1. Introduction
European aspen is popular species common in European temperate climate zones. In Latvia, European aspen growth uses 10% of the area of all forests [
1]. Aspen tree is widely used in production of cellulose, chips, plywood, pellets, etc. [
2]. In most of these processes debarking takes place, and together with targeted products this produces a considerable amount of waste in terms of discarded tree bark. This makes wood bark, and more precisely aspen bark, a widely available lignocellulosic feedstock [
3]. For example, the forestry sector in Latvia produces around 400,000 m
3 of wood bark waste annually [
1]. Most of the produced bark is used as a fuel. Bark as a forestry sector waste product is produced year-round in relatively steady quantities in timber processing plants. However, periods of low heat demand combined with issues related to long-term storage of wood bark in piles outdoors, it is combusted for destruction without any economic gain [
4]. This leads to wood bark being a considerably cheaper lignocellulosic feedstock than for example wood chips or other available resources [
5]. Wood bark is a lignocellulosic resource with a wide range of possible applications. There have been multiple studies beyond the energy utilization of bark as lignocellulosic resource with focus on extractives isolation [
6]. Bark has been extensively studied as a source of biologically active extractives suitable for various pharmaceutical and cosmetological applications [
7,
8,
9]. For example, due to the high content of salicylates, aspen bark water-soluble extracts are commercialized as cosmetics components with skin conditioning and antimicrobial properties [
10]. However, relatively little work has been done to establish biorefinery pathways of holistic bark processing incorporating extractives removal and further valorization of obtained residue by separating its main components: lignin, cellulose, and monomeric carbohydrates [
11,
12,
13].
As a renewable aromatic biopolymer making from 15 to 35% of the bark mass, lignin holds the key for maximizing value extraction from bark biomass. In tree bark lignin content is noticeably higher than in wood, since the bark is the barrier that protects the tree from biotic and abiotic factors. Furthermore, lignin gives the necessary structural strength and together with non-lignin phenolics provides anti-microbial protection [
14,
15,
