
Virtual Screening and Hit Selection of Natural Compounds as Acetylcholinesterase Inhibitors

 , , ,
, , ,  ,
, 

Abstract
:1. Introduction
2. Results
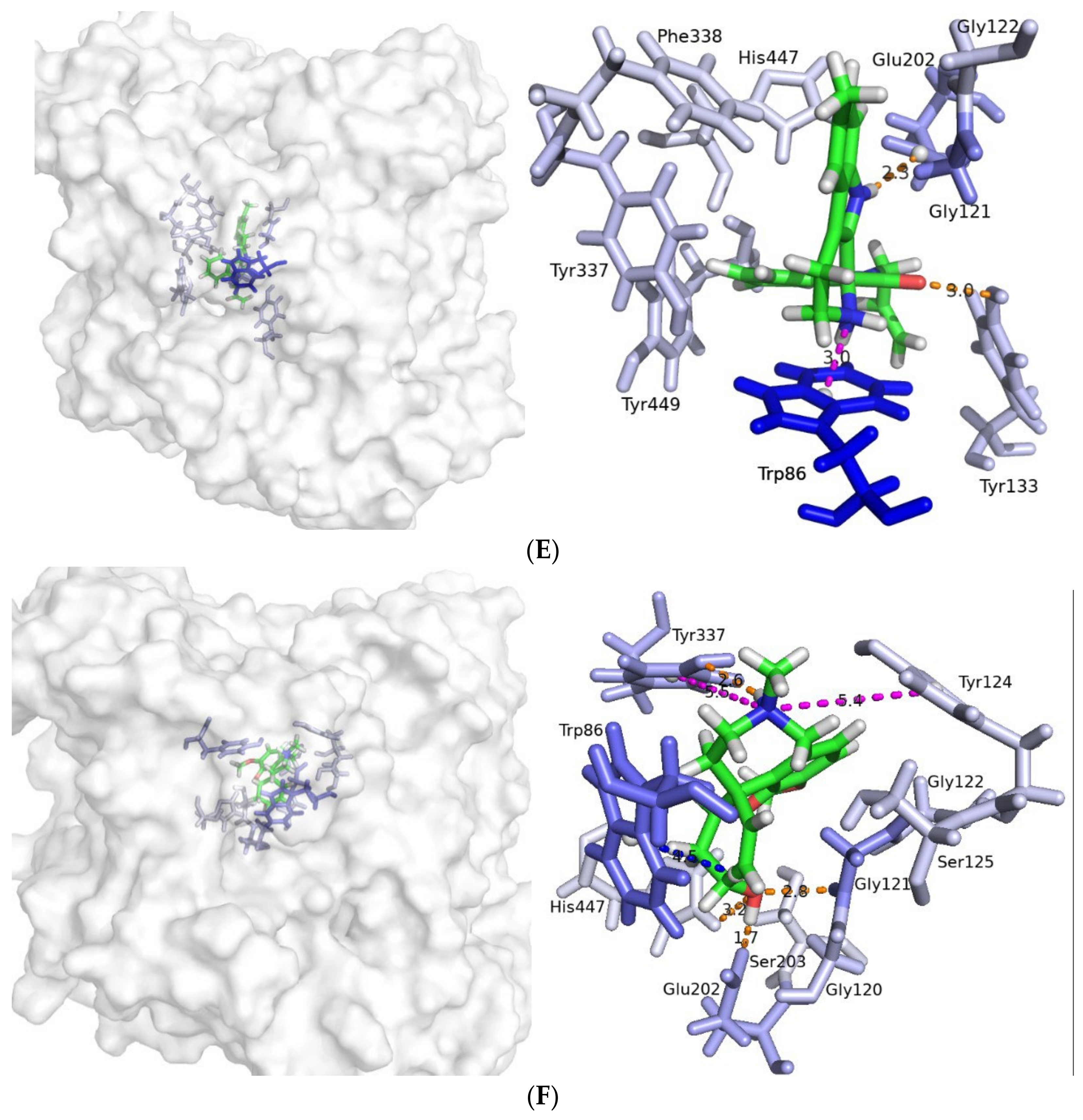
2.1. Virtual Screening by Molecular Docking
2.2. ADME Filtering and Visual Inspection
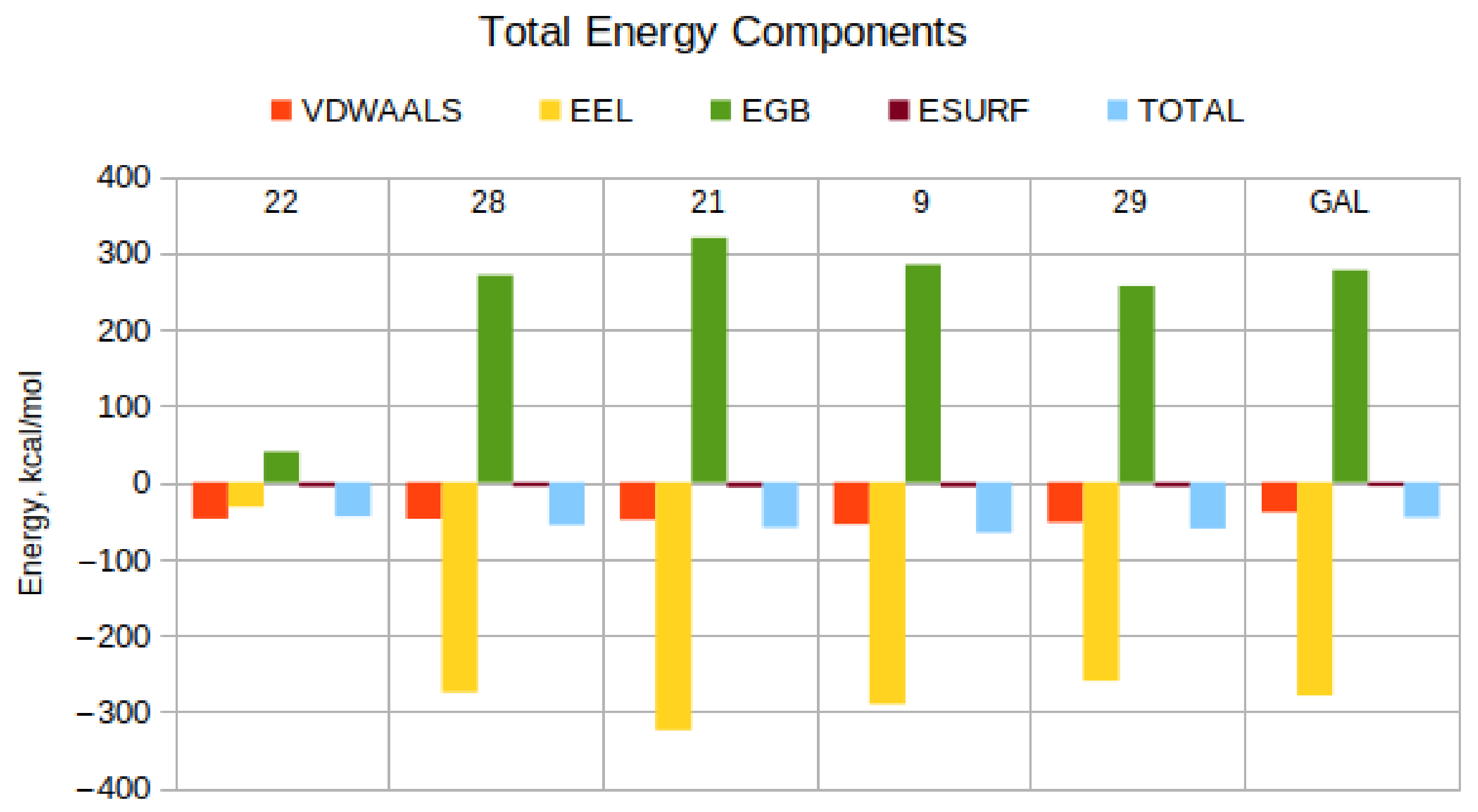
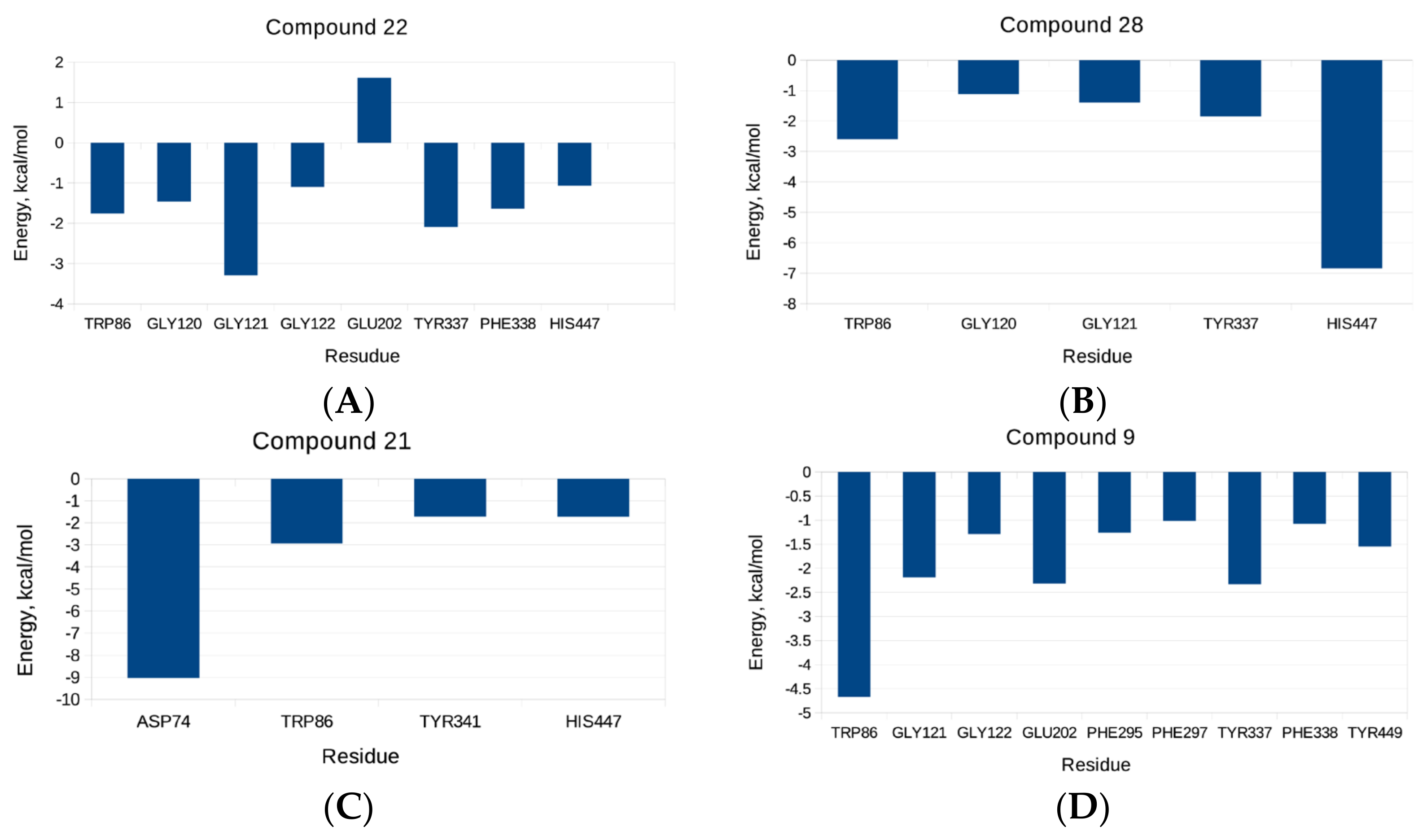
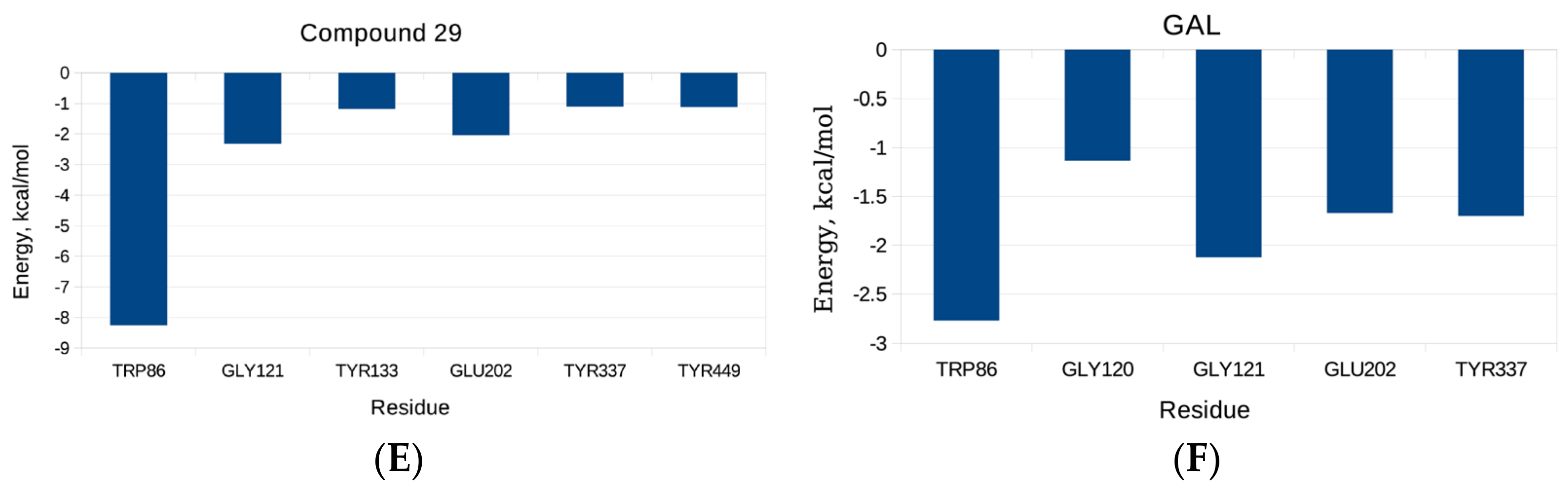
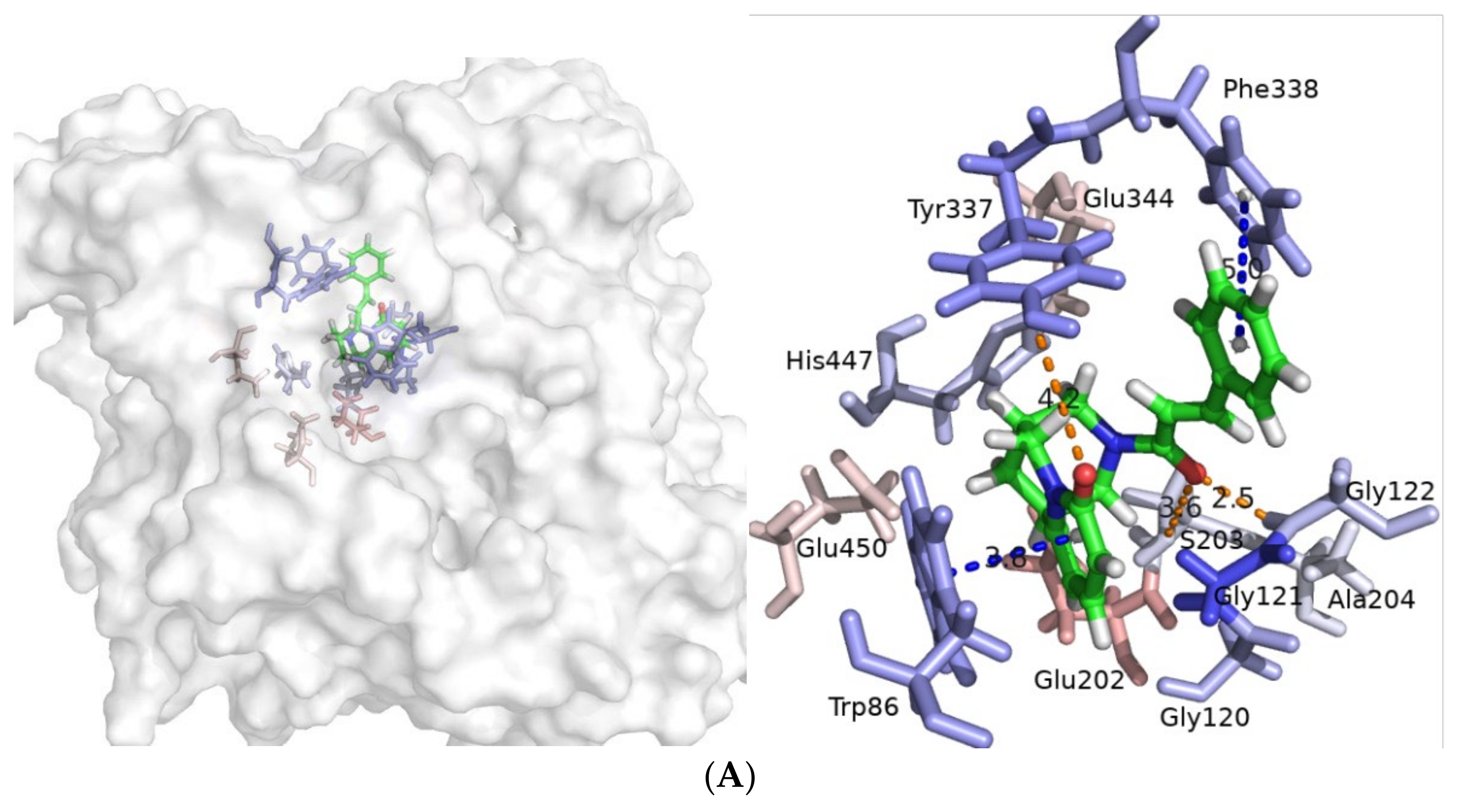
2.3. Molecular Dynamics Simulatioins and Trajectory Analyses
2.4. AChE Inhibitory Activity
2.5. Antioxidant Activity. ABTS Radical Scavenging Activity
3. Discussion
4. Materials and Methods
4.1. Virtual Screening by Molecular Docking
4.2. ADME Filters
4.3. Visual Inspection
4.4. Molecular Dynamic Simulations and Trajectory Analyses
4.4.1. System Preparation
4.4.2. Molecular Dynamic Simulations
4.4.3. Trajectory Processing and MM-GBSA Calculations
4.5. AChE Inhibitory Activity
4.6. Antioxidant Activity. ABTS Radical Scavenging Activity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennet, D.A.; Craft, S. Toward Defining the Preclinical Stages of Alzheimer’s Disease: Recommendations from the National Institute on Aging- Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, S.M.; Atanasova, M.; Dimitrov, I.; Doytchinova, I.A. Cellular Polyamines Condense Hyperphosphorylated Tau, Triggering Alzheimer’s Disease. Sci. Rep. 2020, 10, 10098. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The Diagnosis of Mild Cognitive Impairment Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Focus 2013, 7, 270–279. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimers Dement. 2012, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira-Vieira, H.T.; Guimaraes, M.I.; Silva, R.F.; Ribeiro, M.F. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Cheung, J.; Rudolph, J.M.; Burshteyn, F.; Cassidy, S.M.; Gary, N.E.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary Ligand Binding to Aromatic Residues in the Active-Site Gorge of Acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [Green Version]
- Ordentlich, A.; Barak, D.; Kronman, C.; Flashner, Y.; Leitner, M.; Segall, Y.; Ariel, N.; Cohen, S.; Velan, B.; Shafferman, A. Dissection of the Human Acetylcholinesterase Active Center Determinants of Substrate Specificity. Identification of Residues Constituting the Anionic Site, the Hydrophobic Site, and the Acyl Pocket. J. Biol. Chem. 1993, 268, 17083–17095. [Google Scholar] [CrossRef]
