
The Impact of Retinal Configuration on the Protein–Chromophore Interactions in Bistable Jumping Spider Rhodopsin-1


Abstract
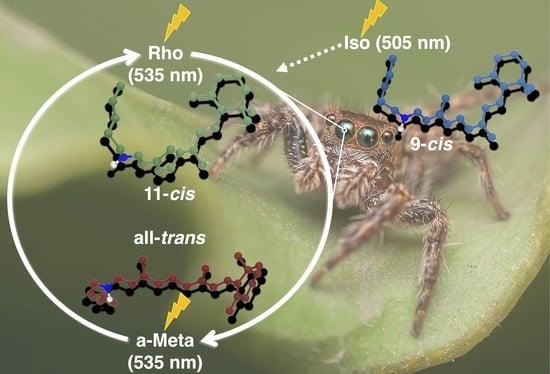
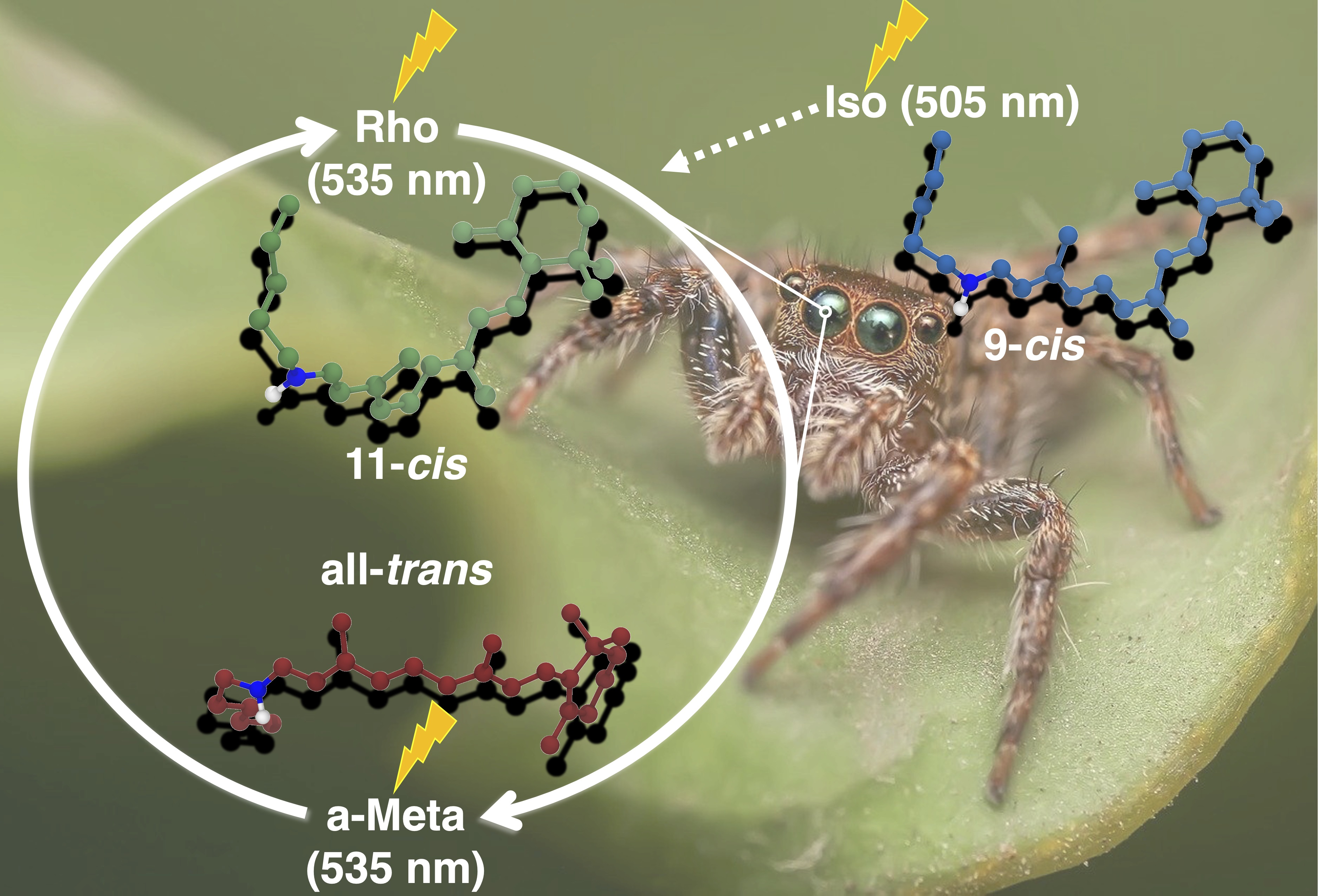
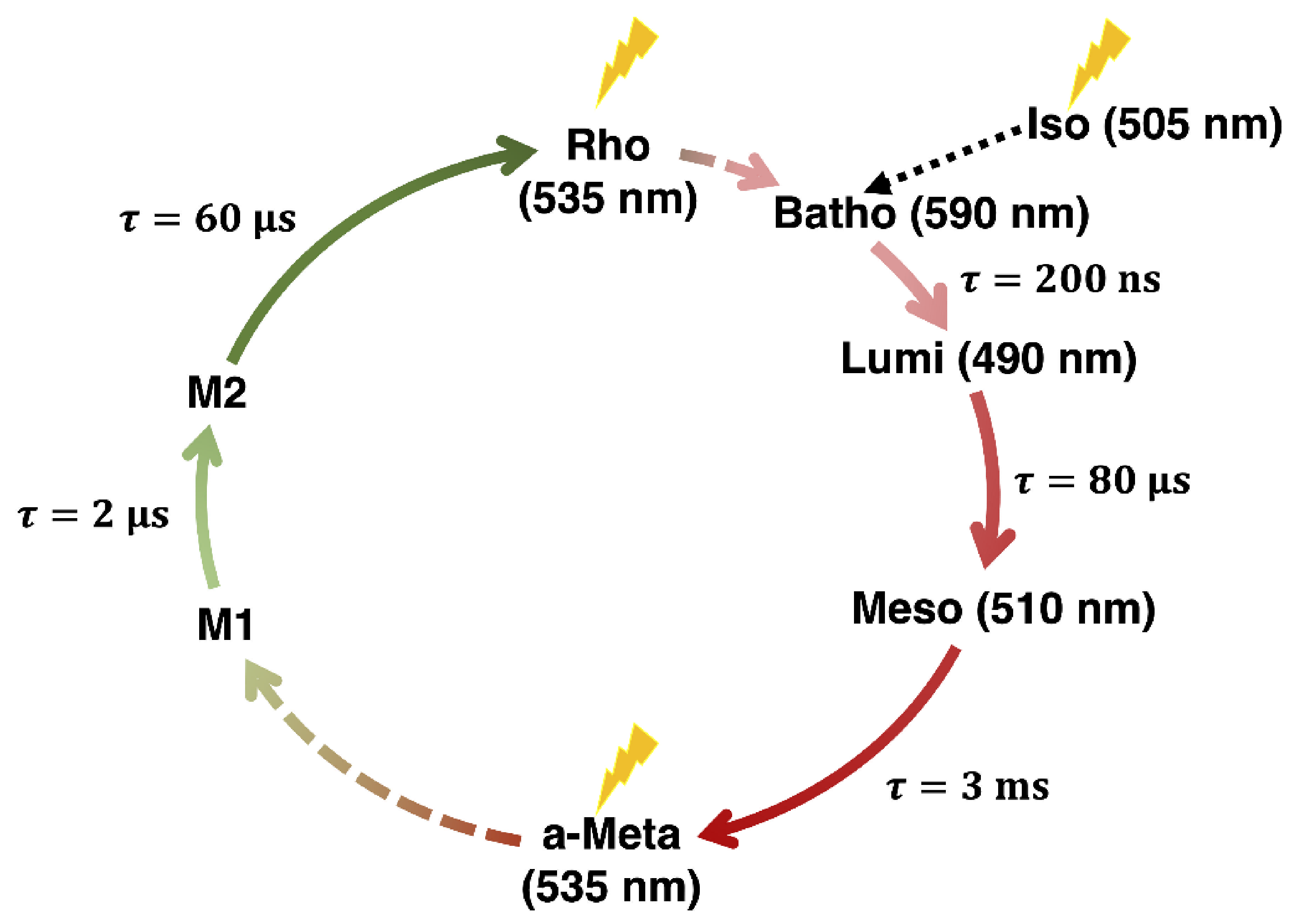
:
1. Introduction
2. Computational Methods
2.1. Model Generation
2.2. Retinal Parameterization
2.3. Classical and QM/MM Molecular Dynamics Simulations
2.4. Spectra Generation
3. Results and Discussion
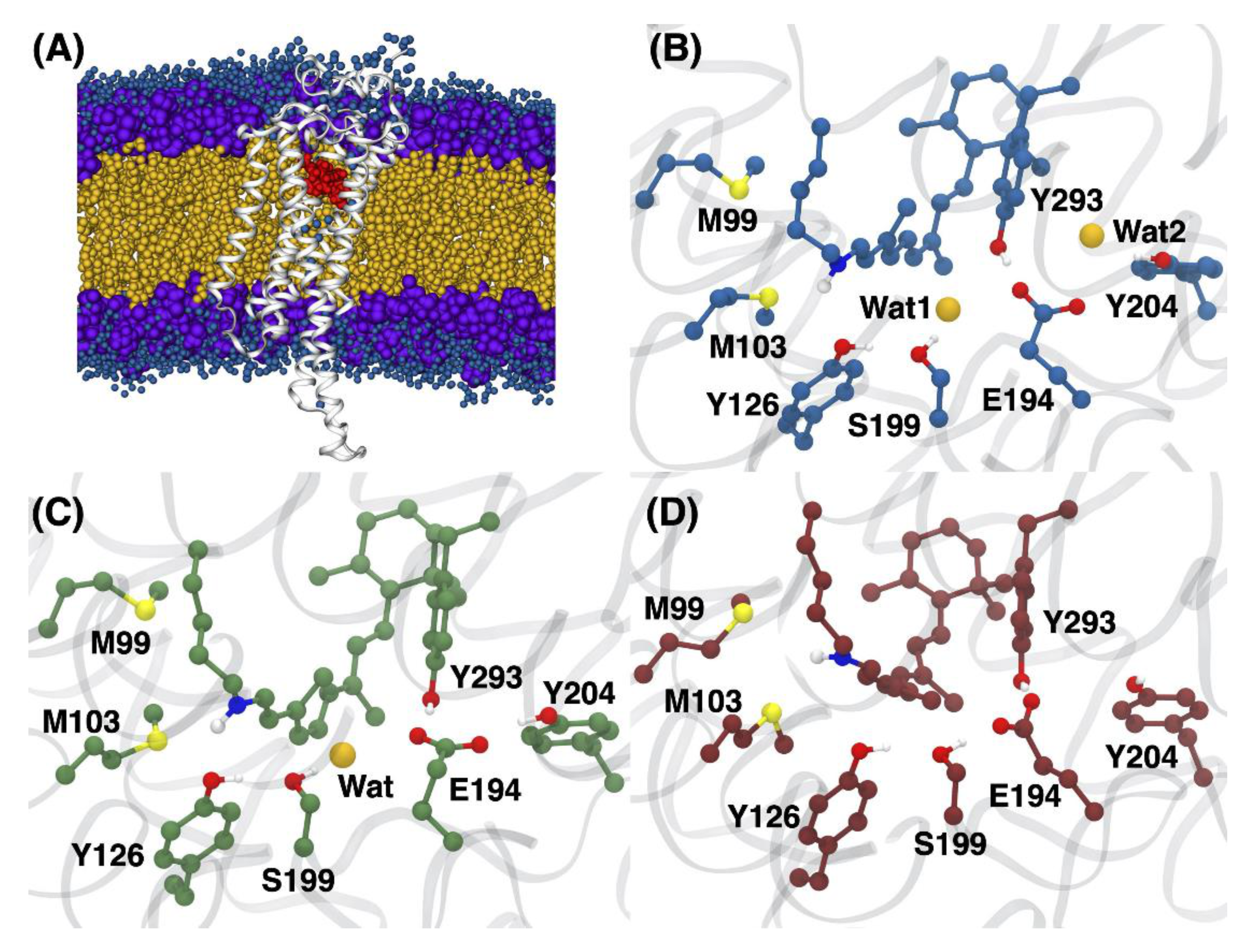
3.1. Structural Differences between the Isomers
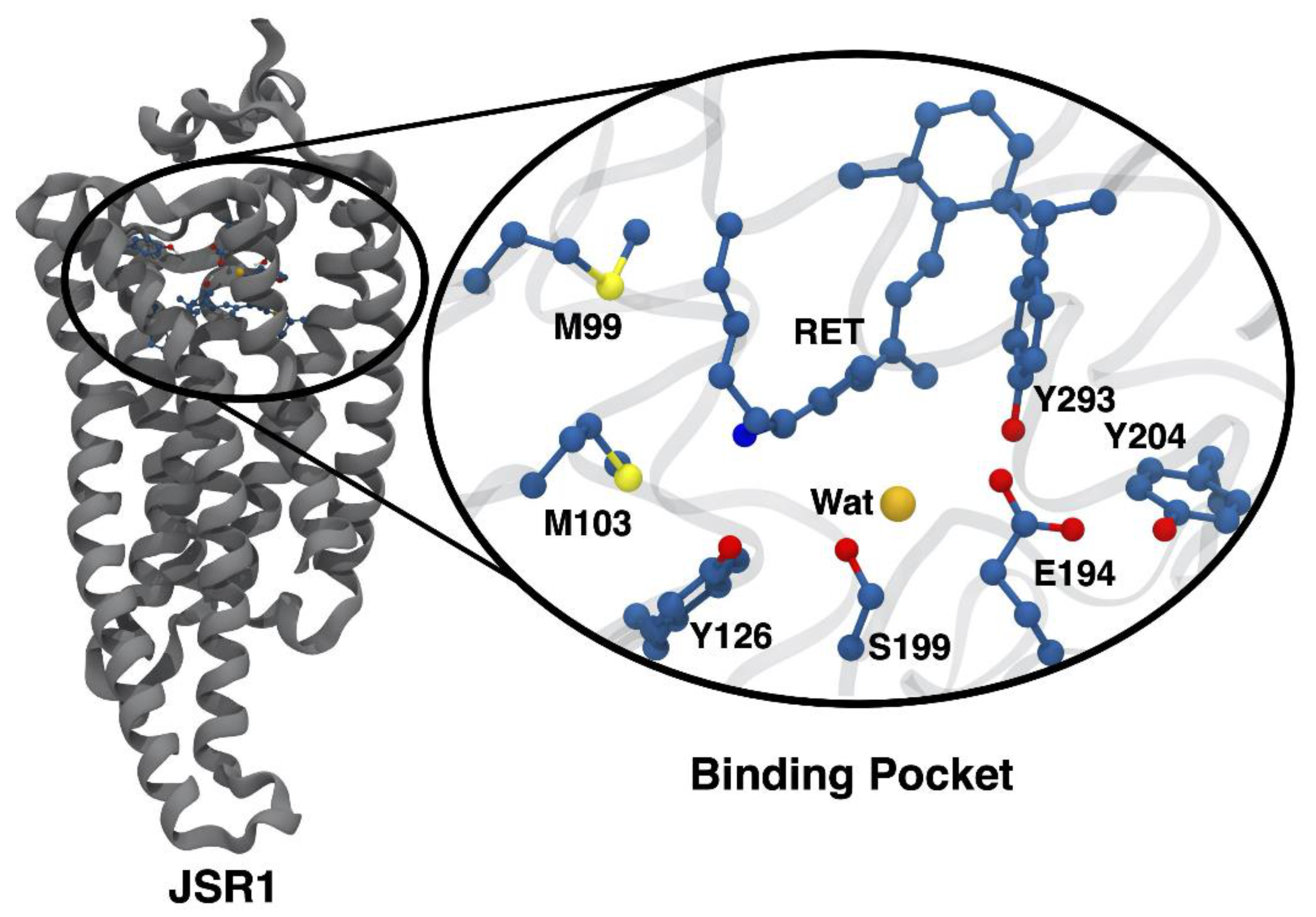
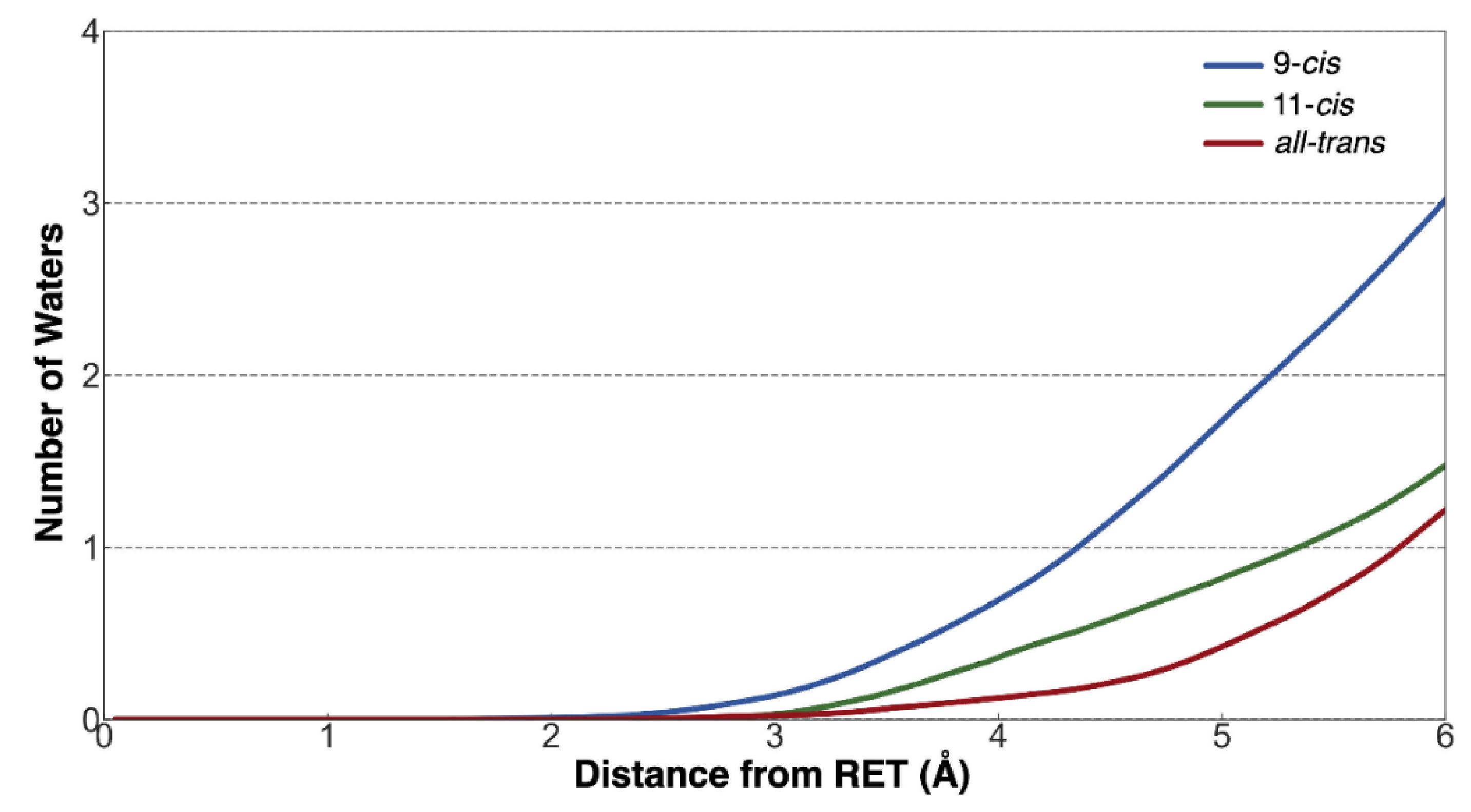
3.2. Changes in the Electrostatic Environment
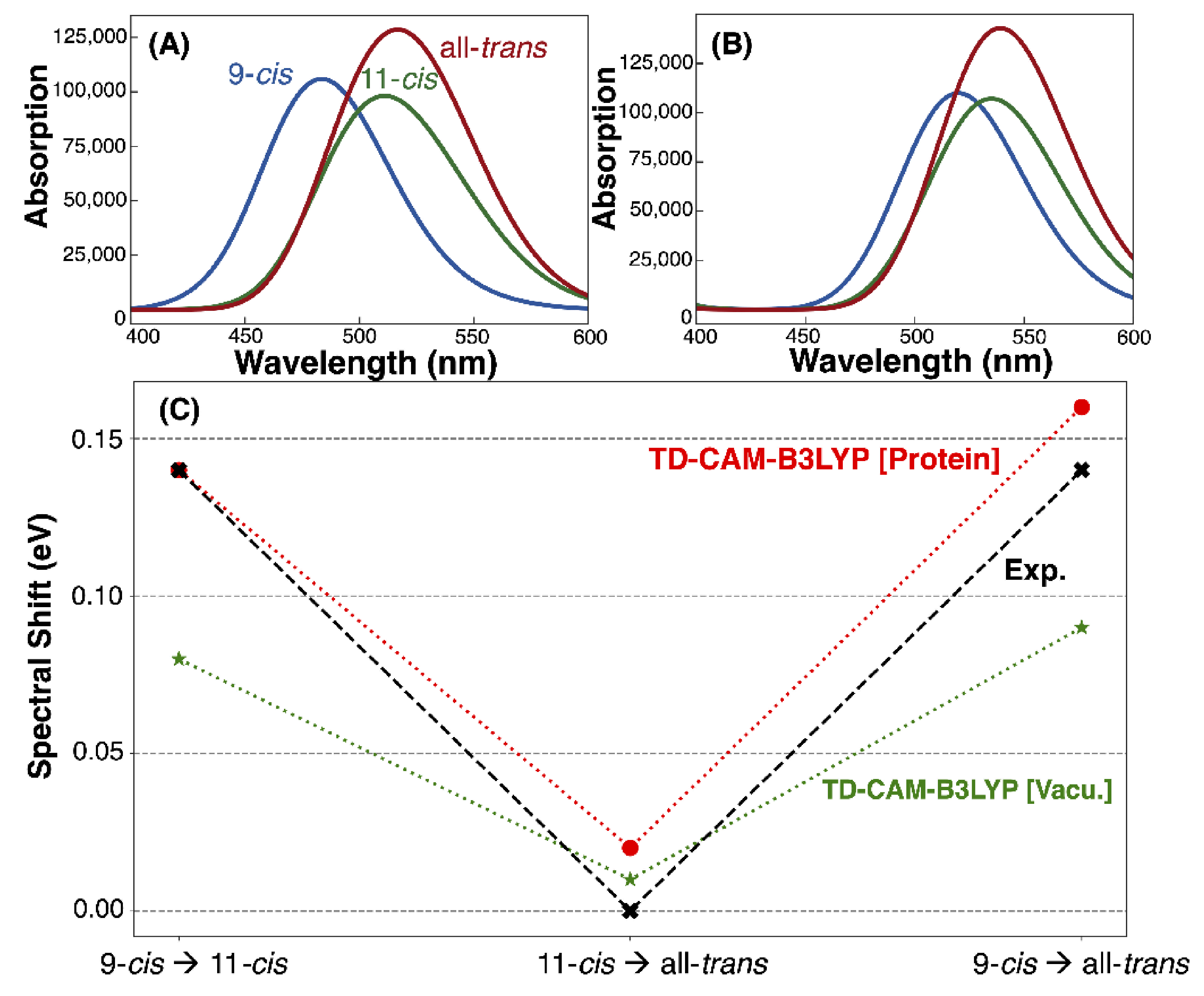
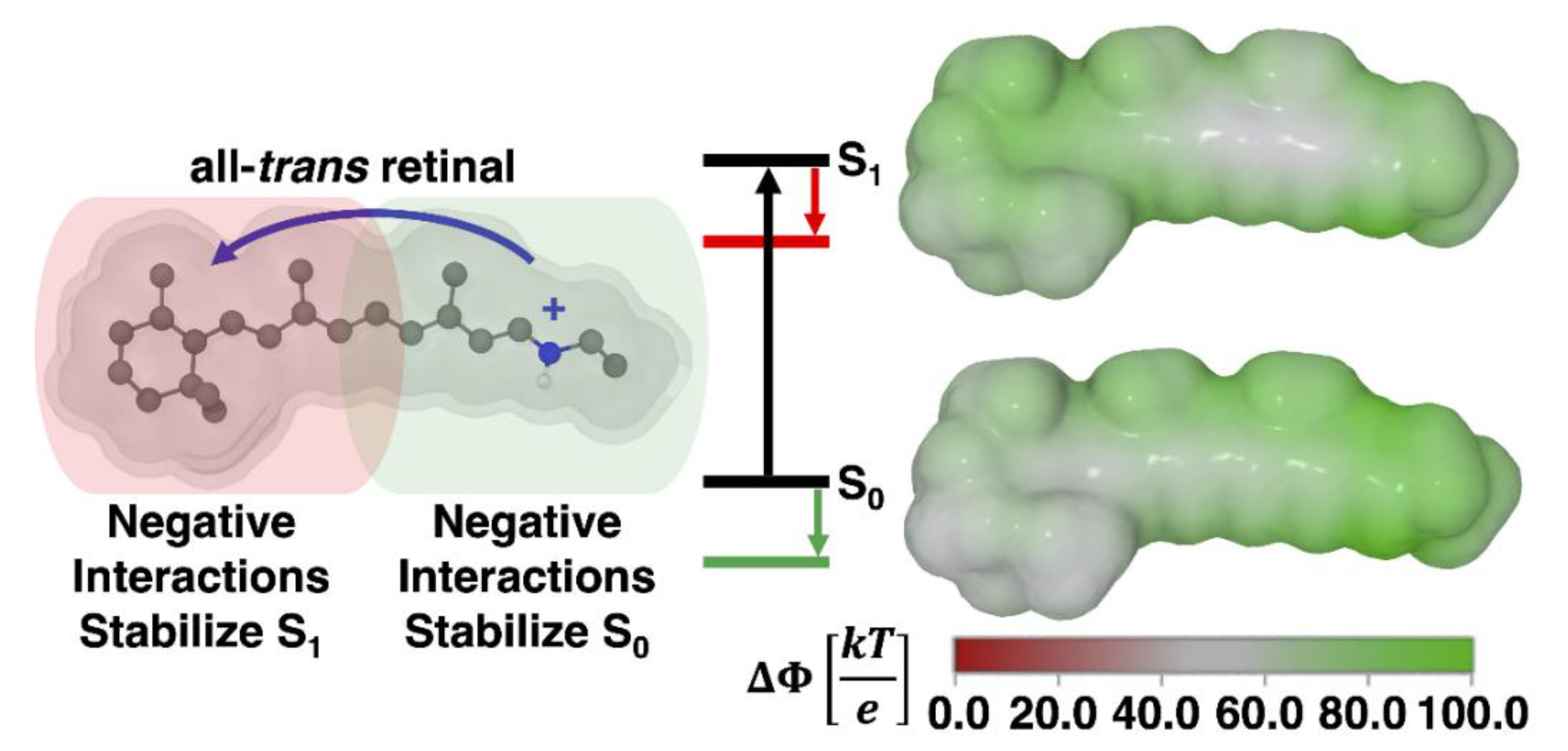
3.3. Absorption Maxima
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palczewski, K. G protein-coupled receptor rhodopsin. Annu. Rev. Biochem. 2006, 75, 743–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenberg, D.; Varma, N.; Deupi, X.; Koyanagi, M.; Terakita, A.; Schertler, G.F.X.; Heberle, J.; Lesca, E. The two-photon reversible reaction of the bistable