
Recent Advances in Catalysis Involving Bidentate N-Heterocyclic Carbene Ligands



Abstract
:1. Introduction
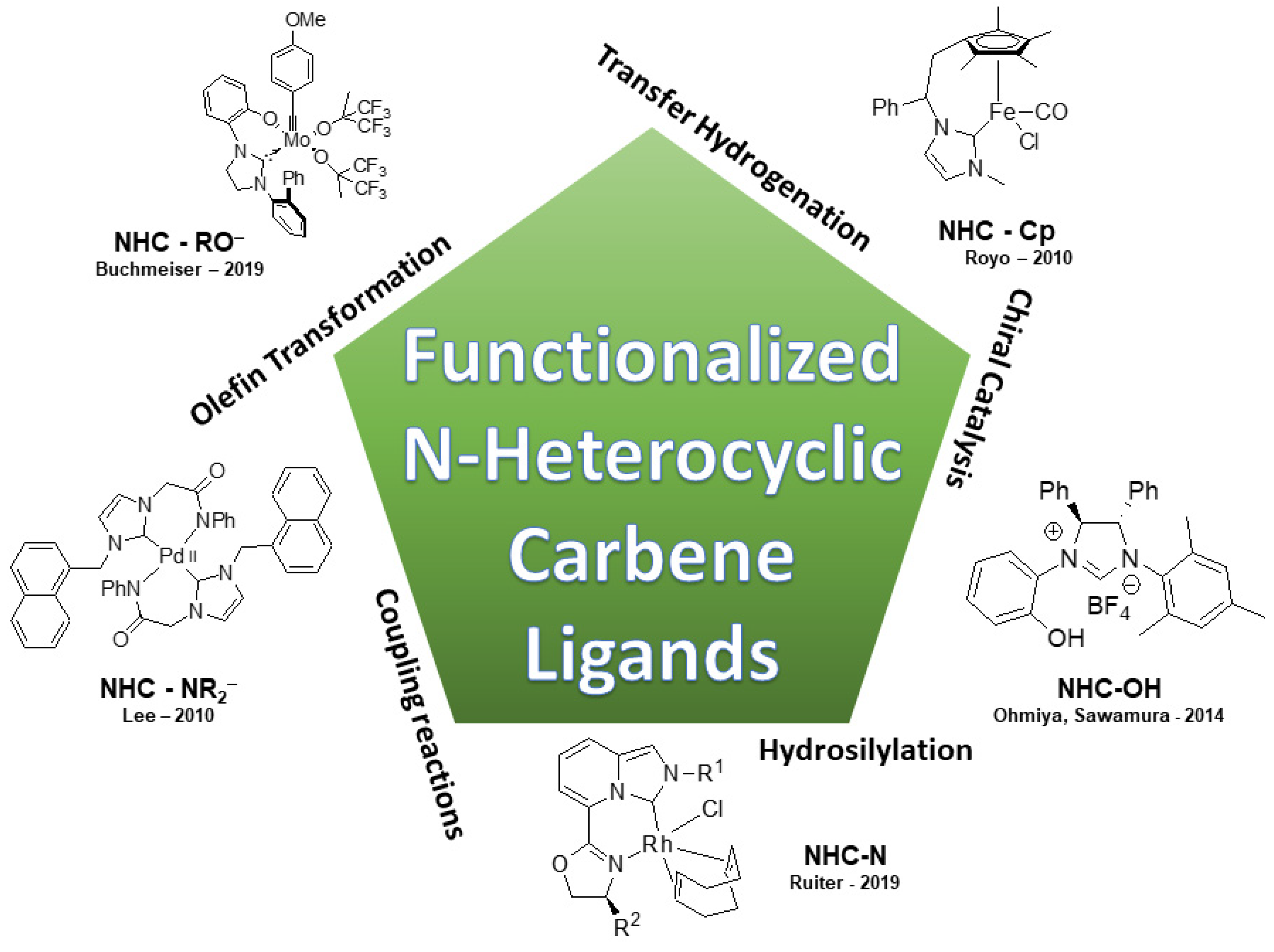
2. Synthesis and Applications of Bidentate Ligands Bearing N-Heterocyclic Carbenes
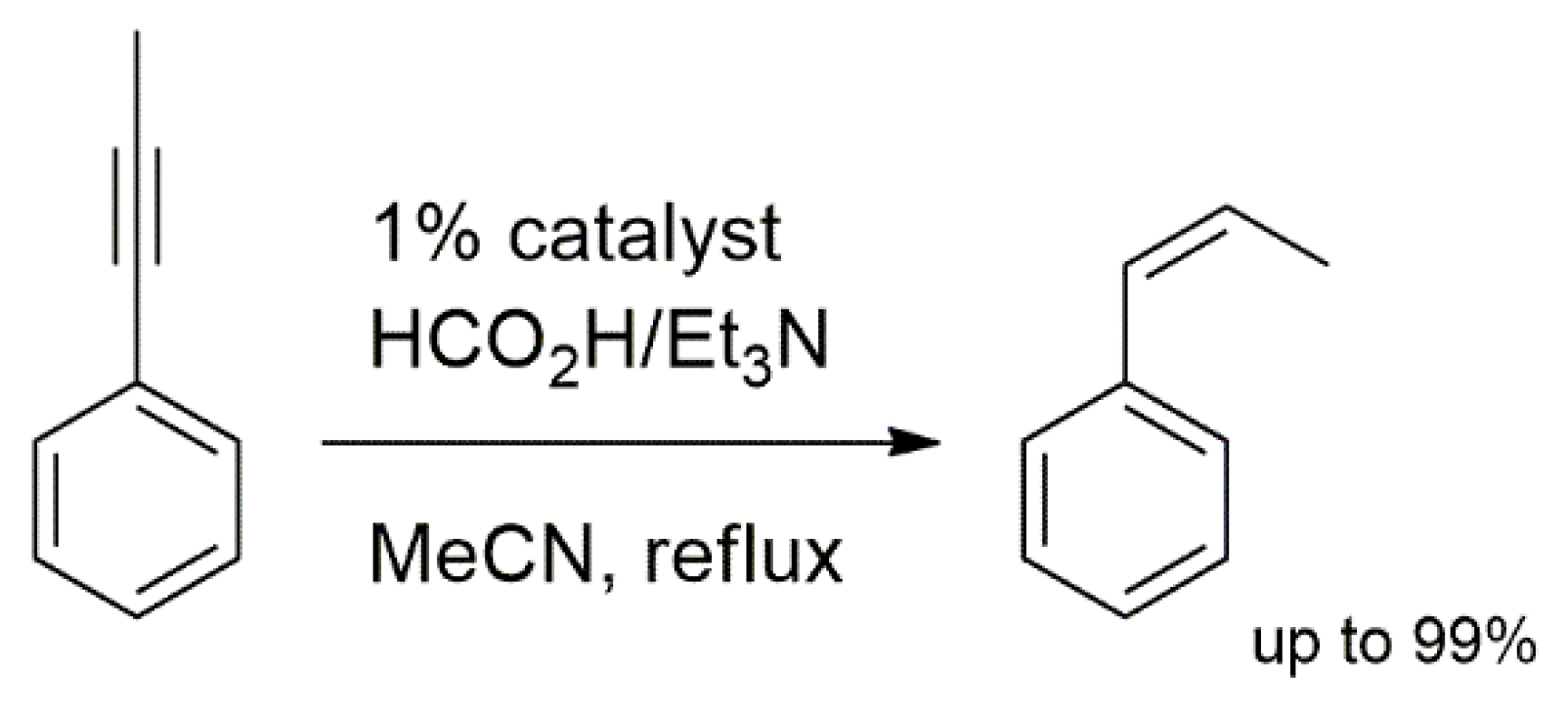
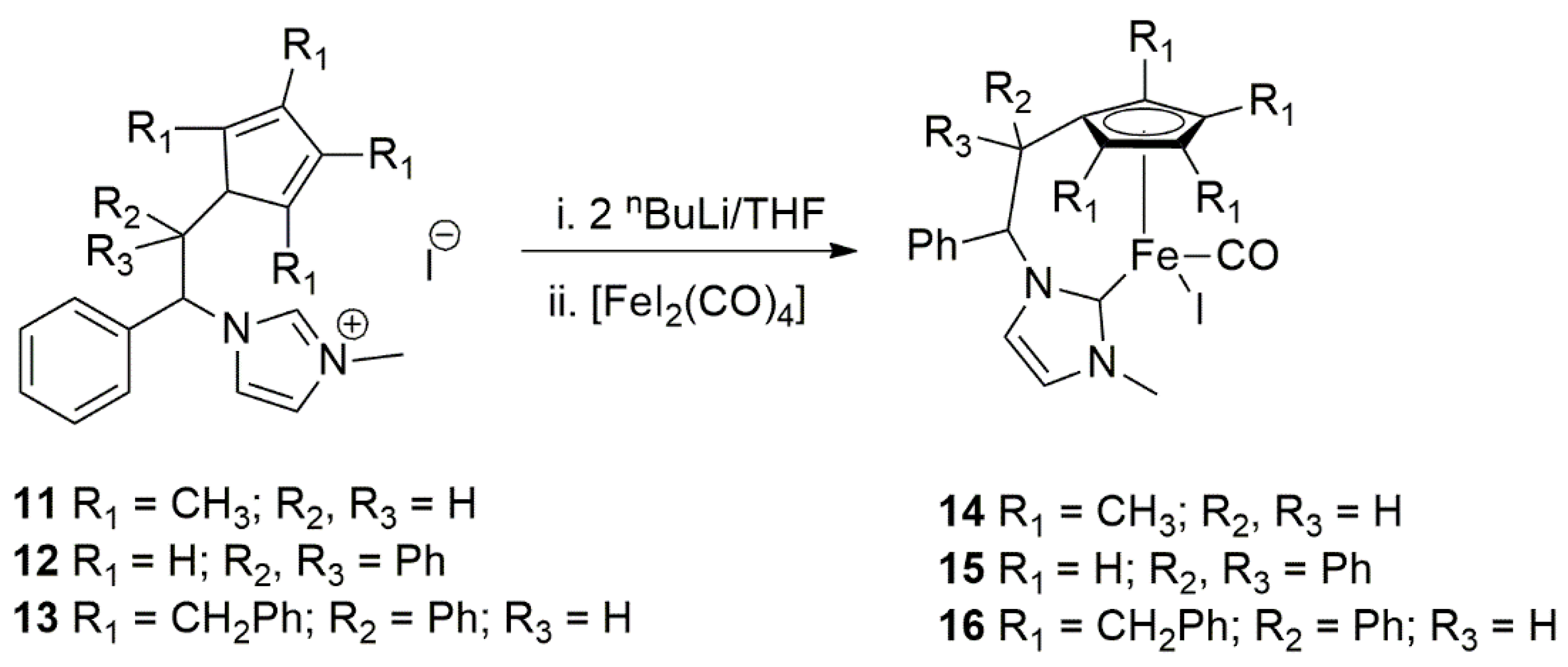
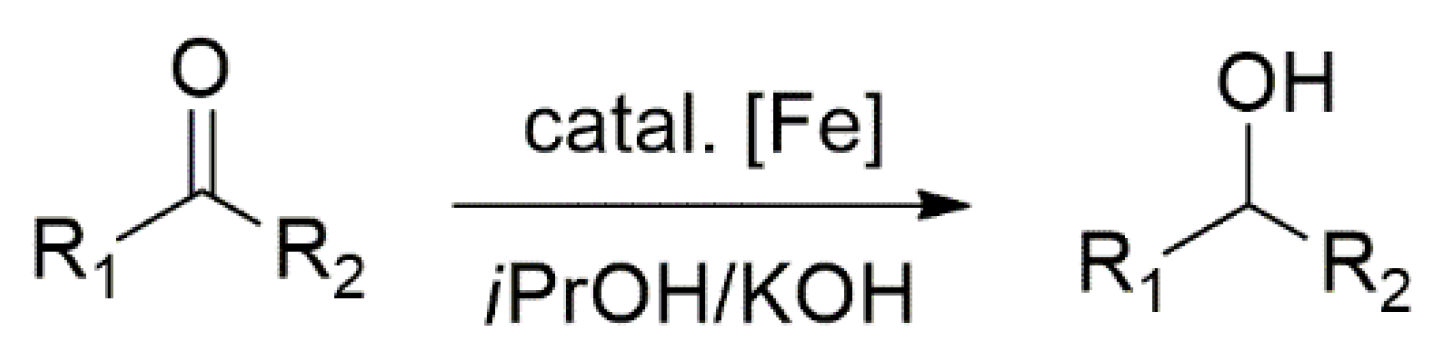
2.1. Hydrogenation Reactions of Carbonyl Compounds, Alkene, and Alkynes
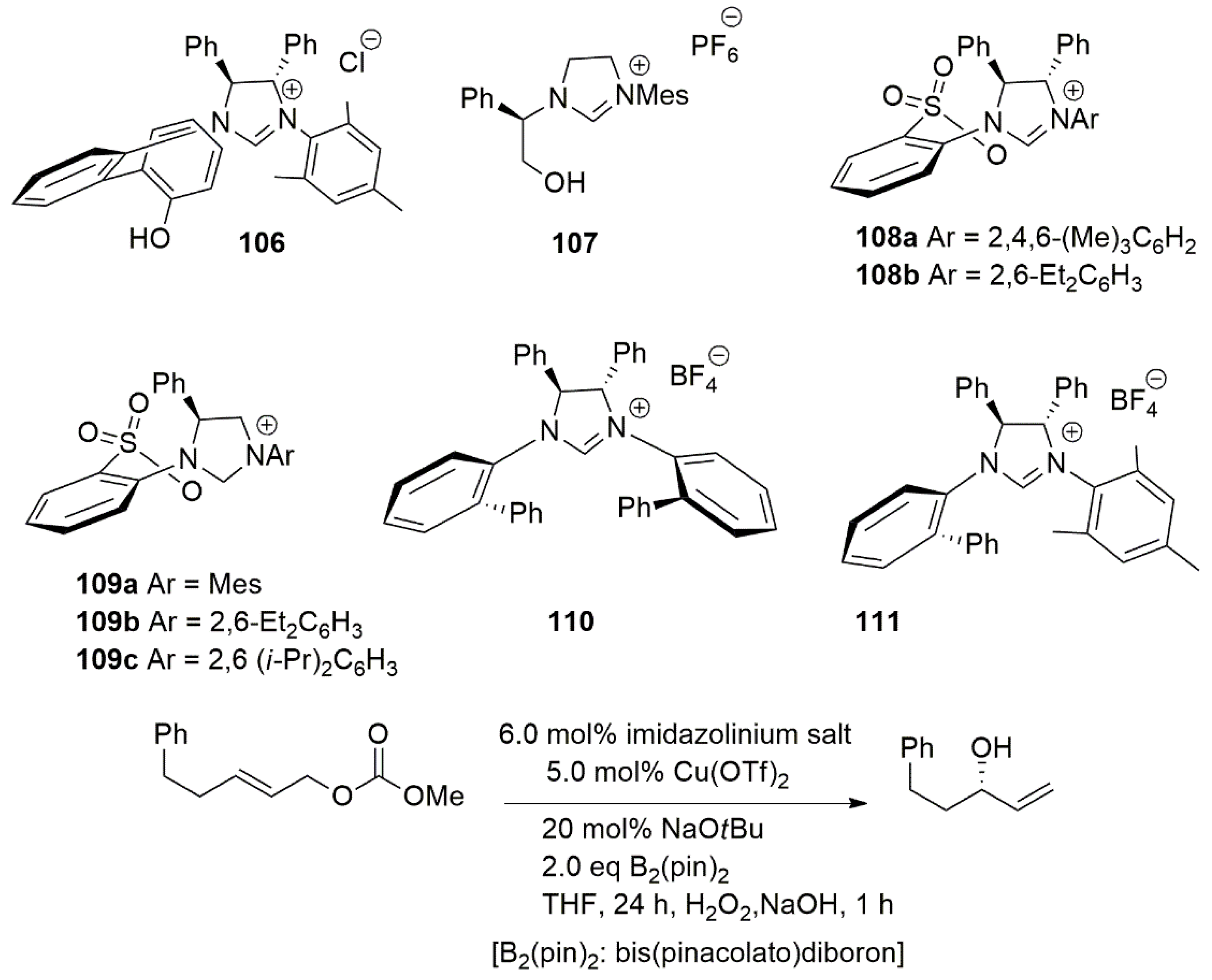
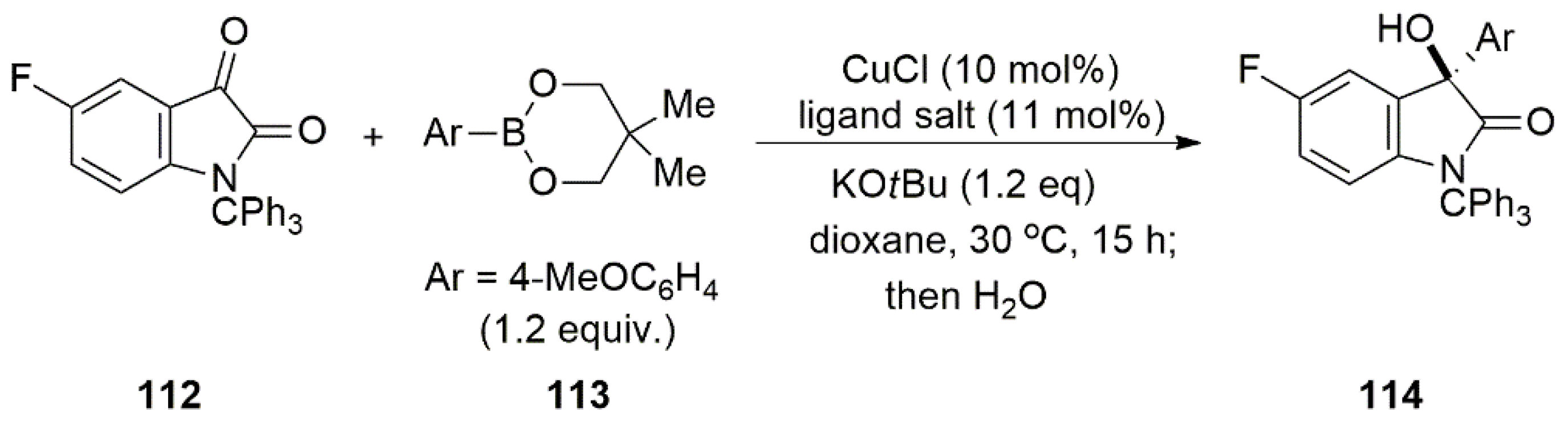
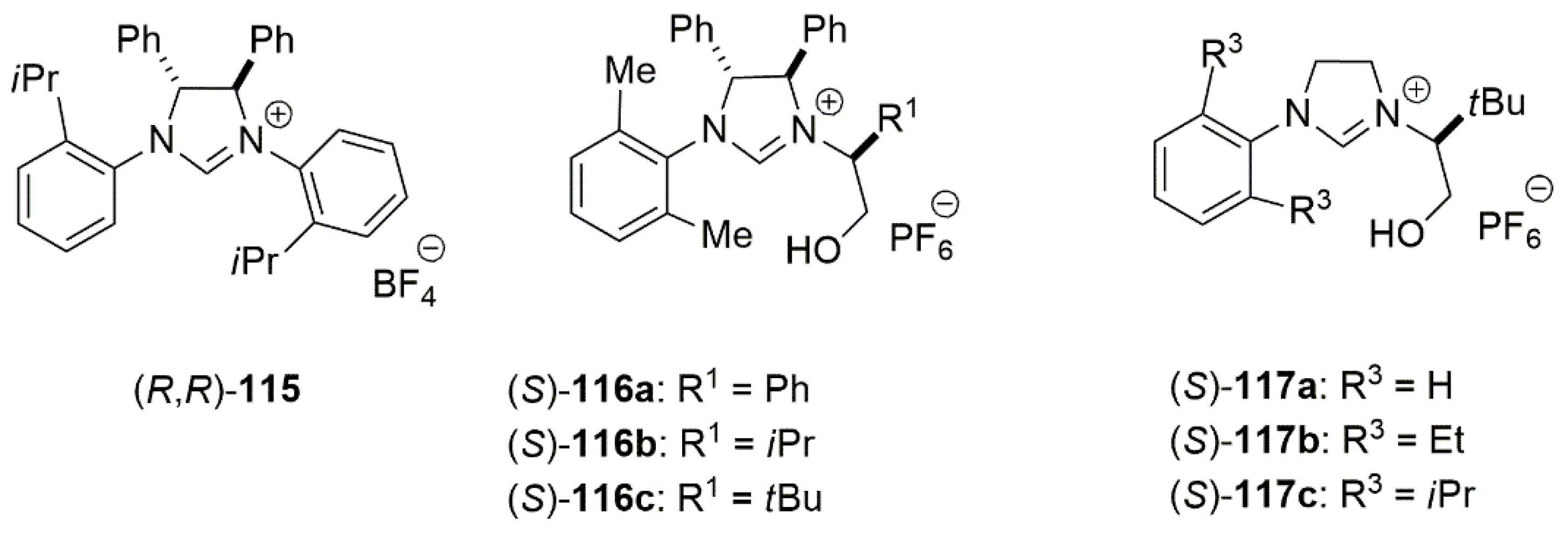
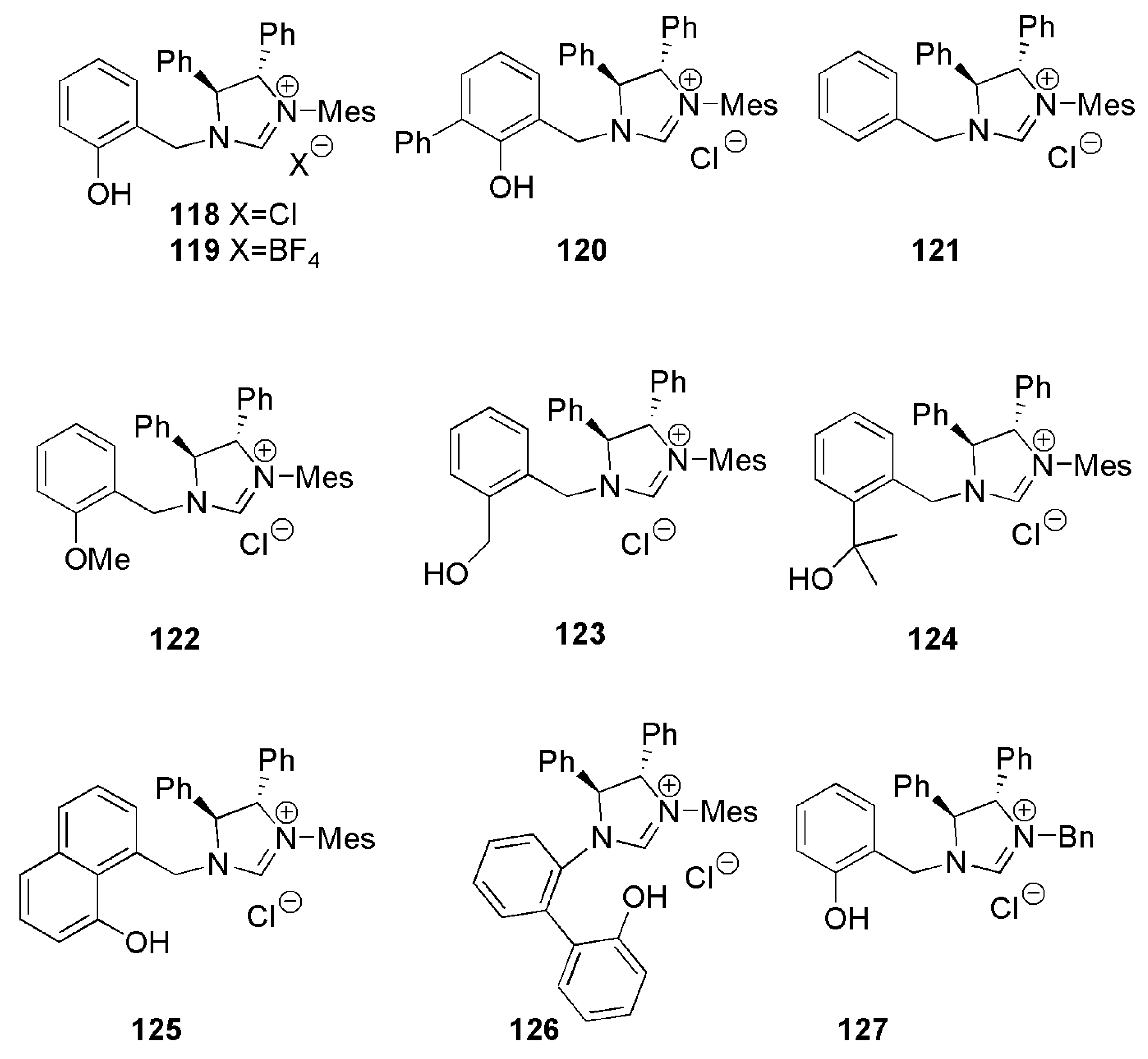
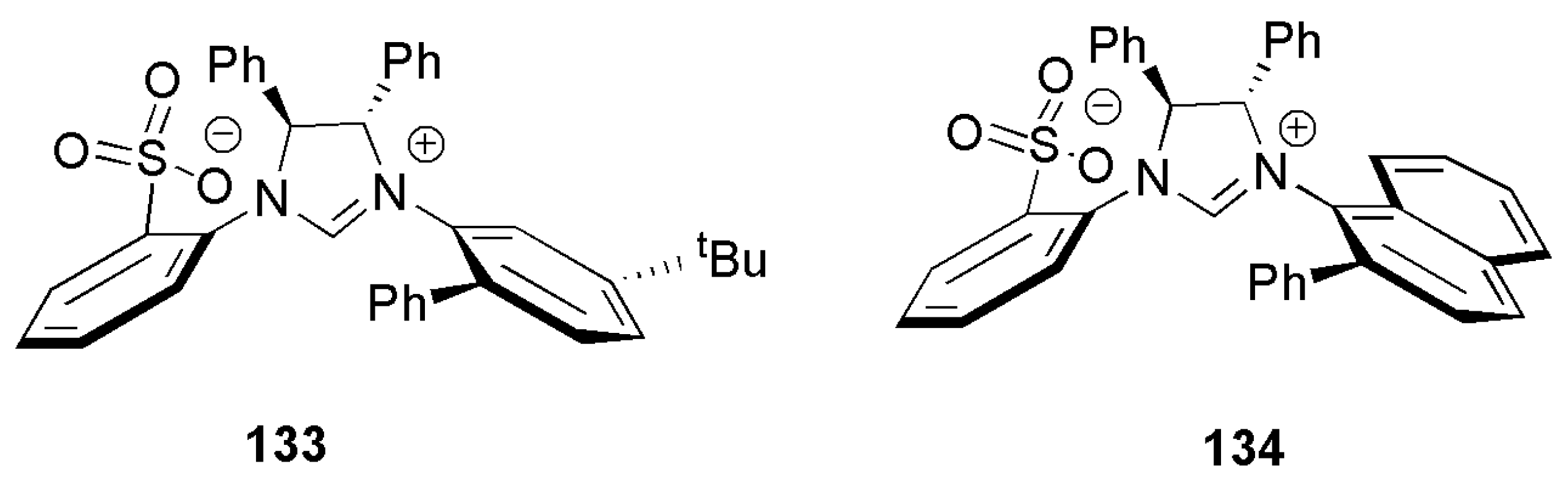
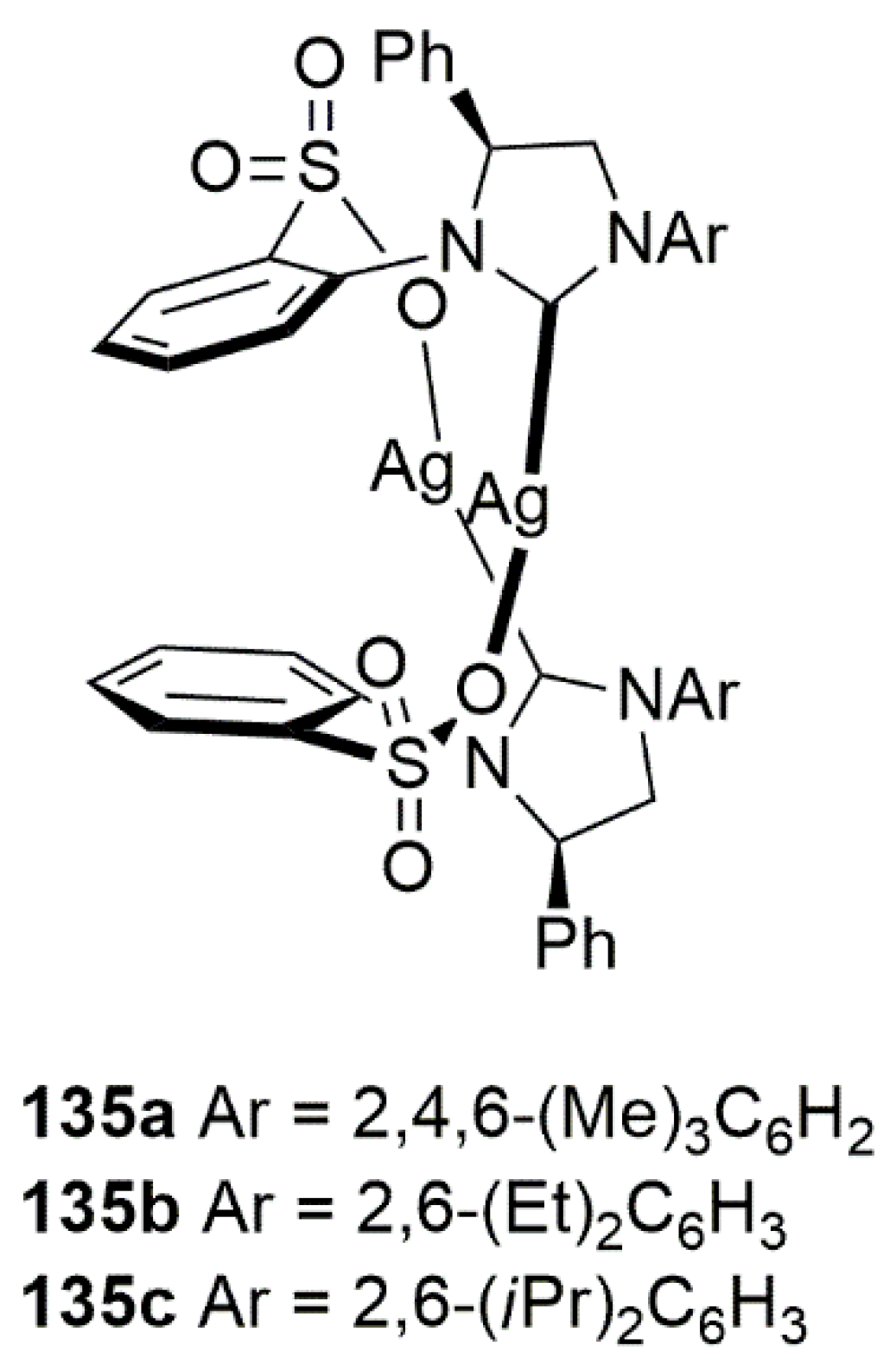
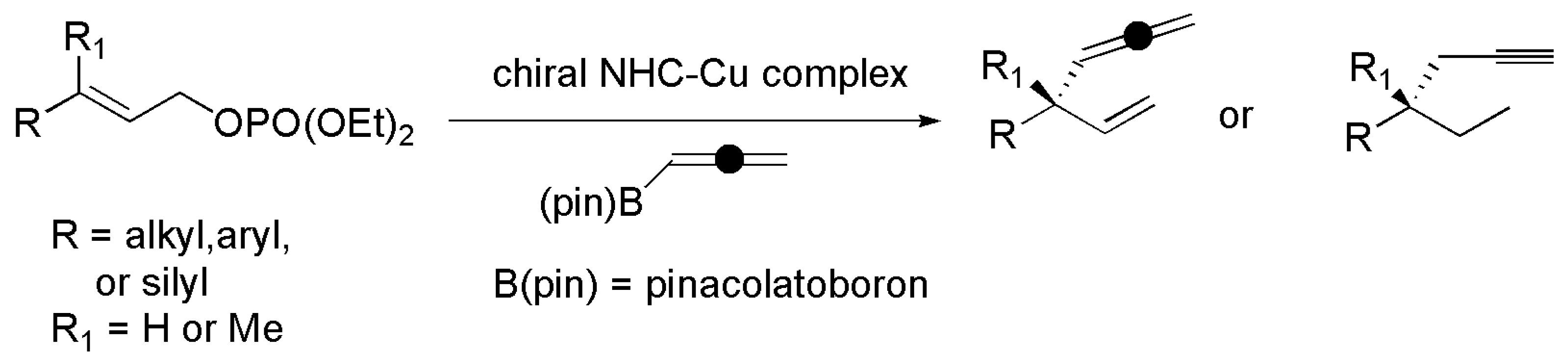
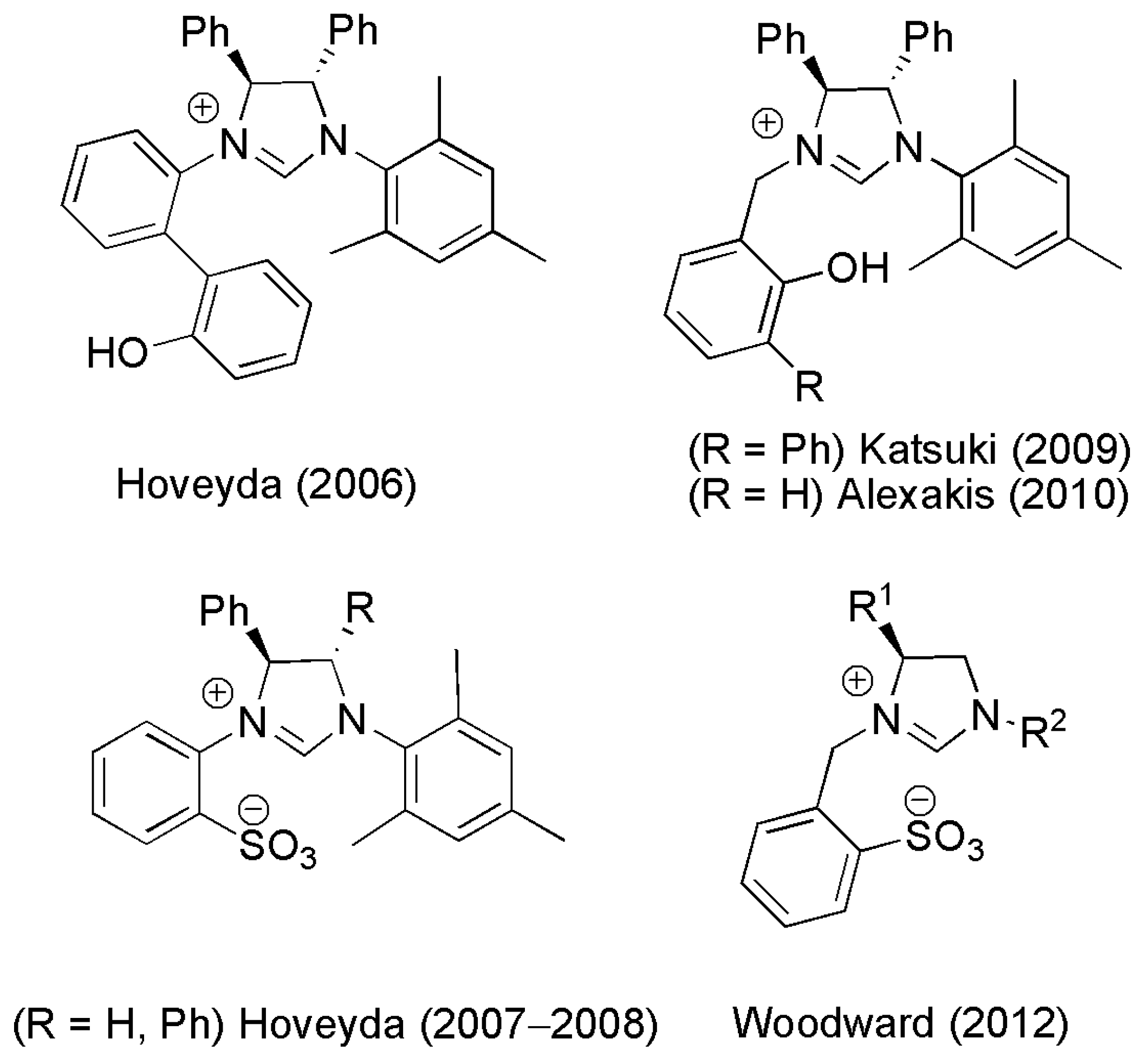
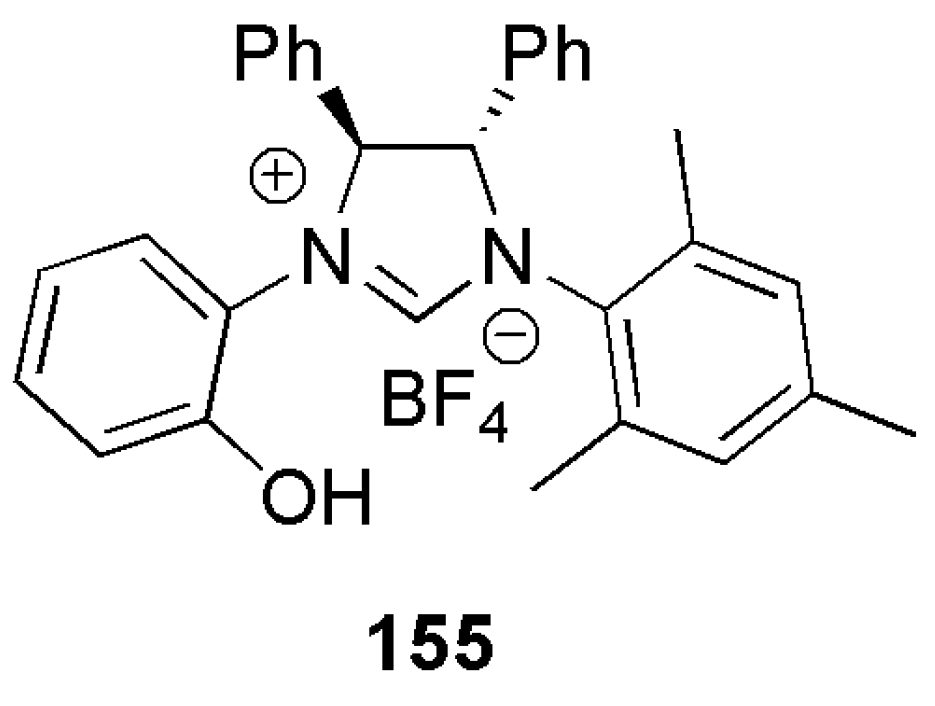
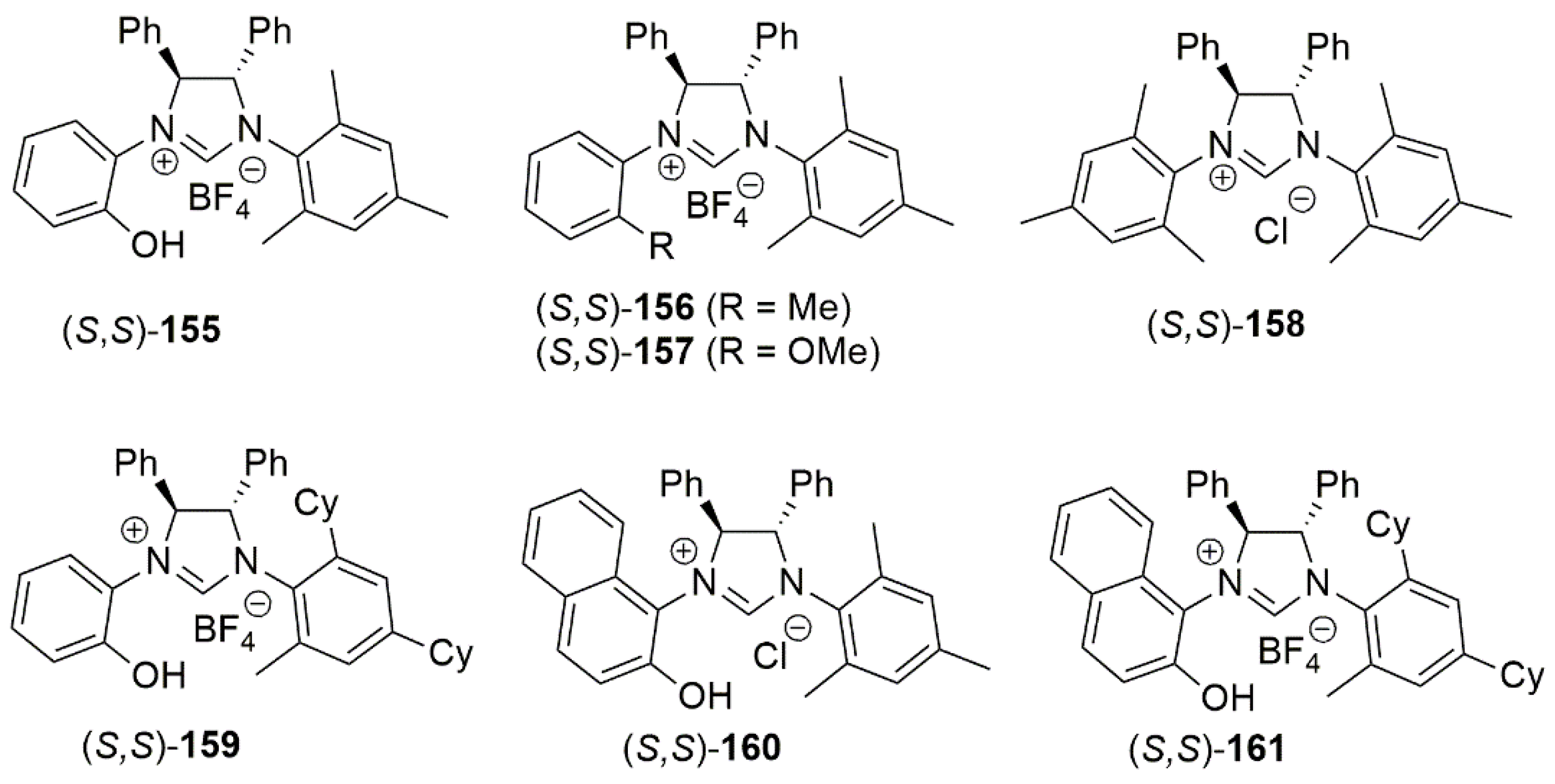
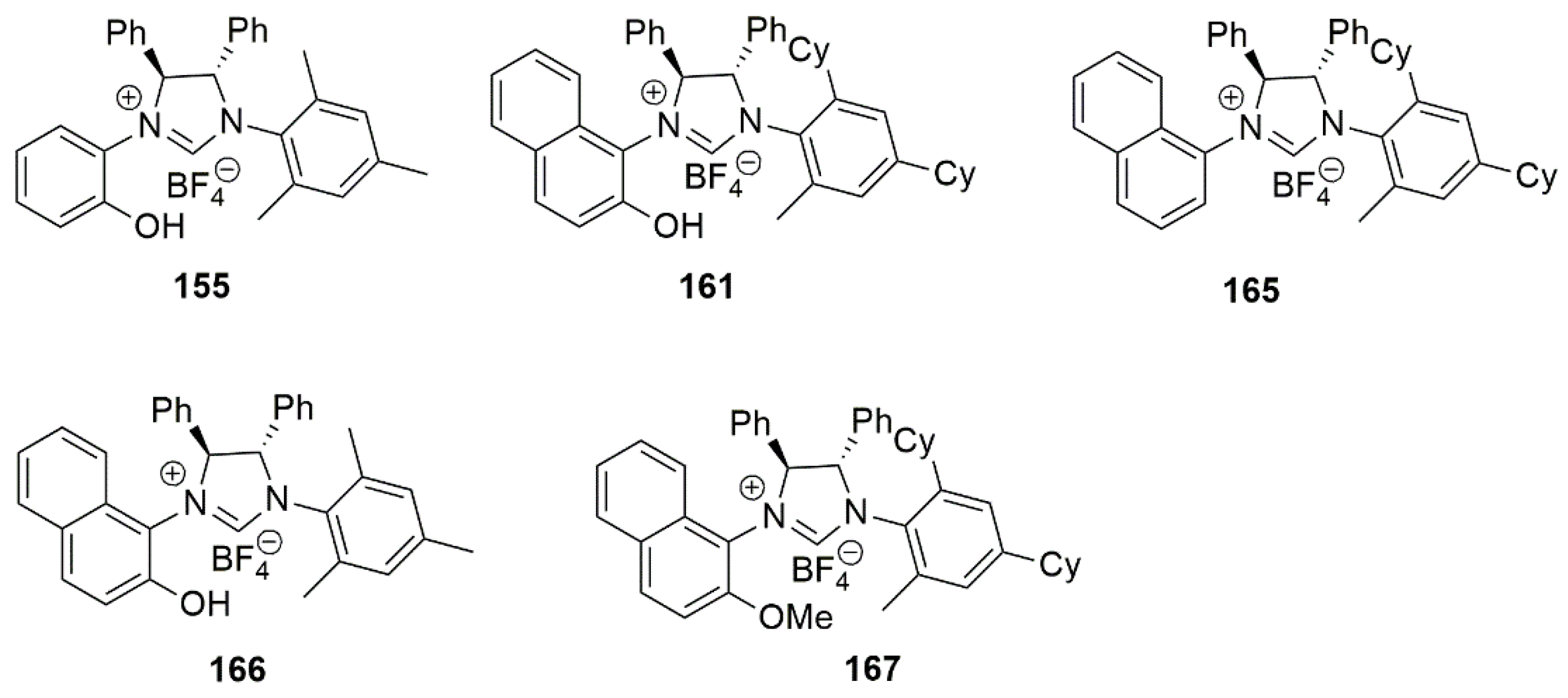
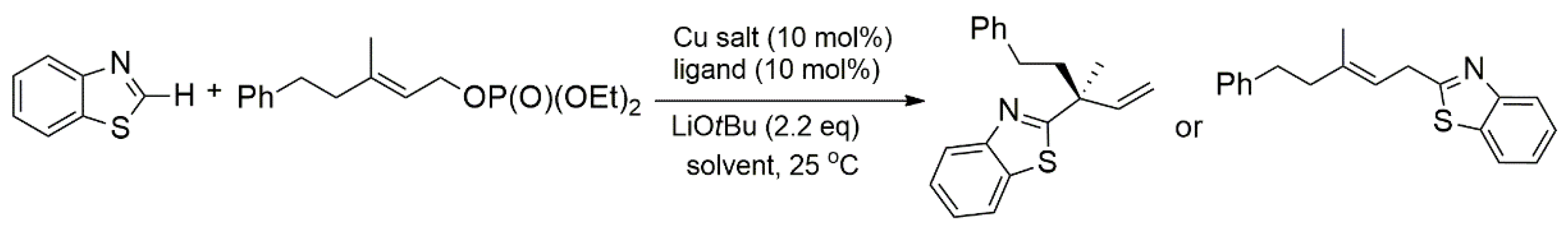
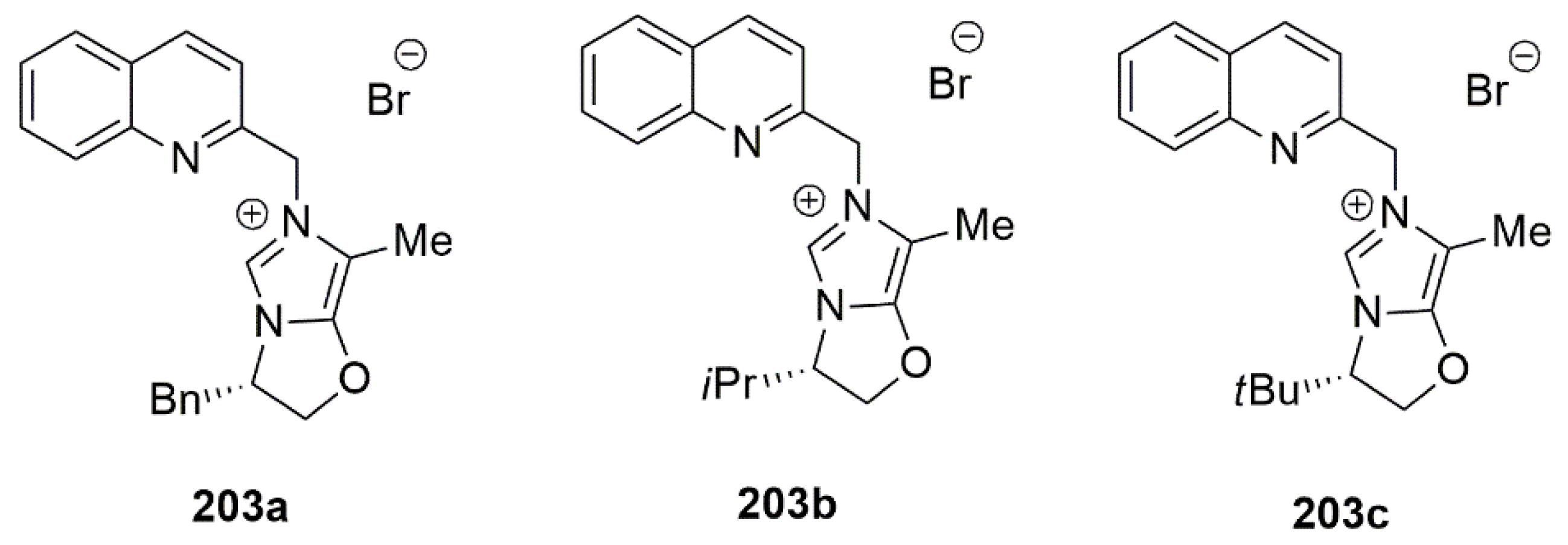
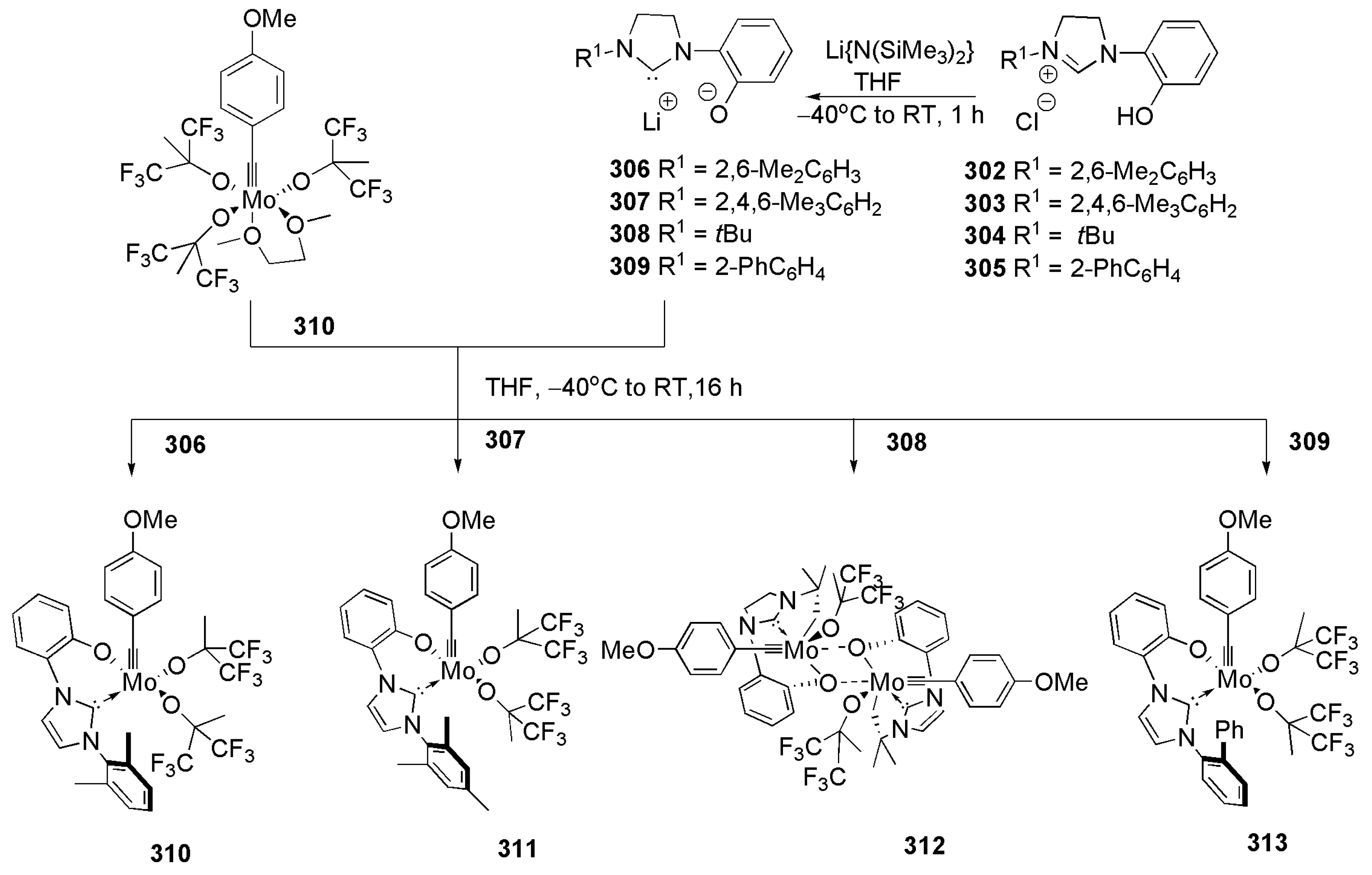
2.2. Chiral Catalysis Involving Bidentate NHC Ligands
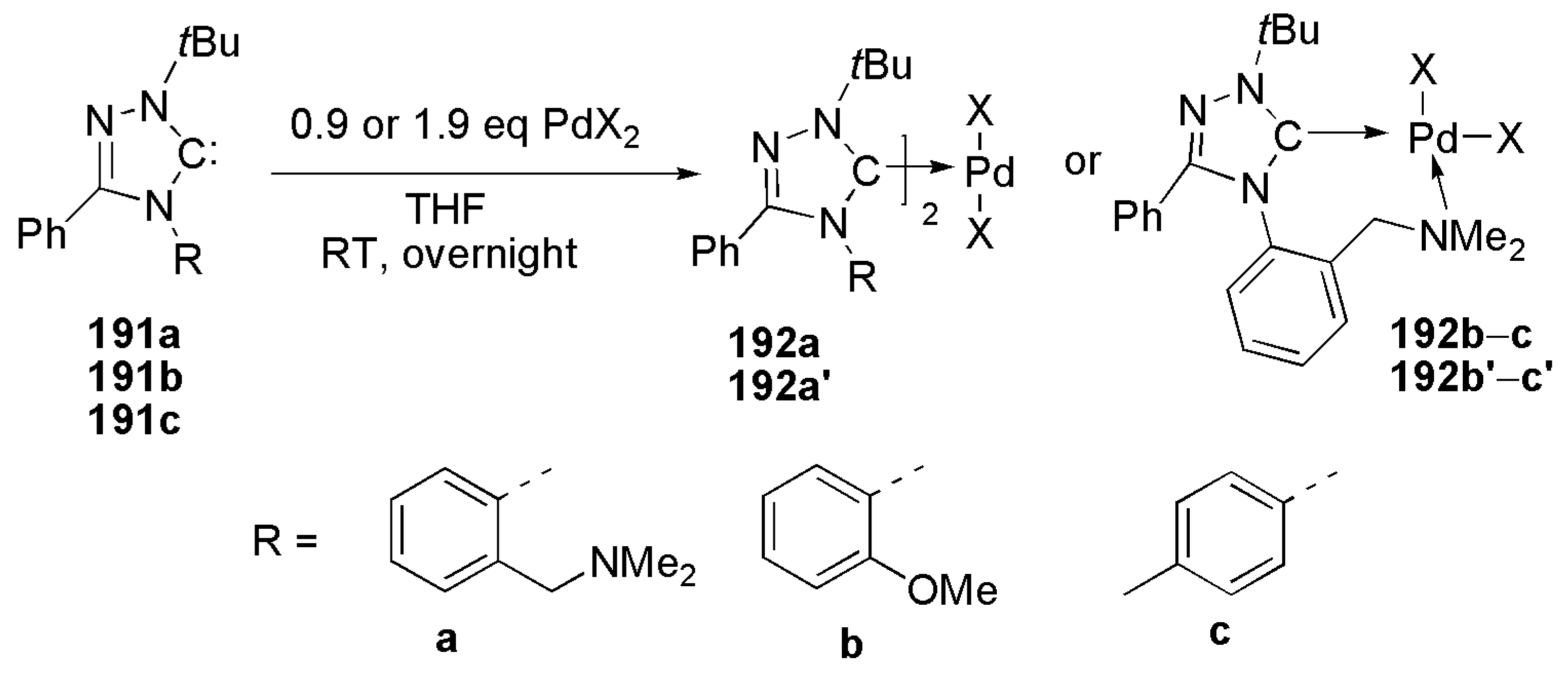
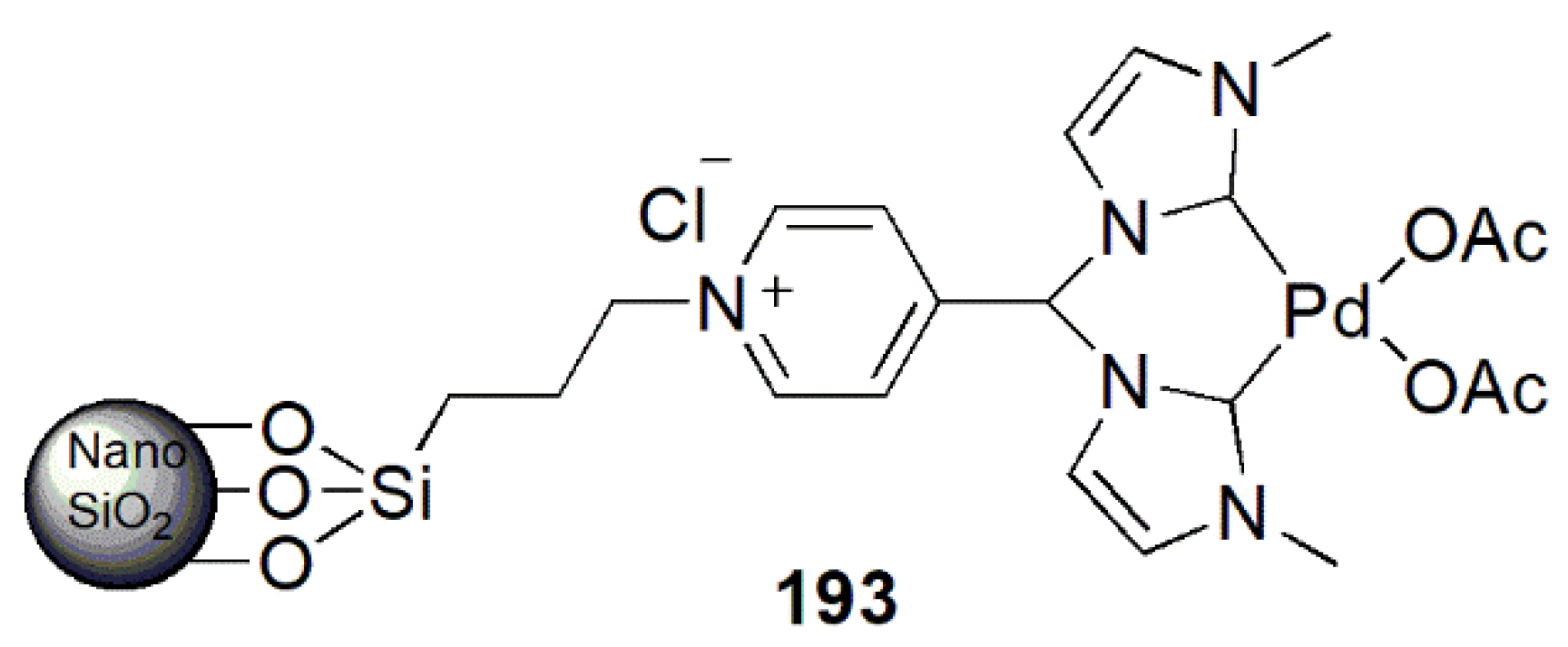
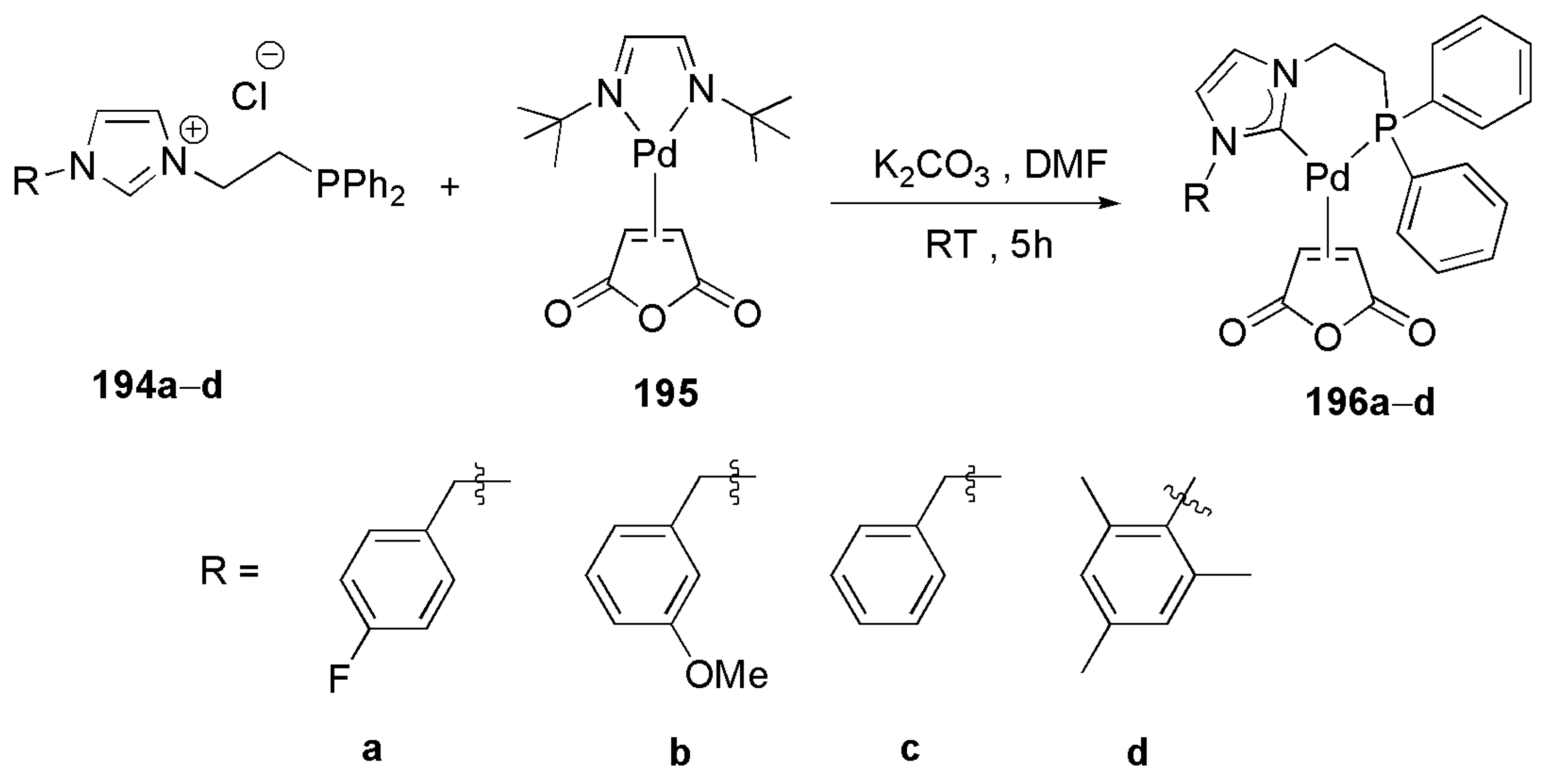
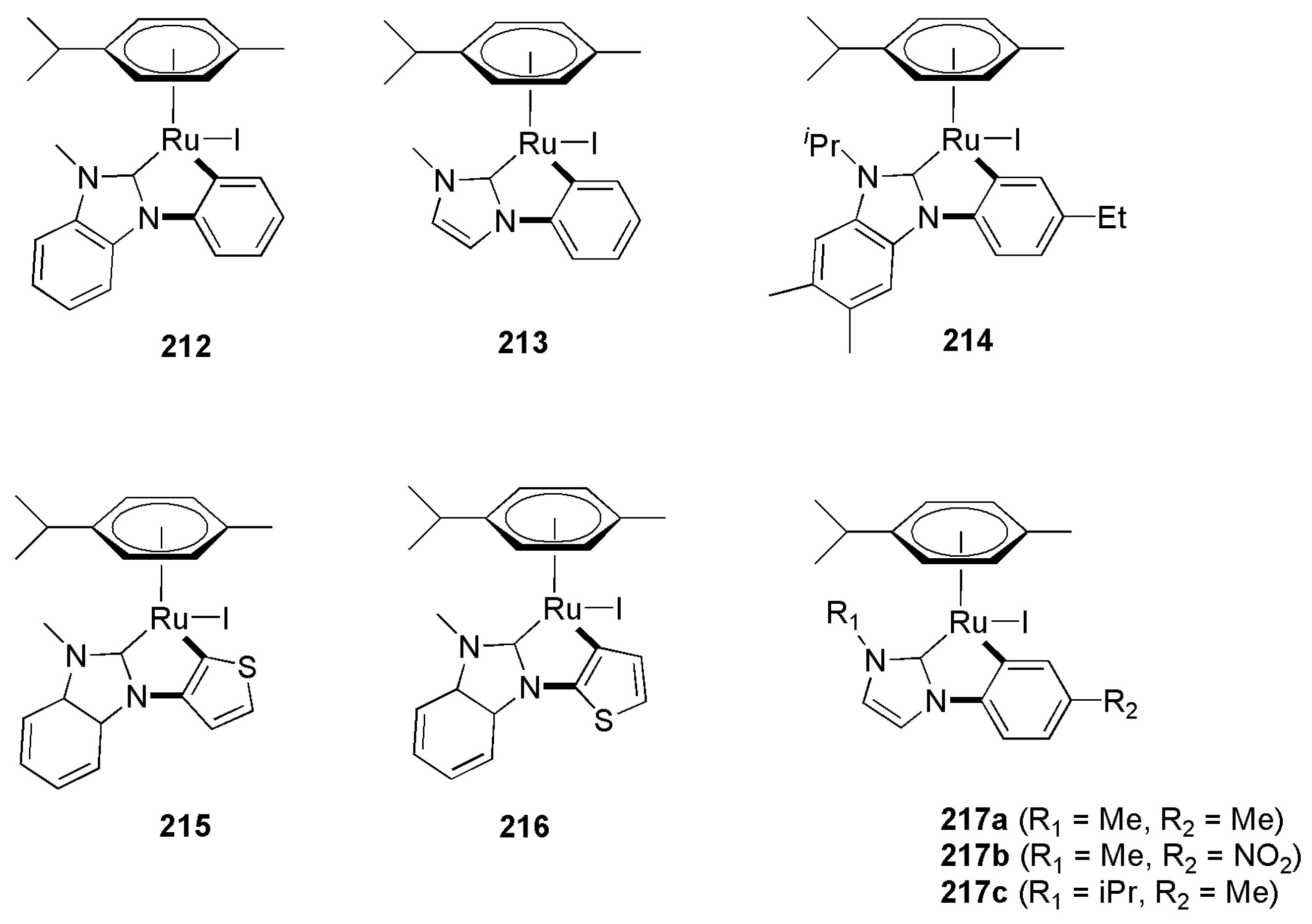
2.3. Coupling Reactions
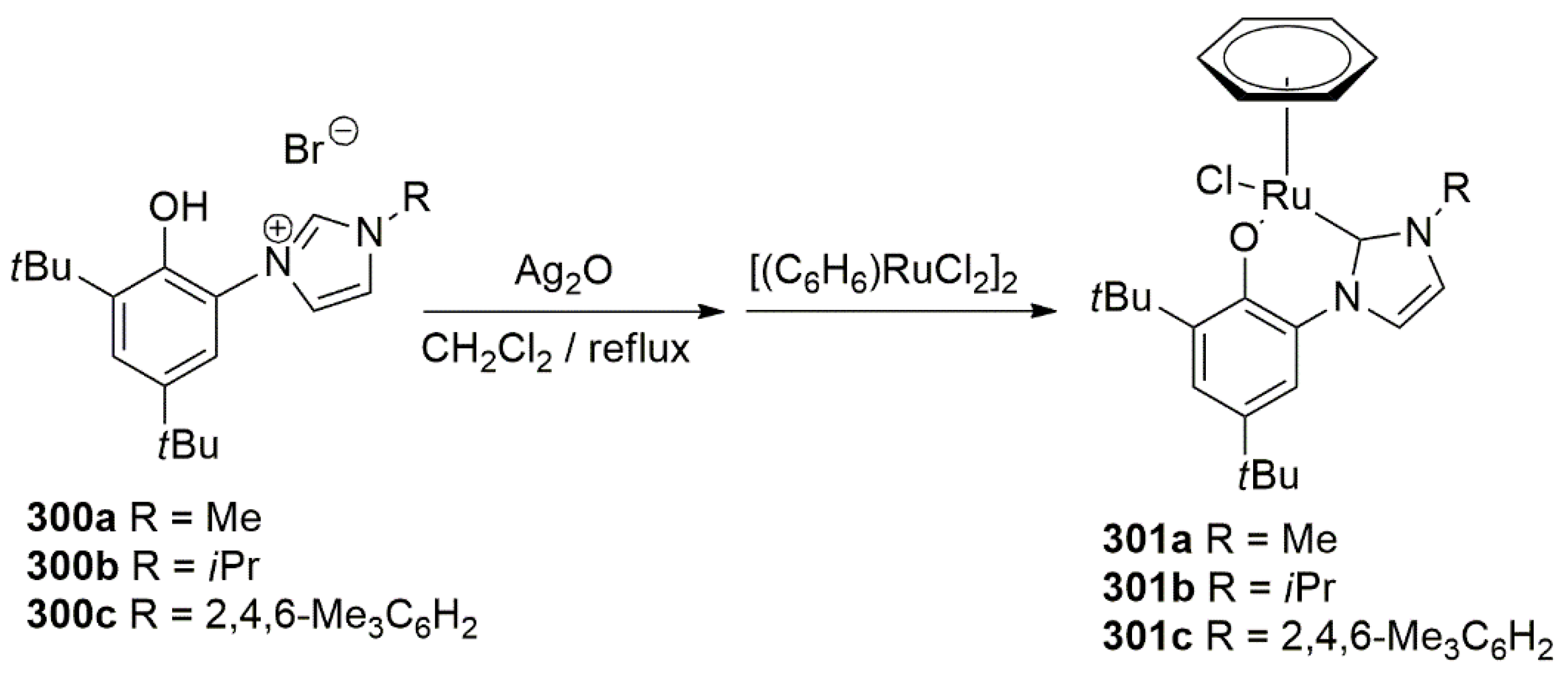
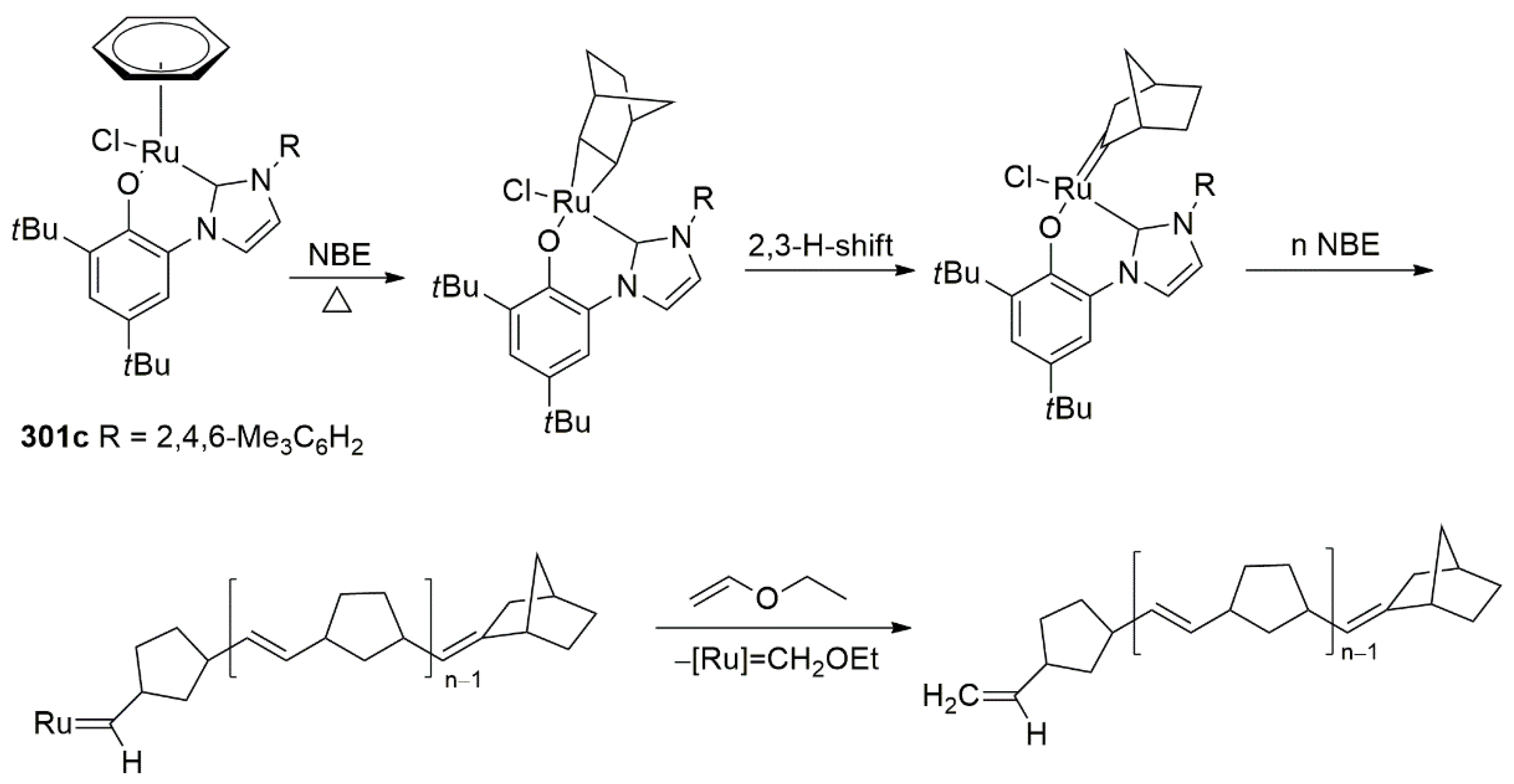
2.4. Ethylene Transformation
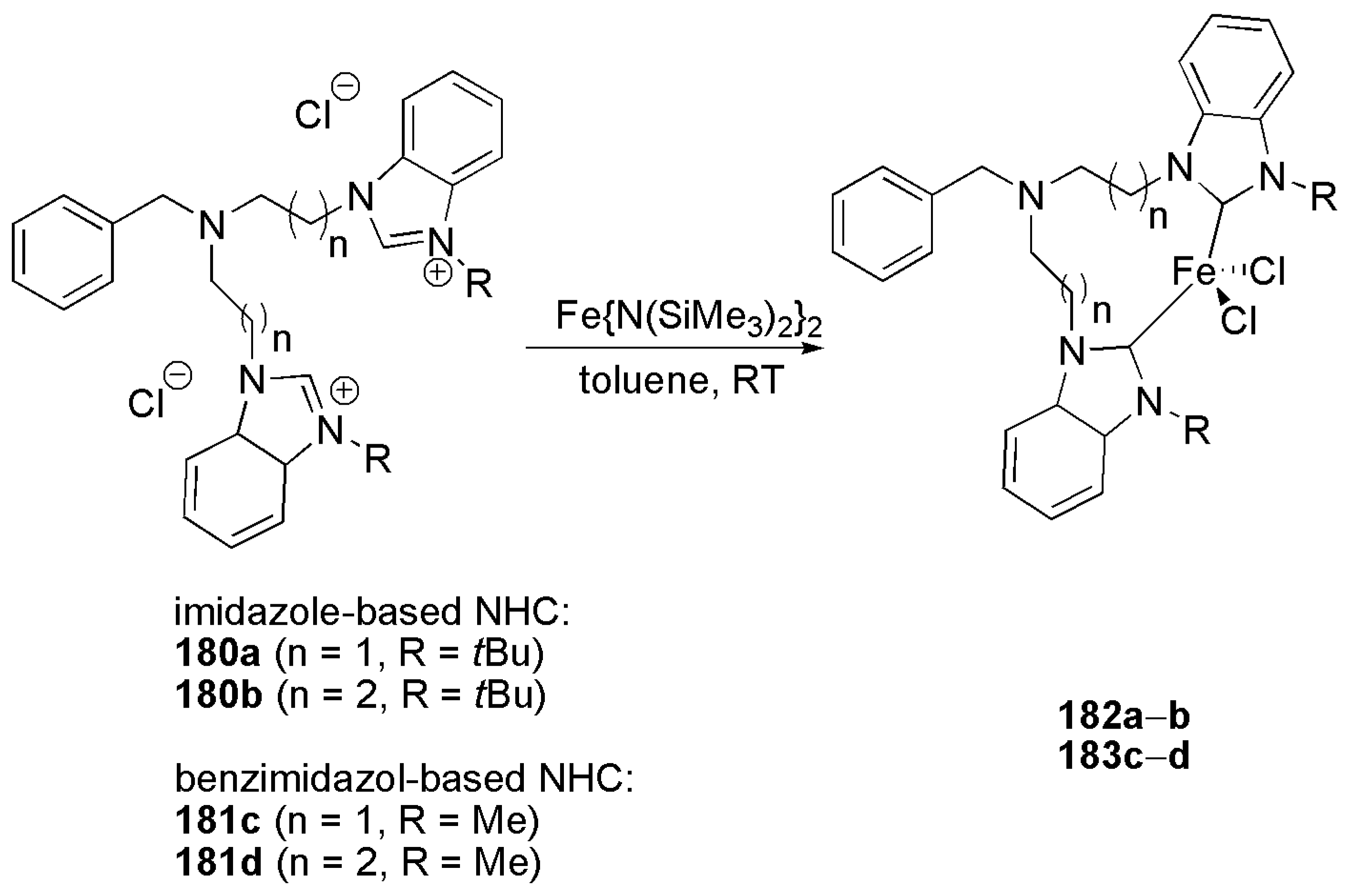
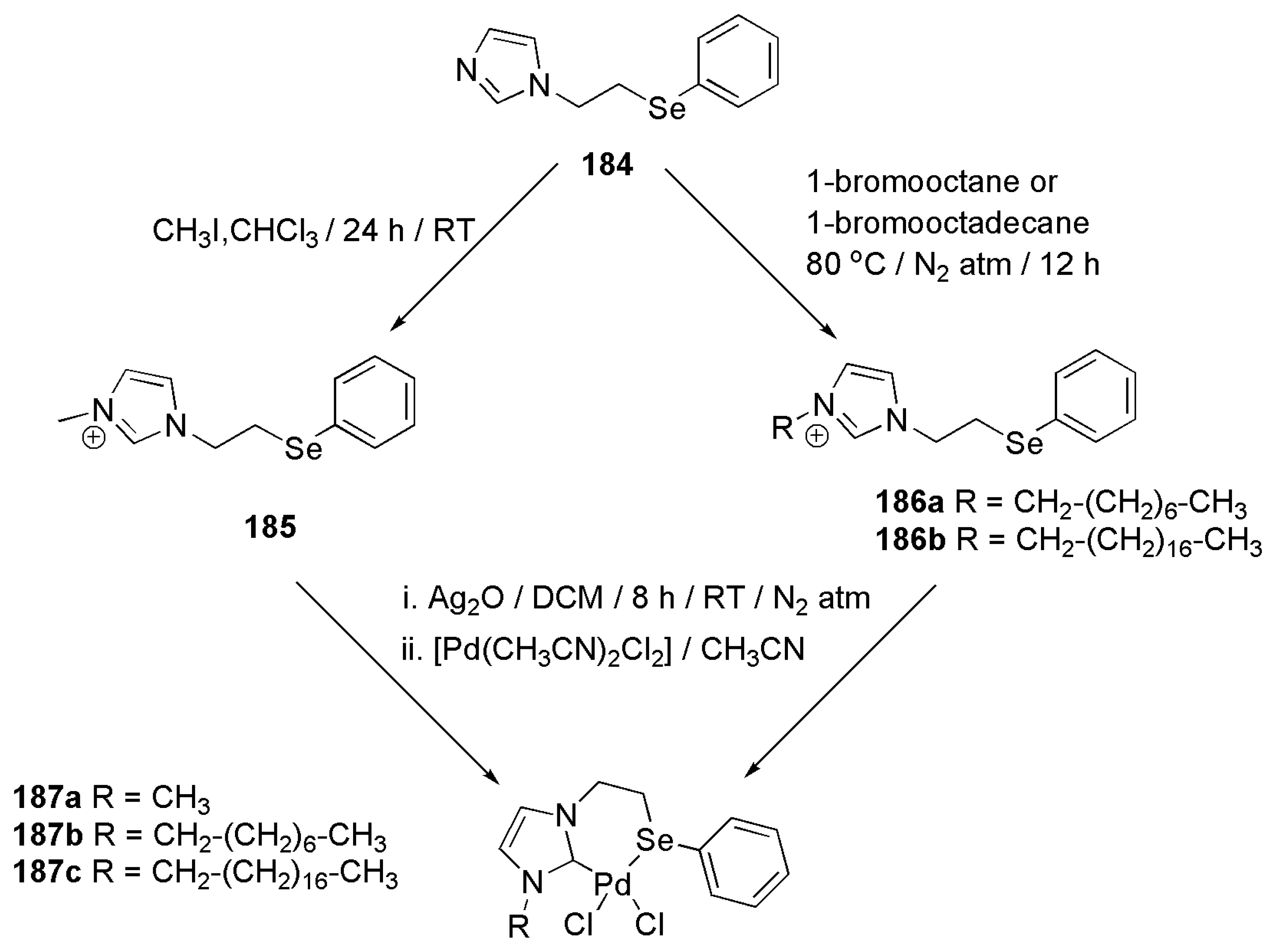
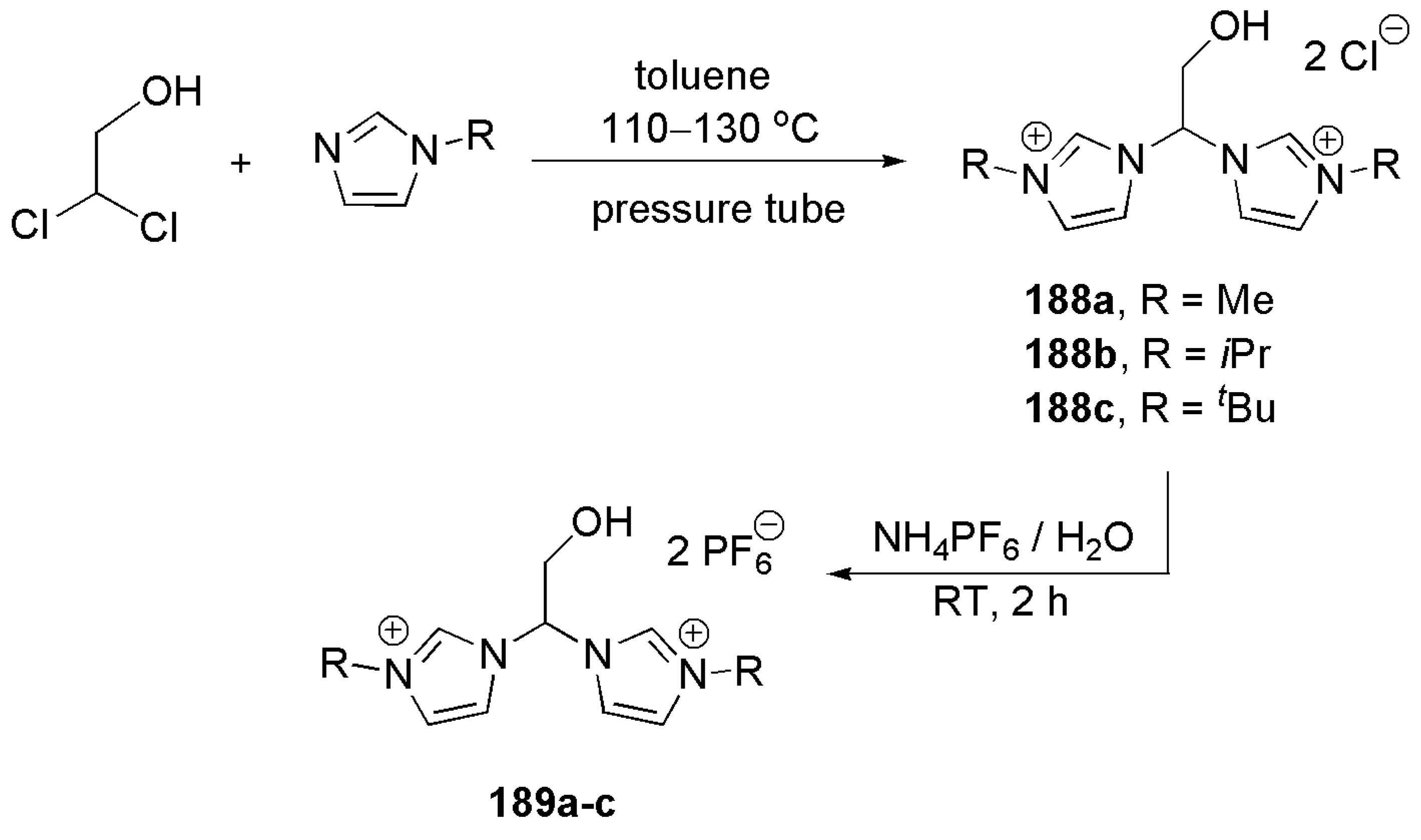
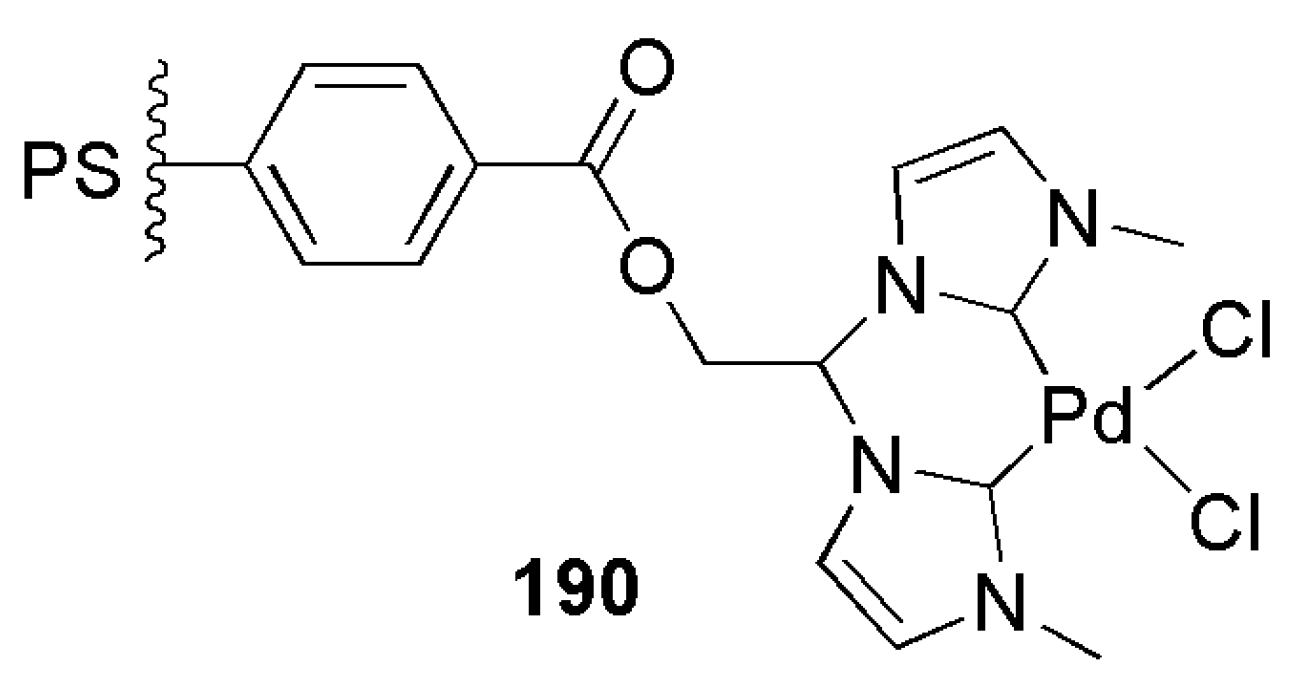
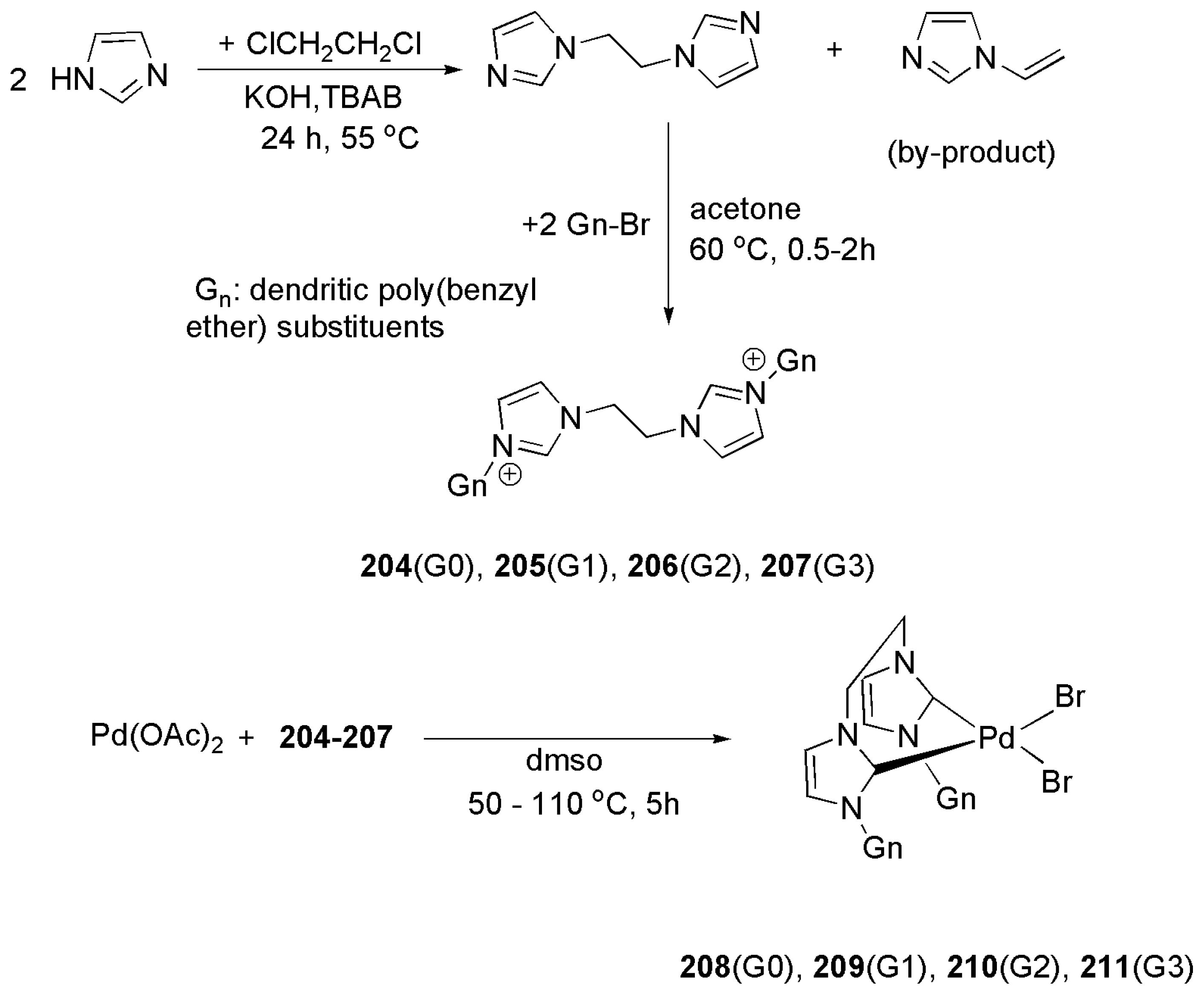
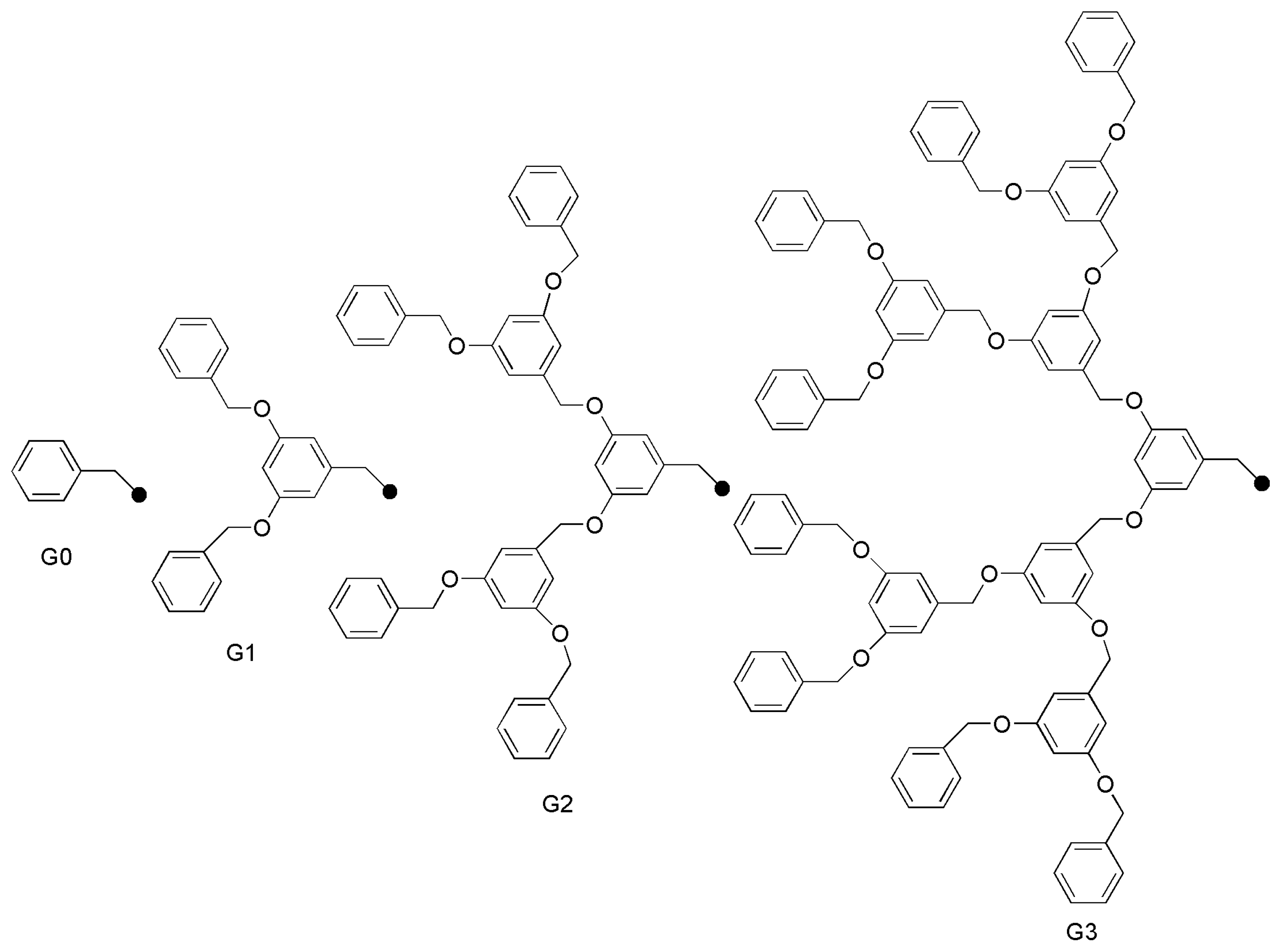
2.5. Other Applications of Bidentate NHC Ligands
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Köcher, C.; Gooßen, L.J.; Artus, G.R.J. Heterocyclic Carbenes: A High-Yielding Synthesis of Novel, Functionalized N-Heterocyclic Carbenes in Liquid Ammonia. Chem. Eur. J. 1996, 2, 1627–1636. [Google Scholar] [CrossRef]
- Edworthy, I.S.; Rodden, M.; Mungur, S.A.; Davis, K.M.; Blake, A.J.; Wilson, C.; Schröder, M.; Arnold, P.L. Silver alkoxide and amino N-heterocyclic carbenes; syntheses and crystal structures. J. Organomet. Chem. 2005, 690, 5710–5719. [Google Scholar] [CrossRef]
- Steinke, T.; Shaw, B.K.; Jong, H.; Patrick, B.O.; Fryzuk, M.D.; Green, J.C. Noninnocent Behavior of Ancillary Ligands: Apparent Trans Coupling of a Saturated N-Heterocyclic Carbene Unit with an Ethyl Ligand Mediated by Nickel. J. Am. Chem. Soc. 2009, 131, 10461–10466. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.; Stingl, V.; Dazinger, G.; Mereiter, K.; Kirchner, K. Migratory Insertion of Acetylene in N-Heterocyclic Carbene Complexes of Ruthenium: Formation of (Ruthenocenylmethyl)imidazolium Salts. Organometallics 2007, 26, 1531–1535. [Google Scholar] [CrossRef]
- Becker, E.; Stingl, V.; Dazinger, G.; Puchberger, M.; Mereiter, K.; Kirchner, K. Facile Migratory Insertion of a N-Heterocyclic Carbene into a Ruthenium−Carbon Double Bond: A New Type of Reaction of a NHC Ligand. J. Am. Chem. Soc. 2006, 128, 6572–6573. [Google Scholar] [CrossRef] [PubMed]
- Hameury, S.; de Frémont, P.; Braunstein, P. Metal complexes with oxygen functionalized NHC ligands: Synthesis and applications. Chem. Soc. Rev. 2017, 46, 632–733. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Müller, D.; Schlepphorst, C.; Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 2017, 46, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, M.C.; Hahn, F.E. Complexes with protic (NH,NH and NH,NR) N-heterocyclic carbene ligands. Coord. Chem. Rev. 2015, 293–294, 95–115. [Google Scholar] [CrossRef]
- Fliedel, C.; Labande, A.; Manoury, E.; Poli, R. Chiral N-heterocyclic carbene ligands with additional chelating group(s) applied to homogeneous metal-mediated asymmetric catalysis. Coord. Chem. Rev. 2019, 394, 65–103. [Google Scholar] [CrossRef]
- Gaillard, S.; Renaud, J.-L. When phosphorus and NHC (N-heterocyclic carbene) meet each other. Dalton Trans. 2013, 42, 7255–7270. [Google Scholar] [CrossRef] [PubMed]
- Naziruddin, A.R.; Hepp, A.; Pape, T.; Hahn, F.E. Synthesis of Rhodium(I) Complexes Bearing Bidentate NH,NR-NHC/Phosphine Ligands. Organometallics 2011, 30, 5859–5866. [Google Scholar] [CrossRef]
- Debono, N.; Labande, A.; Manoury, E.; Daran, J.-C.; Poli, R. Palladium Complexes of Planar Chiral Ferrocenyl Phosphine-NHC Ligands: New Catalysts for the Asymmetric Suzuki−Miyaura Reaction. Organometallics 2010, 29, 1879–1882. [Google Scholar] [CrossRef]
- Miranda-Soto, V.; Grotjahn, D.B.; Cooksy, A.L.; Golen, J.A.; Moore, C.E.; Rheingold, A.L. A Labile and Catalytically Active Imidazol-2-yl Fragment System. Angew. Chem. Int. Ed. 2011, 50, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Soto, V.; Grotjahn, D.B.; DiPasquale, A.G.; Rheingold, A.L. Imidazol-2-yl Complexes of Cp*Ir as Bifunctional Ambident Reactants. J. Am. Chem. Soc. 2008, 130, 13200–13201. [Google Scholar] [CrossRef] [PubMed]
- Clavier, H.; Nolan, S.P. Percent buried volume for phosphine and N-heterocyclic carbene ligands: Steric properties in organometallic chemistry. Chem. Commun. 2010, 46, 841–861. [Google Scholar] [CrossRef] [PubMed]
- Passays, J.; Ayad, T.; Ratovelomanana-Vidal, V.; Gaumont, A.-C.; Jubault, P.; Leclerc, E. Synthesis and evaluation of a broad range of new chiral phosphine–carbene ligands for asymmetric hydrogenation. Tetrahedron Asymmetry 2011, 22, 562–574. [Google Scholar] [CrossRef]
- Loxq, P.; Debono, N.; Gülcemal, S.; Daran, J.-C.; Manoury, E.; Poli, R.; Çetinkaya, B.; Labande, A. Palladium(ii) complexes with planar chiral ferrocenyl phosphane–(benz)imidazol-2-ylidene ligands. New J. Chem. 2014, 38, 338–347. [Google Scholar] [CrossRef]
- Gu, P.; Zhang, J.; Xu, Q.; Shi, M. Synthesis of iridium and rhodium complexes with new chiral phosphine-NHC ligands based on 1,1′-binaphthyl framework and their application in asymmetric hydrogenation. Dalton Trans. 2013, 42, 13599–13606. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Xu, Q.; Shi, M. Synthesis and structural studies on the chiral phosphine-NHC rhodium and palladium complexes for their performances in the metal-catalyzed reactions. Tetrahedron 2014, 70, 7886–7892. [Google Scholar] [CrossRef]
- Zhu, Y.; Khumsubdee, S.; Schaefer, A.; Burgess, K. Asymmetric Syntheses of α-Methyl γ-Amino Acid Derivatives. J. Org. Chem. 2011, 76, 7449–7457. [Google Scholar] [CrossRef]
- Soeta, T.; Hatanaka, Y.; Ishizaka, T.; Ukaji, Y. Chiral NHC ligands bearing a pyridine moiety in copper-catalyzed addition of diethylzinc to nitroalkenes. Tetrahedron 2018, 74, 4601–4605. [Google Scholar] [CrossRef]
- Soeta, T.; Ishizaka, T.; Ukaji, Y. Chiral NHC Ligands Bearing a Pyridine Moiety in Copper-Catalyzed 1,2-Addition of Dialkylzinc Reagents to β-Aryl-α,β-unsaturated N-Tosylaldimines. J. Org. Chem. 2016, 81, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.N.; Haridas, M.; Gangwar, M.K.; Rajakannu, P.; Kalita, A.C.; Ghosh, P. Asymmetric Base-Free Michael Addition at Room Temperature with Nickel-Based Bifunctional Amido-Functionalized N-Heterocyclic Carbene Catalysts. Eur. J. Inorg. Chem. 2015, 2015, 1604–1615. [Google Scholar] [CrossRef]
- Nakano, Y.; Sakaguchi, S. Inversions in asymmetric conjugate addition reaction of cyclic enones catalyzed by the Cu/NHC-AgX system: Factors affecting the stereoselective formation of both enantiomers. J. Organomet. Chem. 2017, 846, 407–416. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nakano, Y.; Shibata, N.; Sakaguchi, S. Enantioselectivity switch in copper-catalyzed conjugate addition reactions under the influence of a chiral N-heterocyclic carbene–silver complex. RSC Adv. 2016, 6, 7755–7759. [Google Scholar] [CrossRef]
- Manabe, Y.; Shinohara, K.; Nakamura, H.; Teramoto, H.; Sakaguchi, S. Chiral N-heterocyclic carbene iridium catalyst for the enantioselective hydrosilane reduction of ketones. J. Mol. Catal. A Chem. 2016, 421, 138–145. [Google Scholar] [CrossRef]
- Kondo, J.; Harano, A.; Dohi, K.; Sakaguchi, S. C2-symmetric functionalized azolium salt from serine ester for Cu-catalyzed asymmetric conjugate addition reaction. J. Mol. Catal. A Chem. 2014, 395, 66–71. [Google Scholar] [CrossRef]
- Yoshimura, M.; Kamisue, R.; Sakaguchi, S. Synthesis of Ru(II) complexes containing N-heterocyclic carbenes functionalized with secondary donor groups: Catalytic activity toward enantioselective transfer hydrogenation. J. Organomet. Chem. 2013, 740, 26–32. [Google Scholar] [CrossRef]
- Wan, K.Y.; Roelfes, F.; Lough, A.J.; Hahn, F.E.; Morris, R.H. Iridium and Rhodium Complexes Containing Enantiopure Primary Amine-Tethered N-Heterocyclic Carbenes: Synthesis, Characterization, Reactivity, and Catalytic Asymmetric Hydrogenation of Ketones. Organometallics 2018, 37, 491–504. [Google Scholar] [CrossRef]