16]. Lignin valorization is currently hindered by the application of delignification methods that significantly alter the native lignin structure to give highly recalcitrant materials [
17,
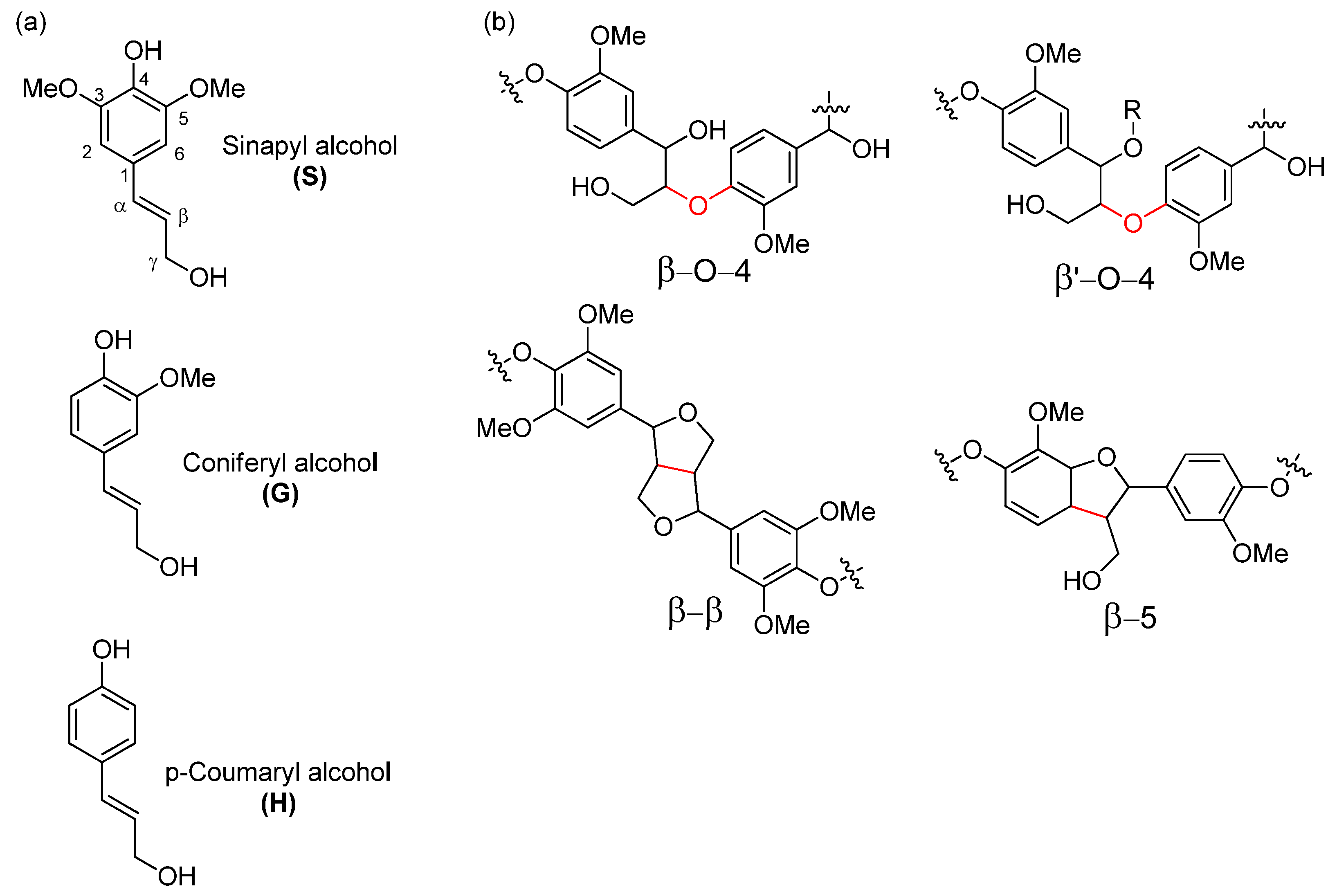
18]. In hardwood such as European aspen, lignin is a complex macromolecule that mostly originates from p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol through radical polymerization, providing a polymer made up of H, G and S units, respectively (
Figure 1a) [
19].
The most abundant lignin linkage motif that the radical polymerization generates is the β–O–4 motif, which can make up to 70% of all linkage motives (
Figure 1b). In addition, lignin also contains native C–C linkage motifs such as β–5, 5–5, β–β. In contrast to the β–O–4 motif, these latter bonds are much more resistant to chemical alteration and generally remain unaltered during pretreatment and fractionation processes.
Lignin in hardwoods such as aspen has more S subunits and less C–C bonds, making it easier to separate than softwood species such as pine [
20,
21,
22,
23]. High-quality lignin in context of its valorization is defined as lignin with abundant aryl−ether linkages, including β–O–4 linkages, for its application in synthesis of versatile polymers as macromonomer [
24,
25] or as a source of renewable monomeric phenolics instead of fossil-based raw materials. These low molecular weight compounds can be used as building block chemicals in different polycondensation reactions, which are versatile in the polymer industry [
26].
A promising approach for obtaining high-quality lignin from lignocellulosic biomass as residual bark is mild organosolv delignification. Relatively little has been done in terms of tree bark organosolv delignification, and to our knowledge European aspen bark organosolv delignification has not been previously studied. Organosolv been described in literature for quite a long time now. Organosolv delignification is a biomass fractionation method using organic solvents, often in the presence of acidic catalyst, that allows efficient fractionation of biomass in its major components (cellulose, lignin, and hemicellulose in soluble monosaccharide fraction) at relatively mild conditions, with temperatures between 100 °C to 200 °C and pressure from atmospheric to 20 bar. In comparison with kraft and sulphite delignification, the organosolv approach is a more environmentally friendly process due to lack of harmful sulphur-containing emissions.