jumping spider rhodopsin-1. Biophys. J. 2019, 116, 1248–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Vachali, P.; Bernstein, P.S.; Photobiol Sci, P.; Tsutsui, K.; Shichida, Y.; Tsukamoto, H.; Terakita, A.; Davies, W.L.; Hankins, M.W.; et al. Diversity and functional properties of bistable pigments. Photochem. Photobiol. Sci. 2010, 9, 1435–1443. [Google Scholar] [CrossRef]
- Hubbard, R.; St George, R.C. The rhodopsin System of the squid. J. Gen. Physiol. 1958, 41, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyanagi, M.; Terakita, A. Diversity of animal opsin-based pigments and their optogenetic potential. Biochim. Biophys. Acta. Bioenerg. 2014, 1837, 710–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, T.; Koyanagi, M.; Tsukamoto, H.; Saeki, S.; Isono, K.; Shichida, Y.; Tokunaga, F.; Kinoshita, M.; Arikawa, K.; Terakita, A. Depth perception from image defocus in a jumping spider. Science 2012, 335, 469–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varma, N.; Mutt, E.; Mühle, J.; Panneels, V.; Terakita, A.; Deupi, X.; Nogly, P.; Schertler, G.F.X.; Lesca, E. Crystal structure of jumping spider rhodopsin-1 as a light sensitive GPCR. Proc. Natl. Acad. Sci. USA 2019, 116, 14547–14556. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, T.; Yamashita, T.; Imamoto, Y.; Shichida, Y. Photochemical properties of mammalian melanopsin. Biochemistry 2012, 51, 5454–5462. [Google Scholar] [CrossRef]
- Hofmann, K.P.; Scheerer, P.; Hildebrand, P.W.; Choe, H.W.; Park, J.H.; Heck, M.; Ernst, O.P. AG protein-coupled receptor at work: The rhodopsin model. Trends Biochem. Sci. 2009, 34, 540–552. [Google Scholar] [CrossRef]
- Nagata, T.; Koyanagi, M.; Tsukamoto, H.; Mutt, E.; Schertler, G.F.X.; Deupi, X.; Terakita, A. The counterion–retinylidene Schiff base interaction of an invertebrate rhodopsin rearranges upon light activation. Commun. Biol. 2019, 2, 1–9. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Murakami, M.; Kouyama, T. Crystal structure of squid rhodopsin. Nature 2008, 453, 363–367. [Google Scholar] [CrossRef]
- Murakami, M.; Kouyama, T. Crystallographic analysis of the primary photochemical reaction of squid rhodopsin. J. Mol. Biol. 2011, 413, 615–627. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Olsson, M.H.M.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Debiec, K.T.; Cerutti, D.S.; Baker, L.R.; Gronenborn, A.M.; Case, D.A.; Chong, L.T. Further along the road less traveled: AMBER ff15ipq, an original protein force field built on a self-consistent physical model. J. Chem. Theory Comput. 2016, 12, 3926–3947. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Adam, S.; Bondar, A.N. Mechanism by which water and protein electrostatic interactions control proton transfer at the active site of channelrhodopsin. PLoS ONE 2018, 13, e0201298. [Google Scholar] [CrossRef]
- Gaus, M.; Cui, Q.; Elstner, M. DFTB3: Extension of the self-consistent-charge density-functional tight-binding method (SCC-DFTB). J. Chem. Theory Comput. 2011, 7, 931–948. [Google Scholar] [CrossRef] [Green Version]
- Adam, S.; Wiebeler, C.; Schapiro, I. Structural factors determining the absorption spectrum of channelrhodopsins: A case study of the chimera C1C2. J. Chem. Theory Comput. 2021, 17, 6302–6313. [Google Scholar] [CrossRef]
