
Hit-to-Lead Short Peptides against Dengue Type 2 Envelope Protein: Computational and Experimental Investigations

 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Molecular Docking Studies
2.2. Molecular Dynamics Studies
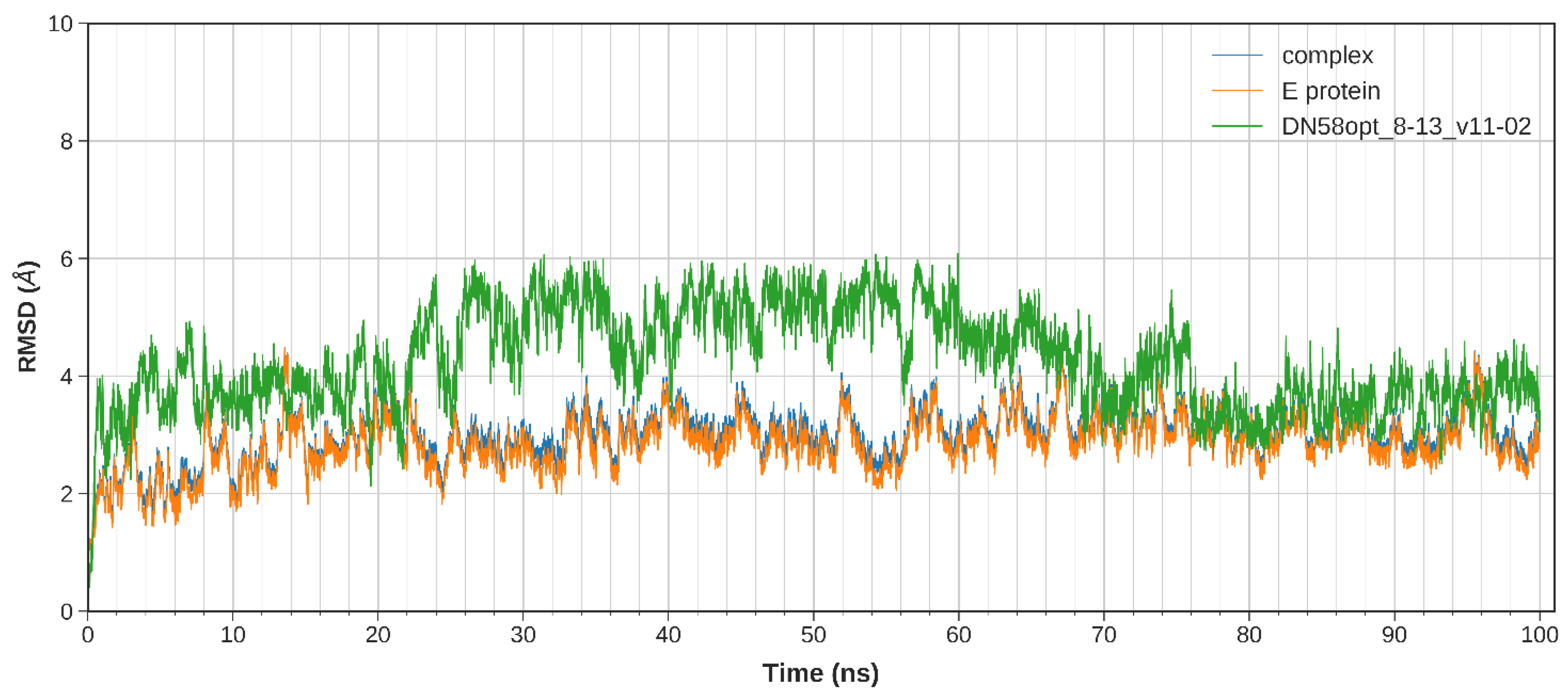
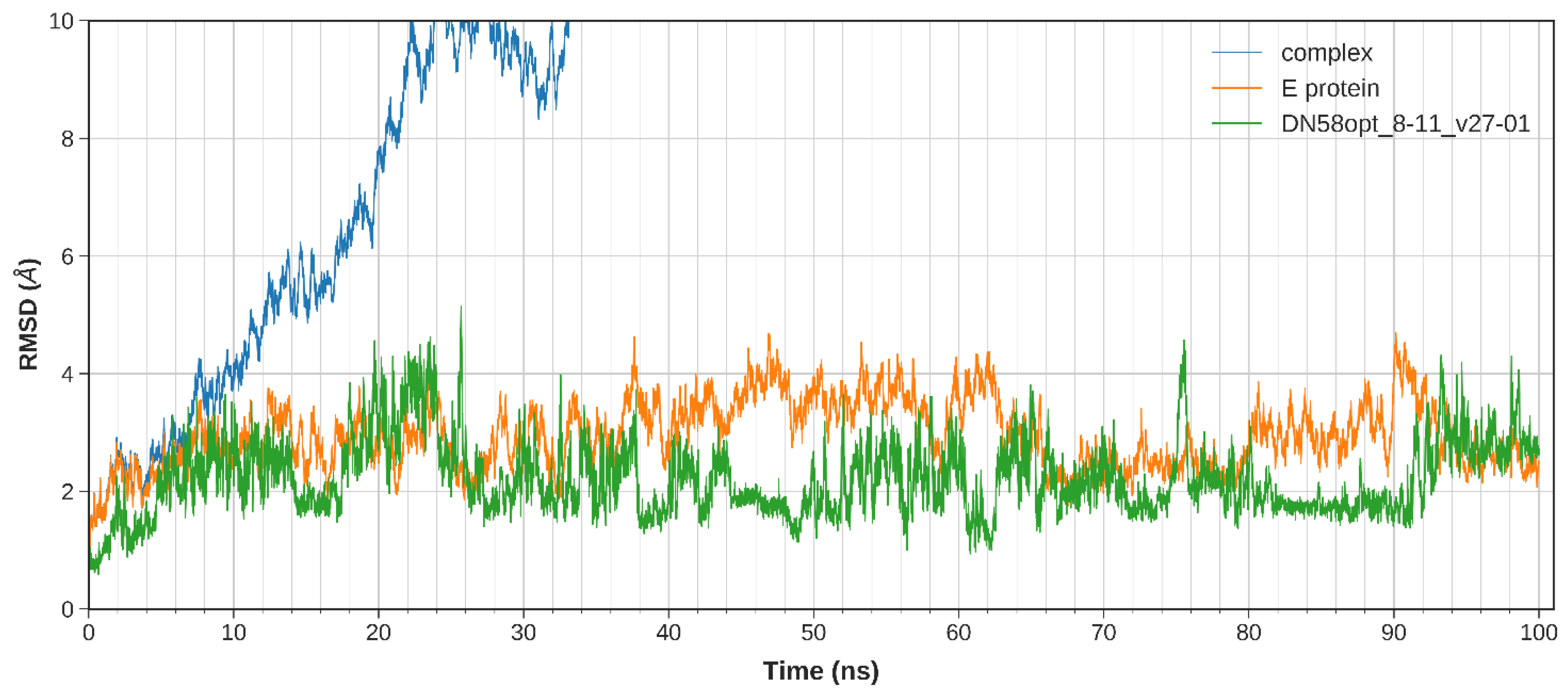