- Ordentlich, A.; Barak, D.; Kronman, C.; Ariel, N.; Segall, Y.; Velan, B.; Shafferman, A. Functional Characteristics of the Oxyanion Hole in Human Acetylcholinesterase. J. Biol. Chem. 1998, 273, 19509–19517. [Google Scholar] [CrossRef] [Green Version]
- Radic, Z.; Pickering, N.A.; Vellom, D.C.; Camp, S.; Taylor, P. Three Distinct Domains in the Cholinesterase Molecule Confer Selectivity for Acetyl- and Butyrylcholinesterase Inhibitors. Biochemistry 1993, 32, 12074–12084. [Google Scholar] [CrossRef]
- Sussman, J.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic Structure of Acetylcholinesterase from Torpedo Californica: A Prototypic Acetylcholine-Binding Protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Kitz, R.J.; Braswell, L.M.; Ginsburg, S. On the Question: Is Acetylcholinesterase an Allosteric Protein? Mol. Pharmacol. 1970, 6, 108–121. [Google Scholar] [PubMed]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A Structural Motif of Acetylcholinesterase That Promotes Amyloid β-Peptide Fibril Formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.; Moore, S. Identification of a Structural Site on Acetylcholinesterase That Promotes Neurite Outgrowth and Binds Laminin-1 and Collagen IV. Biochem. Biophys. Res. Commun. 2004, 319, 448–455. [Google Scholar] [CrossRef]
- Johnson, G.; Moore, S. The Peripheral Anionic Site of Acetylcholinesterase: Structure, Functions and Potential Role in Rational Drug Design. Curr. Pharm. Des. 2006, 12, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Fisher, S.K.; Wonnacott, S. Chapter 13—Acetylcholine. In Basic Neurochemistry; Academic Press: Cambridge, MA, USA, 2012; pp. 258–282. [Google Scholar] [CrossRef]
- Lockridge, O.; Quinn, D.M. 4.14—Esterases. In Comprehensive Toxicology, 2nd ed.; McQueen, C.A., Ed.; Elsevier: Oxford, UK, 2010; Volume 4, pp. 243–273. ISBN 978-0-08-046884-6. [Google Scholar]
- Melnikova, I. Therapies for Alzheimer’s Disease. Nat. Rev. Drug Discov. 2007, 6, 341–342. [Google Scholar] [CrossRef]
- Knapp, M.J.; Knopman, D.S.; Solomon, P.R.; Pendlebury, W.W.; Davis, C.S.; Gracon, S.I. A 30-Week Randomized Controlled Trial of High-Dose Tacrine in Patients with Alzheimer’s Disease. JAMA 1994, 271, 985–991. [Google Scholar] [CrossRef]
- Watkins, P.B. Hepatotoxic Effects of Tacrine Administration in Patients with Alzheimer’s Disease. JAMA 1994, 271, 992. [Google Scholar] [CrossRef]
- Blackard, W.G.; Sood, G.K.; Crowe, D.R.; Fallon, M.B. Tacrine: A Cause of Fatal Hepatotoxicity? J. Clin. Gastroenterol. 1998, 26, 57–59. [Google Scholar] [CrossRef]