- Seabra, G.D.M.; Walker, R.C.; Elstner, M.; Case, D.A.; Roitberg, A.E. Implementation of the SCC-DFTB method for Hybrid QM/MM simulations within the amber molecular dynamics package †. ACS Publ. 2007, 111, 5655–5664. [Google Scholar] [CrossRef] [Green Version]
- Runge, E.; Gross, E.K.U. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Hättig, C. Structure optimizations for excited states with correlated second-order methods: CC2 and ADC(2). Adv. Quantum Chem. 2005, 50, 37–60. [Google Scholar]
- Olsen, J.M.H.; Reine, S.; Vahtras, O.; Kjellgren, E.; Reinholdt, P.; Hjorth Dundas, K.O.; Li, X.; Cukras, J.; Ringholm, M.; Hedegård, E.D.; et al. Dalton project: A python platform for molecular- and electronic-structure simulations of complex systems. J. Chem. Phys. 2020, 152, 214115. [Google Scholar] [CrossRef]
- Aidas, K.; Angeli, C.; Bak, K.L.; Bakken, V.; Bast, R.; Boman, L.; Christiansen, O.; Cimiraglia, R.; Coriani, S.; Dahle, P.; et al. The Dalton quantum chemistry program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 269–284. [Google Scholar] [CrossRef]
