
N-Dealkylation of Amines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Chemical Methods for N-Dealkylation of Amines
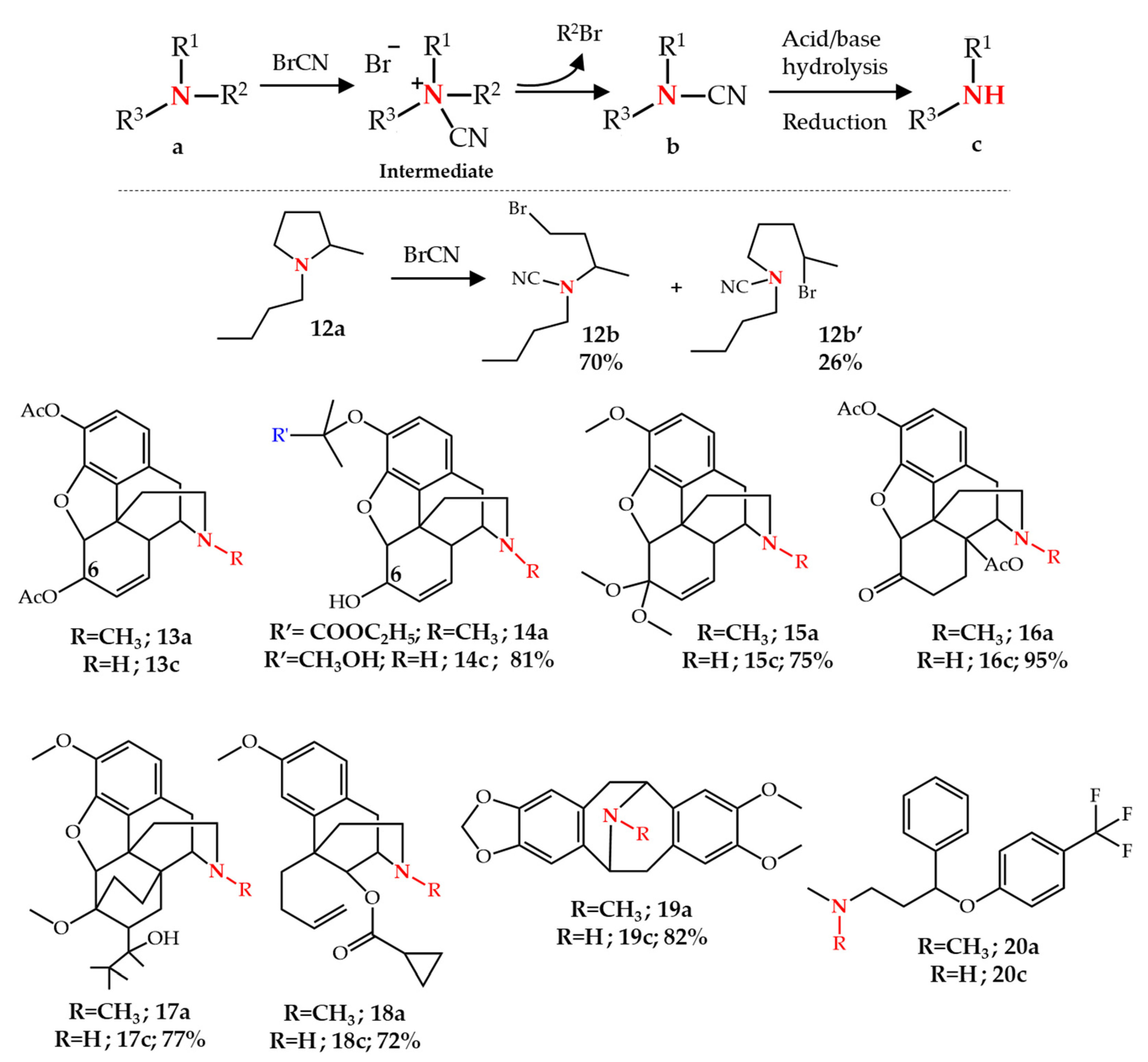
2.1. N-Dealkylation of Amines by the von Braun Reaction