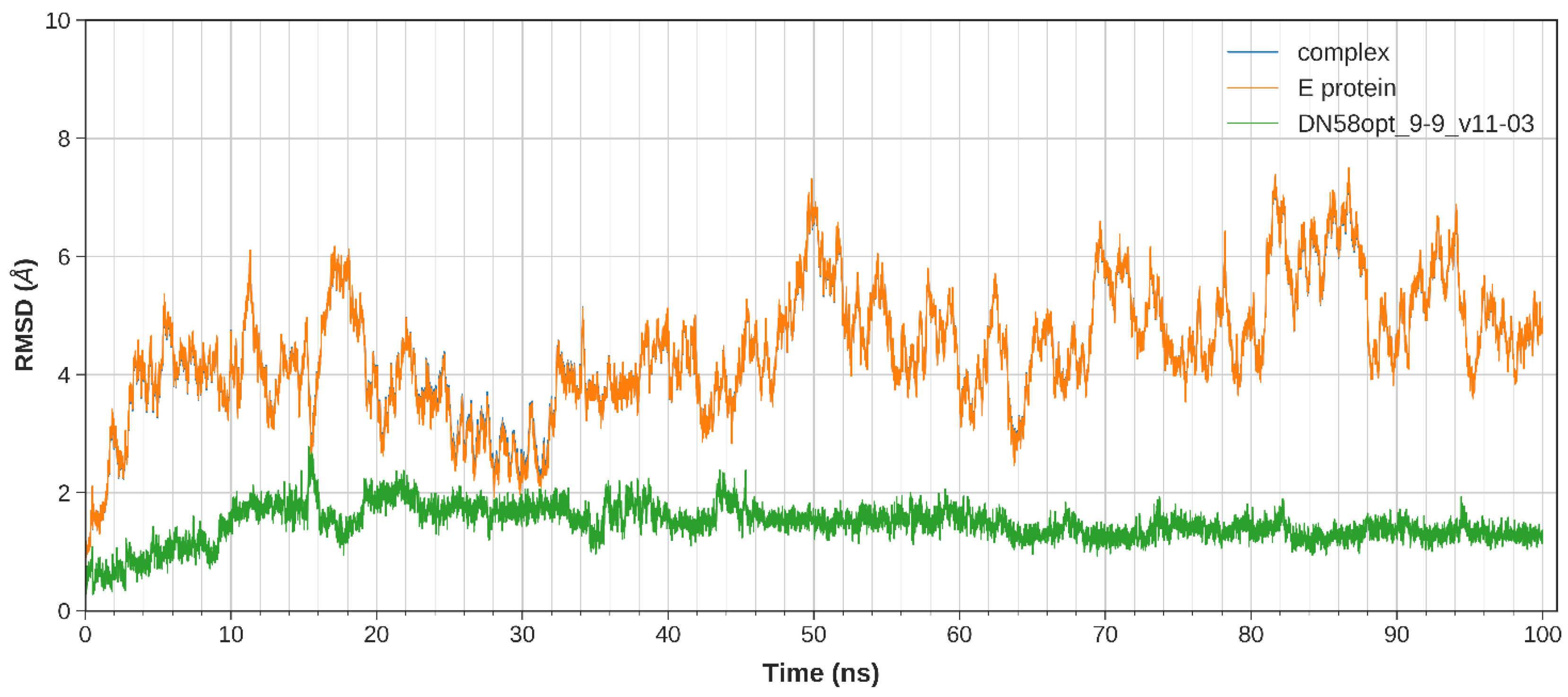
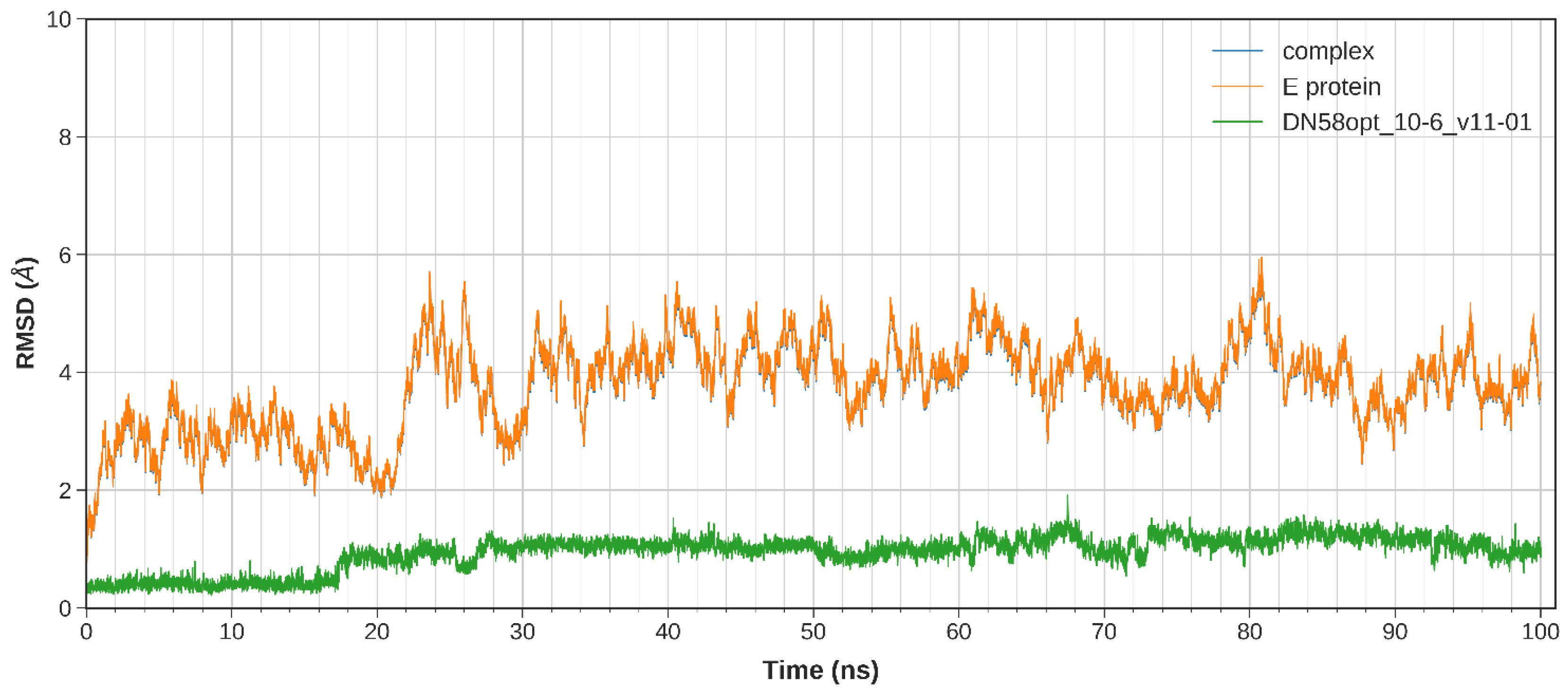
2.2.1. System Stability
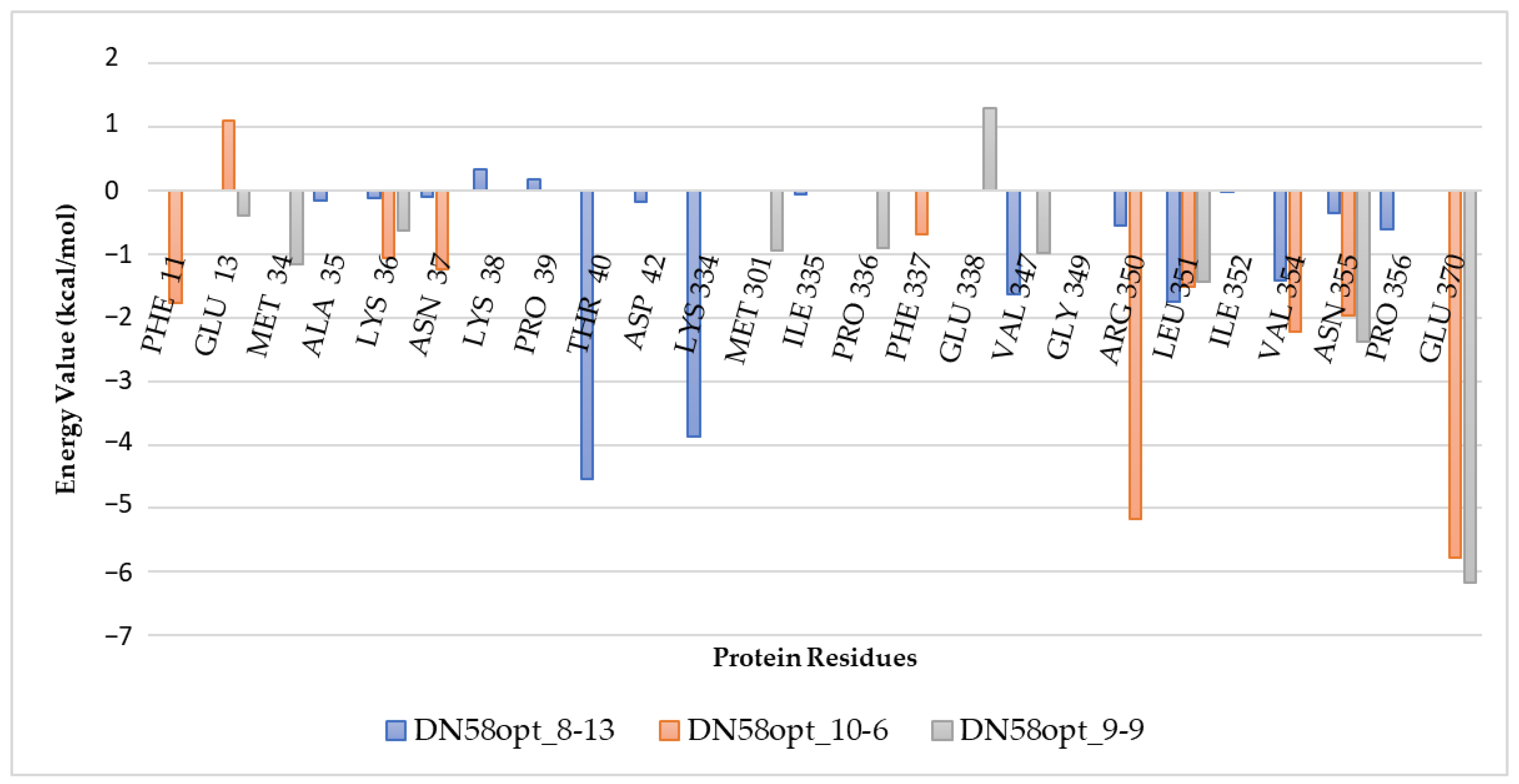
2.2.2. Calculation of Free Energy Binding
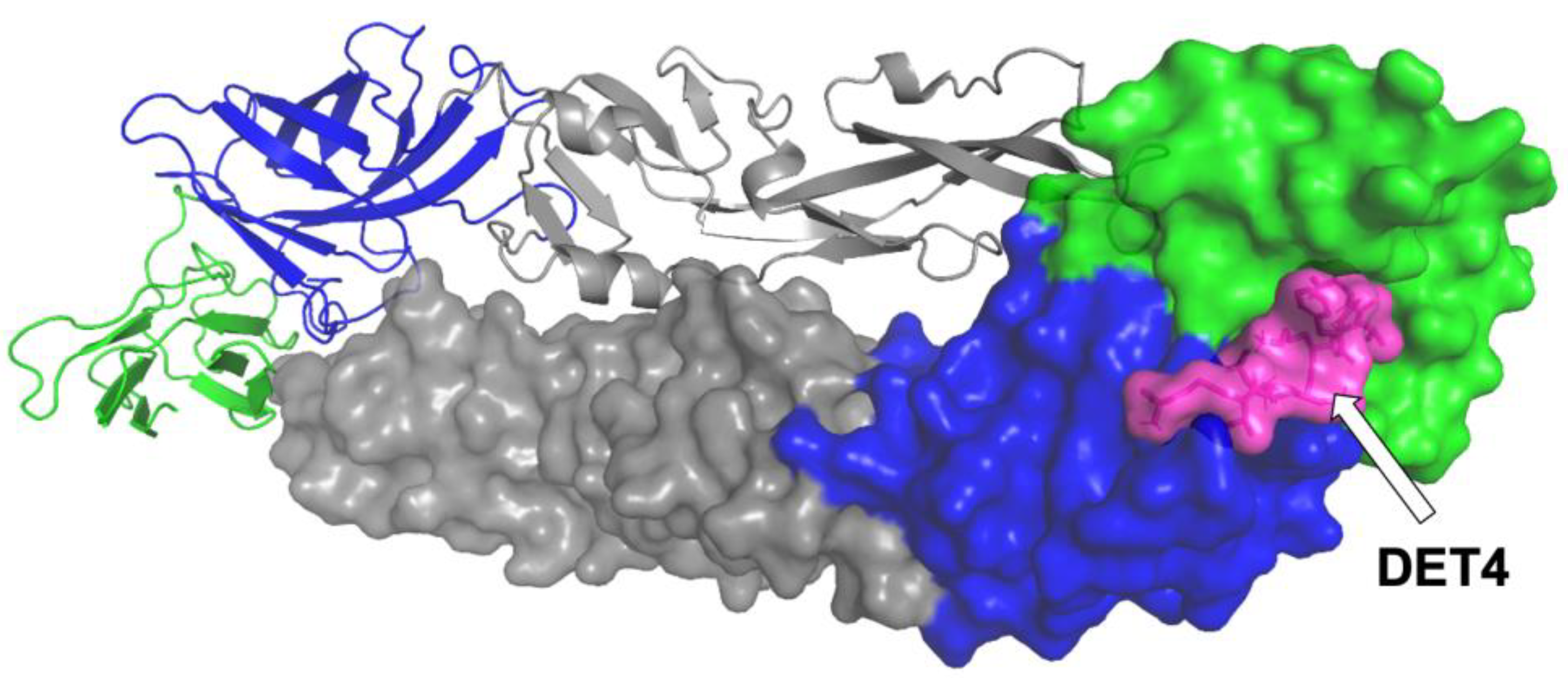
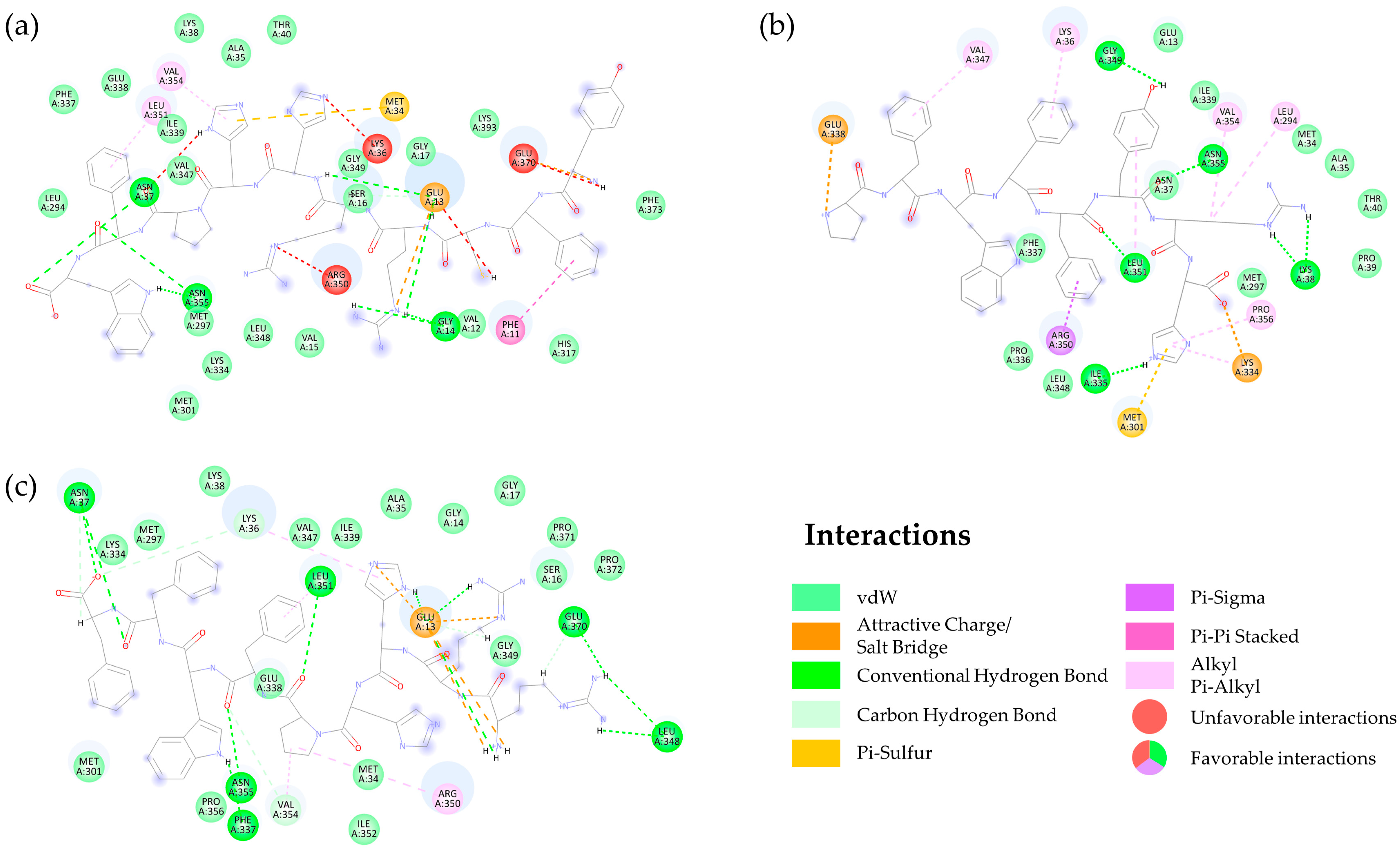