In a typical organosolv delignification process, lignin undergoes partial acid catalyzed depolymerization that can undergo recondensation, resulting in condensed oligomeric lignin fragments with relatively low molecular mass. However, the structural transformation of lignin is highly dependent on the separation conditions. Mild organosolv at temperatures below 120 °C has the advantage that it leaves the more regular β–O–4 intact, and by using specific solvents and extraction setups similar extraction efficiencies can be obtained. Recent work at moderate temperatures (~100–150 °C) and using organic acids or low concentration of mineral acids allows us to obtain relatively low condensed lignin with a high amount of native β-O-4 bonds and good solubility in organic solvents [
27,
28]. Furthermore, extraction efficiency can be further enhanced using flow-through setups that lower the exposure time of released fragments to the reactive medium [
29,
30]. Use of organic solvents in separation allows us to increase the efficiency of the process when compared to dilute acid hydrolysis methods due to the increase of lignin solubility in the used process medium. In addition, the use of alcohols in the organosolv process promotes incorporation of ethers chains into the α position of the phenyl propane units. This structure is called β‘-O-4 and is shown in
Figure 1b. This incorporation protects the native lignin β-O-4 structure from degradation and re-condensation, allowing separation of low condensed lignin more suitable as a feedstock for further valorization pathways while also increasing lignin solubility in organic solvents that can be useful for further processing. [
5,
17,
27,
31,
32,
33,
34]. Therefore, mild organosolv delignification of residual European aspen bark using low molecular weight alcohol solvent systems can be a promising approach for fractionation of this underutilized resource to obtain fractions suitable for further use as starting materials in biorefinery processing clusters.
The scope of this research is to study the process of mild organosolv delignification of extracted aspen using two components mixes of protic and aprotic organic solvents with alcohols and water in terms of the yield and characteristics of precipitated lignin and non-soluble cellulose enriched fraction, to show that used process is suitable for successful aspen bark valorization in biorefinery processing scheme. Process overview is shown in
Figure 2.
2. Results
2.1. Isolation of Extractives
Extractives including lipophilic, non-lignin phenolic, and carbohydrates compounds were separated from aspen bark utilizing the accelerated solvent extraction system (ASE). Extraction conditions were selected based on previous work done in our laboratory to maximize the yield of extractives [
6,
35]. To separate polar, semi-polar and non-polar secondary metabolites, sequential extraction was performed. The first extraction solvent was n-hexane, followed by ethyl acetate, and finally a 40% ethanol water solution (
v/
v). Yields of the separated extractives increased with the increase of the polarity of the utilized extraction solvents. n-hexane soluble extractives made up 5.9 ± 0.7% (wt %) of the biomass, the ethyl acetate fraction 8.0 ± 0.6% and the ethanol soluble fraction 11.9 ± 0.7% of the biomass. The total yield of separated extractives from aspen bark was 25.9 ± 0.9%. Chemical composition and possible directions of practical applications of separated extracts is under development by our group, and since it is not a focal point of this publication it will not be discussed here.
A substantial (70–80%) part of the biomass is left after these steps, of which the components need to be effectively separated for efficient utilization in integrated biorefinery scheme development.
2.2. Characterization of Residual Aspen Bark after Extractives Isolation
The composition of bark before and after extraction was characterized using analytical pyrolysis (Py-GC/MS) by categorizing volatile degradation products formed from biomass components during their thermal decomposition in conditions of analytical pyrolysis. The diagnostic volatile pyrolysis products were classified as:
Carbohydrates-derived compounds represented mainly by aliphatic acids and esters, aliphatic alcohols, aliphatic aldehydes and ketones, furan and pyran derivatives, cyclopentane derivatives, anhydro sugars;
Non-methoxylated aromatic compounds derived mainly from phenolic extractives;
Methoxylated phenols, guaiacyl (G-) and syringyl (S-) derivatives derived from lignin;
Lipophilic extractives-derived compounds (
Table 1).