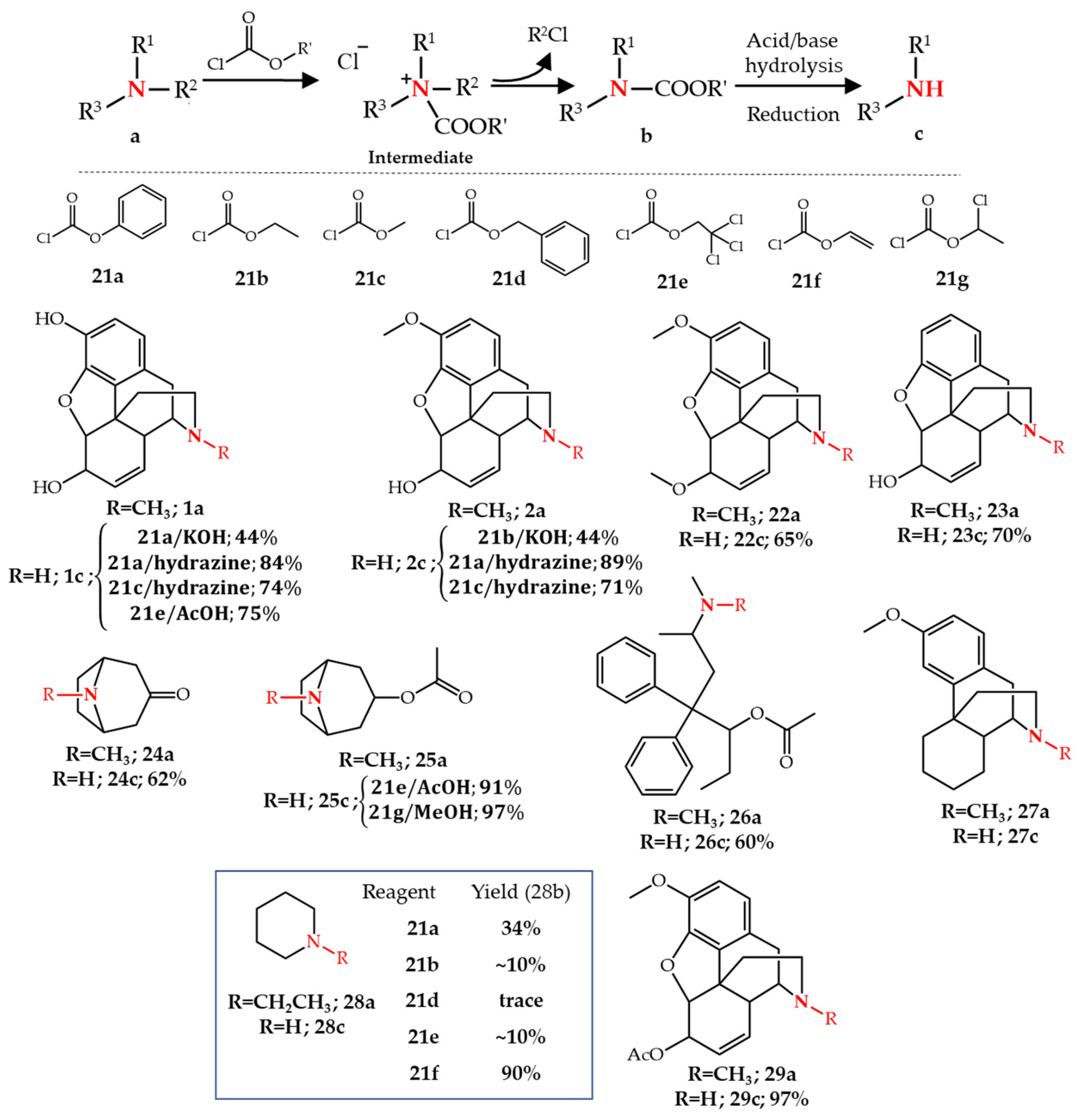
2.2. N-Dealkylation of Amines by Chloroformates
