activity. Lett. Org. Chem. 2016, 13, 171–176. [Google Scholar] [CrossRef]
- Missling, C.U. Anagelsic Therapeutic And Method, 1-(3-4((1R,3S,5S)-Adamantan–1-yl)(phenyl)methyl)propyl)–4-methylpiperazines and Salts Thereof. U.S. Patent 10,195,192, B2, 5 February 2019. [Google Scholar]
- Schmidt, H.R.; Kruse, A.C. The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 2019, 40, 636–654. [Google Scholar] [CrossRef] [PubMed]
- Su, T.P.; Su, T.C.; Nakamura, Y.; Tsai, S.Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef] [Green Version]
- Christ, M.G.; Clement, A.M.; Behl, C. The Sigma-1 Receptor at the Crossroad of Proteostasis, Neurodegeneration, and Autophagy. Trends Neurosci. 2020, 43, 79–81. [Google Scholar] [CrossRef]
- Colabufo, N.A.; Berardi, F.; Contino, M.; Niso, M.; Abate, C.; Perrone, R.; Tortorella, V. Antiproliferative and cytotoxic effects of some σ2 agonists and σ1 antagonists in tumour cell lines. Naunyn. Schmiedebergs. Arch. Pharmacol. 2004, 370, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, M.O.; Karoutzou, O.; Foscolos, A.S.; Papanastasiou, I. Sigma receptor (σR) ligands with antiproliferative and anticancer activity. Molecules 2017, 22, 1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 862. [Google Scholar] [CrossRef] [PubMed]
- Linciano, P.; Rossino, G.; Listro, R.; Rossi, D.; Collina, S. Sigma-1 receptor antagonists: Promising players in fighting neuropathic pain. Pharm. Pat. Anal. 2020, 9, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Colabufo, N.A.; Leopoldo, M.; Ferorelli, S.; Abate, C.; Contino, M.; Perrone, M.G.; Niso, M.; Perrone, R.; Berardi, F. Why PB28 Could Be a Covid 2019 Game Changer? ACS Med. Chem. Lett. 2020, 11, 2048–2050. [Google Scholar] [CrossRef] [PubMed]
- Abate, C.; Niso, M.; Abatematteo, F.S.; Contino, M.; Colabufo, N.A.; Berardi, F. PB28, the Sigma-1 and Sigma-2 Receptors Modulator With Potent Anti–SARS-CoV-2 Activity: A Review About Its Pharmacological Properties and Structure Affinity Relationships. Front. Pharmacol. 2020, 11, 1833. [Google Scholar] [CrossRef]
- Varma, M.V.S.; Kaushal, A.M.; Garg, A.; Garg, S. Factors affecting mechanism and kinetics of drug release from matrix-based oral controlled drug delivery systems. Am. J. Drug Deliv. 2004, 2, 43–57. [Google Scholar] [CrossRef]
- Lamoudi, L.; Chaumeil, J.C.; Daoud, K. Swelling, erosion and drug release characteristics of Sodium Diclofenac from heterogeneous matrix tablets. J. Drug Deliv. Sci. Technol. 2016, 31, 93–100. [Google Scholar] [CrossRef]
- Bruschi, M.L. Main mechanisms to control the drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Cambridge, UK, 2015; pp. 37–62. [Google Scholar]
- Streubel, A.; Siepmann, J.; Dashevsky, A.; Bodmeier, R. pH-independent release of a weakly basic drug from water-insoluble and -soluble matrix tablets. J. Control. Release 2000, 67, 101–110. [Google Scholar] [CrossRef]
- Martínez González, I.; Villafuerte Robles, L. Influence of enteric citric acid on the release profile of 4-aminopyridine from HPMC matrix tablets. Int. J. Pharm. 2003, 251, 183–193. [Google Scholar] [CrossRef]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [Green Version]
- Efentakis, M.; Iliopoyloy, A.; Siamidi, A. Effect of core size and excipients on the lag time and drug release from a pulsatile drug delivery system. Drug Dev. Ind. Pharm. 2011, 37, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.C.; Sheskey, P.J.; Owen, S.C.; American Pharmacists Association. Handbook of Pharmaceutical Excipients; Pharmaceutical Press: Grayslake, IL, USA, 2009; p. 888. [Google Scholar]
- Vlachou, M.; Siamidi, A.; Goula, E.; Georgas, P.; Pippa, N.; Karalis, V.; Sentoukas, T.; Pispas, S. Probing the release of the chronobiotic hormone melatonin from hybrid calcium alginate hydrogel beads. Acta Pharm. 2020, 70, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Vlachou, M.; Tragou, K.; Siamidi, A.; Kikionis, S.; Chatzianagnostou, A.L.; Mitsopoulos, A.; Ioannou, E.; Roussis, V.; Tsotinis, A. Modified in vitro release of the chronobiotic hormone melatonin from matrix tablets based on the marine sulfated polysaccharide ulvan. J. Drug Deliv. Sci. Technol. 2018, 44, 41–48. [Google Scholar] [CrossRef]