According to Py-GC/MS data the extracted bark has significantly lower content of hydrophilic (phenolic) and lipophilic extractives in comparison to the untreated bark (as relative content of corresponding volatiles decreased from 14.90 to 2.15%). The content of lipophilic extractives-derived compounds also decreased from 5.69 to 1.94%. Thus, overall, hydrophilic extractives were efficiently isolated from bark, while a significant part of lipophilic extractives remained in the bark biomass. The relative summary content of lignin-derived G- and S-compounds in untreated aspen bark was 18.18%, while after extraction it was 24.85%. The relative content of carbohydrate-derived compounds in initial bark and extracted residue was 61.23% and 71.06%, correspondingly. So, the isolation of extractives leads to enrichment of both lignin and carbohydrates content in bark biomass, enhancing its potential as feedstock for obtaining these compounds.
2.3. Residual Aspen Bark Organosolv Delignification
To select the optimal solvent system for aspen bark delignification, several options were tested. Delignification conditions remained constant for this set of experiments: 5 h process duration at a temperature of 80 °C and a solid to liquid ratio of 1 to 10. These conditions were selected using previous experience as a starting point for used solvent system optimization [
36]. In total, five different solvent systems were tested. Obtained results in terms of lignin-enriched fraction yield obtained from bark as well as obtained residual fraction yield are shown in
Figure 3.
It was shown that the higher yield was obtained using the 1,4-dioxane/ethanol and n-butanol/water solvent mixtures. For further experiments an n-butanol/water solvent system was being selected as optimal since adequate results can be achieved and butanol is a green solvent. The next aim was to optimize the delignification conditions. In total, four time durations at five different temperatures were tested (1, 3, 5, 20 h and 40, 60, 80, 100, 117 °C (refluxing)). The results of twenty experiments differing by delignification regimes in terms of lignin- and cellulose-enriched fractions yield are presented in
Figure 3 and
Table 2. Input data is shown in
supplementary information (Table S2).
Both temperature and duration of processing enhance the yield of lignin-enriched fractions. The highest yield of 24.03% on DM of extracted biomass was achieved at a temperature of 117 °C and 20 h of duration.
It was shown that increasing temperature and duration of mild organosolv treatment highly promotes the dissolution of biomass components in the n-butanol-water solution, decreasing the yield of the insoluble cellulose-enriched fraction from 90% to 43% while simultaneously increasing the purity of the separated cellulose-enriched fraction in terms of leftover lignin admixtures (
Figure 4).
Using the obtained data, a full factor experimental model was created using DesignExpert 13 software. The following goals were set for the model: maximize the obtained lignin-enriched fraction yield with minimum admixture content while simultaneously obtaining maximum amount of residual cellulose-enriched fraction. The calculations take all these factors into consideration and provide desirability scores for all model variables. The obtained desirability plot for this model is shown in
Figure 5.
The obtained model shows that a more favorable outcome of delignification reaction can be obtained when utilizing conditions with longer treatment time and higher temperature. At the boiling point of butanol (117 °C) and treatment time of 20 h, model desirability was 0.783. From this, these conditions were chosen as most suitable for residual aspen bark delignification. It is important to mention that although higher temperatures do increase the lignin-enriched fraction yield, it can also promote degradation of lignin. Due to this, it vital to perform complete characterization of obtained lignin fractions.
2.4. Characterization of Lignin-Enriched Fraction
2.4.1. Composition of Lignin-Enriched Fraction Isolated from Bark by Solvents of Different Polarity
To compare different solvent systems used in lignin separation, it is vital to perform comprehensive characterization of obtained lignins in terms of admixture content and obtained lignin structure. It was shown that the content of admixtures in precipitated fractions is significantly dependent on the polarity of solvent used for delignification.