3. Transition-Metal Catalyzed N-Dealkylation
3.1. Palladium-Catalyzed
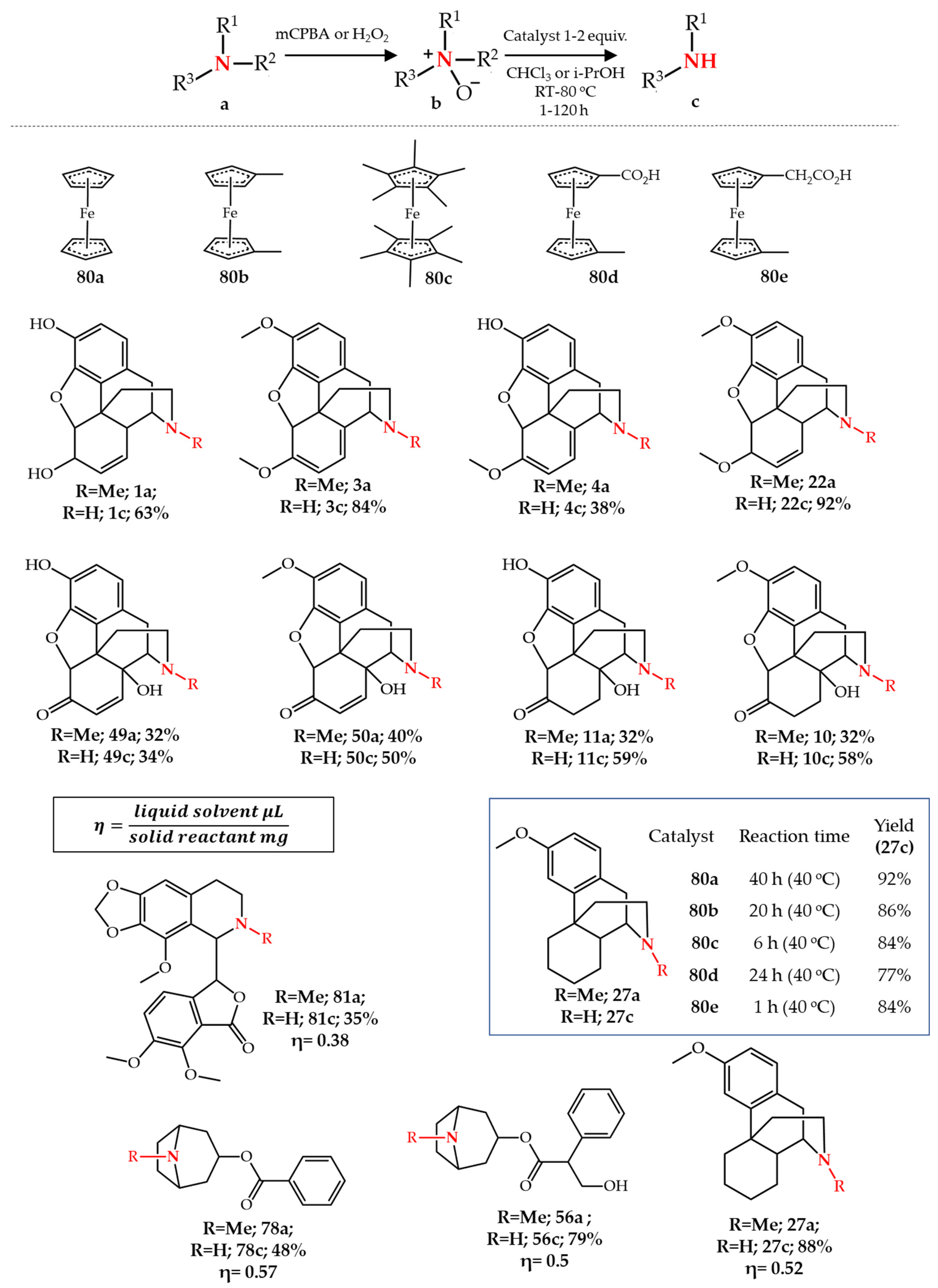