- McGleenon, B.M.; Dynan, K.B.; Passmore, A. Acetylcholinesterase Inhibitors in Alzheimer’s Disease. Br. J. Clin. Pharmacol. 1999, 48, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Batiha, G.E.-S.; Alkazmi, L.M.; Nadwa, E.H.; Rashwan, E.K.; Beshbishy, A.M.; Shaheen, H.; Wasef, L. Physostigmine: A Plant Alkaloid Isolated from Physostigma Venenosum: A Review on Pharmacokinetics, Pharmacological and Toxicological Activities. J. Drug Deliv. Ther. 2020, 10, 187–190. [Google Scholar] [CrossRef] [Green Version]
- Thompson, C.A. FDA Approves Galantamine for Alzheimer’s Disease. Am. J. Health-Syst. Pharm. 2001, 58, 649. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, N.; Chau, S.A.; Kircanski, I.; Lanctôt, K.L. Current and Emerging Drug Treatment Options for Alzheimer’s Disease. Drugs 2011, 71, 2031–2065. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, A.; Piplani, P. Current Pharmacotherapy and Putative Disease-Modifying Therapy for Alzheimer’s Disease. Neurol. Sci. 2016, 37, 1403–1435. [Google Scholar] [CrossRef] [PubMed]
- Loveman, E.; Green, C.; Kirby, J.; Takeda, A.; Picot, J.; Payne, E.; Clegg, A. The Clinical and Cost-Effectiveness of Donepezil, Rivastigmine, Galantamine and Memantine for Alzheimer’s Disease. Health Technol. Assess. 2006, 10, iii–iv. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, A. Natural Products in Drug Discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Kumar, V.; Mal, M.; Houghton, P.J. Acetylcholinesterase Inhibitors from Plants. Phytomedicine 2007, 14, 289–300. [Google Scholar] [CrossRef]
- Williams, P.; Sorribas, A.; Howes, M.-J.R. Natural Products as a Source of Alzheimer’s Drug Leads. Nat. Prod. Rep. 2011, 28, 48–77. [Google Scholar] [CrossRef]
- Houghton, P.J.; Ren, Y.; Howes, M.J. Acetylcholinesterase Inhibitors from Plants and Fungi. Nat. Prod. Rep. 2006, 23, 181–199. [Google Scholar] [CrossRef]
- Murray, A.; Faraoni, M.; Castro, M.; Alza, N.; Cavallaro, V. Natural AChE Inhibitors from Plants and Their Contribution to Alzheimer’s Disease Therapy. Curr. Neuropharmacol. 2013, 11, 388–413. [Google Scholar] [CrossRef] [Green Version]
- Orhan, G.; Orhan, I.; Öztekin-Subutay, N.; Ak, F.; Şener, B. Contemporary Anticholinesterase Pharmaceuticals of Natural Origin and Their Synthetic Analogues for the Treatment of Alzheimer’s Disease. Recent Pat. CNS Drug Discov. 2009, 4, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.Y.; Choi, H. Natural Products from Marine Organisms with Neuroprotective Activity in the Experimental Models of Alzheimer’s Disease, Parkinson’s Disease and Ischemic Brain Stroke: Their Molecular Targets and Action Mechanisms. Arch. Pharmacal Res. 2015, 38, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Scotti, L.; Scotti, M.T. In Silico Studies Applied to Natural Products with Potential Activity against Alzheimer’s Disease. In Computational Modeling of Drugs against Alzheimer’s Disease; Roy, K., Ed.; Humana Press: New York, NY, USA, 2018; Volume 132, pp. 513–531. [Google Scholar]