The FTIR spectra (
Figure 6) of the lignin-enriched fraction showed that the fraction isolated by the most polar ethanol/water solvent contains the least amount of lipophilic admixtures that could originate from fatty acids—glycerol and hydroxyl fatty acid-ferulic acid esters of suberin. This was evidenced by the least absorbance intensity at 2950–2850 cm
−1 attributed to the C-H stretch in methyl and methylene groups as well as the least absorbance at about ~1725 cm
−1 attributed to the presence of unconjugated carbonyls in ester groups. However, the most absorbance intensity in the region of 1100 cm
−1 attributed to the C–O deformation in secondary alcohols and aliphatic ethers indicates that this fraction contains the most of carbohydrates admixtures. In contrast, the fraction isolated by the least polar solvent (diethyl carbonate/ethanol) contained the least amount of carbohydrates admixtures but a significantly higher amount of lipophilic compounds in comparison with fraction isolated by polar ethanol–water solvent. For all solvents except n-butanol-water with decreasing solvent polarity, the content of lipophilic admixtures in precipitated fraction is decreased, increasing the content of carbohydrates admixtures. This dependence was not observed for n-butanol-water solvent. It highly promotes the solubility of lipophilic compounds of bark that were co-precipitated with lignin where water was added as a co-solvent.
The analysis of carbohydrate admixtures after performing hydrolysis of obtained lignin fractions and determining carbohydrates by GC-FID method showed that the only carbohydrates present in lignin-enriched fractions were xylose and arabinose, with content less than 4%. Analytical pyrolysis showed that when using the n-butanol/water solvent system, the obtained lignin-enriched fraction contains a significant (20.8 ± 0.4%) amount of carbohydrate admixtures, which are not detected using GC-FID alditol carbohydrate method of analysis. This can be explained by the formation of hydrolysis-stable n-butanol-carbohydrate ethers during the delignification process, as described in the literature [
30].
The application of analytical pyrolysis allowed obtain additional information concerning composition of lignin enriched fractions. This method is widely used for the characterization of the chemical composition of lignocellulosic biomass by the determination of pyrolysis products referring to the definite groups of biomass substituents [
37]. The volatile pyrolysis products were classified as carbohydrates-derived volatiles, lignin-derived volatiles (consisting of compounds described in
Section 2.2), nitrogen-containing volatiles and aliphatic long-chain and cyclic compounds derived from lipophilic extractives. Data from Py-GC-MS (
Table 3) reveals that dominant non-phenolic admixtures present in the lignin-enriched fraction are lipophilic compounds most likely coming from suberinics present in hardwood tree bark. The lowest amount of lipophilic admixtures was found in the fraction obtained using the n-butanol/water system, reaching 26.4%.
When comparing the FTIR spectra of the lignin-enriched fraction obtained by n-butanol/water mild organosolv delignification with widely available and pure LignoBoost lignin obtained from hardwood biomass, the presence of lipophilic admixtures is clearly confirmed with an increase of peaks at 2950 cm−1 and 1725 cm−1. The amount of lipophilic admixtures in the lignin-enriched fraction is an important factor that needs to be further studied for effective utilization of lignin-enriched fractions, both in terms of admixture effect on lignin fraction usability and possibilities of admixture separation.
2.4.2. Structural Characterization of Lignin-Enriched Fraction
The structure of lignin constituents of separated lignin-enriched fractions, including the distribution of monolignols and dominant linkages between them as determined by semi-quantitative 2D HSQC NMR analysis, are shown in
Table 4.
From obtained data (
Figure 7), it can be seen that by varying used solvent systems, monolignol distribution remains similar, with the exception of the diethyl carbonate/ethanol mixture that promotes lignin condensation and new C–C bond formation of S lignin subunits (
Table 4). In terms of linkage unit content per structural subunit of lignin, the highest amount of β–O–4 and β’–O–4 linkages were obtained by performing delignification with n-butanol- and 1,4-dioxane-containing solvent systems.