- Wan, K.Y.; Lough, A.J.; Morris, R.H. Transition Metal Complexes of an (S,S)-1,2-Diphenylethylamine-Functionalized N-Heterocyclic Carbene: A New Member of the Asymmetric NHC Ligand Family. Organometallics 2016, 35, 1604–1612. [Google Scholar] [CrossRef] [Green Version]
- Khumsubdee, S.; Fan, Y.; Burgess, K. A Comparison between Oxazoline-imidazolinylidene, -imidazolylidine, -benzimidazolylidene Hydrogenation Catalysts. J. Org. Chem. 2013, 78, 9969–9974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Fan, Y.; Burgess, K. Carbene-Metal Hydrides Can Be Much Less Acidic Than Phosphine-Metal Hydrides: Significance in Hydrogenations. J. Am. Chem. Soc. 2010, 132, 6249–6253. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Loudet, A.; Burgess, K. An Asymmetric Hydrogenation Route To (−)-Spongidepsin. Org. Lett. 2010, 12, 4392–4395. [Google Scholar] [CrossRef]
- Khumsubdee, S.; Zhou, H.; Burgess, K. Homo-Roche Ester Derivatives by Asymmetric Hydrogenation and Organocatalysis. J. Org. Chem. 2013, 78, 11948–11955. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Burgess, K. Highly Stereoselective Syntheses of All 1,2,3-Me,OH,Me Triads via Asymmetric Hydrogenation Reactions. Adv. Synth. Catal. 2013, 355, 107–115. [Google Scholar] [CrossRef]
- Zhu, Y.; Burgess, K. Iridium catalyzed enantioselective hydrogenation of α-alkoxy and β-alkoxy vinyl ethers. RSC Adv. 2012, 2, 4728–4735. [Google Scholar] [CrossRef]
- Lund, C.L.; Sgro, M.J.; Cariou, R.; Stephan, D.W. A Cis-Bis-Mixed-Carbene Ruthenium Hydride Complex: An Olefin-Selective Hydrogenation Catalyst. Organometallics 2012, 31, 802–805. [Google Scholar] [CrossRef]
- Dahcheh, F.; Stephan, D.W. Reactions of ruthenium hydrides with ethyl-vinyl sulfide. Dalton Trans. 2014, 43, 3501–3507. [Google Scholar] [CrossRef]
- Busetto, L.; Cassani, M.C.; Femoni, C.; Mancinelli, M.; Mazzanti, A.; Mazzoni, R.; Solinas, G. N-Heterocyclic Carbene-Amide Rhodium(I) Complexes: Structures, Dynamics, and Catalysis. Organometallics 2011, 30, 5258–5272. [Google Scholar] [CrossRef]
- Bartoszewicz, A.; Marcos, R.; Sahoo, S.; Inge, A.K.; Zou, X.; Martín-Matute, B. A Highly Active Bifunctional Iridium Complex with an Alcohol/Alkoxide-Tethered N-Heterocyclic Carbene for Alkylation of Amines with Alcohols. Chem. Eur. J. 2012, 18, 14510–14519. [Google Scholar] [CrossRef] [Green Version]
- Faraji, L.; Jadidi, K.; Notash, B. Synthesis of novel chiral bidentate hydroxyalkyl-N-heterocyclic carbene ligands and their application in palladium-catalyzed Mizoroki–Heck couplings and asymmetric addition of diethylzinc to benzaldehyde. Tetrahedron Lett. 2014, 55, 346–350. [Google Scholar] [CrossRef]
- Napoli, M.; Saturnino, C.; Cianciulli, E.I.; Varcamonti, M.; Zanfardino, A.; Tommonaro, G.; Longo, P. Silver(I) N-heterocyclic carbene complexes: Synthesis, characterization and antibacterial activity. J. Organomet. Chem. 2013, 725, 46–53. [Google Scholar] [CrossRef]
- Yoo, K.S.; O’Neill, J.; Sakaguchi, S.; Giles, R.; Lee, J.H.; Jung, K.W. Asymmetric Intermolecular Boron Heck-Type Reactions via Oxidative Palladium(II) Catalysis with Chiral Tridentate NHC-Amidate-Alkoxide Ligands. J. Org. Chem. 2010, 75, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Eguillor, B.; Esteruelas, M.A.; García-Raboso, J.; Oliván, M.; Oñate, E.; Pastor, I.M.; Peñafiel, I.; Yus, M. Osmium NHC Complexes from Alcohol-Functionalized Imidazoles and Imidazolium Salts. Organometallics 2011, 30, 1658–1667. [Google Scholar] [CrossRef]
- Mangalum, A.; Htet, Y.; Roe, D.A.; McMillen, C.D.; Tennyson, A.G. Net charge effects in N-heterocyclic carbene–ruthenium complexes with similar oxidation states and coordination geometries. Inorg. Chim. Acta 2015, 435, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.-H.; Wang, C.-C.; Chang, I.H.; Liu, Y.-H.; Wang, Y.; Elsevier, C.J.; Liu, S.-T.; Chen, J.-T. Coordination Chemistry of Highly Hemilabile Bidentate Sulfoxide N-Heterocyclic Carbenes with Palladium(II). Chem. Asian J. 2014, 9, 3498–3510. [Google Scholar] [CrossRef]
- Arnold, P.L.; Cadenbach, T.; Marr, I.H.; Fyfe, A.A.; Bell, N.L.; Bellabarba, R.; Tooze, R.P.; Love, J.B. Homo- and heteroleptic alkoxycarbene f-element complexes and their reactivity towards acidic N–H and C–H bonds. Dalton Trans. 2014, 43, 14346–14358. [Google Scholar] [CrossRef] [Green Version]
- Arnold, P.L.; Marr, I.A.; Zlatogorsky, S.; Bellabarba, R.; Tooze, R.P. Activation of carbon dioxide and carbon disulfide by a scandium N-heterocyclic carbene complex. Dalton Trans. 2014, 43, 34–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, S.; Wang, B.; Xie, H.; Yao, C.; Wu, C.; Cui, D. Rare-earth metal alkyl complexes bearing an alkoxy N-heterocyclic carbene ligand: Synthesis, characterization, catalysis for isoprene polymerization. New J. Chem. 2015, 39, 7682–7687. [Google Scholar] [CrossRef]
- Arnold, P.L.; Casely, I.J.; Turner, Z.R.; Carmichael, C.D. Functionalised Saturated-Backbone Carbene Ligands: Yttrium and Uranyl Alkoxy–Carbene Complexes and Bicyclic Carbene–Alcohol Adducts. Chem. Eur. J. 2008, 14, 10415–10422. [Google Scholar] [CrossRef] [PubMed]
- Turner, Z.R.; Bellabarba, R.; Tooze, R.P.; Arnold, P.L. Addition-Elimination Reactions across the M−C Bond of Metal N-Heterocyclic Carbenes. J. Am. Chem. Soc. 2010, 132, 4050–4051. [Google Scholar] [CrossRef] [Green Version]
- Arnold, P.L.; Turner, Z.R.; Germeroth, A.I.; Casely, I.J.; Nichol, G.S.; Bellabarba, R.; Tooze, R.P. Carbon monoxide and carbon dioxide insertion chemistry of f-block N-heterocyclic carbene complexes. Dalton Trans. 2013, 42, 1333–1337. [Google Scholar] [CrossRef] [Green Version]
- Arnold, P.L.; Turner, Z.R.; Germeroth, A.I.; Casely, I.J.; Bellabarba, R.; Tooze, R.P. Lanthanide/actinide differentiation with sterically encumbered N-heterocyclic carbene ligands. Dalton Trans. 2010, 39, 6808–6814. [Google Scholar] [CrossRef] [Green Version]
- Arnold, P.L.; Turner, Z.R.; Kaltsoyannis, N.; Pelekanaki, P.; Bellabarba, R.M.; Tooze, R.P. Covalency in CeIV and UIV Halide and N-Heterocyclic Carbene Bonds. Chem. Eur. J. 2010, 16, 9623–9629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocchino, C.; Napoli, M.; Costabile, C.; Longo, P. Synthesis of octahedral zirconium complex bearing [NHC O] ligands, and its behavior as catalyst in the polymerization of olefins. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 862–870. [Google Scholar] [CrossRef]
- Costabile, C.; Bocchino, C.; Napoli, M.; Longo, P. Group 4 complexes bearing alkoxide functionalized N-heterocyclic carbene ligands as catalysts in the polymerization of olefins. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 3728–3735. [Google Scholar] [CrossRef]
- Tskhovrebov, A.G.; Solari, E.; Scopelliti, R.; Severin, K. Activation of Nitrous Oxide by Dinuclear Ruthenium Complexes. Organometallics 2012, 31, 7235–7240. [Google Scholar] [CrossRef]
- Hameury, S.; de Frémont, P.; Breuil, P.-A.R.; Olivier-Bourbigou, H.; Braunstein, P. Synthesis and Characterization of Palladium(II) and Nickel(II) Alcoholate-Functionalized NHC Complexes and of Mixed Nickel(II)–Lithium(I) Complexes. Inorg. Chem. 2014, 53, 5189–5200. [Google Scholar] [CrossRef]
- Poulten, R.C.; López, I.; Llobet, A.; Mahon, M.F.; Whittlesey, M.K. Stereoelectronic Effects in C–H Bond Oxidation Reactions of Ni(I) N-Heterocyclic Carbene Complexes. Inorg. Chem. 2014, 53, 7160–7169. [Google Scholar] [CrossRef]
- Meyer, A.; Unger, Y.; Poethig, A.; Strassner, T. Platinum(II) Complexes with Tetradentate Dianionic (O∧C*∧C*∧O)-Ligands. Organometallics 2011, 30, 2980–2985. [Google Scholar] [CrossRef]
- Horeglad, P.; Ablialimov, O.; Szczepaniak, G.; Dąbrowska, A.M.; Dranka, M.; Zachara, J. Dialkylgallium Complexes with Alkoxide and Aryloxide Ligands Possessing N-Heterocyclic Carbene Functionalities: Synthesis and Structure. Organometallics 2014, 33, 100–111. [Google Scholar] [CrossRef]
- El-Batta, A.; Waltman, A.W.; Grubbs, R.H. Bis-ligated Ti and Zr complexes of chelating N-heterocyclic carbenes. J. Organomet. Chem. 2011, 696, 2477–2481. [Google Scholar] [CrossRef]
- Miyake, G.M.; Akhtar, M.N.; Fazal, A.; Jaseer, E.A.; Daeffler, C.S.; Grubbs, R.H. Chelating N-heterocyclic carbene group IV complexes for the polymerization of ethylene and styrene. J. Organomet. Chem. 2013, 728, 1–5. [Google Scholar] [CrossRef]
- Khan, R.K.M.; Zhugralin, A.R.; Torker, S.; O’Brien, R.V.; Lombardi, P.J.; Hoveyda, A.H. Synthesis, Isolation, Characterization, and Reactivity of High-Energy Stereogenic-at-Ru Carbenes: Stereochemical Inversion through Olefin Metathesis and Other Pathways. J. Am. Chem. Soc. 2012, 134, 12438–12441. [Google Scholar] [CrossRef]