3.2. Iron-Catalyzed
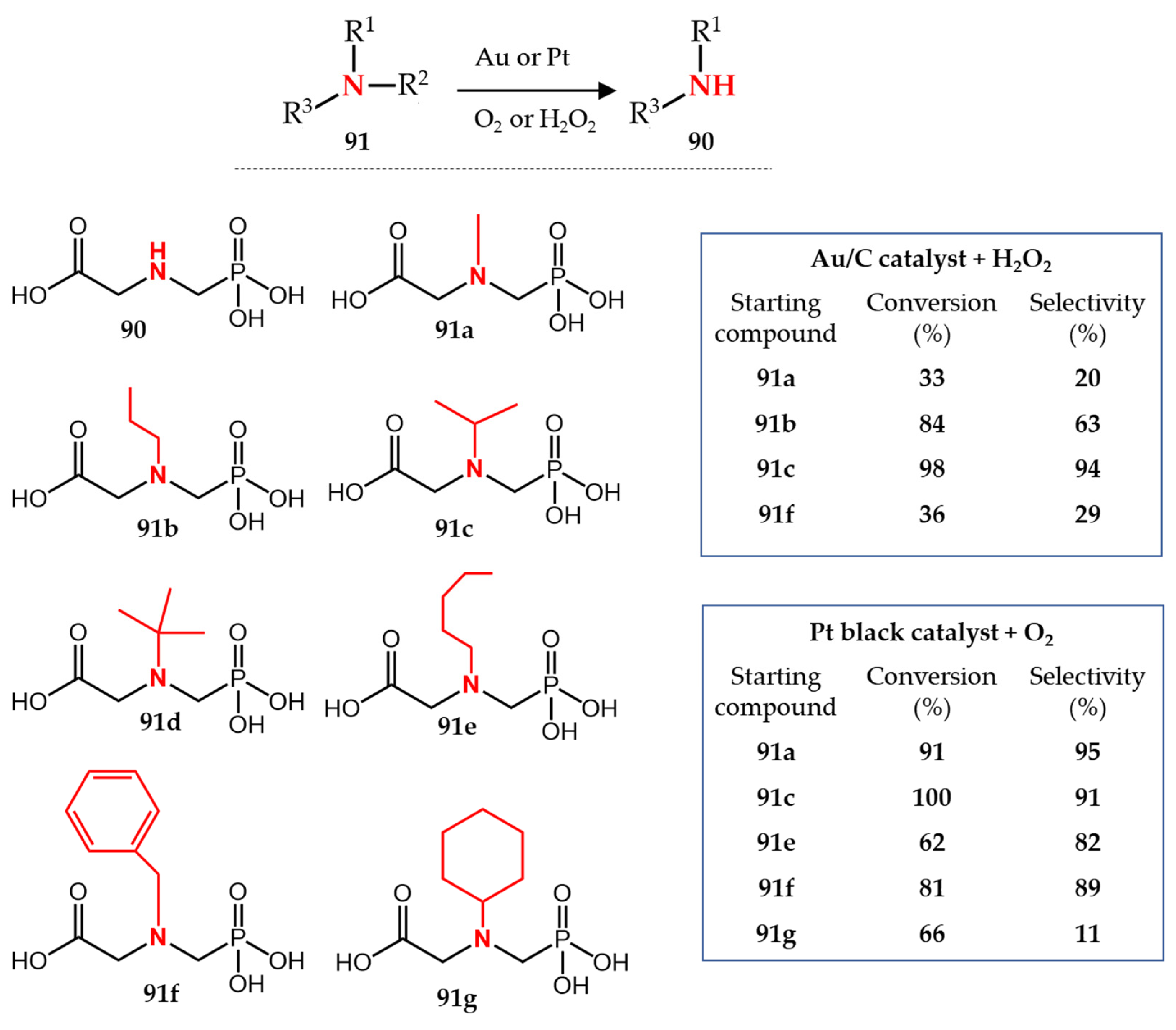
3.3. Gold- and Platinum-Catalyzed