- Huang, L.; Su, T.; Li, X. Natural Products as Sources of New Lead Compounds for the Treatment of Alzheimer’s Disease. Curr. Top. Med. Chem. 2013, 13, 1864–1878. [Google Scholar] [CrossRef] [PubMed]
- Naaz, H.; Singh, S.; Pandey, V.P.; Singh, P.; Dwivedi, U.N. Anti-Cholinergic Alkaloids as Potential Therapeutic Agents for Alzheimer’s Disease: An in Silico Approach. Indian J. Biochem. Biophys. 2013, 50, 120–125. [Google Scholar]
- Awasthi, M.; Upadhyay, A.K.; Singh, S.; Pandey, V.P.; Dwivedi, U.N. Terpenoids as Promising Therapeutic Molecules against Alzheimer’s Disease: Amyloid Beta- and Acetylcholinesterase-Directed Pharmacokinetic and Molecular Docking Analyses. Mol. Simul. 2018, 44, 1–11. [Google Scholar] [CrossRef]
- Ma, D.-L.; Chan, D.S.-H.; Leung, C.-H. Molecular Docking for Virtual Screening of Natural Product Databases. Chem. Sci. 2011, 2, 1656–1665. [Google Scholar] [CrossRef]
- Ambure, P.; Kar, S.; Roy, K. Pharmacophore Mapping-Based Virtual Screening Followed by Molecular Docking Studies in Search of Potential Acetylcholinesterase Inhibitors as Anti-Alzheimer’s Agents. BioSystems 2014, 116, 10–20. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, S.M.; Dimitrov, I.; Doytchinova, I.A. Bridging Solvent Molecules Mediate RNase A—Ligand Binding. PLoS ONE 2019, 14, e0224271. [Google Scholar] [CrossRef]
- Milatovic, D.; Gupta, R.C.; Aschner, M. Anticholinesterase Toxicity and Oxidative Stress. Sci. World J. 2006, 6, 295–310. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Moreno, D.; Soler, F.; Míguez, M.P.; Pérez-López, M. Brain Acetylcholinesterase, Malondialdehyde and Reduced Glutathione as Biomarkers of Continuous Exposure of Tench, Tinca Tinca, to Carbofuran or Deltamethrin. Sci. Total Environ. 2010, 408, 4976–4983. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, M.; Yordanov, N.; Dimitrov, I.; Berkov, S.; Doytchinova, I. Molecular Docking Study on Galantamine Derivatives as Cholinesterase Inhibitors. Mol. Inform. 2015, 34, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, M.; Stavrakov, G.; Philipova, I.; Zheleva, D.; Yordanov, N.; Doytchinova, I. Galantamine Derivatives with Indole Moiety: Docking, Design, Synthesis and Acetylcholinesterase Inhibitory Activity. Bioorg. Med. Chem. 2015, 23, 5382–5389. [Google Scholar] [CrossRef] [PubMed]