2.2.3. Interaction of DN58opt Peptide Fragments in Active Sites of DENV2 E Protein
2.3. Summary and Selection of Lead Peptides
2.4. Safety and Efficacy Evaluation of Peptides against DENV2 E Protein
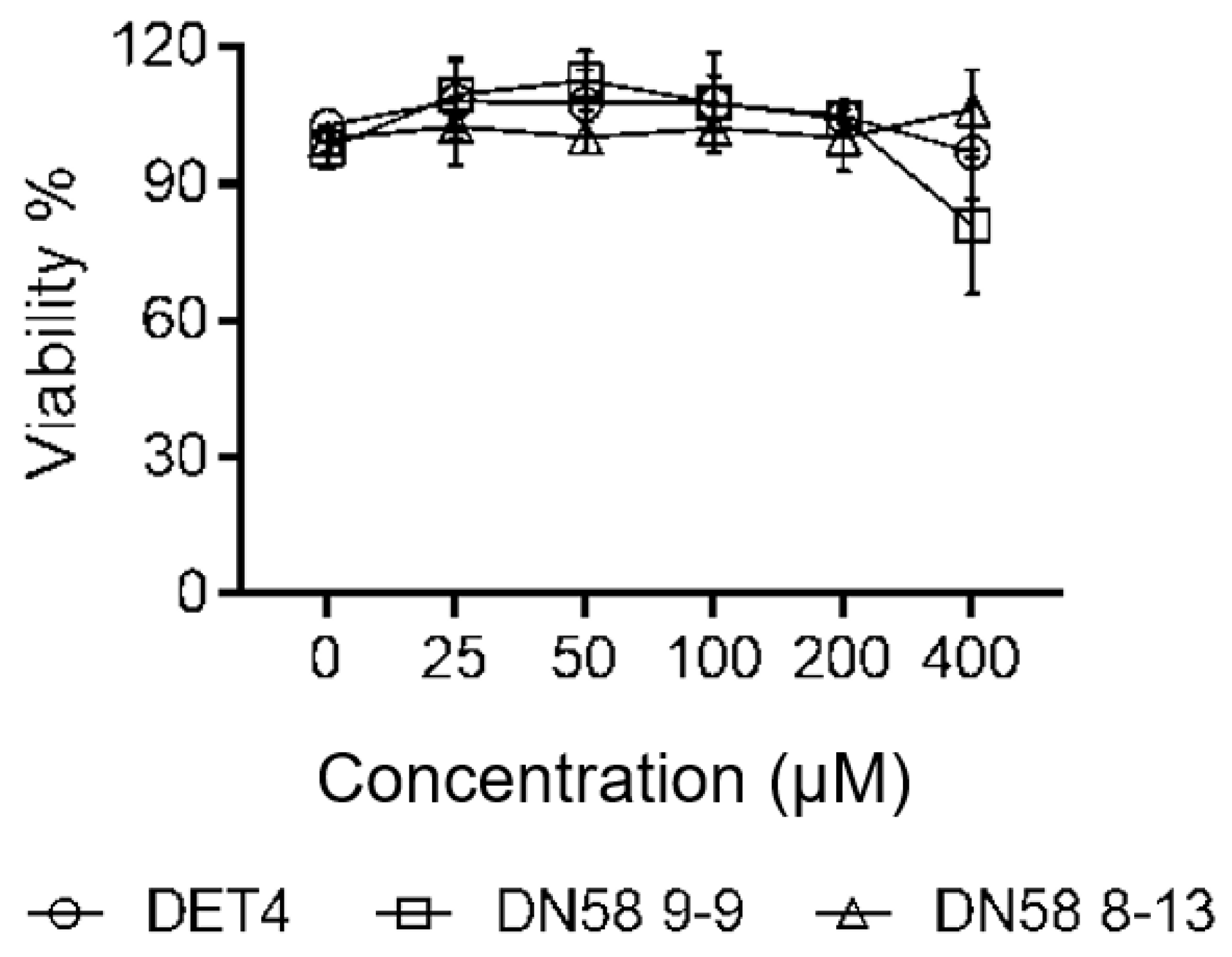
2.4.1. Cytotoxicity Assay (MTS)
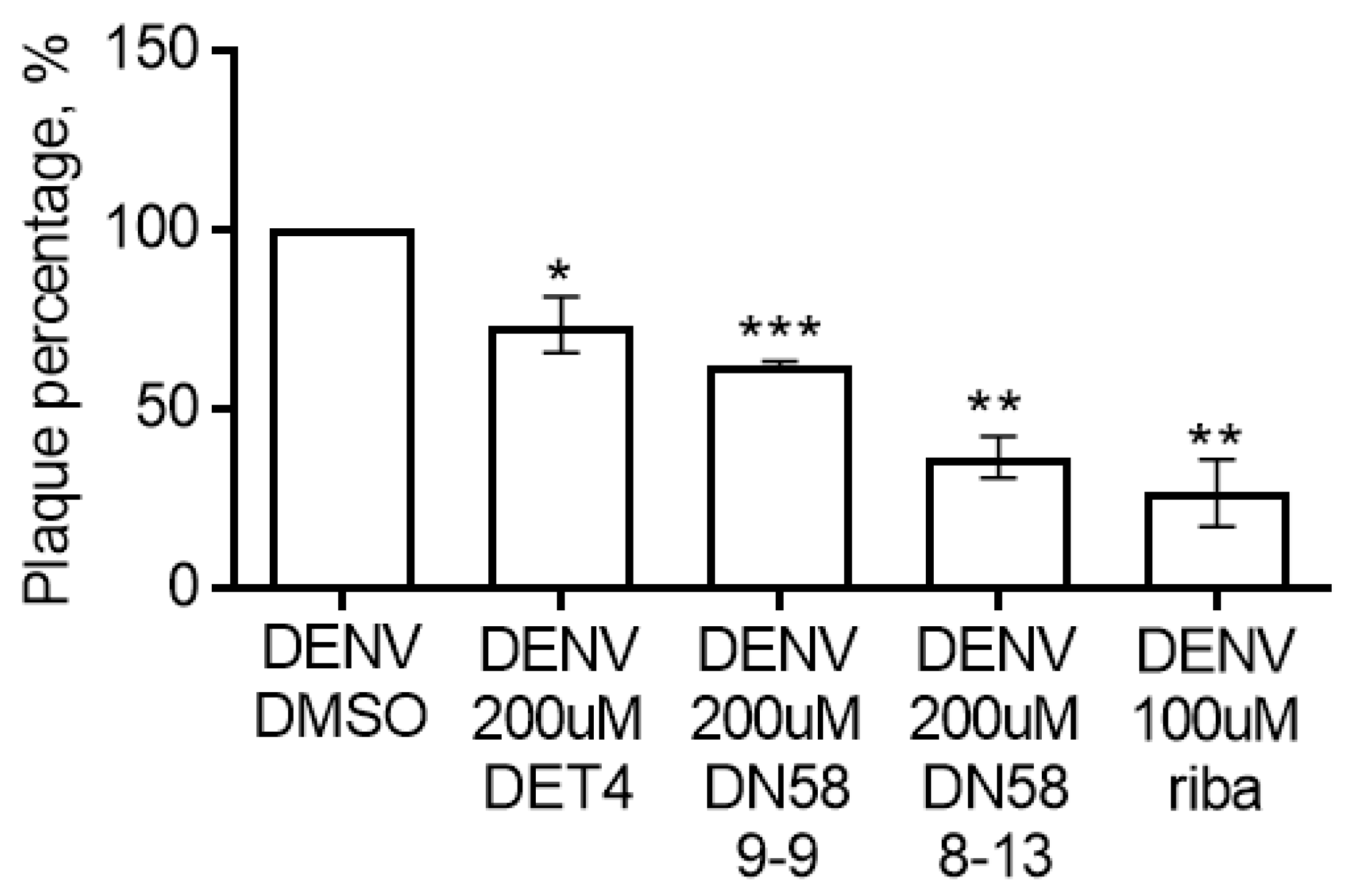