- Tiwari, S.B.; Murthy, T.K.; Pai, M.R.; Mehta, P.R.; Chowdary, P.B. Controlled release formulation of tramadol hydrochloride using hydrophilic and hydrophobic matrix system. AAPS PharmSciTech 2003, 4, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.Q.; Hu, J.H.; Deng, J.X.; Su, H.; Xu, S.; Liu, J.Y. In vitro controlled release of sodium ferulate from Compritol 888 ATO-based matrix tablets. Int. J. Pharm. 2006, 324, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Green, P.G.; Bumgarner, R.E.; Dasgupta, S.; Goddard, W.A.; Blake, G.A. Benzene forms hydrogen bonds with water. Science 1992, 257, 942–945. [Google Scholar] [CrossRef] [PubMed]
- Grassi, M.; Grassi, G. Mathematical Modelling and Controlled Drug Delivery: Matrix Systems. Curr. Drug Deliv. 2005, 2, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Podczeck, F. Comparison of in vitro dissolution profiles by calculating mean dissolution time (MDT) or mean residence time (MRT). Int. J. Pharm. 1993, 97, 93–100. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.A. The concept of dissolution efficiency. J. Pharm. Pharmacol. 1975, 27, 48–49. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Hong, Y.; Chen, H. Development and application of high throughput plasma stability assay for drug discovery. Int. J. Pharm. 2005, 297, 110–119. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. 2002, 98, 6885–6892. [Google Scholar] [CrossRef]
- Lyman, E.; Zuckerman, D.M. Ensemble-based convergence analysis of biomolecular trajectories. Biophys. J. 2006, 91, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | MDT | t20% | t50% | t90% | n | Mean % D.E. | |
|---|---|---|---|---|---|---|---|
| Compound I | 1 | 138.89 | 18 | 137 | 398 | 0.45 | 66.09 |
| 2 | 145.70 | 20 | 144 | 411 | 0.44 | 64.05 | |
| 3 | 139.19 | 21 | 152 | 472 | 0.52 | 59.89 | |
| 4 | 128.36 | 16 | 126 | 338 | 0.44 | 69.83 | |
| 5 | 120.89 | 16 | 119 | 310 | 0.19 | 72.38 | |
| 6 | 114.02 | 15 | 107 | 292 | 0.18 | 74.44 | |
| 7 | 153.95 | 28 | 193 | * | 0.57 | 50.35 | |
| 8 | 157.64 | 35 | 220 | * | 0.59 | 46.10 | |
| 9 | 159.44 | 19 | 175 | 377 | 0.35 | 62.53 | |
| Compound II | 1 | 208.21 | 369 | * | * | 0.99 | 19.33 |
| 2 | 189.56 | 240 | * | * | 0.76 | 14.70 | |
| 3 | 175.24 | 150 | 460 | * | 0.72 | 30.19 | |
| 4 | 175.36 | 116 | 332 | * | 0.78 | 36.96 | |
| 5 | 135.41 | 23 | 201 | * | 0.43 | 51.32 | |
| 6 | 186.95 | 23 | 175 | * | 0.44 | 55.78 |
| Properties | ||
|---|---|---|
| Compound I | Compound II | |
| LogP | 6.0726 | 6.0708 |
| Rotable Bonds | 7 | 7 |
| Hydrogen Bond Acceptors | 2 | 2 |
| Hydrogen Bond Donors | 0 | 0 |
| Surface Area | 200.919 (Å2) | 200.919 |
| Water Solubility | −3.776 (log mol.L−1) | −4.112 |
| Properties | ||
|---|---|---|
| Absorption | ||
| Compound I | Compound II | |
| Caco2 Permeability | 1.02(>0.90) | 0.966(>0.90) |
| Intestinal Absorption (Human) | 100(>30) | 97.152(>30) |
| Skin Permeability | −2.736(<−2.5) | −2.737(<−2.5) |
| P-Glycoprotein Substrate | Yes | Yes |
| P-Glycoprotein I Inhibitor | Yes | Yes |
| P-Glycoprotein II Inhibitor | Yes | Yes |
| Distribution | ||
| VDss (Human) | 0.299(<0.45) | 0.656(>0.45) |
| Fraction Unbound (Human) | 0.193 | 0.164 |
| BBB Permeability | 1.491(>0.3) | 1.471(>0.3) |
| CNS Permeability | −1.552(>−2) | −1.467(>−2) |
| Metabolism | ||
| CYP2D6 Substrate | No | No |
| CYP3A4 Substrate | Yes | Yes |
| CYP1A2 Inhibitior | No | No |
| CYP2C19 Inhibitior | No | No |
| CYP2C9 Inhibitior | No | No |
| CYP2D6 Inhibitior | Yes | Yes |
| CYP3A4 Inhibitior | No | No |
| Excretion | ||
| Total Clearance | 0.283 | 0.404 |
| Renal OCT2 Substrate | No | No |
| Prediction | Probability | Prediction | Probability | |
|---|---|---|---|---|
| Compound I | Compound II | |||
| Organ Toxicity | ||||