From samples under study, the fractions isolated with the diethyl carbonate/ethanol solvents system revealed themselves as most condensed due to lowest content of remaining β–O–4 linkages and appearance of S condensed structures non-typical for native lignin (
Table 4). Among others, lignins isolated by n-butanol/water and ethanol/water solvent systems are most enriched with β–O–4 linkages and therefore can be characterized as the least condensed in comparison with other samples, including commercial hardwood LignoBoost lignin. Moreover, the high content of β’–O–4 structures in isolated lignins, which are absent in commercial lignin, can be estimated as a favorable factor that allowed avoiding undesirable condensation of lignin structures in the pretreatment stages of its chemical modification [
38].
When lignin fractions were obtained at 80 °C and at 117 °C (
Table 5), it was noticeable that an increase in temperature did not dramatically increase unwanted condensation of obtained lignin fractions, indicated by the high number of β-O-4 groups present in lignin that was obtained in harsher conditions.
Obtained lignin-enriched fractions are suitable for further utilization as feedstock for obtaining polymeric materials, including polyurethane and polyepoxides, since they are enriched with reactive -OH groups (both aliphatic and phenolic), as determined with
31P NMR spectroscopy (data shown in
Table 6). In the fractions obtained, the portion of aliphatic groups most reactive in reactions of nucleophilic attachment consisted of 65–81% from total OH content. This can be recognized as a significant advantage of isolated lignins versus commercial lignins, including Alcell, Indulin AT, Sarkanda, Curan-2711P in which the aliphatic groups portion was lower and varied in the range of 27.6–54.2% [
39].
For lignin that was obtained using a n-butanol/water solvent system, at harsher conditions (20 h, 117 °C), aliphatic –OH group content was 2.20 (mmol·g
−1) and total phenolic—OH group content was 0.83 (mmol·g
−1). This indicates that while an increase in temperature does not lead to loss of lignin β–O–4 bonds, it does leads to loss of aliphatic hydroxyl groups to some extent. Nonetheless, obtained lignins, even at harsher conditions, are still enriched with reactive –OH groups and can be successfully used as a natural polymeric feedstock in further valorization. The example of
31P NMR spectra of Butanol/Water organosolv lignin is shown in
Supplementary Materials (Figure S2).
2.5. Characterization of Carbohydrate-Enriched Fraction
The yield of carbohydrates-enriched fraction obtained as residue after delignification varied between 63.6% and 82.4%.
Carbohydrate-enriched fraction was characterized in terms of lignin admixture content by determining Klason lignin content in obtained samples. Obtained results are shown in
Table 7.
When comparing lignin amounts in residuals with starting feedstock it is notable that obtained residues are considerably cleaner than lignin admixtures. With varying solvents Klason lignin content in residuals ranged from 25.5% to 14.9% and a butanol/water solvent system obtained a residual fraction that contained 15.7% of lignin admixtures. By increasing delignification condition harshness, at 117 °C and in a 20 h timeframe, it was possible to obtain purer residual fractions containing only 13.66% lignin admixtures. When considering individual carbohydrate content in obtained residual fractions, obtained results were similar with all used solvent systems. All samples contained predominantly glucose (Glc) with some admixtures of xylose (Xyl) and arabinose (Ara) (
Table 5).
Total carbohydrate content in these samples varied between 39.8% to 51.2%, and when using butanol as a delignification solvent, total carbohydrate content was 42.8%. When performing delignification at harsher conditions with n-butanol as a solvent at 117 °C for 20 h, the residual fraction obtained at this regime was more enriched with carbohydrates and contained the least amount of lignin admixture. Total carbohydrates content in this sample was 61.23%, with a glucose portion of 85%, indicating that this obtained fraction is enriched with cellulose and is promising feedstock in further applications.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}