- Kong, Y.; Xu, S.; Song, H.; Wang, B. Synthesis, Structures, and Norbornene ROMP Behavior of o-Aryloxide-N-Heterocyclic Carbene p-Cymene Ruthenium Complexes. Organometallics 2012, 31, 5527–5532. [Google Scholar] [CrossRef]
- Ruddlesden, A.J.; Mewis, R.E.; Green, G.G.R.; Whitwood, A.C.; Duckett, S.B. Catalytic Transfer of Magnetism Using a Neutral Iridium Phenoxide Complex. Organometallics 2015, 34, 2997–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Cheng, G.; Ma, C.; Guan, X.; Kwok, W.-M.; Chen, Y.; Lu, W.; Che, C.-M. Light-emitting platinum(ii) complexes supported by tetradentate dianionic bis(N-heterocyclic carbene) ligands: Towards robust blue electrophosphors. Chem. Sci. 2013, 4, 2630–2644. [Google Scholar] [CrossRef]
- Nakano, R.; Nozaki, K. Copolymerization of Propylene and Polar Monomers Using Pd/IzQO Catalysts. J. Am. Chem. Soc. 2015, 137, 10934–10937. [Google Scholar] [CrossRef]
- Kong, Y.; Wen, L.; Song, H.; Xu, S.; Yang, M.; Liu, B.; Wang, B. Synthesis, Structures, and Norbornene Polymerization Behavior of Aryloxide-N-Heterocyclic Carbene Ligated Palladacycles. Organometallics 2011, 30, 153–159. [Google Scholar] [CrossRef]
- Li, Z.; Xue, M.; Yao, H.; Sun, H.; Zhang, Y.; Shen, Q. Enol-functionalized N-heterocyclic carbene lanthanide amide complexes: Synthesis, molecular structures and catalytic activity for addition of amines to carbodiimides. J. Organomet. Chem. 2012, 713, 27–34. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, H.; Tao, X.; Shen, Q.; Zhang, Y. Enolate chelating N-heterocyclic carbene complexes of Fe(II): Synthesis, structure and their catalytic activity for ring-opening polymerization of ɛ-caprolactone. Chin. Sci. Bull. 2007, 52, 3193–3199. [Google Scholar] [CrossRef]
- Shanmuganathan, S.; Kühl, O.; Jones, P.G.; Heinicke, J. Nickel and palladium complexes of enolatefunctionalised N-heterocyclic carbenes. Cent. Eur. J. Chem. 2010, 8, 992–998. [Google Scholar] [CrossRef]
- Guernon, H.; Legault, C.Y. N-Acyliminoimidazolium Ylides as Precursors to Anionic N-Heterocyclic Carbene Ligands: Control of Topology and Reactivity. Organometallics 2013, 32, 1988–1994. [Google Scholar] [CrossRef]
- Mangalum, A.; McMillen, C.D.; Tennyson, A.G. Synthesis, coordination chemistry and reactivity of transition metal complexes supported by a chelating benzimidazolylidene carboxylate ligand. Inorg. Chim. Acta 2015, 426, 29–38. [Google Scholar] [CrossRef]
- Gao, F.; McGrath, K.P.; Lee, Y.; Hoveyda, A.H. Synthesis of Quaternary Carbon Stereogenic Centers through Enantioselective Cu-Catalyzed Allylic Substitutions with Vinylaluminum Reagents. J. Am. Chem. Soc. 2010, 132, 14315–14320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Lee, Y.; Mandai, K.; Hoveyda, A.H. Quaternary Carbon Stereogenic Centers through Copper-Catalyzed Enantioselective Allylic Substitutions with Readily Accessible Aryl- or Heteroaryllithium Reagents and Aluminum Chlorides. Angew. Chem. Int. Ed. 2010, 49, 8370–8374. [Google Scholar] [CrossRef] [Green Version]
- Schmid, T.E.; Drissi-Amraoui, S.; Crévisy, C.; Baslé, O.; Mauduit, M. Copper-catalyzed asymmetric conjugate addition of organometallic reagents to extended Michael acceptors. Beilstein J. Org. Chem. 2015, 11, 2418–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehrli, S.; Martin, D.; Rix, D.; Mauduit, M.; Alexakis, A. Formation of Quaternary Chiral Centers by N-Heterocyclic Carbene–Cu-Catalyzed Asymmetric Conjugate Addition Reactions with Grignard Reagents on Trisubstituted Cyclic Enones. Chem. Eur. J. 2010, 16, 9890–9904. [Google Scholar] [CrossRef] [PubMed]
- Jennequin, T.; Wencel-Delord, J.; Rix, D.; Daubignard, J.; Crévisy, C.; Mauduit, M. Chelating Hydroxyalkyl NHC as Efficient Chiral Ligands for Room-Temperature Copper-Catalyzed Asymmetric Allylic Alkylation. Synlett 2010, 2010, 1661–1665. [Google Scholar] [CrossRef]
- Yasuda, Y.; Ohmiya, H.; Sawamura, M. Copper-Catalyzed Enantioselective Allyl–Allyl Coupling between Allylic Boronates and Phosphates with a Phenol/N-Heterocyclic Carbene Chiral Ligand. Angew. Chem. Int. Ed. 2016, 55, 10816–10820. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, Y.; Ohmiya, H.; Sawamura, M. Copper-Catalyzed Enantioselective Coupling between Allyl–boronates and Phosphates Using a Phenol–Carbene Chiral Ligand: Asymmetric Synthesis of Chiral Branched 1,5-Dienes. Synthesis 2018, 50, 2235–2246. [Google Scholar]
- Grassi, D.; Dolka, C.; Jackowski, O.; Alexakis, A. Copper-Free Asymmetric Allylic Alkylation with a Grignard Reagent: Design of the Ligand and Mechanistic Studies. Chem. Eur. J. 2013, 19, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Grassi, D.; Alexakis, A. Improvements and Applications of the Transition Metal-Free Asymmetric Allylic Alkylation using Grignard Reagents and Magnesium Alanates. Adv. Synth. Catal. 2015, 357, 3171–3186. [Google Scholar] [CrossRef]
- Shintani, R.; Takatsu, K.; Hayashi, T. Copper-catalyzed asymmetric addition of arylboronates to isatins: A catalytic cycle involving alkoxocopper intermediates. Chem. Commun. 2010, 46, 6822–6824. [Google Scholar] [CrossRef]
- Gilani, M.A.; Rais, E.; Wilhelm, R. Chiral Imidazolinium Salts with TIPS Groups for the Palladium-Catalyzed α-Arylation and as Chiral Solvating Agents. Synlett 2015, 26, 1638–1641. [Google Scholar]
- Kubota, K.; Osaki, S.; Jin, M.; Ito, H. Copper(I)-Catalyzed Enantioselective Nucleophilic Borylation of Aliphatic Ketones: Synthesis of Enantioenriched Chiral Tertiary α-Hydroxyboronates. Angew. Chem. Int. Ed. 2017, 56, 6646–6650. [Google Scholar] [CrossRef]
- Khan, R.K.M.; O’Brien, R.V.; Torker, S.; Li, B.; Hoveyda, A.H. Z- and Enantioselective Ring-Opening/Cross-Metathesis with Enol Ethers Catalyzed by Stereogenic-at-Ru Carbenes: Reactivity, Selectivity, and Curtin–Hammett Kinetics. J. Am. Chem. Soc. 2012, 134, 12774–12779. [Google Scholar] [CrossRef] [PubMed]
- Hoveyda, A.H. Evolution of Catalytic Stereoselective Olefin Metathesis: From Ancillary Transformation to Purveyor of Stereochemical Identity. J. Org. Chem. 2014, 79, 4763–4792. [Google Scholar] [CrossRef]
- May, T.L.; Dabrowski, J.A.; Hoveyda, A.H. Formation of Vinyl-, Vinylhalide- or Acyl-Substituted Quaternary Carbon Stereogenic Centers through NHC−Cu-Catalyzed Enantioselective Conjugate Additions of Si-Containing Vinylaluminums to β-Substituted Cyclic Enones. J. Am. Chem. Soc. 2011, 133, 736–739. [Google Scholar] [CrossRef] [Green Version]
- Dabrowski, J.A.; Villaume, M.T.; Hoveyda, A.H. Enantioselective Synthesis of Quaternary Carbon Stereogenic Centers through Copper-Catalyzed Conjugate Additions of Aryl- and Alkylaluminum Reagents to Acyclic Trisubstituted Enones. Angew. Chem. Int. Ed. 2013, 52, 8156–8159. [Google Scholar] [CrossRef] [Green Version]
- McGrath, K.P.; Hoveyda, A.H. A Multicomponent Ni-, Zr-, and Cu-Catalyzed Strategy for Enantioselective Synthesis of Alkenyl-Substituted Quaternary Carbons. Angew. Chem. Int. Ed. 2014, 53, 1910–1914. [Google Scholar] [CrossRef]
- Lee, K.-S.; Hoveyda, A.H. Enantioselective Conjugate Silyl Additions to Cyclic and Acyclic Unsaturated Carbonyls Catalyzed by Cu Complexes of Chiral N-Heterocyclic Carbenes. J. Am. Chem. Soc. 2010, 132, 2898–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.M.; Lee, K.-S.; Hoveyda, A.H. Enantioselective Synthesis of Boron-Substituted Quaternary Carbons by NHC−Cu-Catalyzed Boronate Conjugate Additions to Unsaturated Carboxylic Esters, Ketones, or Thioesters. J. Am. Chem. Soc. 2010, 132, 10630–10633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Meng, F.; Torker, S.; Shi, Y.; Hoveyda, A.H. Catalytic Enantioselective Conjugate Additions of (pin)B-Substituted Allylcopper Compounds Generated in situ from Butadiene or Isoprene. Angew. Chem. Int. Ed. 2016, 55, 9997–10002. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T.; Akiyama, Y.; Sawamura, M. Synthesis of a chiral N-heterocyclic carbene bearing a m-terphenyl-based phosphate moiety as an anionic N-substituent and its application to copper-catalyzed enantioselective boron conjugate additions. Tetrahedron Asymmetry 2013, 24, 729–735. [Google Scholar] [CrossRef]
- Latham, C.M.; Blake, A.J.; Lewis, W.; Lawrence, M.; Woodward, S. Short Synthesis of Chiral 4-Substituted (S)-Imidazolinium Salts Bearing Sulfonates and Their Use in γ-Selective Reactions of Allylic Halides with Grignard Reagents. Eur. J. Org. Chem. 2012, 2012, 699–707. [Google Scholar] [CrossRef]
- Dabrowski, J.A.; Gao, F.; Hoveyda, A.H. Enantioselective Synthesis of Alkyne-Substituted Quaternary Carbon Stereogenic Centers through NHC−Cu-Catalyzed Allylic Substitution Reactions with (i-Bu)2(Alkynyl)aluminum Reagents. J. Am. Chem. Soc. 2011, 133, 4778–4781. [Google Scholar] [CrossRef] [Green Version]