| Hepatotoxicity | Inactive | 0.94 | Inactive | 0.93 |
| Toxicity Endpoints | ||||
| Mutagenicity | Inactive | 0.64 | Inactive | 0.56 |
| Carcinogenicity | Inactive | 0.79 | Inactive | 0.82 |
| Cytotoxicity | Inactive | 0.80 | Inactive | 0.77 |
| Immunotoxicity | Inactive | 0.99 | Inactive | 0.92 |
| Toxicological Pathways | ||||
| Aryl hydrocarbon Receptor (AhR) | Inactive | 0.94 | Inactive | 0.94 |
| Andogen Receptor (AR) | Inactive | 0.97 | Inactive | 0.99 |
| Androgen Receptor Ligand Binding Domain (AR-LBD) | Inactive | 0.99 | Inactive | 0.99 |
| Aromatase | Inactive | 0.94 | Inactive | 0.98 |
| Estrogen Receptor Alpha (ER) | Inactive | 0.97 | Inactive | 0.96 |
| Estrogen Receptor Ligand Binding Domain (ER-LBD) | Inactive | 0.98 | Inactive | 0.99 |
| Peroxisome Proliferator Activated Receptor Gamma (PPAR-Gamma) | Inactive | 0.99 | Inactive | 0.99 |
| Nuclear factor (erythroid-derived 2)-like 2/Antioxidant Responsive Element (nrf2/ARE) | Inactive | 0.99 | Inactive | 0.99 |
| Heat Shock Factor Response Element (HSE) | Inactive | 0.99 | Inactive | 0.99 |
| Mitochondrial Membrane Potential (MMP) | Inactive | 0.89 | Inactive | 0.95 |
| Phosphoprotein (Tumor Supressor) p53 | Inactive | 0.94 | Inactive | 0.96 |
| ATPase Family AAA Domain-Containing Protein 5 (ATAD5) | Inactive | 0.99 | Inactive | 0.99 |
| Formulation | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Compound I | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| PEO (7 × 106) | 88 | 88 | 88 | 68 | 68 | 68 | 45 | 45 | 45 |
| HPMC K100 | 45 | 45 | 55 | 45 | 45 | 45 | 88 | 88 | 45 |
| Eudragit L100-55 | 45 | 10 | - | 50 | 50 | 25 | 45 | 45 | 50 |
| Sodium Alginate | 10 | 45 | 45 | 25 | - | - | 10 | - | 48 |
| Ulvan | - | - | - | - | 25 | 50 | - | 10 | - |
| Magnesium Stearate | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Total | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 |
| Formulation | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Compound II | 10 | 10 | 10 | 10 | 10 | 10 |
| PEO (7 × 106) | 100 | 85 | 70 | 55 | 25 | 10 |
| HPMC K100 | 38 | 44 | 50 | 45 | 30 | 30 |
| Eudragit L100-55 | 50 | 44 | 38 | 50 | 95 | 102 |
| Sodium Alginate | - | 15 | 30 | 38 | 38 | 46 |
| Magnesium Stearate | 2 | 2 | 2 | 2 | 2 | 2 |
| Total | 200 | 200 | 200 | 200 | 200 | 200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlachou, M.; Foscolos, A.-S.; Siamidi, A.; Syriopoulou, A.; Georgiou, N.; Dedeloudi, A.; Tsiailanis, A.D.; Tzakos, A.G.; Mavromoustakos, T.; Papanastasiou, I.P. Biophysical Evaluation and In Vitro Controlled Release of Two Isomeric Adamantane Phenylalkylamines with Antiproliferative/Anticancer and Analgesic Activity. Molecules 2022, 27, 7. https://doi.org/10.3390/molecules27010007
Vlachou M, Foscolos A-S, Siamidi A, Syriopoulou A, Georgiou N, Dedeloudi A, Tsiailanis AD, Tzakos AG, Mavromoustakos T, Papanastasiou IP. Biophysical Evaluation and In Vitro Controlled Release of Two Isomeric Adamantane Phenylalkylamines with Antiproliferative/Anticancer and Analgesic Activity. Molecules. 2022; 27(1):7. https://doi.org/10.3390/molecules27010007
Chicago/Turabian StyleVlachou, Marilena, Angeliki-Sofia Foscolos, Angeliki Siamidi, Angeliki Syriopoulou, Nikitas Georgiou, Aikaterini Dedeloudi, Antonis D. Tsiailanis, Andreas G. Tzakos, Thomas Mavromoustakos, and Ioannis P. Papanastasiou. 2022. "Biophysical Evaluation and In Vitro Controlled Release of Two Isomeric Adamantane Phenylalkylamines with Antiproliferative/Anticancer and Analgesic Activity" Molecules 27, no. 1: 7. https://doi.org/10.3390/molecules27010007
APA StyleVlachou, M., Foscolos, A.-S., Siamidi, A., Syriopoulou, A., Georgiou, N., Dedeloudi, A., Tsiailanis, A. D., Tzakos, A. G., Mavromoustakos, T., & Papanastasiou, I. P. (2022). Biophysical Evaluation and In Vitro Controlled Release of Two Isomeric Adamantane Phenylalkylamines with Antiproliferative/Anticancer and Analgesic Activity. Molecules, 27(1), 7. https://doi.org/10.3390/molecules27010007