- Stavrakov, G.; Philipova, I.; Zheleva, D.; Atanasova, M.; Konstantinov, S.; Doytchinova, I. Docking-Based Design of Galantamine Derivatives with Dual-Site Binding to Acetylcholinesterase. Mol. Inform. 2016, 35, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.; Atanasova, M.; Stavrakov, G.; Philipova, I.; Zheleva-Dimitrova, D. Galantamine Derivatives as Acetylcholinesterase Inhibitors: Docking, Design, Synthesis, and Inhibitory Activity. In Computational Modeling of Drugs Against Alzheimer’s Disease. Neuromethods; Roy, K., Ed.; Humana Press: New York, NY, USA, 2018; Volume 132, pp. 163–176. [Google Scholar]
- Doytchinova, I.; Atanasova, M.; Valkova, I.; Stavrakov, G.; Philipova, I.; Zhivkova, Z.; Zheleva-Dimitrova, D.; Konstantinov, S.; Dimitrov, I. Novel Hits for Acetylcholinesterase Inhibition Derived by Docking-Based Screening on ZINC Database. J. Enzym. Inhib. Med. Chem. 2018, 33, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Stavrakov, G.; Philipova, I.; Lukarski, A.; Atanasova, M.; Zheleva, D.; Zhivkova, Z.D.; Ivanov, S.; Atanasova, T.; Konstantinov, S.; Doytchinova, I. Galantamine-Curcumin Hybrids as Dual-Site Binding Acetylcholinesterase Inhibitors. Molecules 2020, 25, 3341. [Google Scholar] [CrossRef]
- Stavrakov, G.; Philipova, I.; Lukarski, A.; Atanasova, M.; Georgiev, B.; Atanasova, T.; Konstantinov, S.; Doytchinova, I. Discovery of a Novel Acetylcholinesterase Inhibitor by Fragment-Based Design and Virtual Screening. Molecules 2021, 26, 2058. [Google Scholar] [CrossRef]
- Ivanov, S.M.; Huber, R.G.; Warwicker, J.; Bond, P.J. Energetics and Dynamics Across the Bcl-2-Regulated Apoptotic Pathway Reveal Distinct Evolutionary Determinants of Specificity and Affinity. Structure 2016, 24, 2024–2033. [Google Scholar] [CrossRef] [Green Version]
- Shamim, A.; Abbasi, S.W.; Azam, S.S. Structural and Dynamical Aspects of Streptococcus Gordonii FabH through Molecular Docking and MD Simulations. J. Mol. Graph. Model. 2015, 60, 180–196. [Google Scholar] [CrossRef]
- Abbasi, S.; Raza, S.; Azam, S.S.; Liedl, K.R.; Fuchs, J.E. Interaction Mechanisms of a Melatonergic Inhibitor in the Melatonin Synthesis Pathway. J. Mol. Liq. 2016, 221, 507–517. [Google Scholar] [CrossRef]
- Azam, S.S.; Abro, A.; Raza, S. Binding Pattern Analysis and Structural Insight into the Inhibition Mechanism of Sterol 24-C Methyltransferase by Docking and Molecular Dynamics Approach. J. Biomol. Struct. Dyn. 2015, 33, 2563–2577. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Azam, S.S. AFD: An Application for Bi-Molecular Interaction Using Axial Frequency Distribution. J. Mol. Modeling 2018, 24, 84. [Google Scholar] [CrossRef] [PubMed]