- Dabrowski, J.A.; Haeffner, F.; Hoveyda, A.H. Combining NHC–Cu and Brønsted Base Catalysis: Enantioselective Allylic Substitution/Conjugate Additions with Alkynylaluminum Reagents and Stereospecific Isomerization of the Products to Trisubstituted Allenes. Angew. Chem. Int. Ed. 2013, 52, 7694–7699. [Google Scholar] [CrossRef]
- Guzman-Martinez, A.; Hoveyda, A.H. Enantioselective Synthesis of Allylboronates Bearing a Tertiary or Quaternary B-Substituted Stereogenic Carbon by NHC-Cu-Catalyzed Substitution Reactions. J. Am. Chem. Soc. 2010, 132, 10634–10637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, B.; Hoveyda, A.H. Site- and Enantioselective Formation of Allene-Bearing Tertiary or Quaternary Carbon Stereogenic Centers through NHC–Cu-Catalyzed Allylic Substitution. J. Am. Chem. Soc. 2012, 134, 1490–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Jung, B.; Torker, S.; Hoveyda, A.H. N-Heterocyclic Carbene–Copper-Catalyzed Group-, Site-, and Enantioselective Allylic Substitution with a Readily Accessible Propargyl(pinacolato)boron Reagent: Utility in Stereoselective Synthesis and Mechanistic Attributes. J. Am. Chem. Soc. 2015, 137, 8948–8964. [Google Scholar] [CrossRef]
- Zhou, Y.; Shi, Y.; Torker, S.; Hoveyda, A.H. SN2″-Selective and Enantioselective Substitution with Unsaturated Organoboron Compounds and Catalyzed by a Sulfonate-Containing NHC-Cu Complex. J. Am. Chem. Soc. 2018, 14, 16842–16854. [Google Scholar] [CrossRef]
- Lee, J.; Torker, S.; Hoveyda, A.H. Versatile Homoallylic Boronates by Chemo-, SN2′-, Diastereo- and Enantioselective Catalytic Sequence of Cu−H Addition to Vinyl-B(pin)/Allylic Substitution. Angew. Chem. Int. Ed. 2017, 56, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Corberán, R.; Mszar, N.W.; Hoveyda, A.H. NHC-Cu-Catalyzed Enantioselective Hydroboration of Acyclic and Exocyclic 1,1-Disubstituted Aryl Alkenes. Angew. Chem. Int. Ed. 2011, 50, 7079–7082. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Jang, H.; Hoveyda, A.H. Exceptionally E- and β-Selective NHC–Cu-Catalyzed Proto-Silyl Additions to Terminal Alkynes and Site- and Enantioselective Proto-Boryl Additions to the Resulting Vinylsilanes: Synthesis of Enantiomerically Enriched Vicinal and Geminal Borosilanes. Chem. Eur. J. 2013, 19, 3204–3214. [Google Scholar] [CrossRef]
- Fliedel, C.; Braunstein, P. Recent advances in S-functionalized N-heterocyclic carbene ligands: From the synthesis of azolium salts and metal complexes to applications. J. Organomet. Chem. 2014, 751, 286–300. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Sasson, Y.; Blum, J. Homogeneous Catalytic Transfer-Hydrogenation of α,β Unsaturated Carbonyl Compounds by Dichlorotris (triphenylphosphine)ruthenium(II). Tetrahedron Lett. 1971, 12, 2167–2170. [Google Scholar] [CrossRef]
- Hillier, A.C.; Lee, H.M.; Stevens, E.D.; Nolan, S.P. Cationic Iridium Complexes Bearing Imidazol-2-ylidene Ligands as Transfer Hydrogenation Catalysts. Organometallics 2001, 20, 4246–4252. [Google Scholar] [CrossRef]
- Albrecht, M.; Miecznikowski, J.R.; Samuel, A.; Faller, J.W.; Crabtree, R.H. Chelated Iridium(III) Bis-carbene Complexes as AirStable Catalysts for Transfer Hydrogenation. Organometallics 2002, 21, 3596–3604. [Google Scholar] [CrossRef]
- da Costa, A.P.; Viciano, M.; Sanau, M.; Merino, S.; Tejeda, J.; Peris, E.; Royo, B. First Cp*-Functionalized N-Heterocyclic Carbene and Its Coordination to Iridium. Study of the Catalytic Properties. Organometallics 2008, 27, 1305–1309. [Google Scholar] [CrossRef]
- Warsink, S.; Drost, R.M.; Lutz, M.; Spek, A.L.; Elsevier, C.J. Modular Synthesis of Bidentate Triazolyl-Functionalized N-Heterocyclic Carbenes and Their Palladium Complexes. Organometallics 2010, 29, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Louie, J.; Grubbs, R.H. Highly active iron imidazolylidene catalysts for atom transfer radical polymerization. Chem. Commun. 2000, 16, 1479–1480. [Google Scholar] [CrossRef] [Green Version]
- Casey, C.P.; Guan, H. An Efficient and Chemoselective Iron Catalyst for the Hydrogenation of Ketones. J. Am. Chem. Soc. 2007, 129, 5816–5817. [Google Scholar] [CrossRef] [PubMed]
- Kandepi, V.V.K.M.; Cardoso, J.M.S.; Peris, E.; Royo, B. Iron(II) Complexes Bearing Chelating Cyclopentadienyl-N-Heterocyclic Carbene Ligands as Catalysts for Hydrosilylation and Hydrogen Transfer Reactions. Organometallics 2010, 29, 2777–2782. [Google Scholar] [CrossRef]
- Cross, W.B.; Daly, C.G.; Boutadla, Y.; Singh, K. Variable coordination of amine functionalised N-heterocyclic carbene ligands to Ru, Rh and Ir: C–H and N–H activation and catalytic transfer hydrogenation. Dalton Trans. 2011, 40, 9722–9730. [Google Scholar] [CrossRef]
- Aupoix, A.; Bournaud, C.; Vo-Thanh, G. Asymmetric Transfer Hydrogenation of Aromatic Ketones Using Rhodium Complexes of Chiral N-Heterocyclic Carbenes Derived from (S)-Pyroglutamic Acid. Eur. J. Org. Chem. 2011, 2011, 2772–2776. [Google Scholar] [CrossRef]
- Chiyojima, H.; Sakaguchi, S. Iridium complex bearing a chiral hydroxy-amide functionalized N-heterocyclic carbene: A catalyst precursor for asymmetric transfer hydrogenation. Tetrahedron Lett. 2011, 52, 6788–6791. [Google Scholar] [CrossRef]
- Li, X.-W.; Wang, G.-F.; Chen, F.; Li, Y.-Z.; Chen, X.-T.; Xue, Z.-L. Ruthenium(II) carbonyl chloride complexes containing pyridine-functionalised bidentate N-heterocyclic carbenes: Synthesis, structures, and impact of the carbene ligands on catalytic activities. Inorg. Chim. Acta 2011, 378, 280–287. [Google Scholar] [CrossRef]
- Fernández, F.E.; Puerta, M.C.; Valerga, P. Half-Sandwich Ruthenium(II) Picolyl-NHC Complexes: Synthesis, Characterization, and Catalytic Activity in Transfer Hydrogenation Reactions. Organometallics 2011, 30, 5793–5802. [Google Scholar] [CrossRef]
- Witt, J.; Pöthig, A.; Kühn, F.E.; Baratta, W. Abnormal N-Heterocyclic Carbene-Phosphine Ruthenium(II) Complexes as Active Catalysts for Transfer Hydrogenation. Organometallics 2013, 32, 4042–4045. [Google Scholar] [CrossRef]
- DePasquale, J.; White, N.J.; Ennis, E.J.; Zeller, M.; Foley, J.P.; Papish, E.T. Synthesis of chiral N-heterocyclic carbene (NHC) ligand precursors and formation of ruthenium(II) complexes for transfer hydrogenation catalysts. Polyhedron 2013, 58, 162–170. [Google Scholar] [CrossRef]
- Sluijter, S.N.; Warsink, S.; Lutz, M.; Elsevier, C.J. Synthesis of palladium(0) and -(ii) complexes with chelating bis(N-heterocyclic carbene) ligands and their application in semihydrogenation. Dalton Trans. 2013, 42, 7365–7372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.-B.; Lee, C.-S.; Lin, W.-J.; Naziruddin, A.R.; Hwang, W.-S. Bis-chelate N-heterocyclic tetracarbene Ru(II) complexes: Synthesis, structure, and catalytic activity toward transfer hydrogenation of ketones. Polyhedron 2013, 53, 243–248. [Google Scholar] [CrossRef]
- DePasquale, J.; Kumar, M.; Zeller, M.; Papish, E.T. Variations on an NHC Theme: Which Features Enhance Catalytic Transfer Hydrogenation with Ruthenium Complexes? Organometallics 2013, 32, 966–979. [Google Scholar] [CrossRef]
- Schumacher, A.; Bernasconi, M.; Pfaltz, A. Chiral N-Heterocyclic Carbene/Pyridine Ligands for the Iridium-Catalyzed Asymmetric Hydrogenation of Olefins. Angew. Chem. Int. Ed. 2013, 52, 7422–7425. [Google Scholar] [CrossRef] [PubMed]
- Semwal, S.; Ghorai, D.; Choudhury, J. Wingtip-Dictated Cyclometalation of N-Heterocyclic Carbene Ligand Framework and Its Implication toward Tunable Catalytic Activity. Organometallics 2014, 33, 7118–7124. [Google Scholar] [CrossRef]
- Sluijter, S.N.; Elsevier, C.J. Synthesis and Reactivity of Heteroditopic Dicarbene Rhodium(I) and Iridium(I) Complexes Bearing Chelating 1,2,3-Triazolylidene–Imidazolylidene Ligands. Organometallics 2014, 33, 6389–6397. [Google Scholar] [CrossRef]
- Delgado-Rebollo, M.; Canseco-Gonzalez, D.; Hollering, M.; Mueller-Bunz, H.; Albrecht, M. Synthesis and catalytic alcohol oxidation and ketone transfer hydrogenation activity of donor-functionalized mesoionic triazolylidene ruthenium(ii) complexes. Dalton Trans. 2014, 43, 4462–4473. [Google Scholar] [CrossRef] [Green Version]
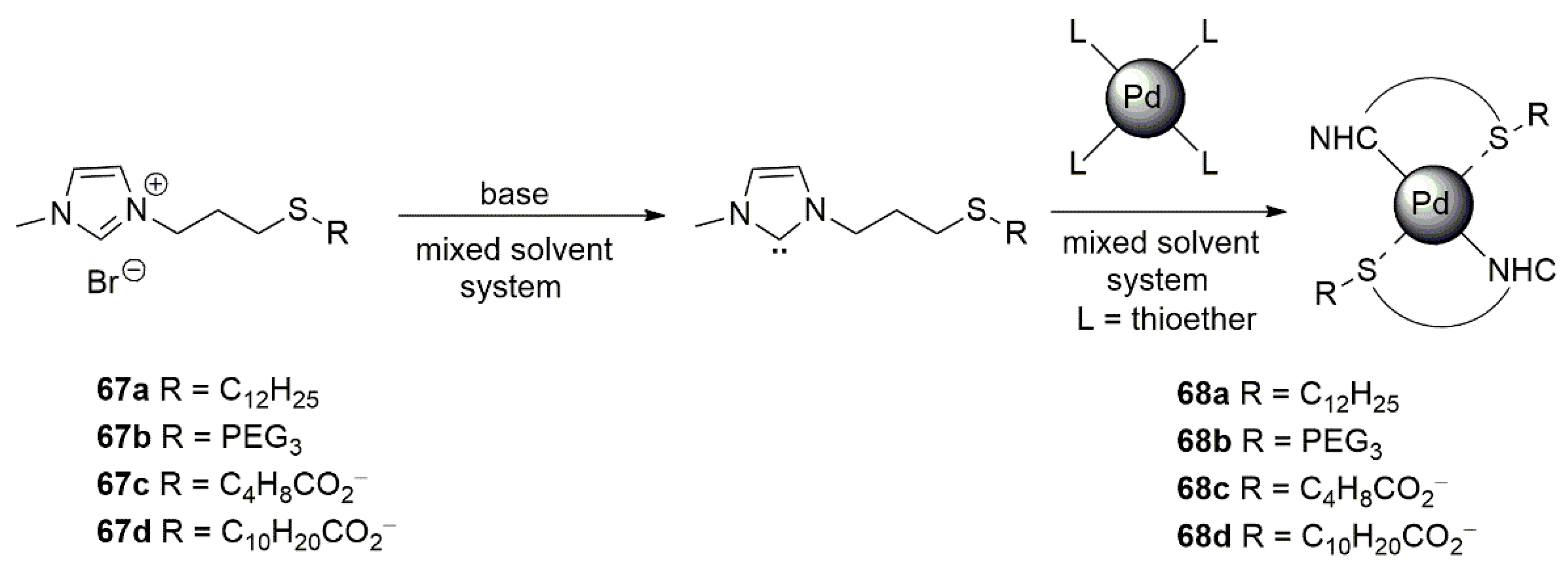
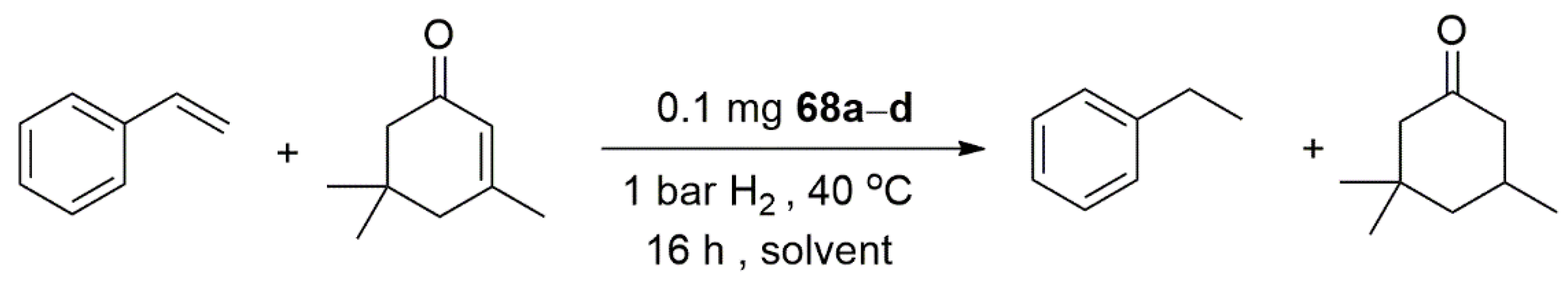
- Rühling, A.; Schaepe, K.; Rakers, L.; Vonhören, B.; Tegeder, P.; Ravoo, B.J.; Glorius, F. Modular Bidentate Hybrid NHC-Thioether Ligands for the Stabilization of Palladium Nanoparticles in Various Solvents. Angew. Chem. Int. Ed. 2016, 55, 5856–5860. [Google Scholar] [CrossRef] [PubMed]
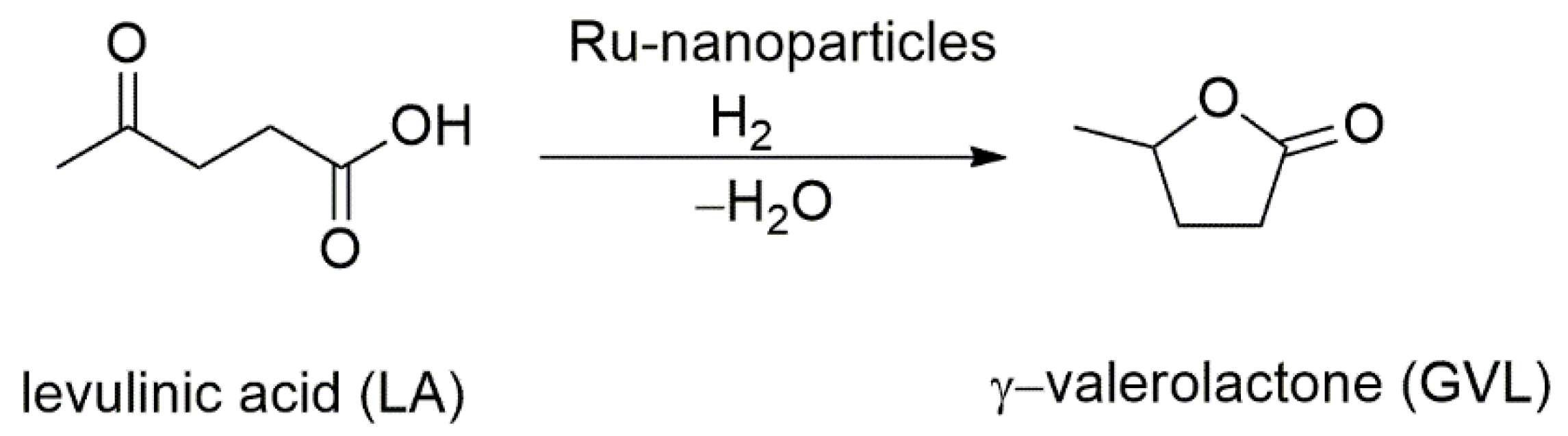
- Tay, B.Y.; Wang, C.; Phua, P.H.; Stubbs, L.P.; Huynh, H.V. Selective hydrogenation of levulinic acid to γ-valerolactone using in situ generated ruthenium nanoparticles derived from Ru–NHC complexes. Dalton Trans. 2016, 45, 3558–3563. [Google Scholar] [CrossRef] [Green Version]