2.4.2. Plaque Formation Assay
3. Materials and Methods
3.1. Molecular Docking Studies
3.1.1. Model of Envelope Protein and Its Known Inhibitor
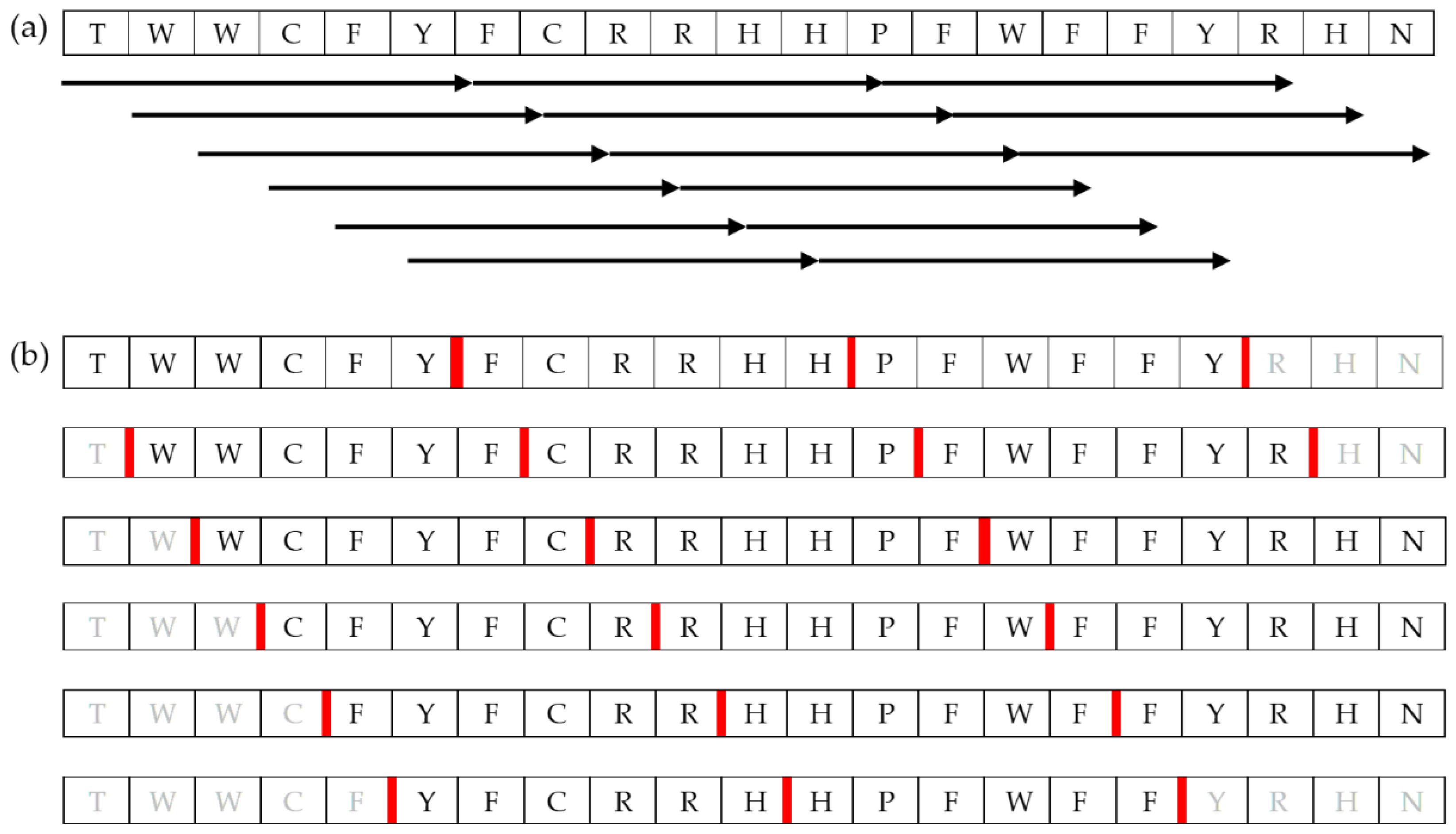
3.1.2. Construction of Short-Chain Peptide Library
3.1.3. Virtual Screening of the Peptide Library via Molecular Docking
3.1.4. Analyses of the Virtual Screening Results
3.2. Molecular Dynamics Studies
3.2.1. Molecular Dynamics Simulation of the E Protein–Peptide Complexes
3.2.2. Analyses of the Molecular Dynamic Trajectories
3.2.3. Calculation of Free Energy of Binding
3.3. Peptide Synthesis
3.4. Safety and Efficacy Evaluation of Peptides against DENV2 E Protein
3.4.1. MTS Cytotoxicity Assay
3.4.2. Plaque Formation Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Dengue and Severe Dengue. Available online: https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (accessed on 1 April 2022).