- Tran-Nguyen, V.-K.; Jacquemard, C.; Rognan, D. LIT-PCBA: An Unbiased Data Set for Machine Learning and Virtual Screening. J. Chem. Inf. Modeling 2020, 60, 4263–4273. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Rozengart, E.; Basova, N.E. Ammonium Compounds with Localized and Delocalized Charge as Reversible Inhibitors of Cholinesterases of Different Origin. J. Evol. Biochem. Physiol. 2001, 37, 604–610. [Google Scholar] [CrossRef]
- Walker, N.; Howe, C.; Glover, M.; McRobbie, H.; Barnes, J.; Nosa, V.; Parag, V.; Bassett, B.; Bullen, C. Cytisine versus Nicotine for Smoking Cessation. N. Engl. J. Med. 2014, 371, 2353–2362. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.; Naz, Q.; Kartal, M.; Tosun, F.; Sener, B.; Choudhary, M.I. In Vitro Anticholinesterase Activity of Various Alkaloids. Z. Nat. C 2007, 62, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Schmeller, T.; Sauerwein, M.; Sporer, F.; Wink, M.; Müller, W.E. Binding of Quinolizidine Alkaloids to Nicotinic and Muscarinic Acetylcholine Receptors. J. Nat. Prod. 1994, 57, 1316–1319. [Google Scholar] [CrossRef]
- Tang, X.C.; He, X.C.; Bai, D.L. Huperzine A: A Novel Acetylcholinesterase Inhibitor. Drugs Future 1999, 24, 647. [Google Scholar] [CrossRef]
- Wang, R.; Yan, H.; Tang, X. Progress in Studies of Huperzine A, a Natural Cholinesterase Inhibitor from Chinese Herbal Medicine1. Acta Pharmacol. Sin. 2006, 27, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, H.; Wei, Z.; Song, Y.; Zhang, L.; Chen, H. Efficacy and Safety of Natural Acetylcholinesterase Inhibitor Huperzine A in the Treatment of Alzheimer’s Disease: An Updated Meta-Analysis. J. Neural Transm. 2009, 116, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Coleman, B.R.; Ratcliffe, R.H.; Oguntayo, S.A.; Shi, X.; Doctor, B.P.; Gordon, R.K.; Nambiar, M.P. [+]-Huperzine A Treatment Protects against N-Methyl-d-Aspartate-Induced Seizure/Status Epilepticus in Rats. Chem. Biol. Interact. 2008, 175, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anouhe, J.-B.S.; Adima, A.A.; Niamké, F.B.; Stien, D.; Amian, B.K.; Blandinières, P.-A.; Virieux, D.; Pirat, J.-L.; Kati-Coulibaly, S.; Amusant, N. Dicorynamine and Harmalan-N-Oxide, Two New β-Carboline Alkaloids from Dicorynia Guianensis Amsh Heartwood. Phytochem. Lett. 2015, 12, 158–163. [Google Scholar] [CrossRef]