- Balamurugan, G.; Ramesh, R.; Malecki, J.G. Cyclometalated Ru(II)-NHC Complexes as Effective Catalysts for Transfer Hydrogenation: Influence of Wingtip Group on Catalytic Outcome. ChemistrySelect 2017, 2, 10603–10608. [Google Scholar] [CrossRef]
- Wylie, W.N.O.; Lough, A.J.; Morris, R.H. Transmetalation of a Primary Amino-Functionalized N-Heterocyclic Carbene Ligand from an Axially Chiral Square-Planar Nickel(II) Complex to a Ruthenium-(II) Precatalyst for the Transfer Hydrogenation of Ketones. Organometallics 2009, 28, 6755–6761. [Google Scholar]
- Wylie, W.N.O.; Lough, A.J.; Morris, R.H. The hydrogenation of molecules with polar bonds catalyzed by a ruthenium(II) complex bearing a chelating N-heterocyclic carbene with a primary amine donor. Chem. Commun. 2009, 46, 8240–8242. [Google Scholar]
- Wylie, W.N.O.; Lough, A.J.; Morris, R.H. Mechanistic Investigation of the Hydrogenation of Ketones Catalyzed by a Ruthenium(II) Complex Featuring an N-Heterocyclic Carbene with a Tethered Primary Amine Donor: Evidence for an Inner Sphere Mechanism. Organometallics 2011, 30, 1236–1252. [Google Scholar]
- Wylie, W.N.O.; Lough, A.J.; Morris, R.H. Factors Favoring Efficient Bifunctional Catalysis. Study of a Ruthenium(II) Hydrogenation Catalyst Containing an N-Heterocyclic Carbene with a Primary Amine Donor. Organometallics 2012, 31, 2137–2151. [Google Scholar]
- Wylie, W.N.O.; Lough, A.J.; Morris, R.H. Primary Amine Functionalized N-Heterocyclic Carbene Complexes of Iridium: Synthesis, Structure, and Catalysis. Organometallics 2013, 32, 3808–3818. [Google Scholar]
- Wylie, W.N.O.; Morris, R.H. Ester Hydrogenation Catalyzed by a Ruthenium(II) Complex Bearing an N-Heterocyclic Carbene Tethered with an “NH2” Group and a DFT Study of the Proposed Bifunctional Mechanism. ACS Catal. 2013, 3, 32–40. [Google Scholar]
- Wan, K.Y.; Sung, M.M.H.; Lough, A.J.; Morris, R.H. Half Sandwich Ruthenium Catalyst Bearing an Enantiopure Primary Amine Tethered to an N-Heterocyclic Carbene for Ketone Hydrogenation. ACS Catal. 2017, 7, 6827–6842. [Google Scholar] [CrossRef]
- Song, G.; Wang, X.; Li, Y.; Li, X. Iridium Abnormal N-Heterocyclic Carbene Hydrides via Highly Selective C−H Activation. Organometallics 2008, 27, 1187–1192. [Google Scholar] [CrossRef]
- Franco, F.; Pinto, M.F.; Royo, B.; Lloret-Fillol, J. A Highly Active N-Heterocyclic Carbene Manganese(I) Complex for Selective Electrocatalytic CO2 Reduction to CO. Angew. Chem. Int. Ed. 2018, 57, 4603–4606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.; Friães, S.; Franco, F.; Lloret-Fillol, J.; Royo, B. Manganese N-Heterocyclic Carbene Complexes for Catalytic Reduction of Ketones with Silanes. ChemCatChem 2018, 10, 2734–2740. [Google Scholar] [CrossRef]
- Buhaibeh, R.; Filippov, O.A.; Bruneau-Voisine, A.; Willot, J.; Duhayon, C.; Valyaev, D.A.; Lugan, N.; Canac, Y.; Sortais, J.-B. Phosphine-NHC Manganese Hydrogenation Catalyst Exhibiting a Non-Classical Metal-Ligand Cooperative H2 Activation Mode. Angew. Chem. Int. Ed. 2019, 58, 6727–6731. [Google Scholar] [CrossRef] [Green Version]
- van Putten, R.; Benschop, J.; de Munck, V.J.; Weber, M.; Müller, C.; Filonenko, G.A.; Pidko, E.A. Efficient and Practical Transfer Hydrogenation of Ketones Catalyzed by a Simple Bidentate Mn−NHC Complex. ChemCatChem 2019, 11, 5232–5235. [Google Scholar] [CrossRef]
- Malan, F.P.; Singleton, E.; van Rooyen, P.H.; Landman, M. Tandem transfer hydrogenation–epoxidation of ketone substrates catalysed by alkene-tethered Ru(ii)–NHC complexes. New J. Chem. 2019, 43, 8472–8481. [Google Scholar] [CrossRef]
- Bauri, S.; Mallik, A.; Rit, A. Naphthyl-Derived Orthometalated RuII-NHC Complexes: Effect of the NHC Donors and/or Substitution Pattern on Their Synthesis and Catalytic Activity. Organometallics 2020, 39, 3362–3374. [Google Scholar] [CrossRef]
- Illam, P.M.; Singh, V.K.; Priya, R.A. Modulating the electronics of orthometalated RuII-NHC complexes via substitution patterns or NHC donors: Studies towards the impacts in catalysis. J. Organomet. Chem. 2021, 951, 122008. [Google Scholar] [CrossRef]
- Wei, Z.; Wang, Y.; Li, Y.; Ferraccioli, R.; Liu, Q. Bidentate NHC-Cobalt Catalysts for the Hydrogenation of Hindered Alkenes. Organometallics 2020, 39, 3082–3087. [Google Scholar] [CrossRef]
- Quan, X.; Kerdphon, S.; Peters, B.B.C.; Rujirawanich, J.; Krajangsri, S.; Jongcharoenkamol, J.; Andersson, P.G. Cationic NHC-Phosphine Iridium Complexes: Highly Active Catalysts for Base-Free Hydrogenation of Ketones. Chem. Eur. J. 2020, 26, 13311–13316. [Google Scholar] [CrossRef]
- Zhao, D.; Candish, L.; Paul, D.; Glorius, F. N-Heterocyclic Carbenes in Asymmetric Hydrogenation. ACS Catal. 2016, 6, 5978–5988. [Google Scholar] [CrossRef]
- Ito, H.; Ito, S.; Sasaki, Y.; Matsuura, K.; Sawamura, M. Copper-Catalyzed Enantioselective Substitution of Allylic Carbonates with Diboron: An Efficient Route to Optically Active α-Chiral Allylboronates. J. Am. Chem. Soc. 2007, 129, 14856–14857. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Hall, D.G. 3-Boronoacrolein as an Exceptional Heterodiene in the Highly Enantio- and Diastereoselective Cr(III)-Catalyzed Three-Component [4+2]/Allylboration. J. Am. Chem. Soc. 2003, 125, 9308–9309. [Google Scholar] [CrossRef] [PubMed]
- Jackowski, O.; Alexakis, A. Copper-Free Asymmetric Allylic Alkylation with Grignard Reagents. Angew. Chem. Int. Ed. 2010, 49, 3346–3350. [Google Scholar] [CrossRef]
- Shintani, R.; Takatsu, K.; Takeda, M.; Hayashi, T. Copper-Catalyzed Asymmetric Allylic Substitution of Allyl Phosphates with Aryl- and Alkenylboronates. Angew. Chem. Int. Ed. 2011, 50, 8656–8659. [Google Scholar] [CrossRef] [PubMed]
- Takatsu, K.; Shintani, R.; Hayashi, T. Copper-Catalyzed 1,4-Addition of Organoboronates to Alkylidene Cyanoacetates: Mechanistic Insight and Application to Asymmetric Catalysis. Angew. Chem. Int. Ed. 2011, 50, 5548–5552. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Tang, W.; Dagneau, P.; Faraoni, R. A Catalytic Asymmetric Three-Component 1,4-Addition/Aldol Reaction: Enantioselective Synthesis of the Spirocyclic System of Vannusal A. Angew. Chem. Int. Ed. 2005, 44, 3874–3879. [Google Scholar] [CrossRef]
- Shintani, R.; Ichikawa, Y.; Hayashi, T.; Chen, J.; Nakao, Y.; Hiyama, T. Catalytic Asymmetric Synthesis of Allylsilanes through Rhodium/Chiral Diene-Catalyzed 1,4-Addition of Alkenyl[2-(hydroxymethyl)phenyl]dimethylsilanes. Org. Lett. 2007, 9, 4643–4645. [Google Scholar] [CrossRef]
- Brown, M.K.; May, T.L.; Baxter, C.A.; Hoveyda, A.H. All-Carbon Quaternary Stereogenic Centers by Enantioselective Cu-Catalyzed Conjugate Additions Promoted by a Chiral N-Heterocyclic Carbene. Angew. Chem. Int. Ed. 2007, 46, 1097–1100. [Google Scholar] [CrossRef]
- Lee, Y.; Akiyama, K.; Gillingham, D.G.; Brown, M.K.; Hoveyda, A.H. Highly Site- and Enantioselective Cu-Catalyzed Allylic Alkylation Reactions with Easily Accessible Vinylaluminum Reagents. J. Am. Chem. Soc. 2008, 130, 446–447. [Google Scholar] [CrossRef]
- Lee, Y.; Hoveyda, A.H. Lewis Base Activation of Grignard Reagents with N-Heterocyclic Carbenes. Cu-Free Catalytic Enantioselective Additions to γ-Chloro-α,β-Unsaturated Esters. J. Am. Chem. Soc. 2006, 128, 15604–15605. [Google Scholar] [CrossRef]
- Uchida, T.; Katsuki, T. Construction of a new type of chiral bidentate NHC ligands: Copper-catalyzed asymmetric conjugate alkylation. Tetrahedron Lett. 2009, 50, 4741–4743. [Google Scholar] [CrossRef]
- Grassi, D.; Alexakis, A. Copper-Free Asymmetric Allylic Alkylation Using Grignard Reagents on Bifunctional Allylic Bromides. Org. Lett. 2012, 14, 1568–1571. [Google Scholar] [CrossRef] [PubMed]
- Magrez, M.; Le Guen, Y.; Baslé, O.; Crévisy, C.; Mauduit, M. Bidentate Hydroxyalkyl NHC Ligands for the Copper-Catalyzed Asymmetric Allylic Substitution of Allyl Phosphates with Grignard Reagents. Chem. Eur. J. 2013, 19, 1199–1203. [Google Scholar] [CrossRef]
- Queval, P.; Jahier, C.; Rouen, M.; Artur, I.; Legeay, J.-C.; Falivene, L.; Toupet, L.; Crévisy, C.; Cavallo, L.; Baslé, O.; et al. Multicomponent Synthesis of Unsymmetrical Unsaturated N-Heterocyclic Carbene Precursors and Their Related Transition-Metal Complexes. Angew. Chem. Int. Ed. 2013, 52, 14103–14107. [Google Scholar] [CrossRef] [PubMed]
- Altenhoff, G.; Goddard, R.; Lehmann, C.W.; Glorius, F. Sterically Demanding, Bioxazoline-Derived N-Heterocyclic Carbene Ligands with Restricted Flexibility for Catalysis. J. Am. Chem. Soc. 2004, 126, 15195–15201. [Google Scholar] [CrossRef]
- Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Stable Cyclic (Alkyl)(Amino)Carbenes as Rigid or Flexible, Bulky, Electron-Rich Ligands for Transition-Metal Catalysts: A Quaternary Carbon Atom Makes the Difference. Angew. Chem. Int. Ed. 2005, 44, 5705–5709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahier-Diallo, C.; Morin, M.S.T.; Queval, P.; Rouen, M.; Artur, I.; Querard, P.; Toupet, L.; Crévisy, C.; Baslé, O.; Mauduit, M. Multicomponent Synthesis of Chiral Bidentate Unsymmetrical Unsaturated N-Heterocyclic Carbenes: Copper-Catalyzed Asymmetric C–C Bond Formation. Chem. Eur. J. 2015, 21, 993–997. [Google Scholar] [CrossRef]
- Harada, A.; Makida, Y.; Sato, T.; Ohmiya, H.; Sawamura, M. Copper-Catalyzed Enantioselective Allylic Alkylation of Terminal Alkyne Pronucleophiles. J. Am. Chem. Soc. 2014, 136, 13932–13939. [Google Scholar] [CrossRef]
- Hamilton, J.Y.; Sarlah, D.; Carreira, E.M. Iridium-Catalyzed Enantioselective Allylic Alkynylation. Angew. Chem. Int. Ed. 2013, 52, 7532–7535. [Google Scholar] [CrossRef]
- Hojoh, K.; Shido, Y.; Ohmiya, H.; Sawamura, M. Construction of Quaternary Stereogenic Carbon Centers through Copper-Catalyzed Enantioselective Allylic Cross-Coupling with Alkylboranes. Angew. Chem. Int. Ed. 2014, 53, 4954–4958. [Google Scholar] [CrossRef]
- Drissi-Amraoui, S.; Morin, M.S.T.; Crévisy, C.; Baslé, O.; Marcia de Figueiredo, R.; Mauduit, M.; Campagne, J.-M. Copper-Catalyzed Asymmetric Conjugate Addition of Dimethylzinc to Acyl-N-methylimidazole Michael Acceptors: A Powerful Synthetic Platform. Angew. Chem. Int. Ed. 2015, 54, 11830–11834. [Google Scholar] [CrossRef]
- Drissi-Amraoui, S.; Schmid, T.E.; Lauberteaux, J.; Crévisy, C.; Baslé, O.; de Figueiredo, R.M.; Halbert, S.; Gérard, H.; Mauduit, M.; Campagne, J.-M. Copper-Catalyzed Asymmetric Conjugate Addition of Dimethylzinc to Acyl-N-methylimidazole Michael Acceptors: Scope, Limitations and Iterative Reactions. Adv. Synth. Catal. 2016, 358, 2519–2540. [Google Scholar] [CrossRef]
- Ohmiya, H.; Zhang, H.; Shibata, S.; Harada, A.; Sawamura, M. Construction of Quaternary Stereogenic Carbon Centers through Copper-Catalyzed Enantioselective Allylic Alkylation of Azoles. Angew. Chem. Int. Ed. 2016, 55, 4777–4780. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Huynh, H.V. Sulfur-Functionalized N-Heterocyclic Carbene Complexes of Pd(II): Syntheses, Structures and Catalytic Activities. Molecules 2012, 17, 2491–2517. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Cheng, P.-Y.; Tsai, Y.-H.; Lin, G.-R.; Liu, S.-P.; Sie, M.-H.; Lee, H.M. Efficient Heck Reactions Catalyzed by Palladium(0) and -(II) Complexes Bearing N-Heterocyclic Carbene and Amide Functionalities. Organometallics 2010, 29, 3901–3911. [Google Scholar] [CrossRef]
- Yuan, D.; Huynh, H.V. Dinuclear and Tetranuclear Palladium(II) Complexes of a Thiolato-Functionalized, Benzannulated N-Heterocyclic Carbene Ligand and Their Activities toward Suzuki−Miyaura Coupling. Organometallics 2010, 29, 6020–6027. [Google Scholar] [CrossRef]
- Fliedel, C.; Braunstein, P. Thioether-Functionalized N-Heterocyclic Carbenes: Mono- and Bis-(S,CNHC) Palladium Complexes, Catalytic C−C Coupling, and Characterization of a Unique Ag4I4(S,CNHC)2 Planar Cluster. Organometallics 2010, 29, 5614–5626. [Google Scholar] [CrossRef]
- Meyer, S.; Orben, C.M.; Demeshko, S.; Dechert, S.; Meyer, F. Synthesis and Characterization of Di- and Tetracarbene Iron(II) Complexes with Chelating N-Heterocyclic Carbene Ligands and Their Application in Aryl Grignard–Alkyl Halide Cross-Coupling. Organometallics 2011, 30, 6692–6702. [Google Scholar] [CrossRef]
- Simpson, P.V.; Skelton, B.W.; Brown, D.H.; Baker, M.V. Synthesis and Characterisation of Mono- and Bidentate Alkoxybenzimidazolin-2-ylidene Palladium Complexes: Interesting Solution Behaviour and Application in Catalysis. Eur. J. Inorg. Chem. 2011, 2011, 1937–1952. [Google Scholar] [CrossRef]
- Chen, C.; Zheng, Q.; Ni, S.; Wang, H. Facile one-pot nanocatalysts encapsulation of palladium–NHC complexes for aqueous Suzuki–Miyaura couplings. New J. Chem. 2018, 42, 4624–4630. [Google Scholar] [CrossRef]
- Liu, X.; Xu, W.; Xiang, D.; Zhang, Z.; Chen, D.; Hu, Y.; Li, Y.; Ouyang, Y.; Lin, H. Palladium immobilized on functionalized hypercrosslinked polymers: A highly active and recyclable catalyst for Suzuki–Miyaura coupling reactions in water. New J. Chem. 2019, 43, 12206–12210. [Google Scholar] [CrossRef]
- Wu, J.; Dai, W.; Farnaby, J.H.; Hazari, N.; Le Roy, J.J.; Mereacre, V.; Murugesu, M.; Powell, A.K.; Takase, M.K. Synthesis and catalytic activity of iron complexes with bidentate NHC ligands. Dalton Trans. 2013, 42, 7404–7413. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.N.; Joshi, H.; Sharma, A.K.; Prakash, O.; Singh, A.K. Selenium-Containing N-Heterocyclic Carbenes and Their First Palladium(II) Complexes: Synthesis, Structure, and Pendent Alkyl Chain Length Dependent Catalytic Activity for Suzuki–Miyaura Coupling. Organometallics 2013, 32, 2443–2451. [Google Scholar] [CrossRef]
- Li, K.; Guan, X.; Ma, C.-W.; Lu, W.; Chen, Y.; Che, C.-M. Blue electrophosphorescent organoplatinum(ii) complexes with dianionic tetradentate bis(carbene) ligands. Chem. Commun. 2011, 47, 9075–9077. [Google Scholar] [CrossRef]
- Azua, A.; Sanz, S.; Peris, E. Water-Soluble IrIII N-Heterocyclic Carbene Based Catalysts for the Reduction of CO2 to Formate by Transfer Hydrogenation and the Deuteration of Aryl Amines in Water. Chem. Eur. J. 2011, 17, 3963–3967. [Google Scholar] [CrossRef]
- Li, P.; Herrmann, W.A.; Kühn, F.E. Unsaturated NHC Complexes Immobilized by the Backbone: Synthesis and Application. ChemCatChem 2013, 5, 3324–3329. [Google Scholar] [CrossRef]
- Zhong, R.; Pöthig, A.; Haslinger, S.; Hofmann, B.; Raudaschl-Sieber, G.; Herdtweck, E.; Herrmann, W.A.; Kühn, F.E. Toward Tunable Immobilized Molecular Catalysts: Functionalizing the Methylene Bridge of Bis(N-heterocyclic carbene) Ligands. ChemPlusChem 2014, 79, 1294–1303. [Google Scholar] [CrossRef]
- Yagyu, T.; Tsubouchi, A.; Nishimura, Y. Syntheses and characterization of palladium(II) complexes with a bidentate bis-NHC ligand having methyl and aryl substituents on terminal nitrogen atoms. J. Organomet. Chem. 2014, 767, 1–5. [Google Scholar] [CrossRef]
- Turek, J.; Panov, I.; Semler, M.; Štěpnička, P.; De Proft, F.; Padělková, Z.; Růžička, A. Palladium(II) Complexes of 1,2,4-Triazole-Based N-Heterocyclic Carbenes: Synthesis, Structure, and Catalytic Activity. Organometallics 2014, 33, 3108–3118. [Google Scholar] [CrossRef]
- Jhou, Y.-M.; Nandi, D.; Lee, J.-Y.; Tzeng, R.-J.; Lee, H.M. Palladium(0) complexes of N-heterocyclic carbene ligands with dangling NMeCO functionalities: Synthesis, reactivity and application in Mizoroki–Heck reactions. Polyhedron 2015, 100, 28–35. [Google Scholar] [CrossRef]
- Pahlevanneshan, Z.; Moghadam, M.; Mirkhani, V.; Tangestaninejad, S.; Mohammadpoor–Baltork, I.; Loghmani-Khouzani, H. A new N–heterocyclic carbene palladium complex immobilized on nano silica: An efficient and recyclable catalyst for Suzuki–Miyaura CC coupling reaction. J. Organomet. Chem. 2016, 809, 31–37. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Shen, J.-S.; Tzeng, R.-J.; Lu, I.C.; Lii, J.-H.; Hu, C.-H.; Lee, H.M. Well-defined palladium(0) complexes bearing N-heterocyclic carbene and phosphine moieties: Efficient catalytic applications in the Mizoroki–Heck reaction and direct C–H functionalization. Dalton Trans. 2016, 45, 10375–10388. [Google Scholar] [CrossRef]
- Liang, Q.; Janes, T.; Gjergji, X.; Song, D. Iron complexes of a bidentate picolyl-NHC ligand: Synthesis, structure and reactivity. Dalton Trans. 2016, 45, 13872–13880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazari, S.H.; Bourdeau, J.E.; Talley, M.R.; Valdivia-Berroeta, G.A.; Smith, S.J.; Michaelis, D.J. Nickel-Catalyzed Suzuki Cross Couplings with Unprotected Allylic Alcohols Enabled by Bidentate N-Heterocyclic Carbene (NHC)/Phosphine Ligands. ACS Catal. 2018, 8, 86–89. [Google Scholar] [CrossRef]
- Tang, J.; He, Y.; Yu, J.; Zhang, D. N,N′-Bisaryl Substituted Chiral Linker-Bridged Bis(N-Heterocyclic Carbene) Palladium Complexes: Design, Synthesis, and Catalytic Properties. Organometallics 2017, 36, 1372–1382. [Google Scholar] [CrossRef]
- Ingalls, E.L.; Holtzen, G.A.; Kaminsky, W.; Michael, F.E. Synthesis and structural characterization of palladium(II) complexes of chiral bidentate N-heterocyclic carbene-quinoline ligands. J. Organomet. Chem. 2017, 832, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.; Gómez-Sal, P.; Flores, J.C.; de Jesús, E. Highly Recoverable Pd(II) Catalysts for the Mizoroki–Heck Reaction Based on N-Heterocyclic Carbenes and Poly(benzyl ether) Dendrons. Organometallics 2018, 37, 3598–3610. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Tang, X.-S.; Yang, Z.-Q.; Yu, B.-Y.; Wang, H.-J.; Sang, W.; Yuan, Y.; Chen, C.; Verpoort, F. Highly active bidentate N-heterocyclic carbene/ruthenium complexes performing dehydrogenative coupling of alcohols and hydroxides in open air. Chem. Commun. 2019, 55, 8591–8594. [Google Scholar] [CrossRef]