- Ebi, K.L.; Nealon, J. Dengue in a changing climate. Environ. Res. 2016, 151, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Robinson, A.W.T. Vector Control Interventions to Prevent Dengue: Current Situation and Strategies for Future Improvements to Management of Aedes in India. J. Emerg. Infect. Dis. 2017, 2, 1000123. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Bambrick, H.; Pongsumpun, P.; Dhewantara, P.W.; Toan, D.T.T.; Hu, W. Dengue outbreaks in the COVID-19 era: Alarm raised for Asia. PLoS Negl. Trop. Dis. 2021, 15, e0009778. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.S.; Rasotgi, V.; Jain, S.; Gupta, V. Discovery of fifth serotype of dengue virus (DENV-5): A new public health dilemma in dengue control. Med. J. Armed Forces India 2015, 71, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Dorigatti, I.; Donnelly, C.A.; Laydon, D.J.; Small, R.; Jackson, N.; Coudeville, L.; Ferguson, N.M. Refined efficacy estimates of the Sanofi Pasteur dengue vaccine CYD-TDV using machine learning. Nat. Commun. 2018, 9, 3644. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, S.S.; Durbin, A.P.; Pierce, K.K.; Elwood, D.; McElvany, B.D.; Fraser, E.A.; Carmolli, M.P.; Tibery, C.M.; Hynes, N.A.; Jo, M.; et al. In a randomized trial, the live attenuated tetravalent dengue vaccine TV003 is well-tolerated and highly immunogenic in subjects with flavivirus exposure prior to vaccination. PLoS Negl. Trop. Dis. 2017, 11, e0005584. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Yoon, I.K. A review of Dengvaxia(R): Development to deployment. Hum. Vaccin. Immunother. 2019, 15, 2295–2314. [Google Scholar] [CrossRef] [Green Version]
- Ellan, K.; Thayan, R.; Raman, J.; Hidari, K.; Ismail, N.; Sabaratnam, V. Anti-viral activity of culinary and medicinal mushroom extracts against dengue virus serotype 2: An in-vitro study. BMC Complement. Altern. Med. 2019, 19, 260. [Google Scholar] [CrossRef]
- Pujar, G.V.; Sethu, A.K.; Bhagyalalitha, M.; Singh, M. Dengue structural proteins as antiviral drug targets: Current status in the drug discovery & development. Eur. J. Med. Chem. 2021, 221, 113527. [Google Scholar] [CrossRef]
- Liao, M.; Kielian, M. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus membrane fusion. J. Cell Biol. 2005, 171, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Rahman, N.A.; Othman, R.; Hu, P.; Huang, M. Computational identification of self-inhibitory peptides from envelope proteins. Proteins 2012, 80, 2154–2168. [Google Scholar] [CrossRef] [PubMed]
- Baharuddin, A.; Amir Hassan, A.; Othman, R.; Xu, Y.; Huang, M.; Ario Tejo, B.; Yusof, R.; Abdul Rahman, N.; Othman, S. Dengue envelope domain III-peptide binding analysis via tryptophan fluorescence quenching assay. Chem. Pharm. Bull. 2014, 62, 947–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA Discovery Studio; Version 4.0; Dassault Systèmes: San Diego, CA, USA; Available online: https://www.accelrys.com (accessed on 6 October 2021).