- Koenig, X.; Hilber, K. The Anti-Addiction Drug Ibogaine and the Heart: A Delicate Relation. Molecules 2015, 20, 2208–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, H.R.; Targowska-Duda, K.M.; Feuerbach, D.; Jozwiak, K. Coronaridine Congeners Inhibit Human A3β4 Nicotinic Acetylcholine Receptors by Interacting with Luminal and Non-Luminal Sites. Int. J. Biochem. Cell Biol. 2015, 65, 81–90. [Google Scholar] [CrossRef]
- Arias, H.R.; Lykhmus, O.; Uspenska, K.; Skok, M. Coronaridine Congeners Modulate Mitochondrial A3β4* Nicotinic Acetylcholine Receptors with Different Potency and through Distinct Intra-Mitochondrial Pathways. Neurochem. Int. 2018, 114, 26–32. [Google Scholar] [CrossRef]
- Arias, H.R.; Tae, H.-S.; Micheli, L.; Yousuf, A.; Ghelardini, C.; Adams, D.J.; di Cesare Mannelli, L. Coronaridine Congeners Decrease Neuropathic Pain in Mice and Inhibit A9α10 Nicotinic Acetylcholine Receptors and CaV2.2 Channels. Neuropharmacology 2020, 175, 108194. [Google Scholar] [CrossRef]
- Arias, H.R.; do Rego, J.L.; do Rego, J.C.; Chen, Z.; Anouar, Y.; Scholze, P.; Gonzales, E.B.; Huang, R.; Chagraoui, A. Coronaridine Congeners Potentiate GABAA Receptors and Induce Sedative Activity in Mice in a Benzodiazepine-Insensitive Manner. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 101, 109930. [Google Scholar] [CrossRef]
- Wiart, C. Lead Compounds from Medicinal Plants for the Treatment of Cancer; Elsevier: London, UK, 2013; ISBN 9780123983718. [Google Scholar]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings 1PII of Original Article: S0169-409X(96)00423-1. The Article Was Originally Published in Advanced Drug Delivery Reviews 23 (1997). Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Baell, B.J.; Holloway, A.G. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The Design of Leadlike Combinatorial Libraries. Angew. Chem. Int. Ed. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Adelman, S.A.; Doll, J.D. Generalized Langevin Equation Approach for Atom/Solid-surface Scattering: Collinear Atom/Harmonic Chain Model. J. Chem. Phys. 1974, 61, 4242–4245. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- López, S.; Bastida, J.; Viladomat, F.; Codina, C. Acetylcholinesterase Inhibitory Activity of Some Amaryllidaceae Alkaloids and Narcissus Extracts. Life Sci. 2002, 71, 2521–2529. [Google Scholar] [CrossRef]
- Grochowski, D.M.; Uysal, S.; Aktumsek, A.; Granica, S.; Zengin, G.; Ceylan, R.; Locatelli, M.; Tomczyk, M. In Vitro Enzyme Inhibitory Properties, Antioxidant Activities, and Phytochemical Profile of Potentilla Thuringiaca. Phytochem. Lett. 2017, 20, 365–372. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compund | ChemPLP | ΔHsolv, avrg, kcal/mol | ChemPLP + |ΔHsolv, avrg| | IC50, mM | ABTS (%) |