- Wu, X.-J.; Wang, H.-J.; Yang, Z.-Q.; Tang, X.-S.; Yuan, Y.; Su, W.; Chen, C.; Verpoort, F. Efficient and phosphine-free bidentate N-heterocyclic carbene/ruthenium catalytic systems for the dehydrogenative amidation of alcohols and amines. Org. Chem. Front. 2019, 6, 563–570. [Google Scholar] [CrossRef]
- Romine, A.M.; Demer, M.J.; Gembicky, M.; Rheingold, A.L.; Engle, K.M. Ligand Rearrangement Leads to Tetrahydrothiophene-Functionalized N,S-Heterocyclic Carbene Palladium(II) Complexes. Organometallics 2021, 40, 2311–2319. [Google Scholar] [CrossRef]
- Lahneche, Y.D.; Lachguar, A.; Mouton, C.; Daran, J.-C.; Manoury, E.; Poli, R.; Benslimane, M.; Labande, A.; Deydier, E. Phosphine/N-heterocyclic carbene palladium complex for Suzuki–Miyaura cross-coupling reactions: The role of water on activity. Inorg. Chim. Acta 2019, 492, 91–97. [Google Scholar] [CrossRef]
- Waltman, A.W.; Grubbs, R.H. A New Class of Chelating N-Heterocyclic Carbene Ligands and Their Complexes with Palladium. Organometallics 2004, 23, 3105–3107. [Google Scholar] [CrossRef] [Green Version]
- Khlebnikov, V.; Meduri, A.; Mueller-Bunz, H.; Montini, T.; Fornasiero, P.; Zangrando, E.; Milani, B.; Albrecht, M. Palladium Carbene Complexes for Selective Alkene Di- and Oligomerization. Organometallics 2012, 31, 976–986. [Google Scholar] [CrossRef]
- Elser, I.; Frey, W.; Wurst, K.; Buchmeiser, M.R. Molybdenum Imido Alkylidene Complexes Containing N- and C-Chelating N-Heterocyclic Carbenes. Organometallics 2016, 35, 4106–4111. [Google Scholar] [CrossRef]
- Buchmeiser, M.R.; Sen, S.; Lienert, C.; Widmann, L.; Schowner, R.; Herz, K.; Hauser, P.; Frey, W.; Wang, D. Molybdenum Imido Alkylidene N-Heterocyclic Carbene Complexes: Structure–Productivity Correlations and Mechanistic Insights. ChemCatChem 2016, 8, 2710–2723. [Google Scholar] [CrossRef]
- Noda, S.; Nakamura, A.; Kochi, T.; Chung, L.W.; Morokuma, K.; Nozaki, K. Mechanistic Studies on the Formation of Linear Polyethylene Chain Catalyzed by Palladium Phosphine−Sulfonate Complexes: Experiment and Theoretical Studies. J. Am. Chem. Soc. 2009, 131, 14088–14100. [Google Scholar] [CrossRef]
- Tao, W.; Akita, S.; Nakano, R.; Ito, S.; Hoshimoto, Y.; Ogoshi, S.; Nozaki, K. Copolymerisation of ethylene with polar monomers by using palladium catalysts bearing an N-heterocyclic carbene–phosphine oxide bidentate ligand. Chem. Commun. 2017, 53, 2630–2633. [Google Scholar] [CrossRef] [PubMed]
- Postigo, L.; Royo, B. N-Heterocyclic Carbene Complexes of Nickel as Efficient Catalysts for Hydrosilylation of Carbonyl Derivatives. Adv. Synth. Catal. 2012, 354, 2613–2618. [Google Scholar] [CrossRef]
- van der Ende, M.; Hauser, P.M.; Lienert, C.; Wang, D.; Frey, W.; Buchmeiser, M.R. Ti (IV) Complexes with Bidentate O-Chelating N-Heterocyclic Carbenes for Use in the Homopolymerization of Ethylene and Its Copolymerization with Cyclic Olefins. ChemCatChem 2019, 11, 744–752. [Google Scholar] [CrossRef]
- Quadri, C.C.; Lalrempuia, R.; Frøystein, N.Å.; Törnroos, K.W.; Le Roux, E. Steric factors on unsymmetrical O-hydroxyaryl N-Heterocyclic carbene ligands prevailing the stabilization of single stereoisomer of bis-ligated titanium complexes. J. Organomet. Chem. 2018, 860, 106–116. [Google Scholar] [CrossRef]
- Tao, W.; Wang, X.; Ito, S.; Nozaki, K. Palladium complexes bearing an N-heterocyclic carbene–sulfonamide ligand for cooligomerization of ethylene and polar monomers. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 474–477. [Google Scholar] [CrossRef]
- Ren, X.; Wesolek, M.; Bailly, C.; Karmazin, L.; Braunstein, P. Silver(I) and Nickel(II) Complexes with Oxygen- or Nitrogen-Functionalized NHC Ditopic Ligands and Catalytic Ethylene Oligomerization. Eur. J. Inorg. Chem. 2020, 2020, 1073–1087. [Google Scholar] [CrossRef]
- Mo, Z.; Liu, Y.; Deng, L. Anchoring of Silyl Donors on a N-Heterocyclic Carbene through the Cobalt-Mediated Silylation of Benzylic C–H Bonds. Angew. Chem. Int. Ed. 2013, 52, 10845–10849. [Google Scholar] [CrossRef]
- Liang, Q.; Liu, N.J.; Song, D. Constructing reactive Fe and Co complexes from isolated picolyl-functionalized N-heterocyclic carbenes. Dalton Trans. 2018, 47, 9889–9896. [Google Scholar] [CrossRef]
- Lopes, R.; Cardoso, J.M.S.; Postigo, L.; Royo, B. Reduction of Ketones with Silanes Catalysed by a Cyclopentadienyl-Functionalised N-Heterocyclic Iron Complex. Catal. Lett. 2013, 143, 1061–1066. [Google Scholar] [CrossRef]
- Debono, N.; Daran, J.-C.; Poli, R.; Labande, A. A rhodium(I) dicarbonyl complex with a redox-active ferrocenyl phosphine-NHC ligand: Enhanced reactivity of the metal centre through ferrocene oxidation. Polyhedron 2015, 86, 57–63. [Google Scholar] [CrossRef]
- Swamy, C.A.P.; Varenikov, A.; Ruiter, G.d. Chiral Imidazo[1,5-a]pyridine–Oxazolines: A Versatile Family of NHC Ligands for the Highly Enantioselective Hydrosilylation of Ketones. Organometallics 2020, 39, 247–257. [Google Scholar]
- Bertini, S.; Albrecht, M. O-Functionalised NHC Ligands for Efficient Nickel-catalysed C–O Hydrosilylation. CHIMIA Int. J. Chem. 2020, 74, 483–488. [Google Scholar] [CrossRef]
- da Costa, A.P.; Mata, J.A.; Royo, B.; Peris, E. Preparation of Cp-Functionalized N-Heterocyclic Carbene Complexes of Ruthenium. Resolution of Chiral Complexes and Catalytic Studies. Organometallics 2010, 29, 1832–1838. [Google Scholar] [CrossRef]
- Ammar, H.B.; Hassine, B.B.; Fischmeister, C.; Dixneuf, P.H.; Bruneau, C. Imidazolium-Oxazoline Salts in Ruthenium-Catalyzed Allylic Substitution and Cross Metathesis of Formed Branched Isomers. Eur. J. Inorg. Chem. 2010, 2010, 4752–4756. [Google Scholar] [CrossRef]
- Peters, M.; Breinbauer, R. A simple synthesis of functionalized 3-methyl-1-pyridinyl-1H-imidazolium salts as bidentate N-heterocyclic-carbene precursors and their application in Ir-catalyzed arene borylation. Tetrahedron Lett. 2010, 51, 6622–6625. [Google Scholar] [CrossRef]
- Pubill-Ulldemolins, C.; Poyatos, M.; Bo, C.; Fernández, E. Rhodium–NHC complexes mediate diboration versus dehydrogenative borylation of cyclic olefins: A theoretical explanation. Dalton Trans. 2013, 42, 746–752. [Google Scholar] [CrossRef]
- Demir, S.; Özdemir, İ.; Çetinkaya, B.; Arslan, H.; Van Derveer, D. Synthesis and characterization of bidentate NHC–Pd complexes and their role in amination reactions. Polyhedron 2011, 30, 195–200. [Google Scholar] [CrossRef]
- Cardoso, J.M.S.; Royo, B. Unprecedented synthesis of iron–NHC complexes by C–H activation of imidazolium salts. Mild catalysts for reduction of sulfoxides. Chem. Commun. 2012, 48, 4944–4946. [Google Scholar] [CrossRef] [PubMed]
- Naziruddin, A.R.; Zhuang, C.-S.; Lin, W.-J.; Hwang, W.-S. Donor functionalized ruthenium N-heterocyclic carbene complexes in alcohol oxidation reactions. Dalton Trans. 2014, 43, 5335–5342. [Google Scholar] [CrossRef]
- Fujita, K.-i.; Tamura, R.; Tanaka, Y.; Yoshida, M.; Onoda, M.; Yamaguchi, R. Dehydrogenative Oxidation of Alcohols in Aqueous Media Catalyzed by a Water-Soluble Dicationic Iridium Complex Bearing a Functional N-Heterocyclic Carbene Ligand without Using Base. ACS Catal. 2017, 7, 7226–7230. [Google Scholar] [CrossRef]
- Stanton, C.J.; Machan, C.W.; Vandezande, J.E.; Jin, T.; Majetich, G.F.; Schaefer, H.F.; Kubiak, C.P.; Li, G.; Agarwal, J. Re(I) NHC Complexes for Electrocatalytic Conversion of CO2. Inorg. Chem. 2016, 55, 3136–3144. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Mehmood, F.; Lam, T.L.; Chan, S.L.-F.; Wu, Y.; Yeung, C.-S.; Guan, X.; Li, K.; Chung, C.Y.-S.; Zhou, C.-Y.; et al. Stable luminescent iridium(iii) complexes with bis(N-heterocyclic carbene) ligands: Photo-stability, excited state properties, visible-light-driven radical cyclization and CO2 reduction, and cellular imaging. Chem. Sci. 2016, 7, 3123–3136. [Google Scholar] [CrossRef] [Green Version]
- Siek, S.; Burks, D.B.; Gerlach, D.L.; Liang, G.; Tesh, J.M.; Thompson, C.R.; Qu, F.; Shankwitz, J.E.; Vasquez, R.M.; Chambers, N.; et al. Iridium and Ruthenium Complexes of N-Heterocyclic Carbene- and Pyridinol-Derived Chelates as Catalysts for Aqueous Carbon Dioxide Hydrogenation and Formic Acid Dehydrogenation: The Role of the Alkali Metal. Organometallics 2017, 36, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, D.L.; Siek, S.; Burks, D.B.; Tesh, J.M.; Thompson, C.R.; Vasquez, R.M.; White, N.J.; Zeller, M.; Grotjahn, D.B.; Papish, E.T. Ruthenium (II) and iridium (III) complexes of N-heterocyclic carbene and pyridinol derived bidentate chelates: Synthesis, characterization, and reactivity. Inorg. Chim. Acta 2017, 466, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Dong, J.; Wang, B. Homo- and copolymerization of norbornene with tridentate nickel complexes bearing o-aryloxide-N-heterocyclic carbene ligands. Dalton Trans. 2018, 47, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Tang, Y.; Wang, Z.; Xu, S.; Song, H.; Wang, B. Ruthenium Ring-Opening Metathesis Polymerization Catalysts Bearing o- Aryloxide-N-Heterocyclic Carbenes. Macromol. Chem. Phys. 2013, 214, 492–498. [Google Scholar] [CrossRef]