- Westbrook, J.; Feng, Z.; Chen, L.; Yang, H.; Berman, H.M. The Protein Data Bank and structural genomics. Nucleic Acids Res. 2003, 31, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.J.; Hsieh, M.T.; Young, M.J.; Kao, C.L.; King, C.C.; Chang, W. An external loop region of domain III of dengue virus type 2 envelope protein is involved in serotype-specific binding to mosquito but not mammalian cells. J. Virol. 2004, 78, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, R.J.; Capener, C.; Baaden, M.; Bond, P.J.; Campbell, J.; Patargias, G.; Arinaminpathy, Y.; Sansom, M.S. Membrane protein structure quality in molecular dynamics simulation. J. Mol. Graph. Model. 2005, 24, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tai, W.; Wang, N.; Li, X.; Jiang, S.; Debnath, A.K.; Du, L.; Chen, S. Identification of Novel Natural Products as Effective and Broad-Spectrum Anti-Zika Virus Inhibitors. Viruses 2019, 11, 1091. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, A.C.; Agharbaoui, F.E.; Khazali, A.S.; Yusof, R.; Abd Rahman, N.; Ahmad Fuaad, A.A.H. Computational-aided design: Minimal peptide sequence to block dengue virus transmission into cells. J. Biomol. Struct. Dyn. 2020, 31, 1–10. [Google Scholar] [CrossRef]
- de la Guardia, C.; Quijada, M.; Lleonart, R. Phage-Displayed Peptides Selected to Bind Envelope Glycoprotein Show Antiviral Activity against Dengue Virus Serotype 2. Adv. Virol. 2017, 2017, 1827341. [Google Scholar] [CrossRef]
- Rothan, H.A.; Bahrani, H.; Mohamed, Z.; Teoh, T.C.; Shankar, E.M.; Rahman, N.A.; Yusof, R. A combination of doxycycline and ribavirin alleviated chikungunya infection. PLoS ONE 2015, 10, e0126360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhoot, M.A.; Rathinam, A.K.; Wang, S.M.; Manikam, R.; Sekaran, S.D. Inhibition of dengue virus entry into target cells using synthetic antiviral peptides. Int. J. Med. Sci. 2013, 10, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF Chimera, MODELLER, and IMP: An integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.A.; Liu, L.; Ghaderi, N.; Johns, A.; Hatcher, M.E.; Mueller, L.J. The Amide Rotational Barriers in Picolinamide and Nicotinamide: NMR and ab Initio Studies. J. Am. Chem. Soc. 2003, 125, 10125–10132. [Google Scholar] [CrossRef]
- Lim, S.K.; Othman, R.; Yusof, R.; Heh, C.H. Rational Drug Discovery of HCV Helicase Inhibitor: Improved Docking Accuracy with Multiple Seeding in AutoDock Vina and In Situ Minimization. Curr. Comput. Aided Drug Des. 2017, 13, 160–169. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Lieb, E.H.; Thirring, W.E. Universal nature of van der Waals forces for Coulomb systems. Phys. Rev. A 1986, 34, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Fuaad, A.A.; Skwarczynski, M.; Toth, I. The Use of Microwave-Assisted Solid-Phase Peptide Synthesis and Click Chemistry for the Synthesis of Vaccine Candidates Against Hookworm Infection. Methods Mol. Biol. 2016, 1403, 639–653. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Vina Affinity (kcal/mol) | MMGBSA (kcal/mol) | MMPBSA (kcal/mol) |
|---|---|---|---|---|
| DET4 (standard) | AGVKDGKLDF | −6.6 | −41.3249 | −40.9460 |
| DN58opt_8-13 | PFWFFYRH | −9.0 | −41.8317 | −39.4506 |
| DN58opt_7-12 | HHPFWFFY | −8.7 | −54.4044 | −44.3665 |
| DN58opt_6-1 | TWWCFY | −8.6 | −29.4070 | −27.2727 |
| DN58opt_6-11 | HHPFWF | −8.6 | −23.2421 | −23.3695 |
| DN58opt_7-10 | RRHHPFWF | −8.6 | −35.1696 | −31.8476 |
| DN58opt_6-10 | RHHPFW | −8.4 | −11.9393 | −16.1549 |
| DN58opt_6-13 | PFWFFY | −8.4 | −35.0189 | −27.4746 |
| DN58opt_7-9 | CRRHHPFW | −8.4 | −37.2646 | −38.2679 |
| DN58opt_9-9 | RRHHPFWFF | −8.4 | −40.1397 | −44.0312 |
| DN58opt_10-6 | YFCRRHHPFW | −8.4 | −82.5097 | −77.5954 |
| DN58opt_6-2 | WWCFYF | −8.3 | −39.4207 | −31.8778 |
| DN58opt_6-12 | HPFWFF | −8.3 | −34.4456 | −32.1598 |
| DN58opt_6-15 | WFFYRH | −8.3 | −38.8074 | −36.8260 |
| DN58opt_9-11 | HHPFWFFYR | −8.2 | −52.0706 | −46.9053 |
| DN58opt_8-2 | WWCFYFCR | −8.1 | −48.6330 | −44.6472 |
| DN58opt_8-11 | HHPFWFFY | −8.1 | −0.5408 | −2.2644 |
| DN58opt_9-1 | TWWCFYFCR | −8.1 | −20.8016 | −20.7366 |
| DN58opt_6-16 | FFYRHN | −8.0 | −17.8920 | −20.2634 |
| DN58opt_7-2 | WWCFYFC | −8.0 | −36.8728 | −31.1389 |
| DN58opt_7-11 | RHHPFWFF | −8.0 | −29.0445 | −24.6885 |
| Peptide | vdW | EEL | EPB | EN POLAR | ΔEbinding (kcal/mol) |
|---|---|---|---|---|---|
| DN58opt_8-13 | −50.4578 ± 0.3663 | −146.8887 ± 1.2591 | 163.4684 ± 1.2054 | −5.5725 ± 0.0259 | −39.4506 ± 0.4436 |
| DN58opt_10-6 | −93.4038 ± 0.3287 | −457.2968 ± 1.9056 | 482.7020 ± 1.7063 | −9.5968 ± 0.0205 | −77.5954 ± 0.5297 |
| DN58opt_9-9 | −58.7485 ± 0.3692 | −358.8480 ± 1.5831 | 381.1478 ± 1.4254 | −7.5825 ± 0.0244 | −44.0312 ± 0.5104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaidi, N.J.; Abdullah, A.A.; Heh, C.H.; Lin, C.-H.; Othman, R.; Ahmad Fuaad, A.A.H. Hit-to-Lead Short Peptides against Dengue Type 2 Envelope Protein: Computational and Experimental Investigations. Molecules 2022, 27, 3233. https://doi.org/10.3390/molecules27103233
Zaidi NJ, Abdullah AA, Heh CH, Lin C-H, Othman R, Ahmad Fuaad AAH. Hit-to-Lead Short Peptides against Dengue Type 2 Envelope Protein: Computational and Experimental Investigations. Molecules. 2022; 27(10):3233. https://doi.org/10.3390/molecules27103233
Chicago/Turabian StyleZaidi, Norburhanuddin Johari, Adib Afandi Abdullah, Choon Han Heh, Chun-Hung Lin, Rozana Othman, and Abdullah Al Hadi Ahmad Fuaad. 2022. "Hit-to-Lead Short Peptides against Dengue Type 2 Envelope Protein: Computational and Experimental Investigations" Molecules 27, no. 10: 3233. https://doi.org/10.3390/molecules27103233
APA StyleZaidi, N. J., Abdullah, A. A., Heh, C. H., Lin, C.-H., Othman, R., & Ahmad Fuaad, A. A. H. (2022). Hit-to-Lead Short Peptides against Dengue Type 2 Envelope Protein: Computational and Experimental Investigations. Molecules, 27(10), 3233. https://doi.org/10.3390/molecules27103233