|---|---|---|---|---|---|
| 5 | 85.0500 | −30.8296 | 115.8796 | >10 | na |
| 9 | 85.9159 | −65.8063 | 151.7222 | 1.8 ± 0.75 | 6.43 ± 0.85 |
| 16 | 76.7413 | −45.8669 | 122.6082 | na | 95.82 ± 0.21 |
| 17 | 82.7631 | −34.3182 | 117.0813 | >10 | na |
| 18 | 79.0189 | −41.3130 | 120.3319 | na | na |
| 21 | 82.7599 | −59.1422 | 141.9021 | 1.2 ± 0.19 | 34.68 ± 1.27 |
| 22 | 84.6421 | −44.4386 | 129.0807 | 0.39 ± 0.16 | na |
| 25 | 77.6389 | −42.3505 | 119.9894 | Na | na |
| 28 | 73.5909 | −55.8388 | 129.4297 | 0.62 ± 0.14 | 70.55 ± 0.85 |
| 29 | 77.5505 | −60.2032 | 137.7537 | 5.7 ± 3.50 | 80.94 ± 0.94 |
| GAL | 72.1100 | −45.5914 | 117.7014 | 0.002 ± 0.0003 | |
| BHT | 92.38 ± 0.21 |
| Compound | EVDW Kcal/Mol | EEL Kcal/Mol | EGB Kcal/Mol | ESURF Kcal/Mol | TOTAL Kcal/Mol | EEL + EGB, Kcal/Mol |
|---|---|---|---|---|---|---|
| 22 | −47.15 | −31.53 | 40.20 | −5.96 | −44.44 | 8.67 |
| 28 | −47.40 | −274.18 | 271.14 | −5.39 | −55.84 | −3.05 |
| 21 | −48.46 | −324.22 | 320.09 | −6.55 | −59.14 | −4.13 |
| 9 | −54.68 | −289.90 | 284.96 | −6.18 | −65.81 | −4.95 |
| 29 | −52.21 | −259.20 | 257.11 | −5.91 | −60.20 | −2.08 |
| GAL | −39.18 | −278.77 | 277.42 | −5.06 | −45.59 | −1.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atanasova, M.; Dimitrov, I.; Ivanov, S.; Georgiev, B.; Berkov, S.; Zheleva-Dimitrova, D.; Doytchinova, I. Virtual Screening and Hit Selection of Natural Compounds as Acetylcholinesterase Inhibitors. Molecules 2022, 27, 3139. https://doi.org/10.3390/molecules27103139
Atanasova M, Dimitrov I, Ivanov S, Georgiev B, Berkov S, Zheleva-Dimitrova D, Doytchinova I. Virtual Screening and Hit Selection of Natural Compounds as Acetylcholinesterase Inhibitors. Molecules. 2022; 27(10):3139. https://doi.org/10.3390/molecules27103139
Chicago/Turabian StyleAtanasova, Mariyana, Ivan Dimitrov, Stefan Ivanov, Borislav Georgiev, Strahil Berkov, Dimitrina Zheleva-Dimitrova, and Irini Doytchinova. 2022. "Virtual Screening and Hit Selection of Natural Compounds as Acetylcholinesterase Inhibitors" Molecules 27, no. 10: 3139. https://doi.org/10.3390/molecules27103139
APA StyleAtanasova, M., Dimitrov, I., Ivanov, S., Georgiev, B., Berkov, S., Zheleva-Dimitrova, D., & Doytchinova, I. (2022). Virtual Screening and Hit Selection of Natural Compounds as Acetylcholinesterase Inhibitors. Molecules, 27(10), 3139. https://doi.org/10.3390/molecules27103139