- Ricardi, N.; González-Espinoza, C.E.; Adam, S.; Church, J.; Schapiro, I.; Wesolowski, T.A. Embedding non-rigid solutes in an averaged environment: A case study on rhodopsins. ChemRxiv 2021, Version 1. 1–22. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bertalan, É.; Lesca, E.; Schertler, G.F.X.; Bondar, A.N. C-Graphs tool with graphical user interface to dissect conserved hydrogen-bond networks: Applications to visual rhodopsins. J. Chem. Inf. Model. 2021, 61, 5692–5707. [Google Scholar] [CrossRef]
- Chibani, S.; Jacquemin, D.; Laurent, A.D. Modelling solvent effects on the absorption and emission spectra of constrained cyanines with both implicit and explicit QM/EFP models. Comput. Theor. Chem. 2014, 1040, 321–327. [Google Scholar] [CrossRef]
- Ziegler, T.; Krykunov, M.; Cullen, J. The implementation of a self-consistent constricted variational density functional theory for the description of excited states. J. Chem. Phys. 2012, 136, 124107. [Google Scholar] [CrossRef] [Green Version]
- Demoulin, B.; El-Tahawy, M.M.T.; Nenov, A.; Garavelli, M.; Le Bahers, T. Intramolecular photo-induced charge transfer in visual retinal chromophore mimics: Electron density-based indices at the TD-DFT and post-HF levels. Theor. Chem. Acc. 2016, 135, 1–10. [Google Scholar] [CrossRef]