- Yang, D.; Tang, Y.; Song, H.; Wang, B. Synthesis, Structures, and Norbornene Polymerization Behavior of Palladium Complexes Bearing Tridentate o-Aryloxide-N-heterocyclic Carbene Ligands. Organometallics 2016, 35, 1392–1398. [Google Scholar] [CrossRef]
- Li, J.-Q.; Andersson, P.G. Room temperature and solvent-free iridium-catalyzed selective alkylation of anilines with alcohols. Chem. Commun. 2013, 49, 6131–6133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thongpaen, J.; Schmid, T.E.; Toupet, L.; Dorcet, V.; Mauduit, M.; Baslé, O. Directed ortho C–H borylation catalyzed using Cp*Rh(iii)–NHC complexes. Chem. Commun. 2018, 54, 8202–8205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, P.L.; Kerr, R.W.F.; Weetman, C.; Docherty, S.R.; Rieb, J.; Cruickshank, F.L.; Wang, K.; Jandl, C.; McMullon, M.W.; Pöthig, A.; et al. Selective and catalytic carbon dioxide and heteroallene activation mediated by cerium N-heterocyclic carbene complexes. Chem. Sci. 2018, 9, 8035–8045. [Google Scholar] [CrossRef] [Green Version]
- Gatus, M.R.D.; McBurney, R.T.; Bhadbhade, M.; Messerle, B.A. Enhancements in catalytic reactivity and selectivity of homobimetallic complexes containing heteroditopic ligands. Dalton Trans. 2017, 46, 7457–7466. [Google Scholar] [CrossRef]
- Munz, D.; Poethig, A.; Tronnier, A.; Strassner, T. ortho-Phenylene bridged palladium bis-N-heterocyclic carbene complexes: Synthesis, structure and catalysis. Dalton Trans. 2013, 42, 7297–7304. [Google Scholar] [CrossRef]
- Munz, D.; Strassner, T. Propane Activation by Palladium Complexes with Chelating Bis(NHC) Ligands and Aerobic Cooxidation. Angew. Chem. Int. Ed. 2014, 53, 2485–2488. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.; Yang, J.; Song, H.-B.; Tang, L.-F. Synthesis and structural characterization of N-heterocyclic carbene silver complexes derived from N-ferrocenylmethyl-N′-(pyridylmethyl)imidazolium iodides. Polyhedron 2013, 59, 29–37. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Li, B.; Jiang, S.; Sheng, L. Synthesis, characterization, electrochemical properties and catalytic reactivity of N-heterocyclic carbene-containing diiron complexes. RSC Adv. 2015, 5, 29022–29031. [Google Scholar] [CrossRef]
- Dutta, B.; Schwarz, R.; Omar, S.; Natour, S.; Abu-Reziq, R. Homogeneous and Semi-Heterogeneous Magnetically Retrievable Bis-N-Heterocyclic Carbene Rhodium(I) Based Catalysts for Selective Hydroaminomethylation Reactions. Eur. J. Org. Chem. 2015, 2015, 1961–1969. [Google Scholar] [CrossRef]
- Liu, H.-J.; Gil-Sepulcre, M.; Francàs, L.; Nolis, P.; Parella, T.; Benet-Buchholz, J.; Fontrodona, X.; García-Antón, J.; Romero, N.; Llobet, A.; et al. Mononuclear ruthenium compounds bearing N-donor and N-heterocyclic carbene ligands: Structure and oxidative catalysis. Dalton Trans. 2017, 46, 2829–2843. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Li, Y.; Liu, J.; Lan, X.-B.; Liu, Y.; Zhao, C.; Ke, Z. A bifunctional strategy for N-heterocyclic carbene-stabilized iridium complex-catalyzed N-alkylation of amines with alcohols in aqueous media. Green Chem. 2019, 21, 219–224. [Google Scholar] [CrossRef]
- Elser, I.; Groos, J.; Hauser, P.M.; Koy, M.; van der Ende, M.; Wang, D.; Frey, W.; Wurst, K.; Meisner, J.; Ziegler, F.; et al. Molybdenum and Tungsten Alkylidyne Complexes Containing Mono-, Bi-, and Tridentate N-Heterocyclic Carbenes. Organometallics 2019, 38, 4133–4146. [Google Scholar] [CrossRef]
- Weetman, C.; Notman, S.; Arnold, P.L. Destruction of chemical warfare agent simulants by air and moisture stable metal NHC complexes. Dalton Trans. 2018, 47, 2568–2574. [Google Scholar] [CrossRef] [Green Version]
- Manguin, R.; Pichon, D.; Tarrieu, R.; Vives, T.; Roisnel, T.; Dorcet, V.; Crévisy, C.; Miqueu, K.; Favereau, L.; Crassous, J.; et al. A kinetic resolution strategy for the synthesis of chiral octahedral NHC–iridium(iii) catalysts. Chem. Commun. 2019, 55, 6058–6061. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Ghosh, D.; Kuo, Y.-T.; Lee, H.M. Dimetallic Palladium-NHC Complexes: Synthesis, Characterization, and Catalytic Application for Direct C−H Arylation Reaction of Heteroaromatics with Aryl Chlorides. Adv. Synth. Catal. 2020, 362, 648–658. [Google Scholar] [CrossRef]
- Gupta, S.K.; Choudhury, J. A Mixed N-Heterocyclic Carbene/2,2′-Bipyridine-Supported Robust Ruthenium(II) Oxidation Precatalyst for Benzylic C−H Oxidation. ChemCatChem 2017, 9, 1979–1984. [Google Scholar] [CrossRef]
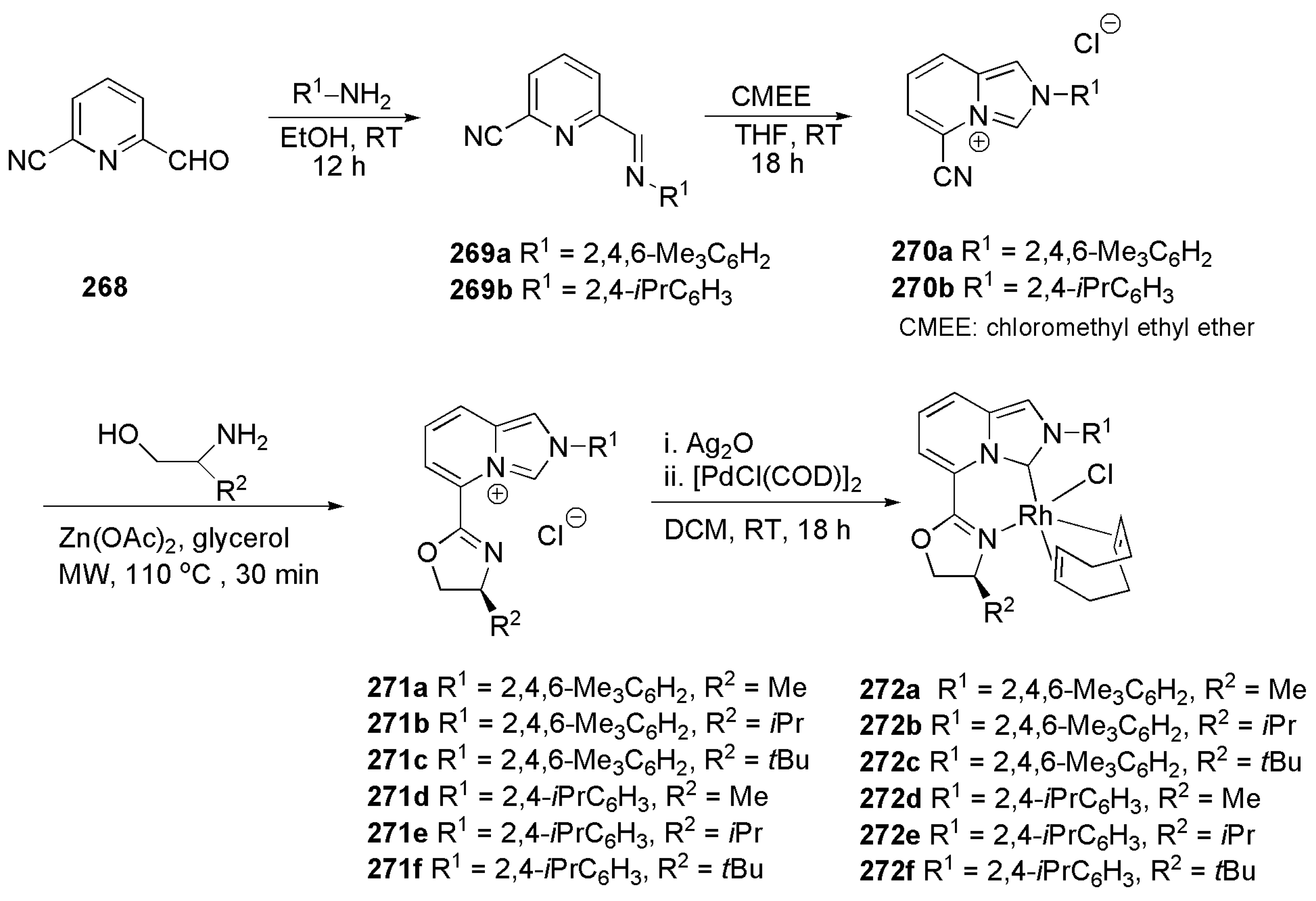
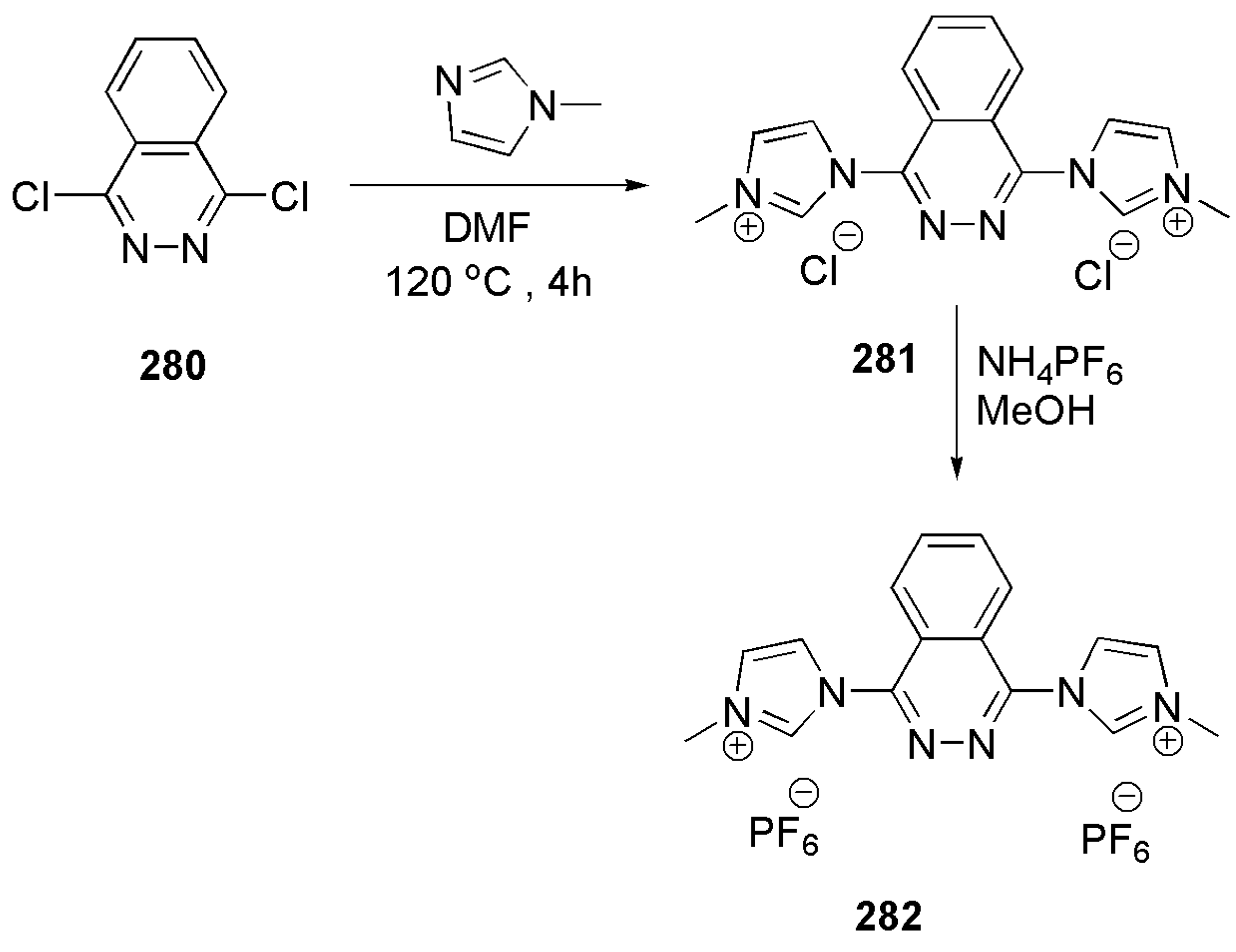
- Pham, P.; Hilty, C. Tunable iridium catalyst designs with bidentate N-heterocyclic carbene ligands for SABRE hyperpolarization of sterically hindered substrates. Chem. Commun. 2020, 56, 15466–15469. [Google Scholar] [CrossRef] [PubMed]
- Labande, A.; Debono, N.; Sournia-Saquet, A.; Daran, J.-C.; Poli, R. Oxidation-promoted activation of a ferrocene C–H bond by a rhodium complex. Dalton Trans. 2013, 42, 6531–6537. [Google Scholar] [CrossRef]
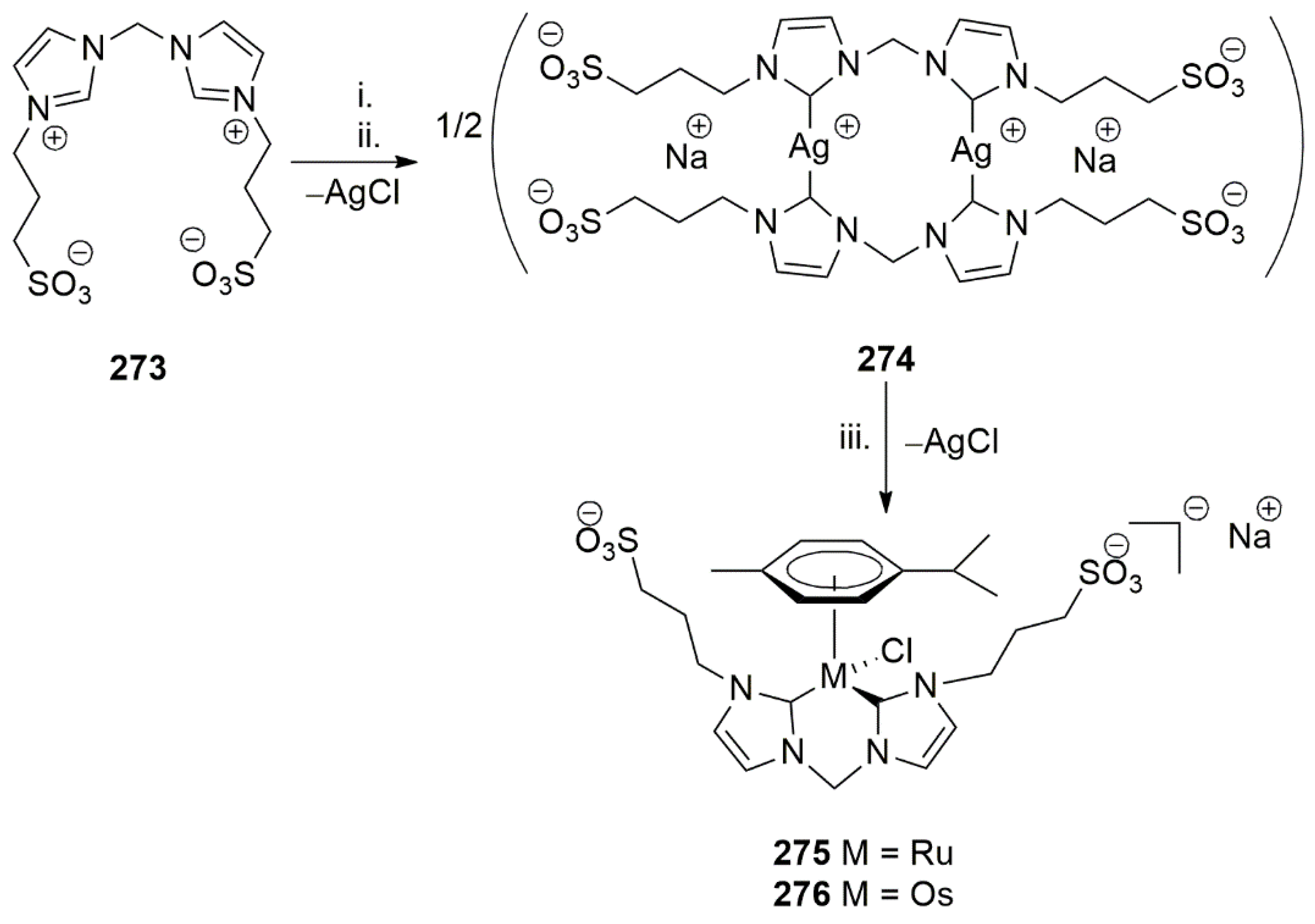
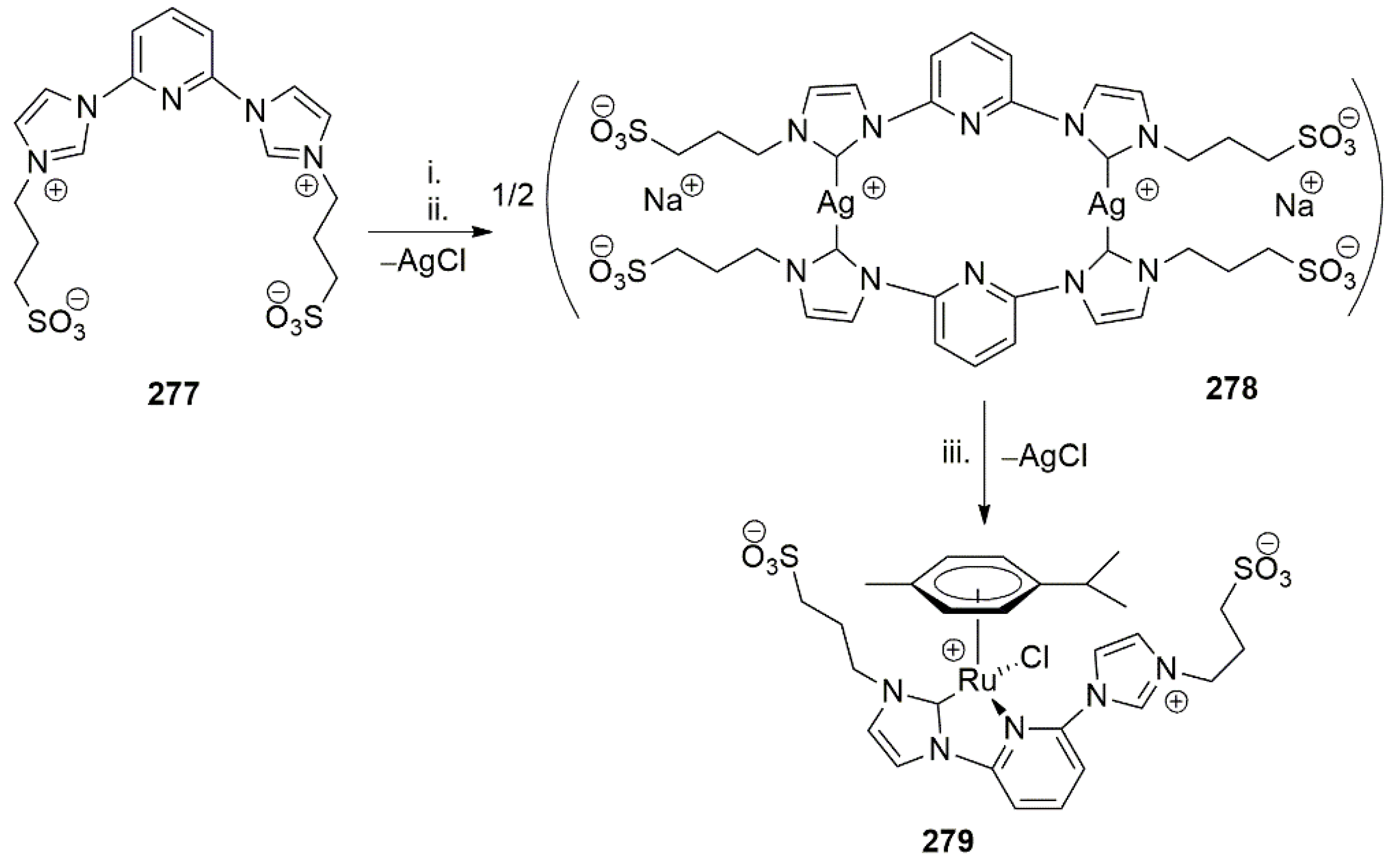
- Jantke, D.; Cokoja, M.; Pöthig, A.; Herrmann, W.A.; Kühn, F.E. Synthesis and Characterization of Highly Water Soluble Ruthenium(II) and Osmium(II) Complexes Bearing Chelating Sulfonated N-Heterocyclic Carbene Ligands. Organometallics 2013, 32, 741–744. [Google Scholar] [CrossRef]
- Papini, G.; Pellei, M.; Gioia Lobbia, G.; Burini, A.; Santini, C. Sulfonate- or carboxylate-functionalized N-heterocyclic bis-carbene ligands and related water soluble silver complexes. Dalton Trans. 2009, 35, 6985–6990. [Google Scholar] [CrossRef] [PubMed]
- Tonzetich, Z.J.; Lam, Y.C.; Müller, P.; Schrock, R.R. Facile Synthesis of a Tungsten Alkylidyne Catalyst for Alkyne Metathesis. Organometallics 2007, 26, 475–477. [Google Scholar] [CrossRef]
- Scattolin, T.; Nolan, S.P. Synthetic Routes to Late Transition Metal–NHC Complexes. Trends Chem. 2020, 2, 721–736. [Google Scholar] [CrossRef]
- Bellotti, P.; Koy, M.; Hopkinson, M.N.; Glorius, F. Recent advances in the chemistry and applications of N-heterocyclic carbenes. Nat. Rev. Chem. 2021, 5, 711–725. [Google Scholar] [CrossRef]



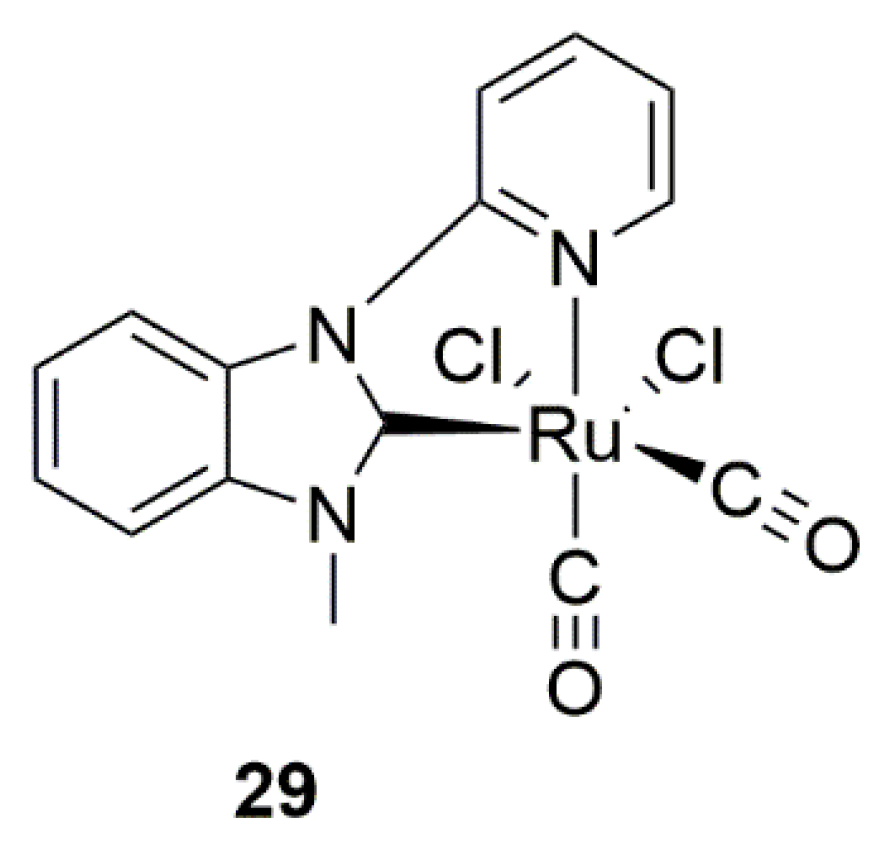
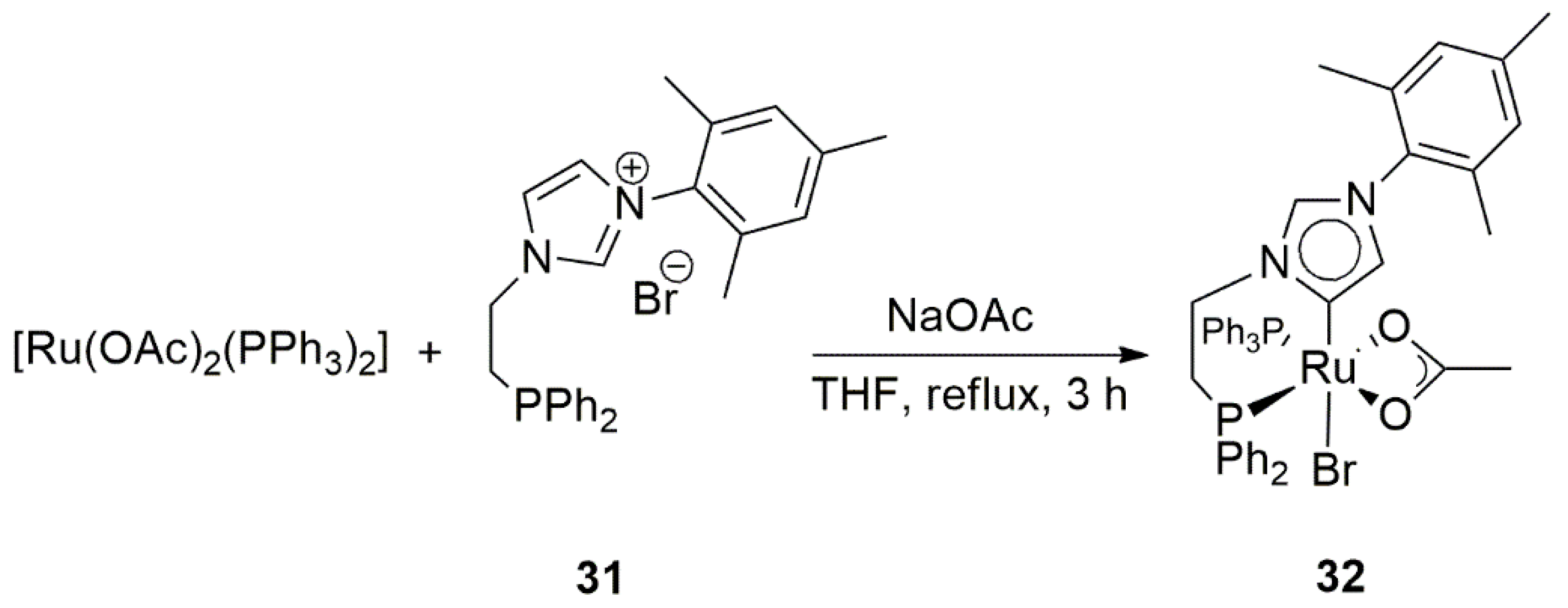
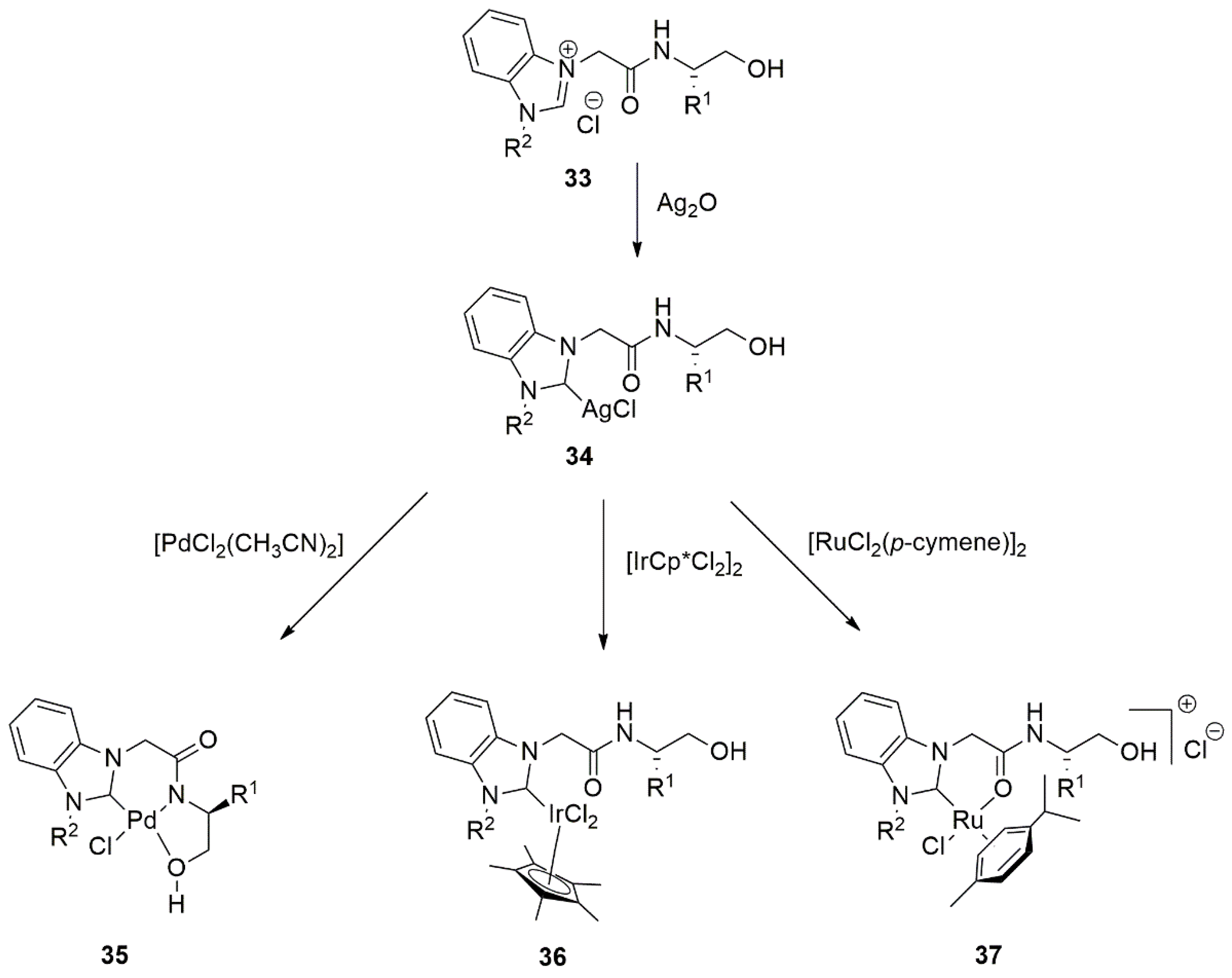
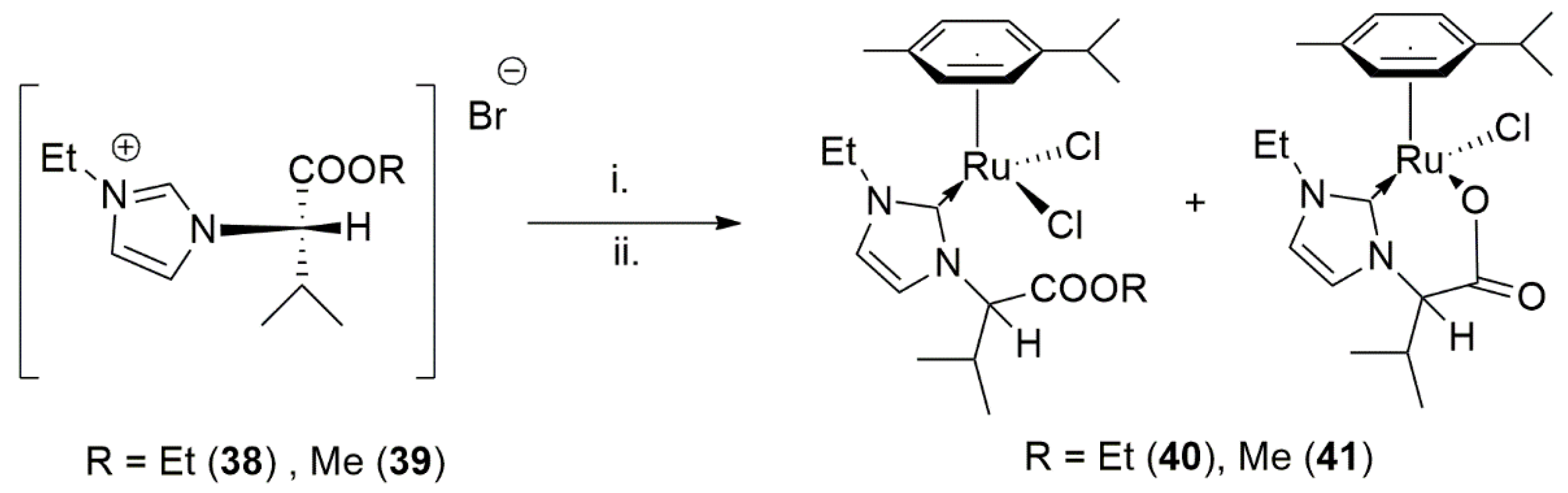




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


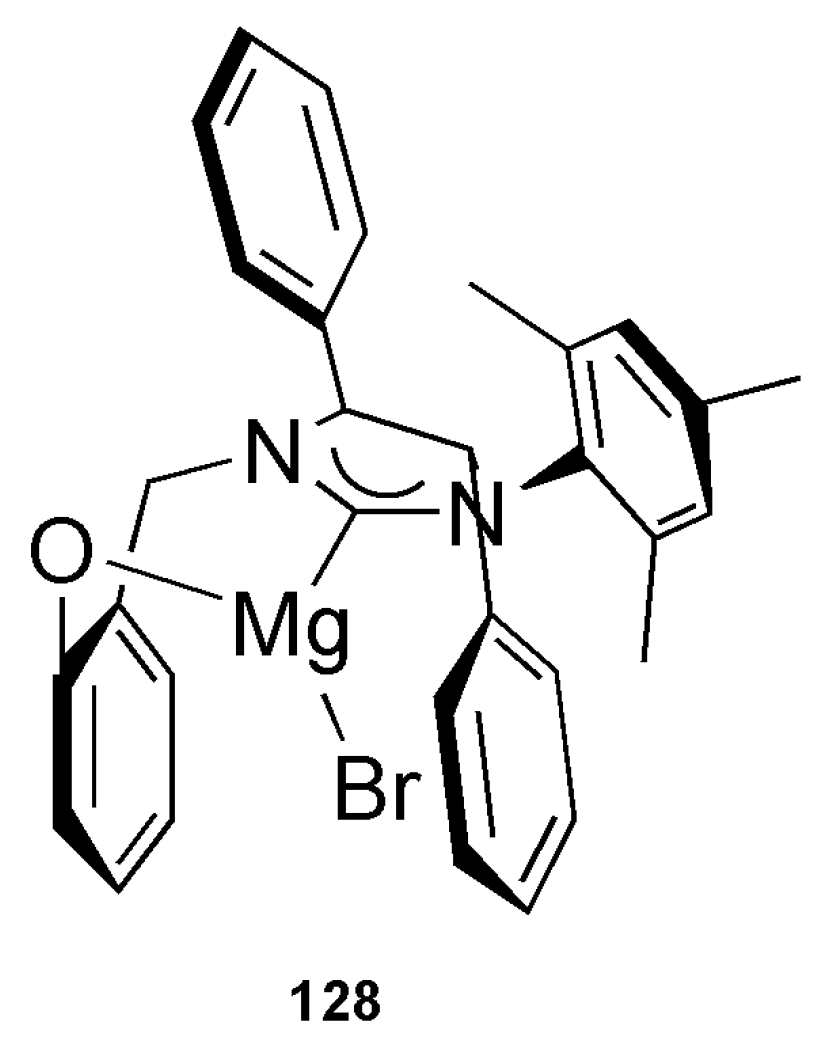
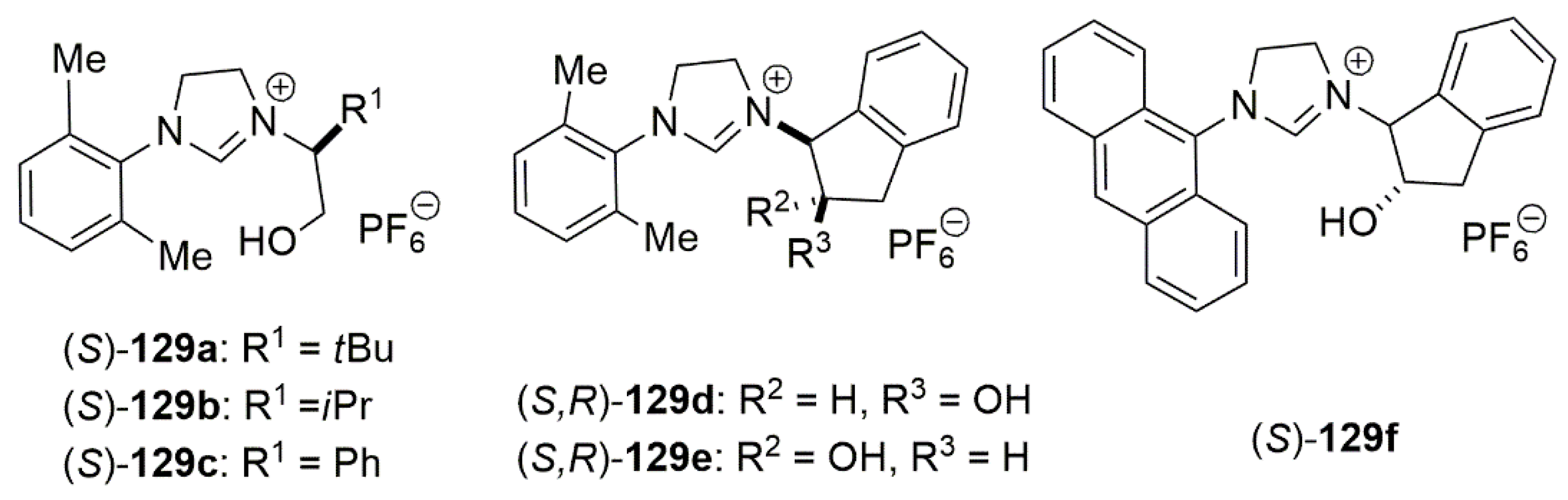
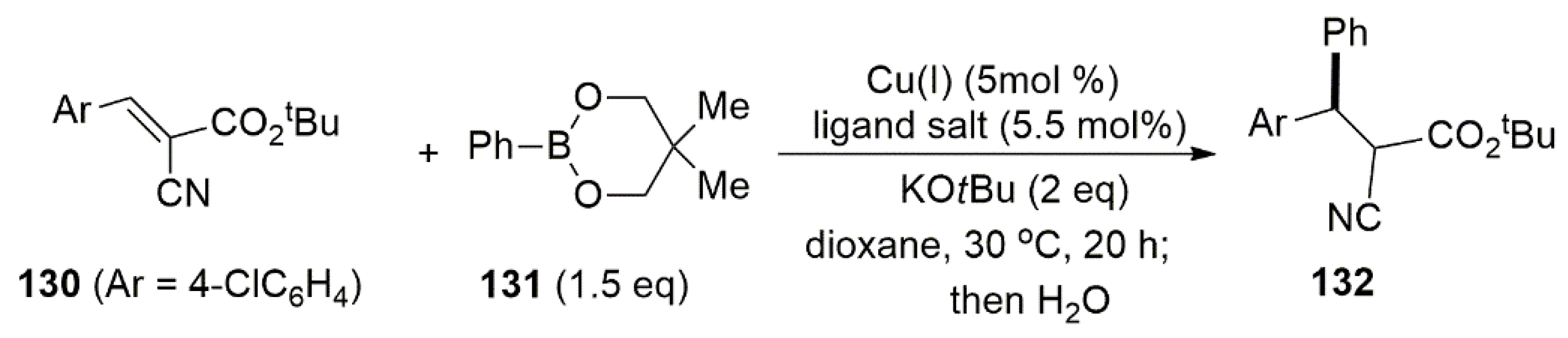
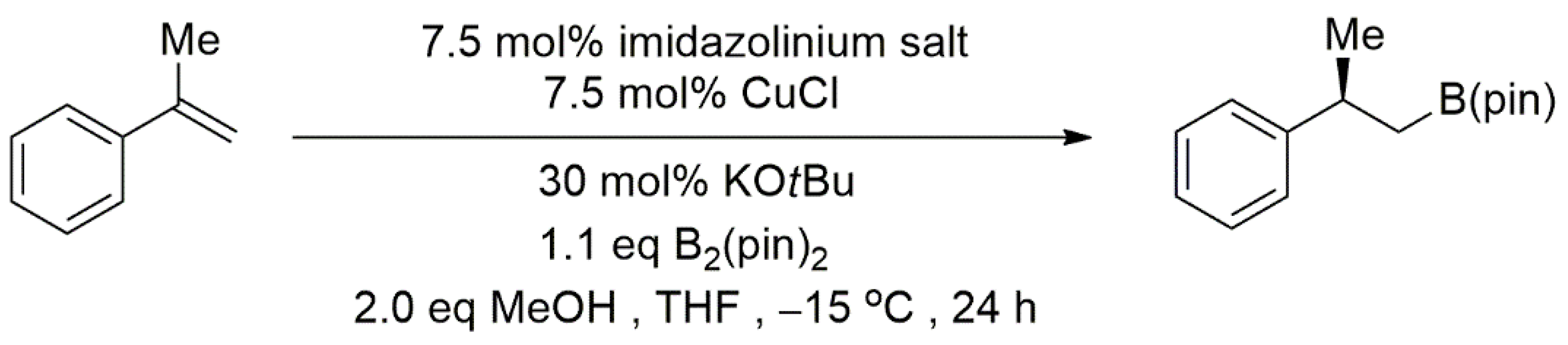


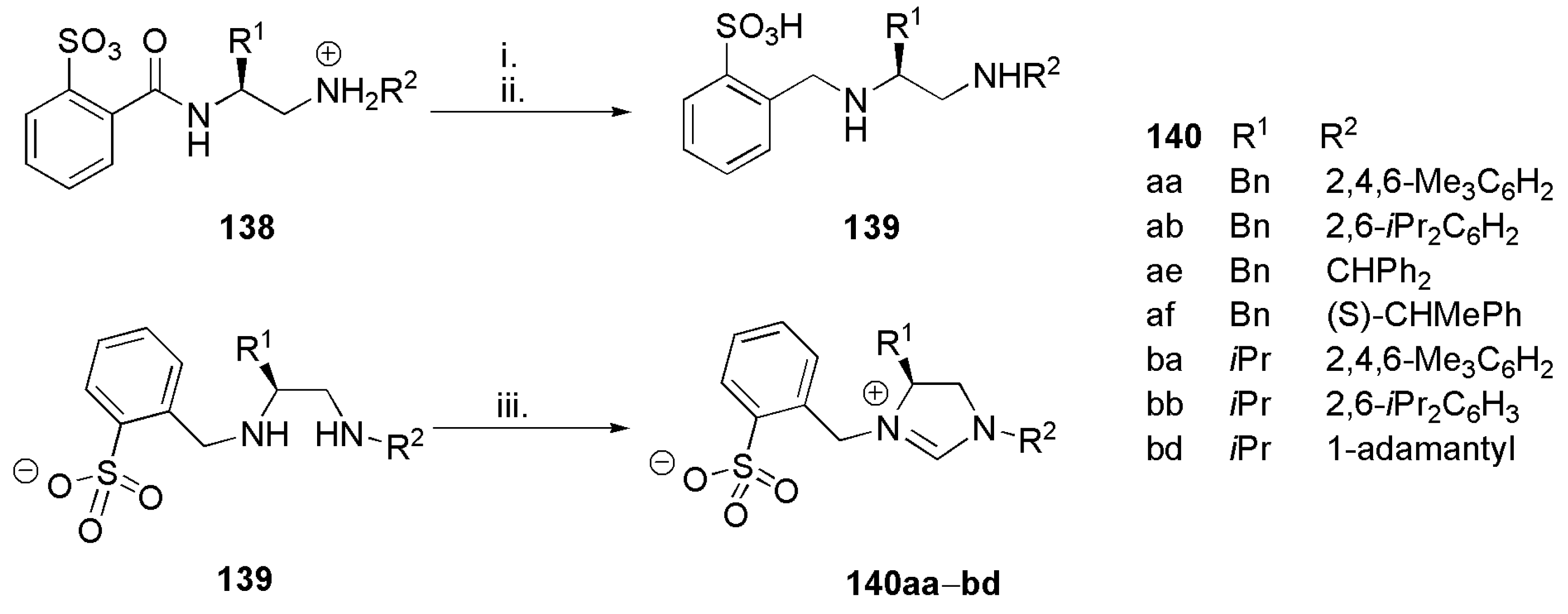
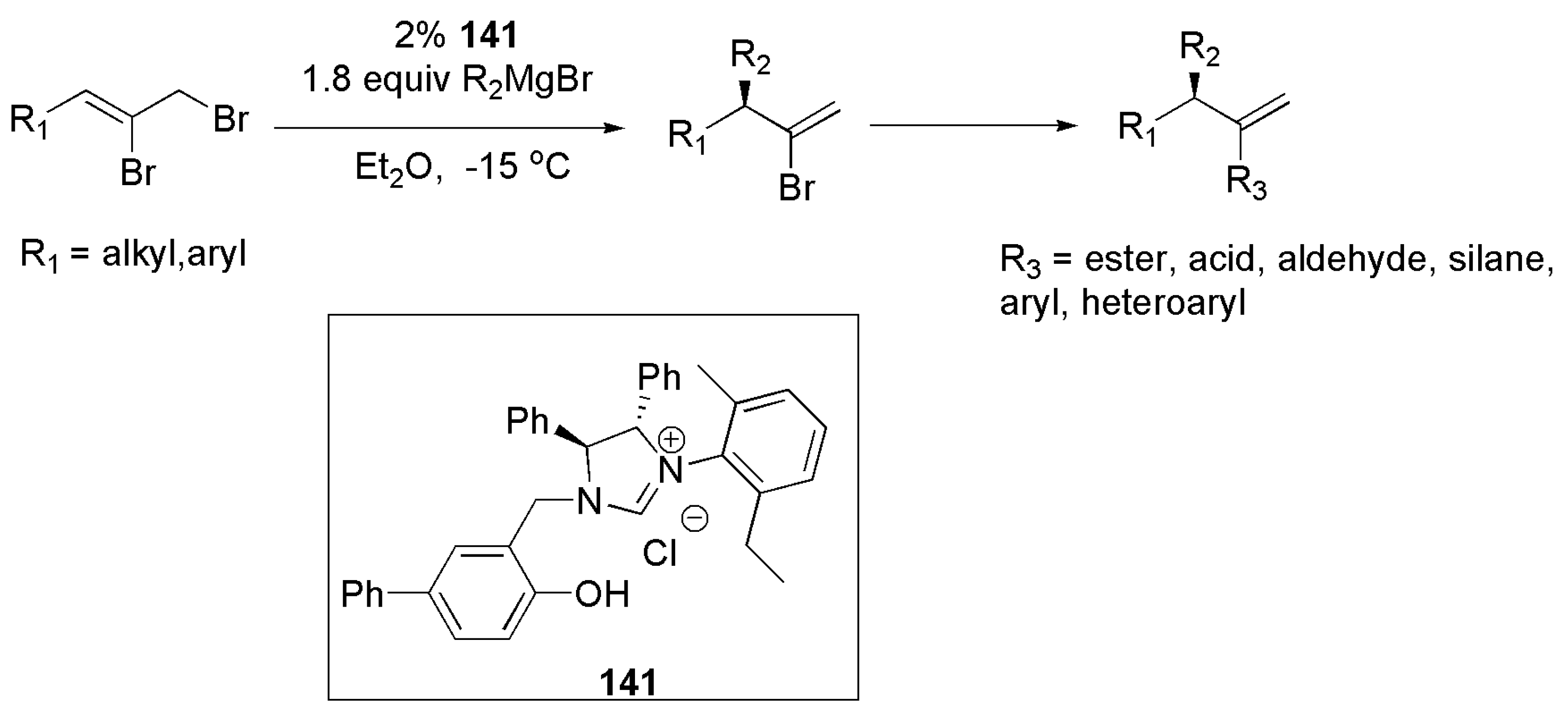

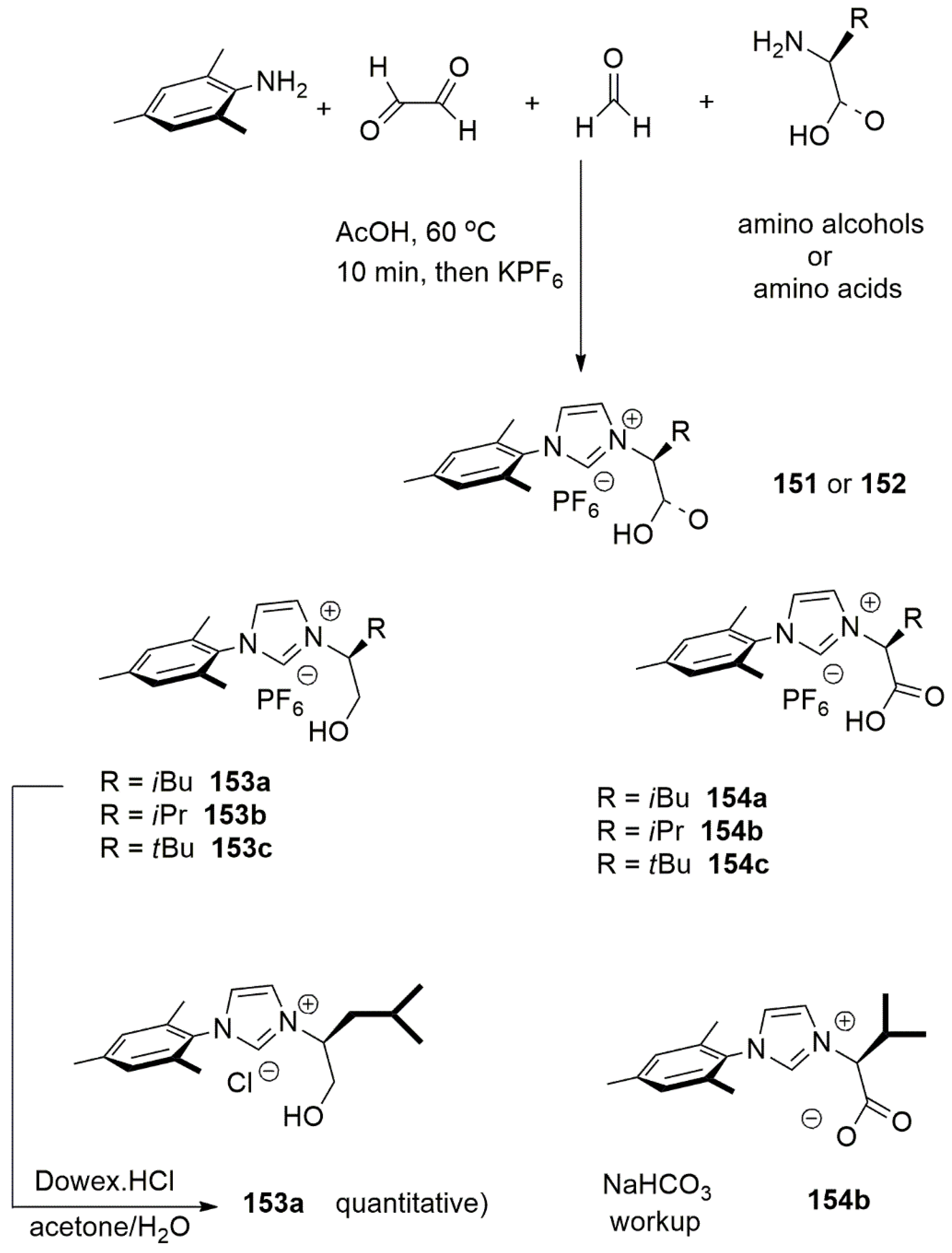
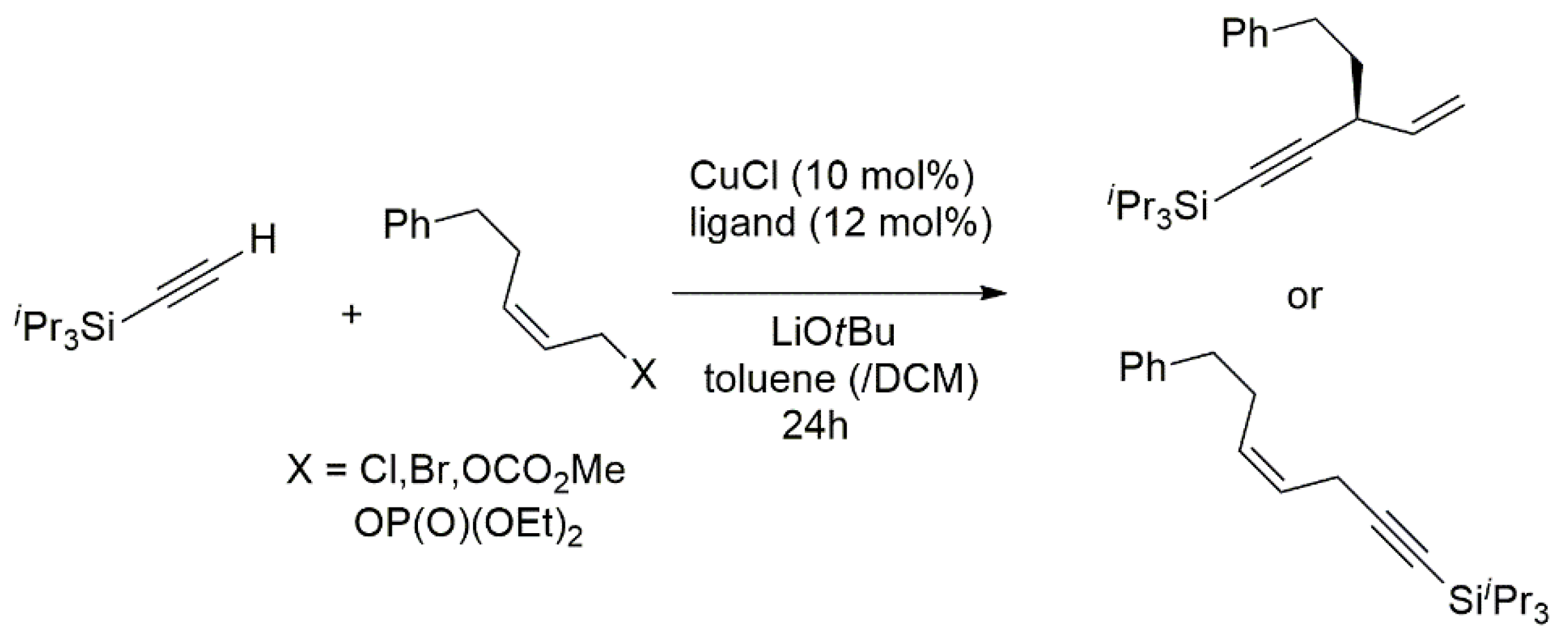



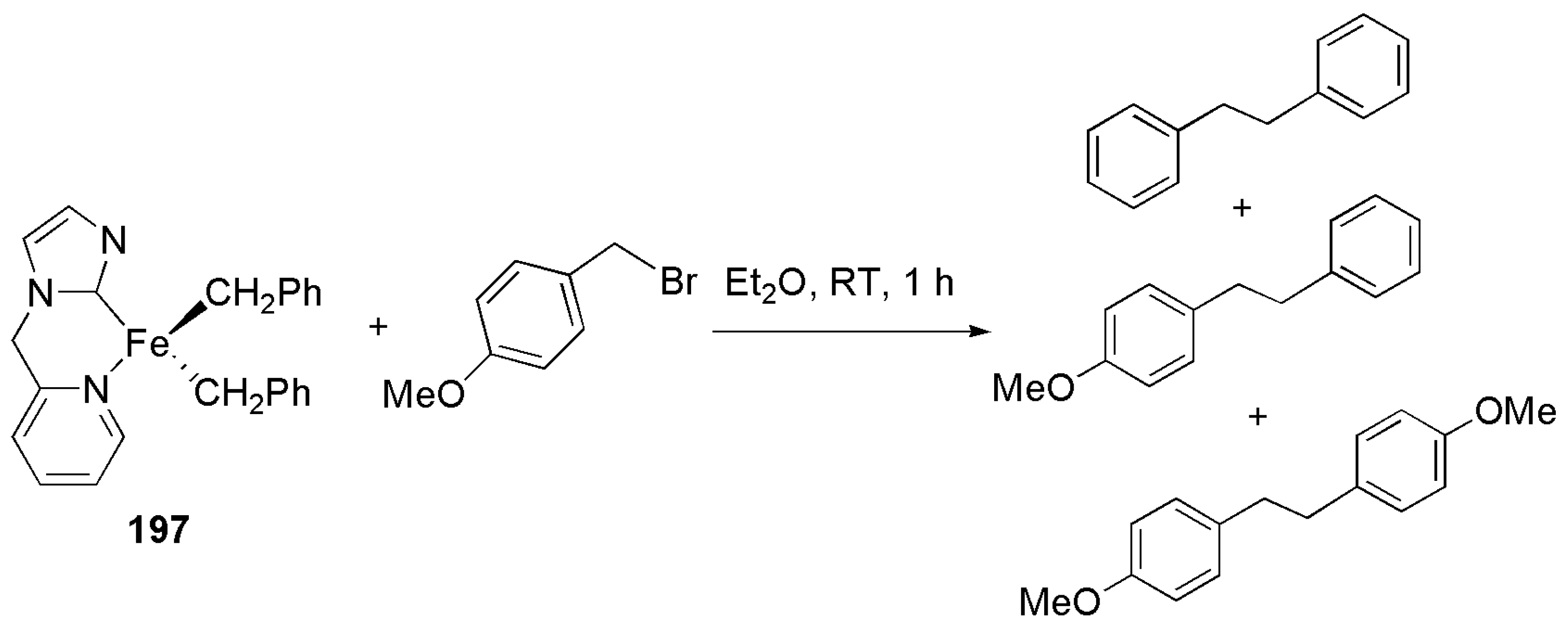
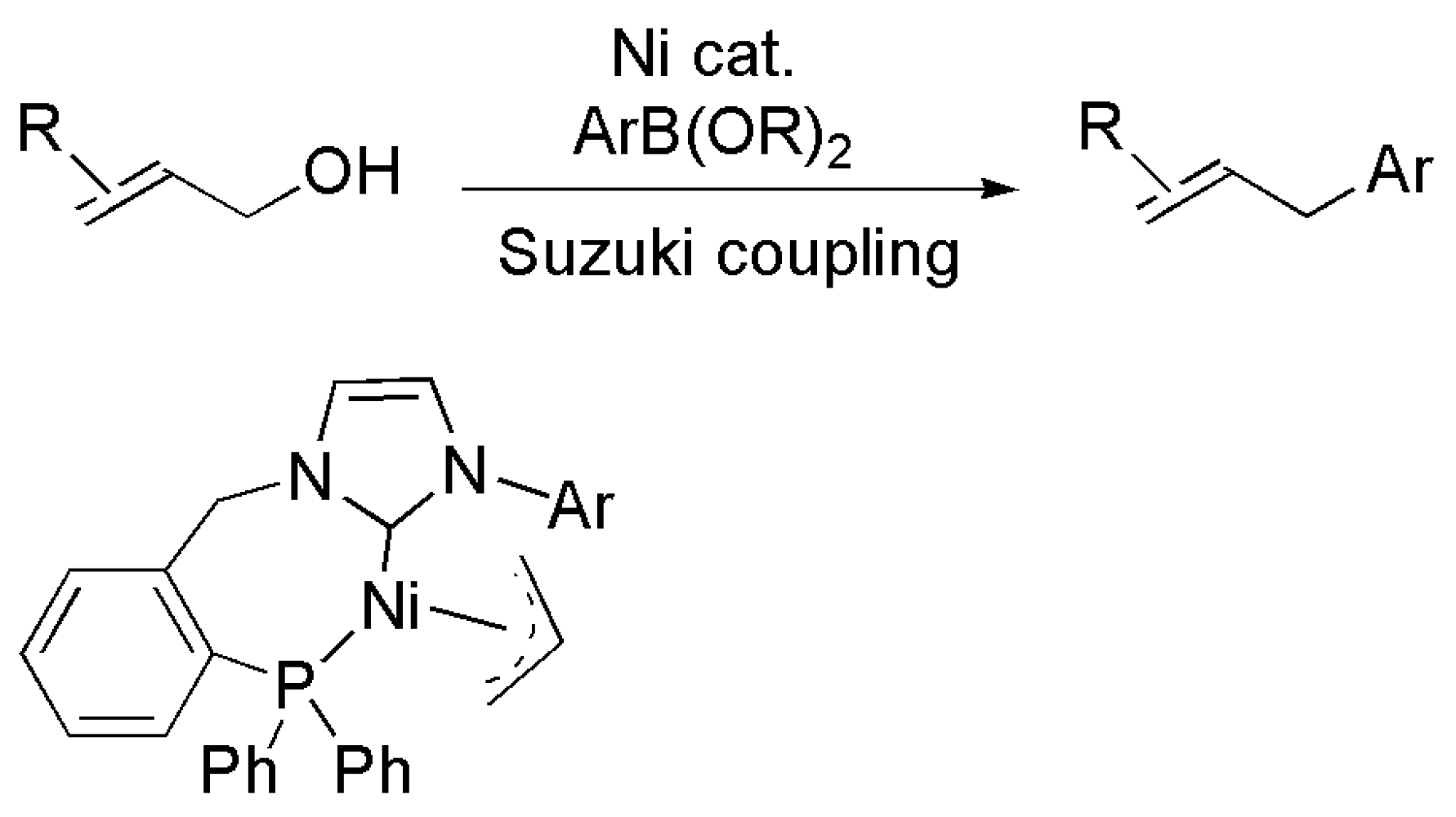
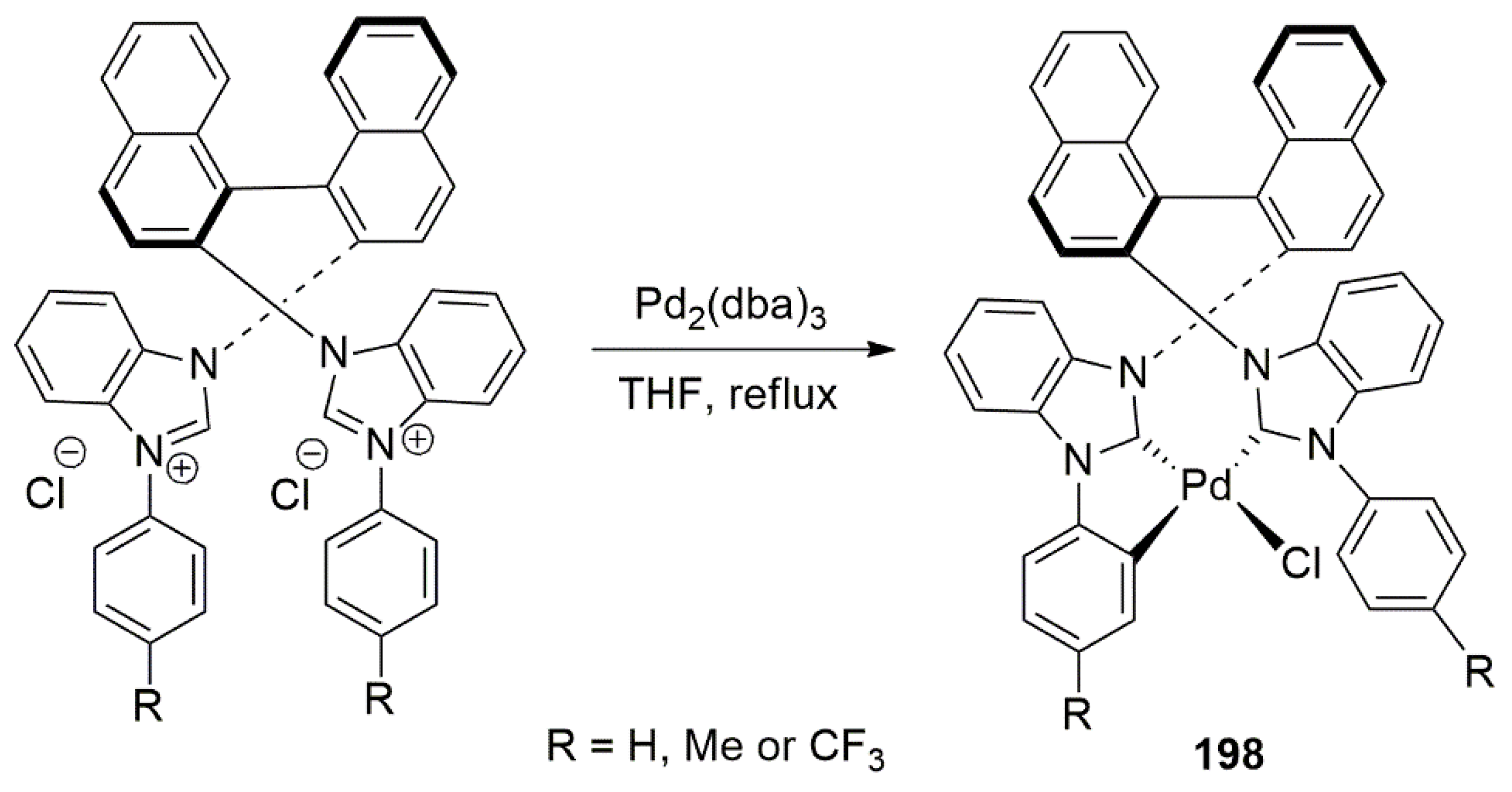

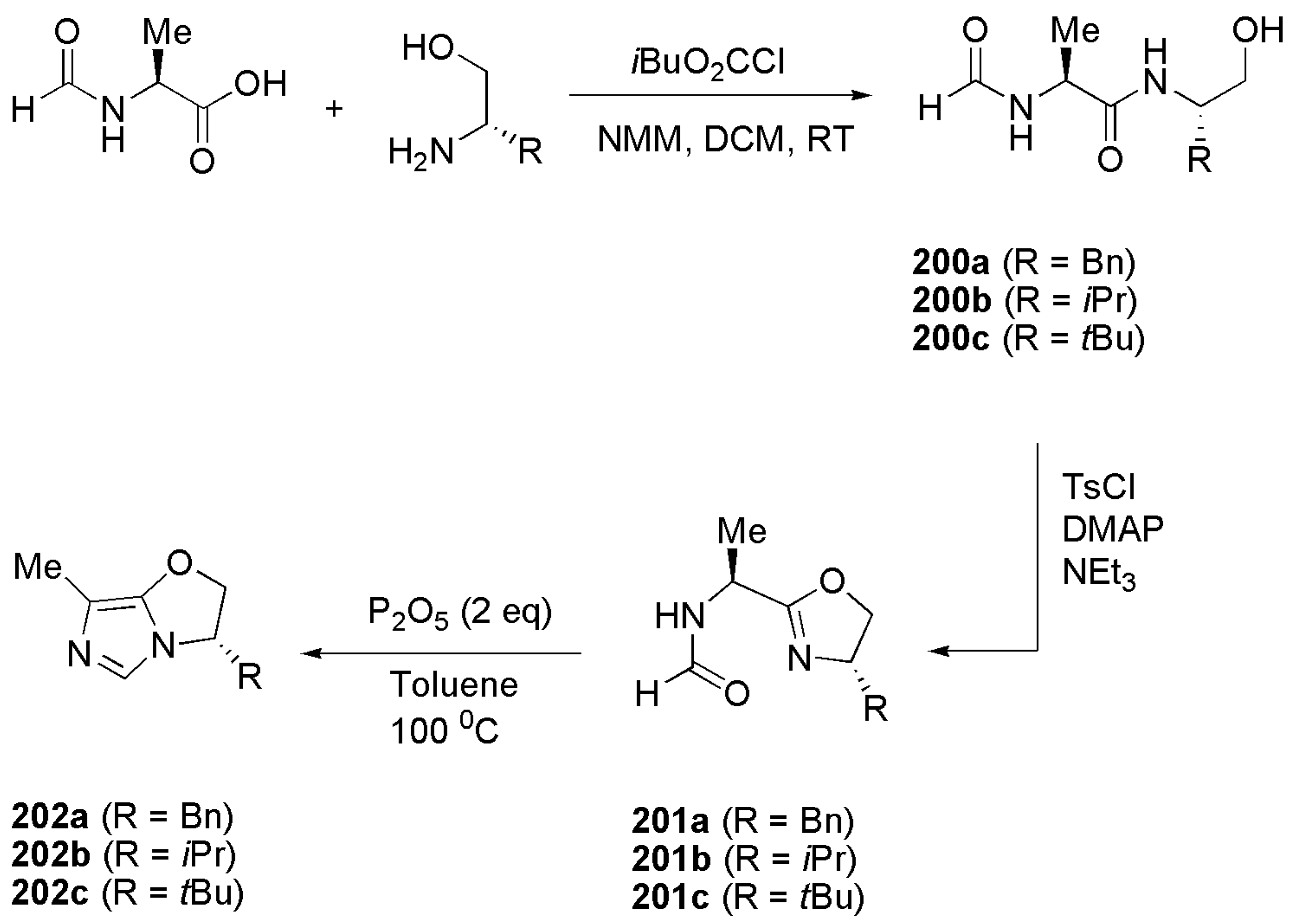
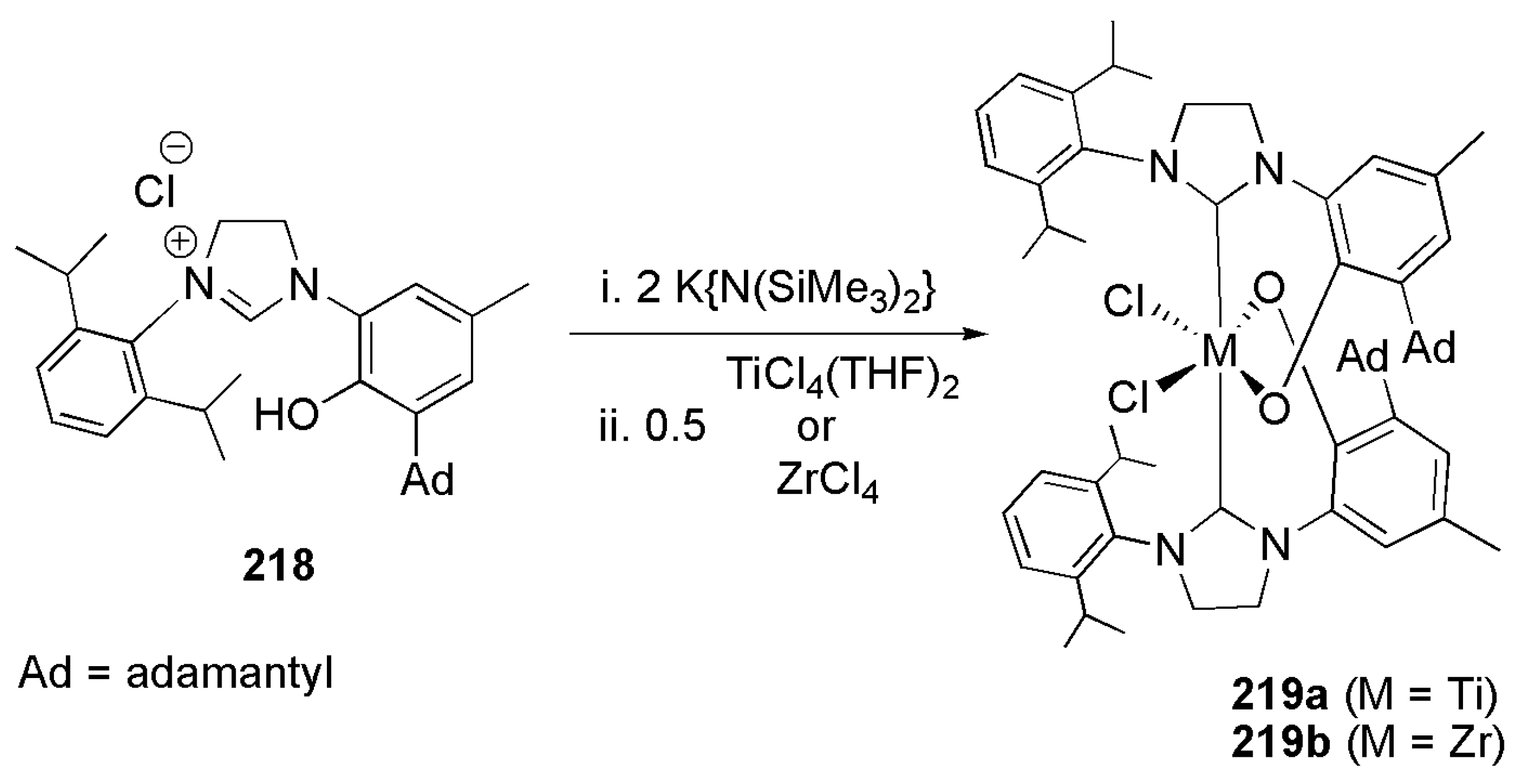
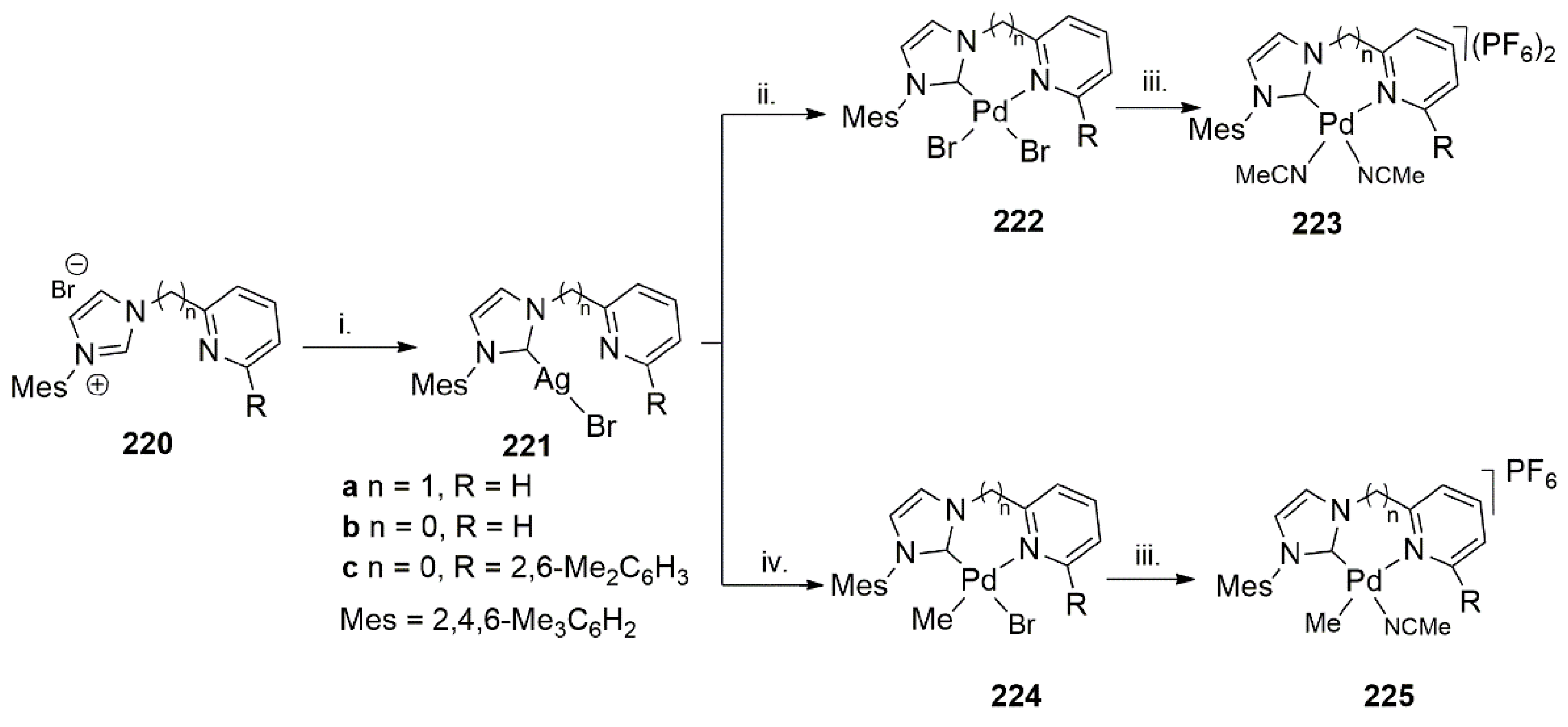
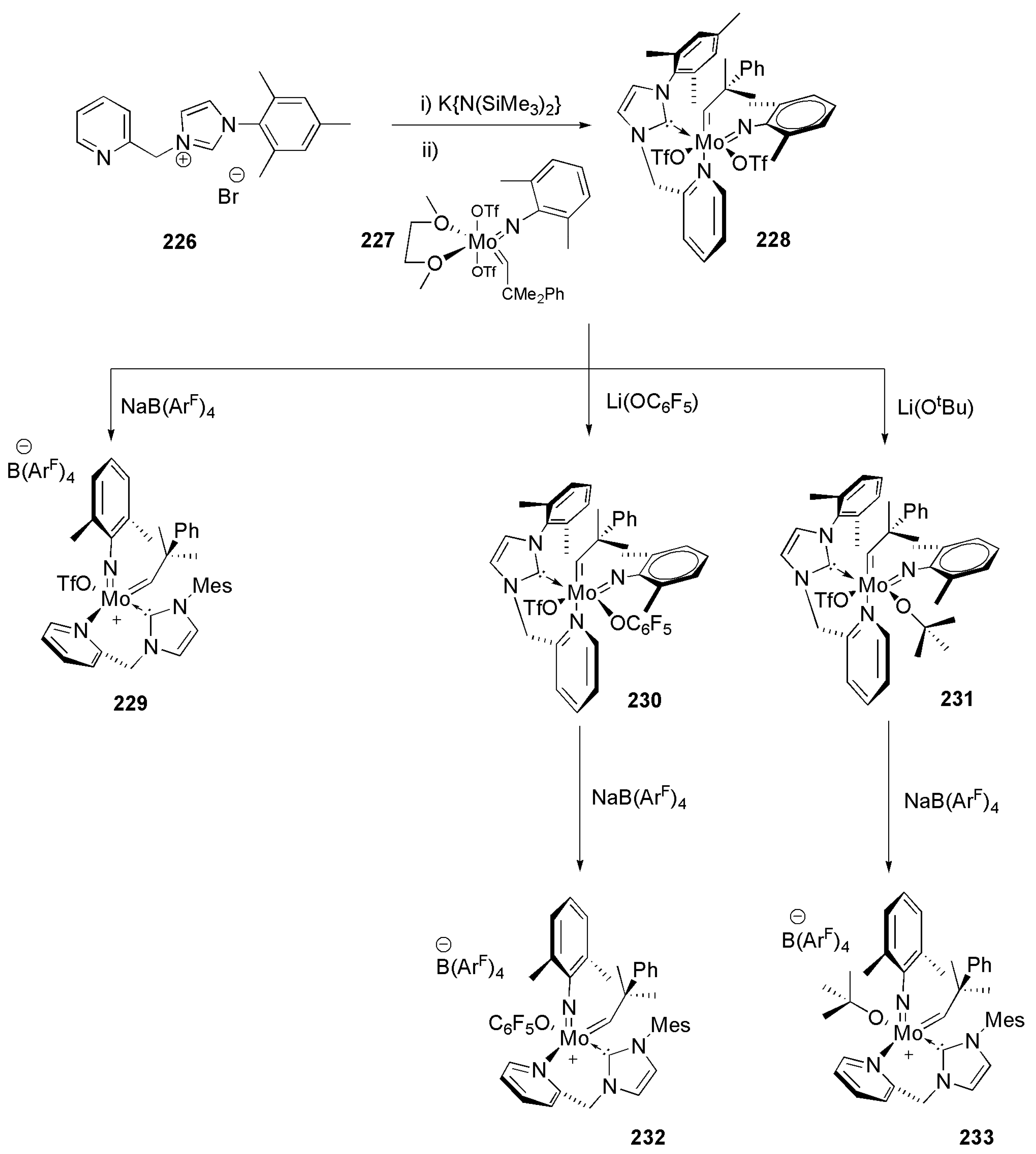
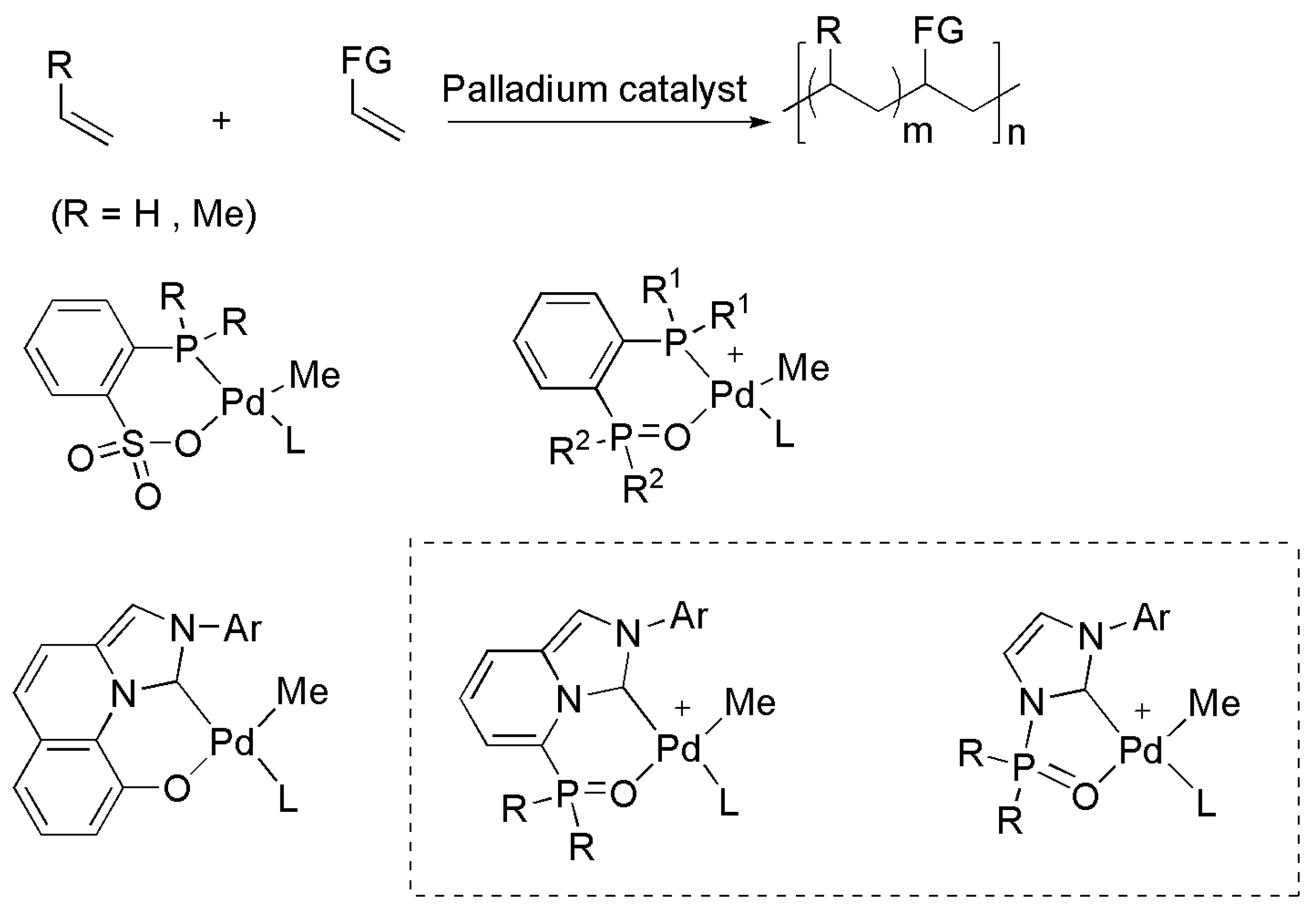
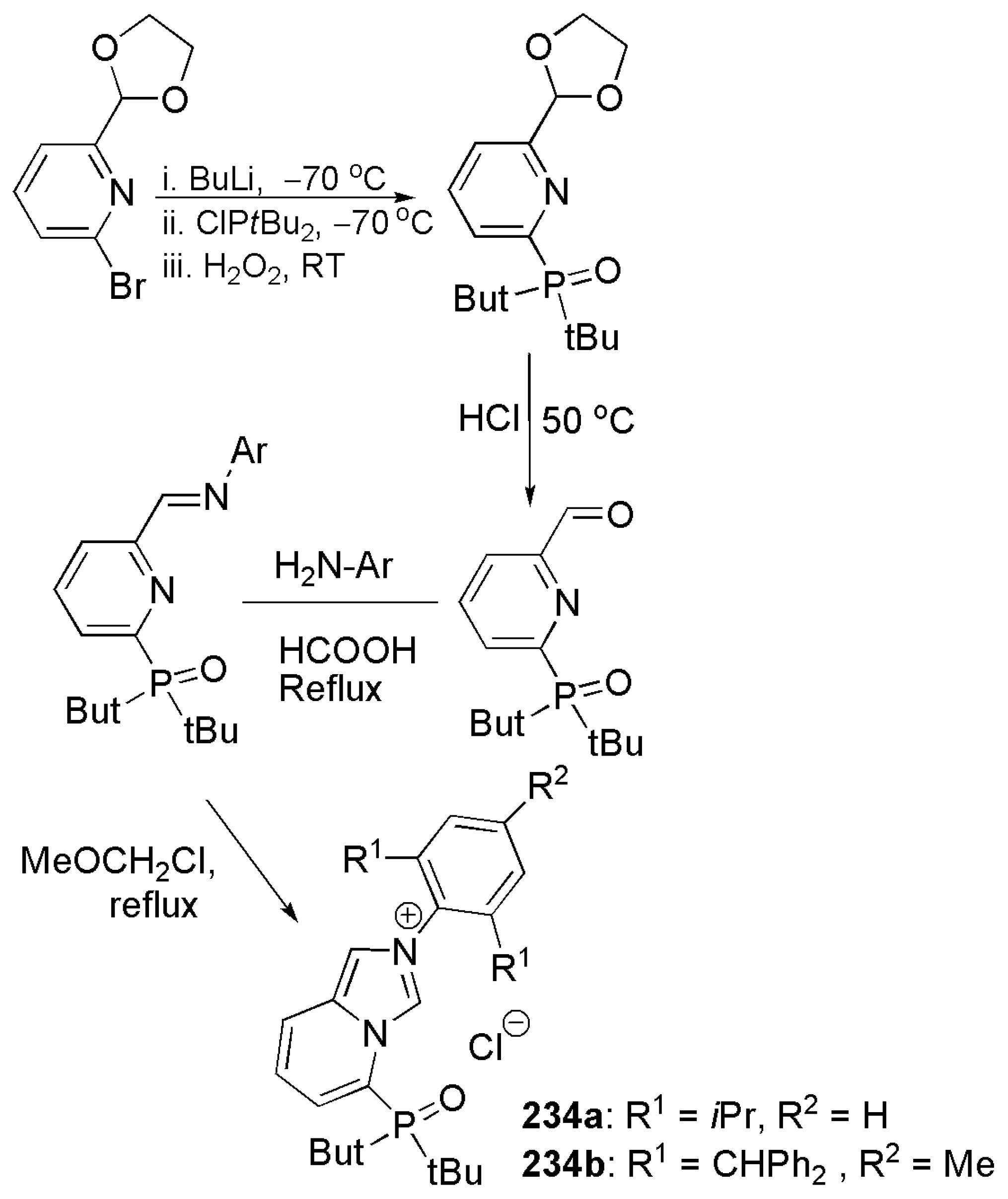
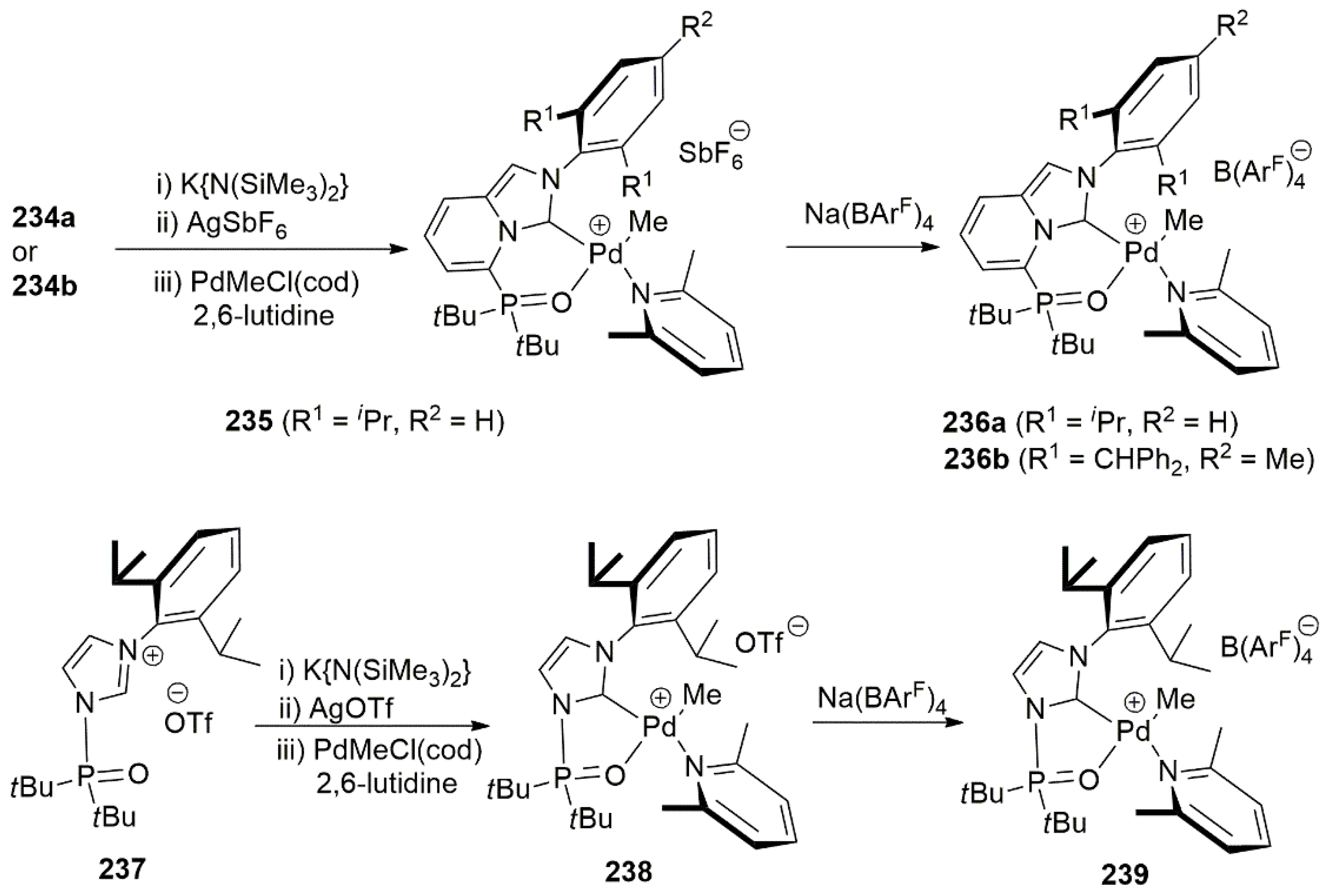
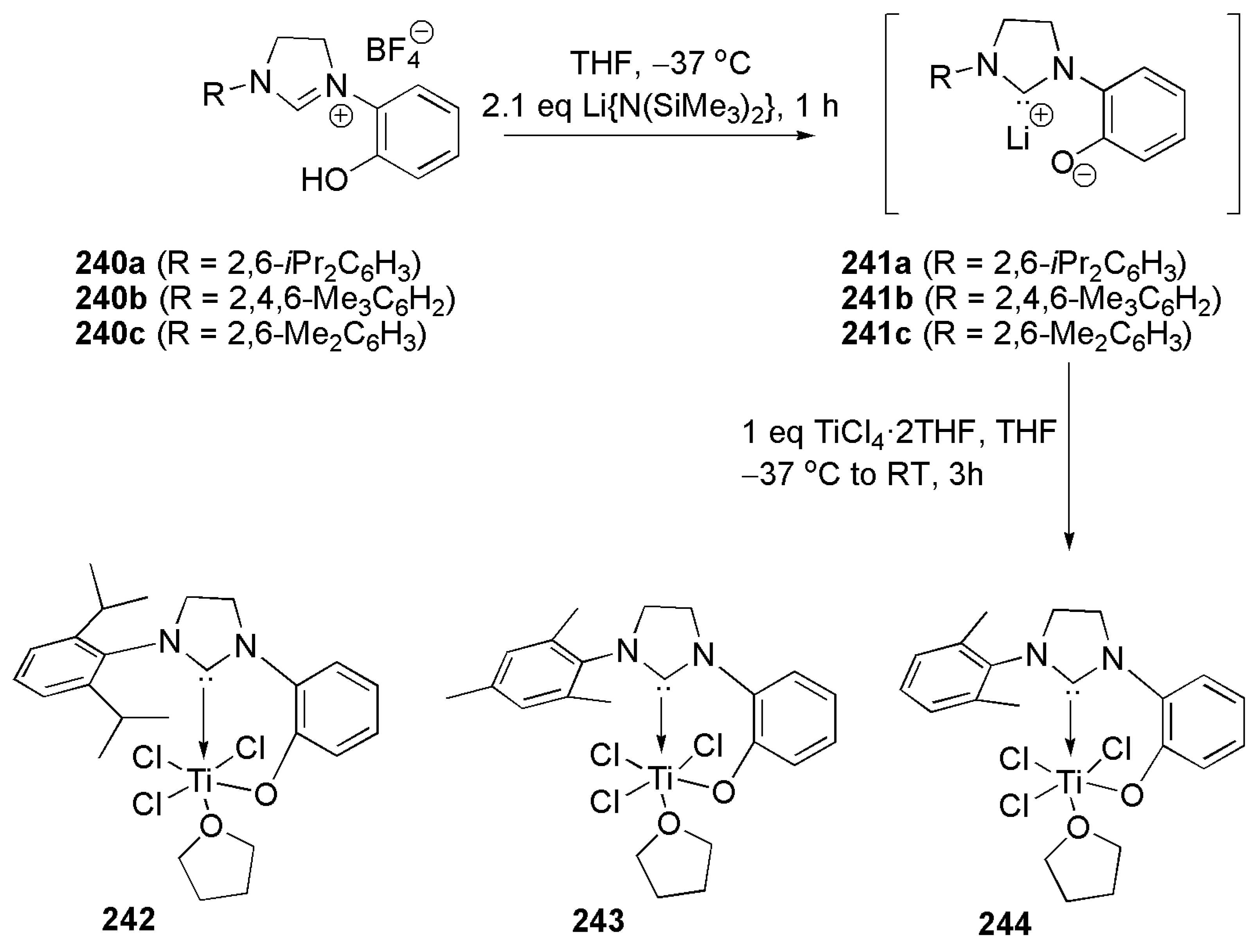


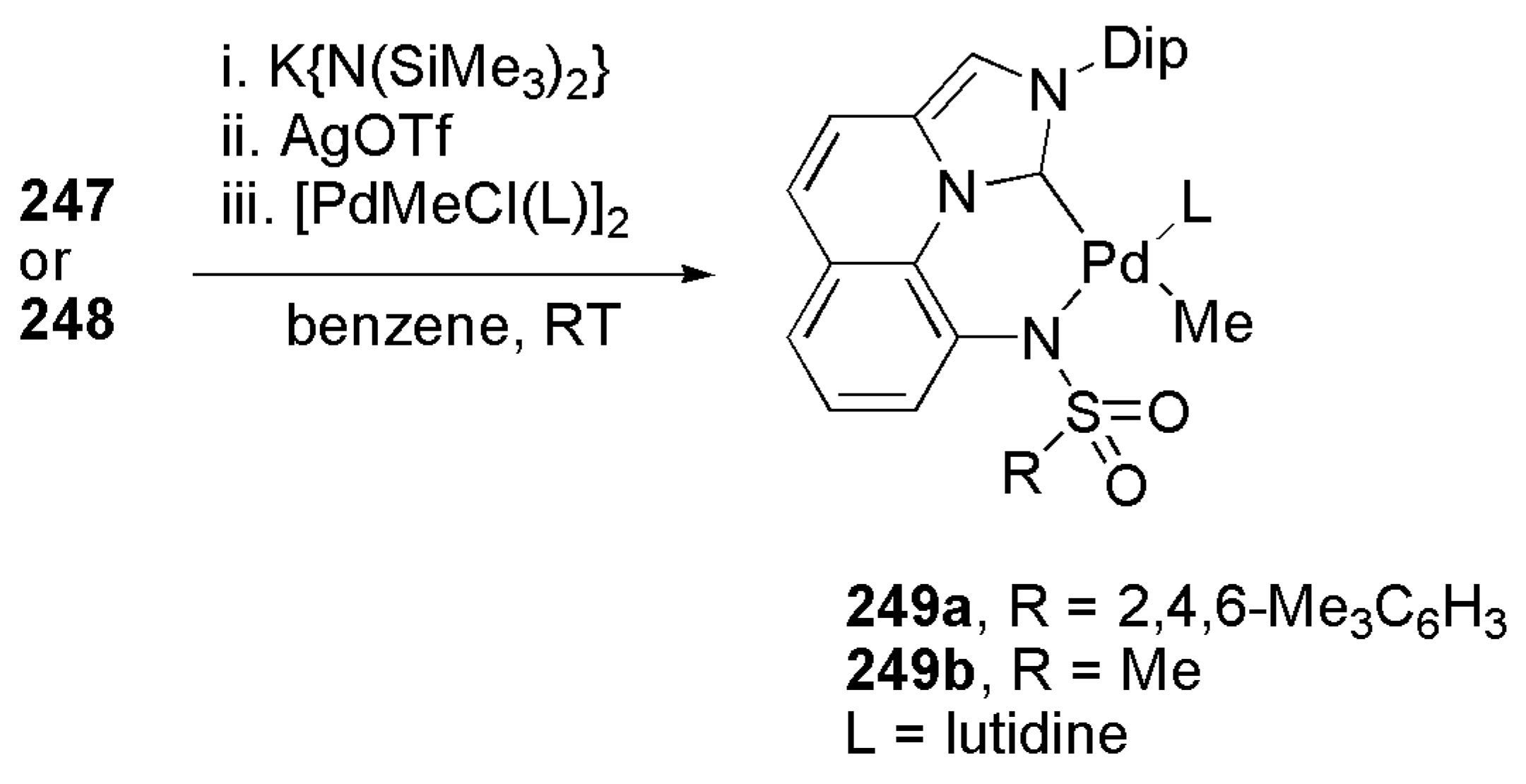
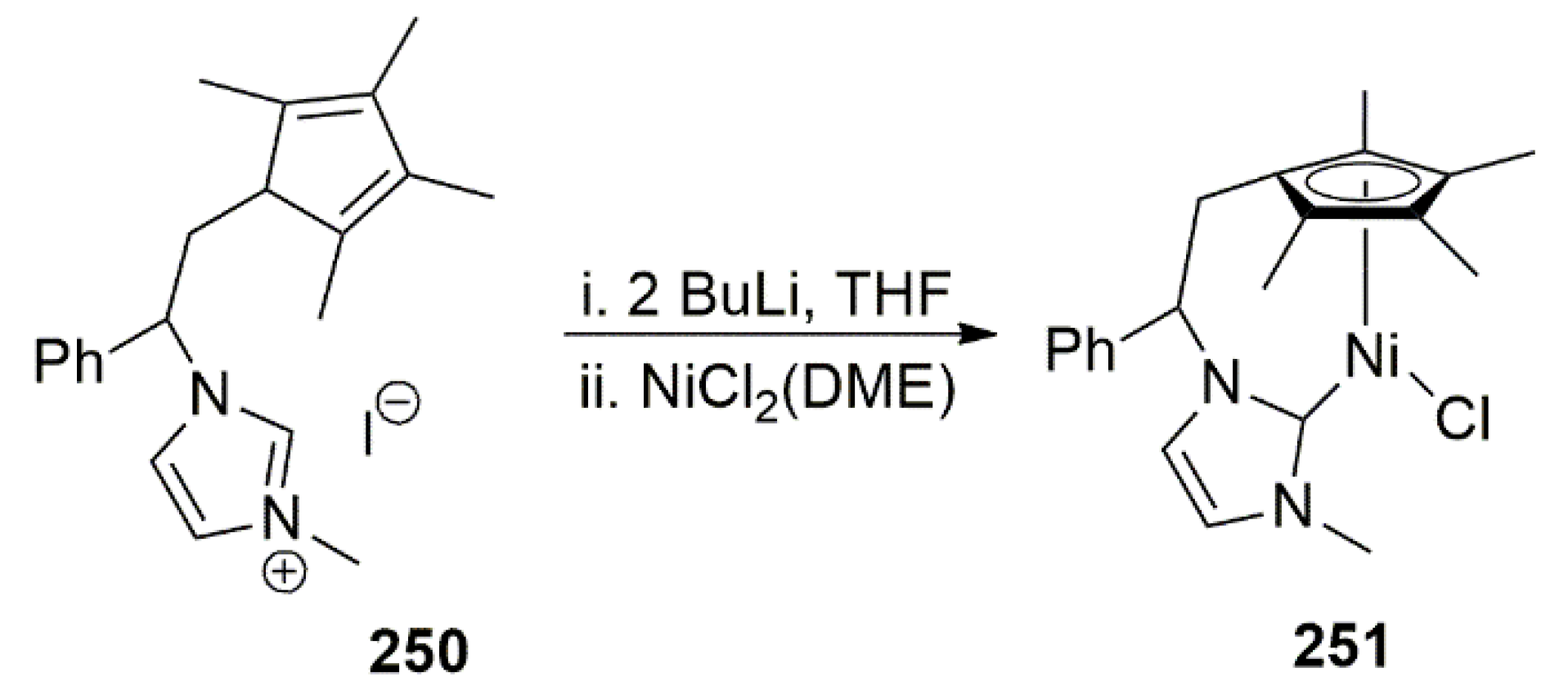
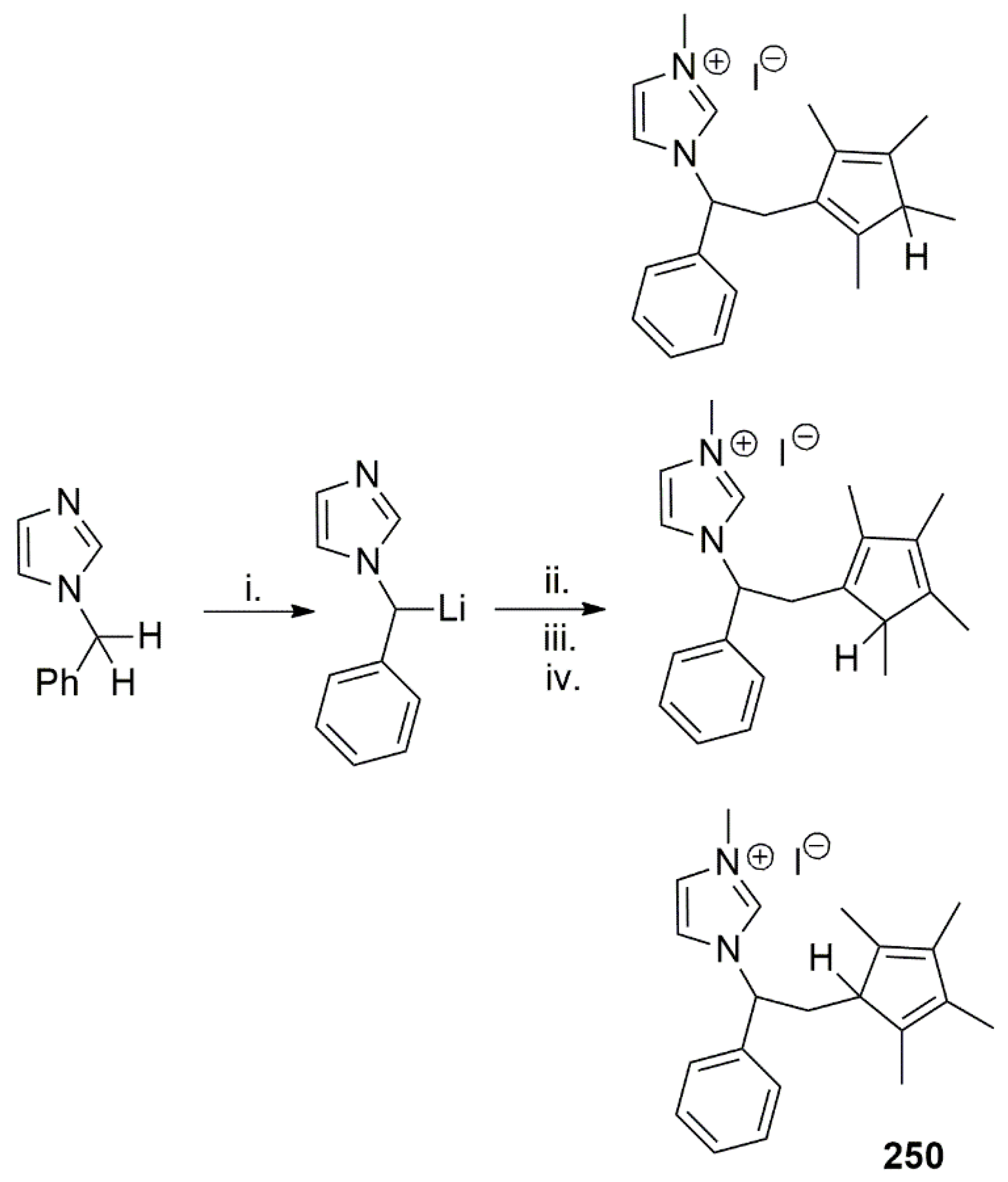



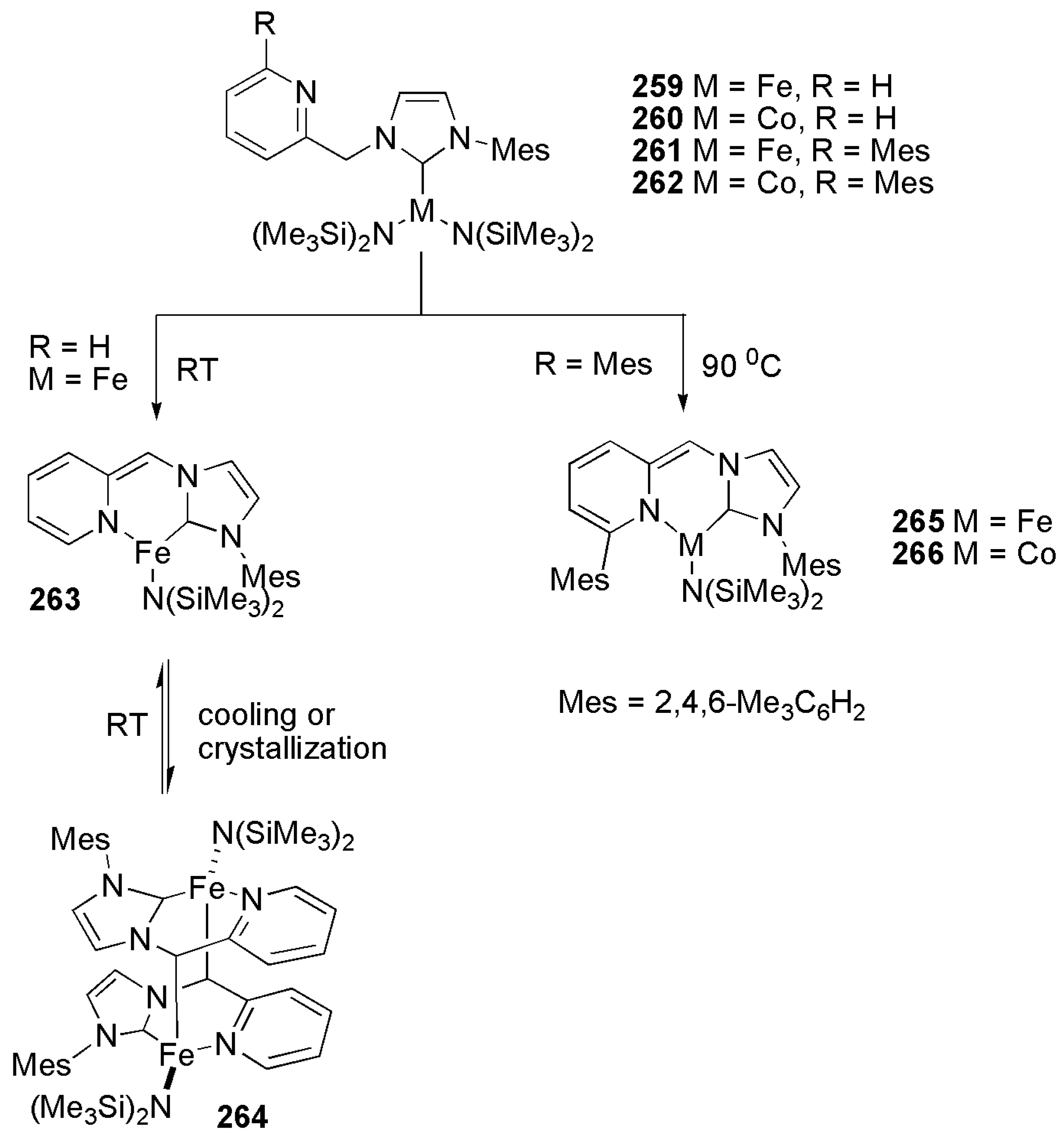
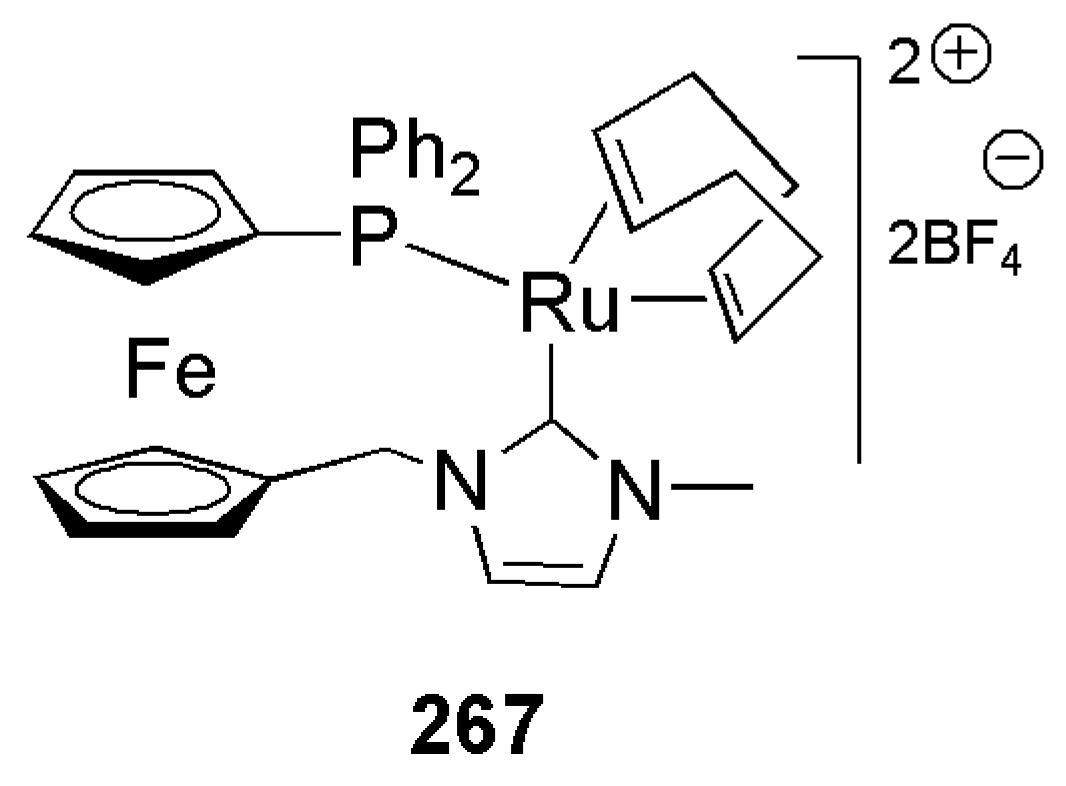




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neshat, A.; Mastrorilli, P.; Mousavizadeh Mobarakeh, A. Recent Advances in Catalysis Involving Bidentate N-Heterocyclic Carbene Ligands. Molecules 2022, 27, 95. https://doi.org/10.3390/molecules27010095
Neshat A, Mastrorilli P, Mousavizadeh Mobarakeh A. Recent Advances in Catalysis Involving Bidentate N-Heterocyclic Carbene Ligands. Molecules. 2022; 27(1):95. https://doi.org/10.3390/molecules27010095
Chicago/Turabian StyleNeshat, Abdollah, Piero Mastrorilli, and Ali Mousavizadeh Mobarakeh. 2022. "Recent Advances in Catalysis Involving Bidentate N-Heterocyclic Carbene Ligands" Molecules 27, no. 1: 95. https://doi.org/10.3390/molecules27010095
APA StyleNeshat, A., Mastrorilli, P., & Mousavizadeh Mobarakeh, A. (2022). Recent Advances in Catalysis Involving Bidentate N-Heterocyclic Carbene Ligands. Molecules, 27(1), 95. https://doi.org/10.3390/molecules27010095