- Fujimoto, K.; Hayashi, S.; Hasegawa, J.Y.; Nakatsuji, H. Theoretical studies on the color-tuning mechanism in retinal proteins. J. Chem. Theory Comput. 2007, 3, 605–618. [Google Scholar] [CrossRef]
- Motto, M.G.; Sheves, M.; Tsujimoto, K.; Balogh-Nair, V.; Nakanishi, K. Opsin shifts in bovine rhodopsin and bacteriorhodopsin. comparison of two external point-charge models. J. Am. Chem. Soc. 1980, 102, 7947–7949. [Google Scholar] [CrossRef]
- Nakanishi, K.; Balogh-Nair, V.; Amaboldi, M.; Tsujimoto, K.; Honig, B. An external point-charge model for bacteriorhodopsin to Account for its purple color. J. Am. Chem. Soc. 1980, 102, 7945–7947. [Google Scholar] [CrossRef]
- Barry, H.; Uri, D.; Koji, N.; Valeria, B.N.; Gawinowicz, M.A.; Maria, A.; Motto, M.G. An external point-charge model for wavelength regulation in visual pigments. J. Am. Chem. Soc. 1979, 101, 7084–7086. [Google Scholar] [CrossRef]
- Mordechai, S.; Koji, N.; Barry, H. Through-space electrostatic effects in electronic spectra. experimental Evidence for the external point-charge model of visual pigments. J. Am. Chem. Soc. 1979, 101, 7086–7088. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Vacuum (nm, eV) | TD-CAM-B3LYP (nm, eV) | Exp. (nm, eV) [2,7] |
|---|---|---|---|
| 9-cis | 520, 2.38 | 484, 2.56 | 505, 2.46 |
| 11-cis | 537, 2.31 | 512, 2.42 | 535, 2.32 |
| all-trans | 540, 2.30 | 517, 2.40 | 535, 2.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Church, J.R.; Olsen, J.M.H.; Schapiro, I. The Impact of Retinal Configuration on the Protein–Chromophore Interactions in Bistable Jumping Spider Rhodopsin-1. Molecules 2022, 27, 71. https://doi.org/10.3390/molecules27010071
Church JR, Olsen JMH, Schapiro I. The Impact of Retinal Configuration on the Protein–Chromophore Interactions in Bistable Jumping Spider Rhodopsin-1. Molecules. 2022; 27(1):71. https://doi.org/10.3390/molecules27010071
Chicago/Turabian StyleChurch, Jonathan R., Jógvan Magnus Haugaard Olsen, and Igor Schapiro. 2022. "The Impact of Retinal Configuration on the Protein–Chromophore Interactions in Bistable Jumping Spider Rhodopsin-1" Molecules 27, no. 1: 71. https://doi.org/10.3390/molecules27010071