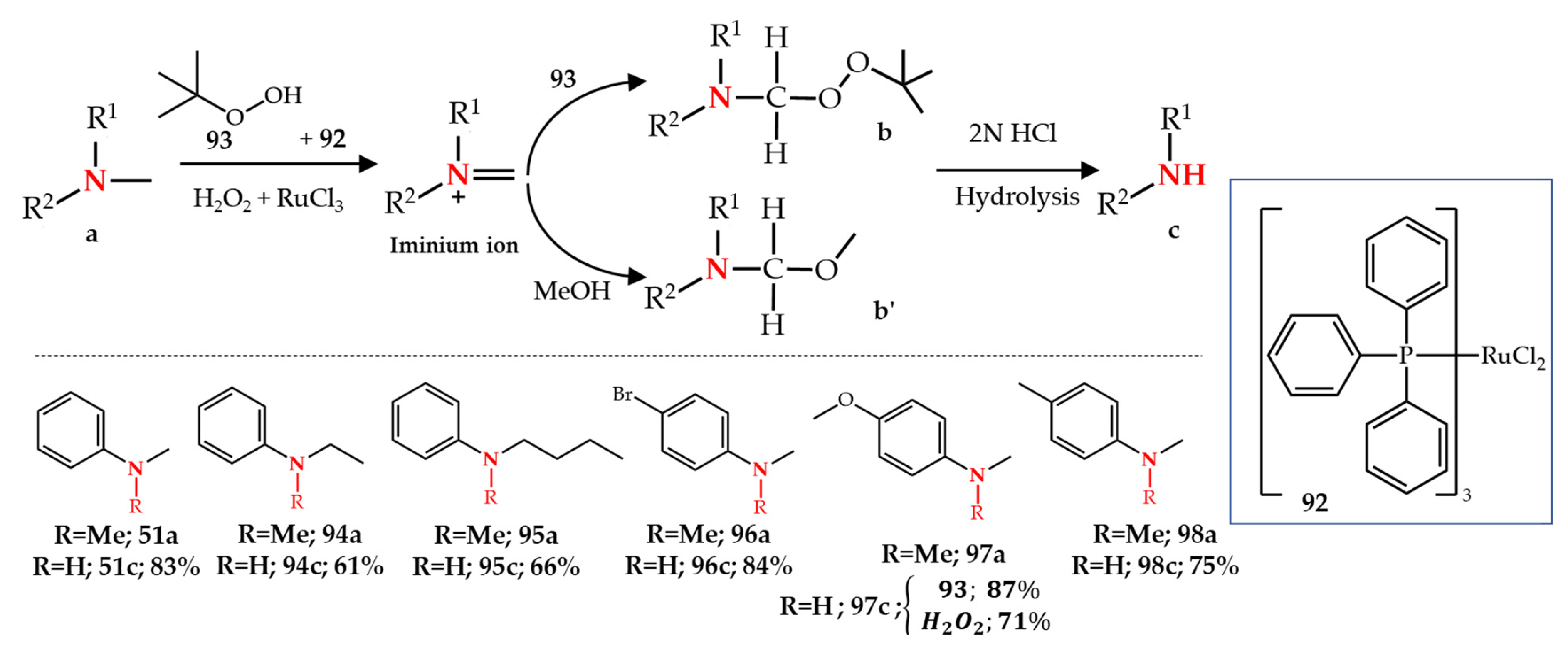
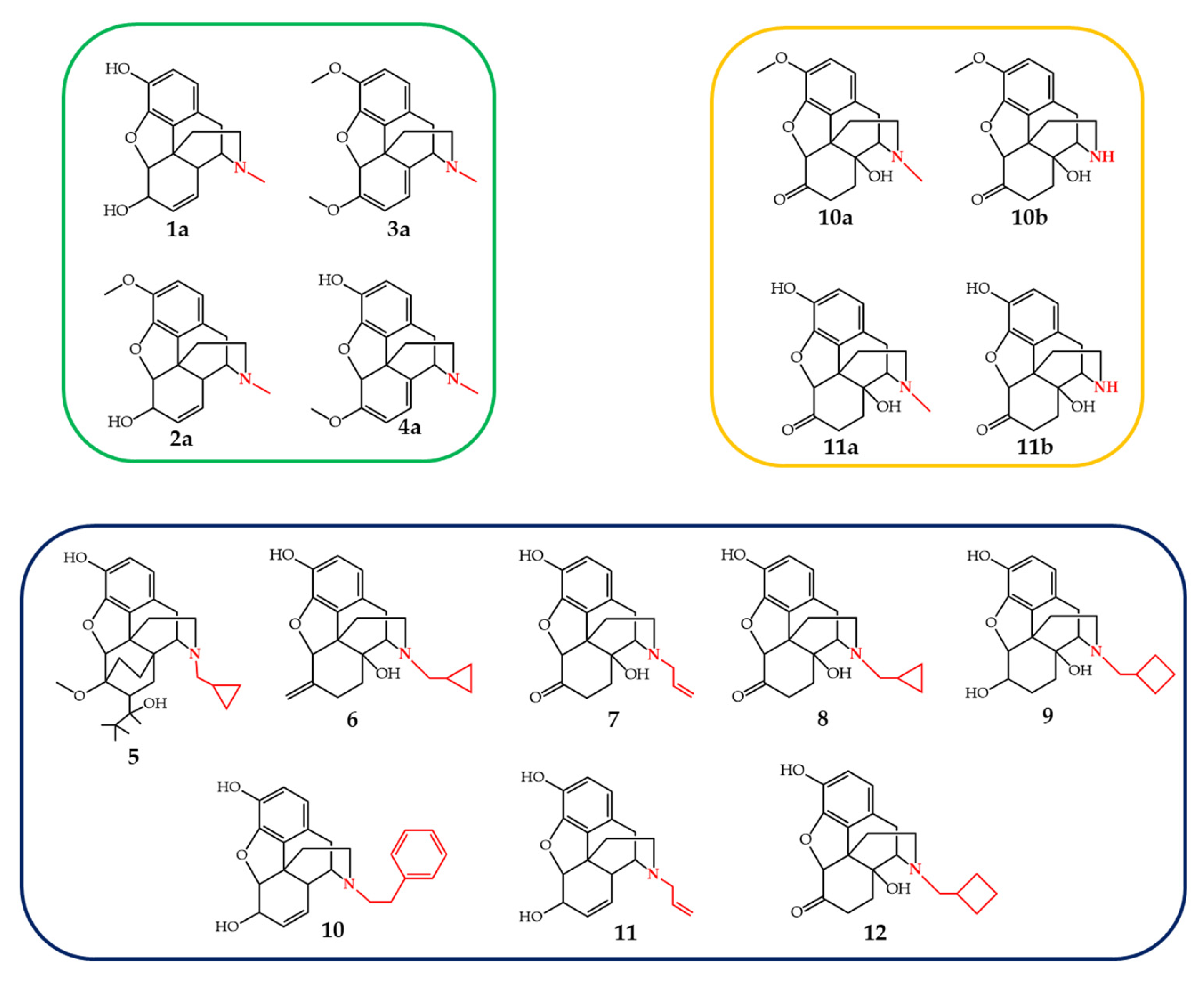
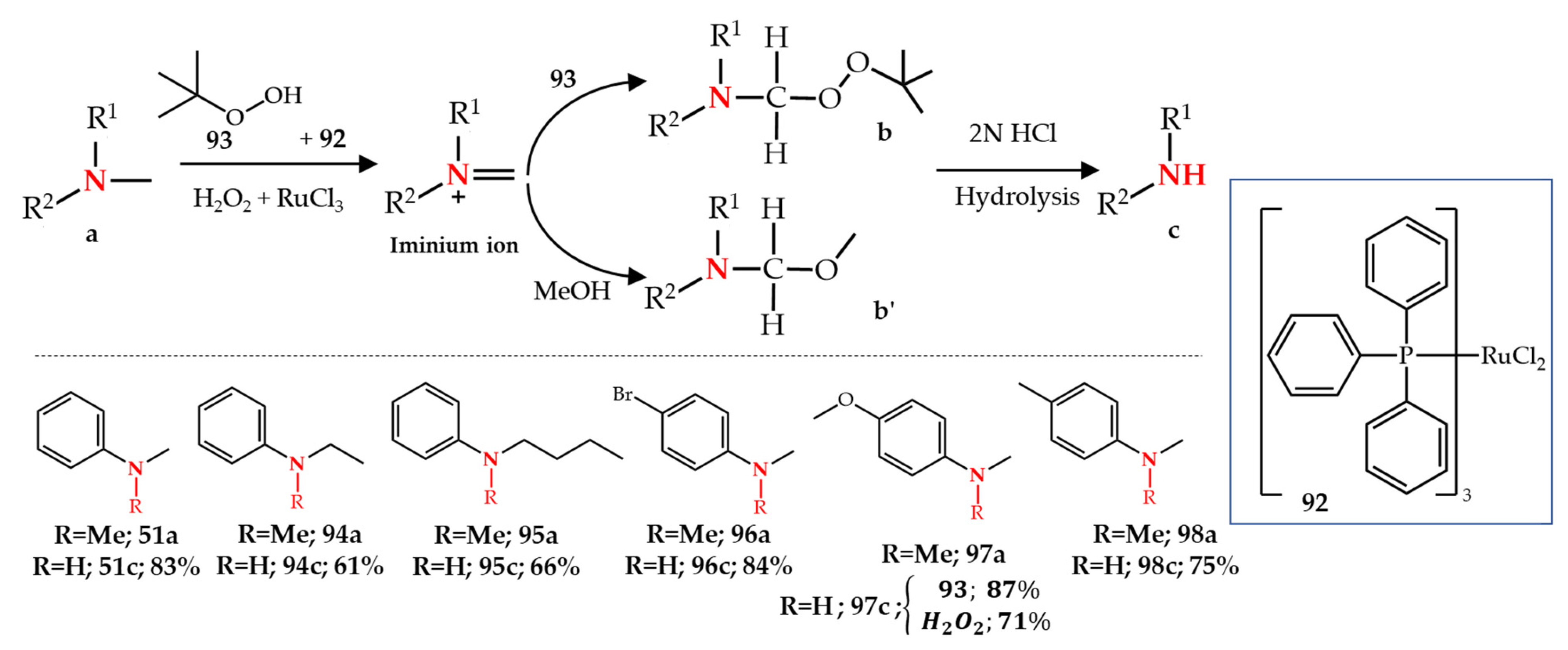
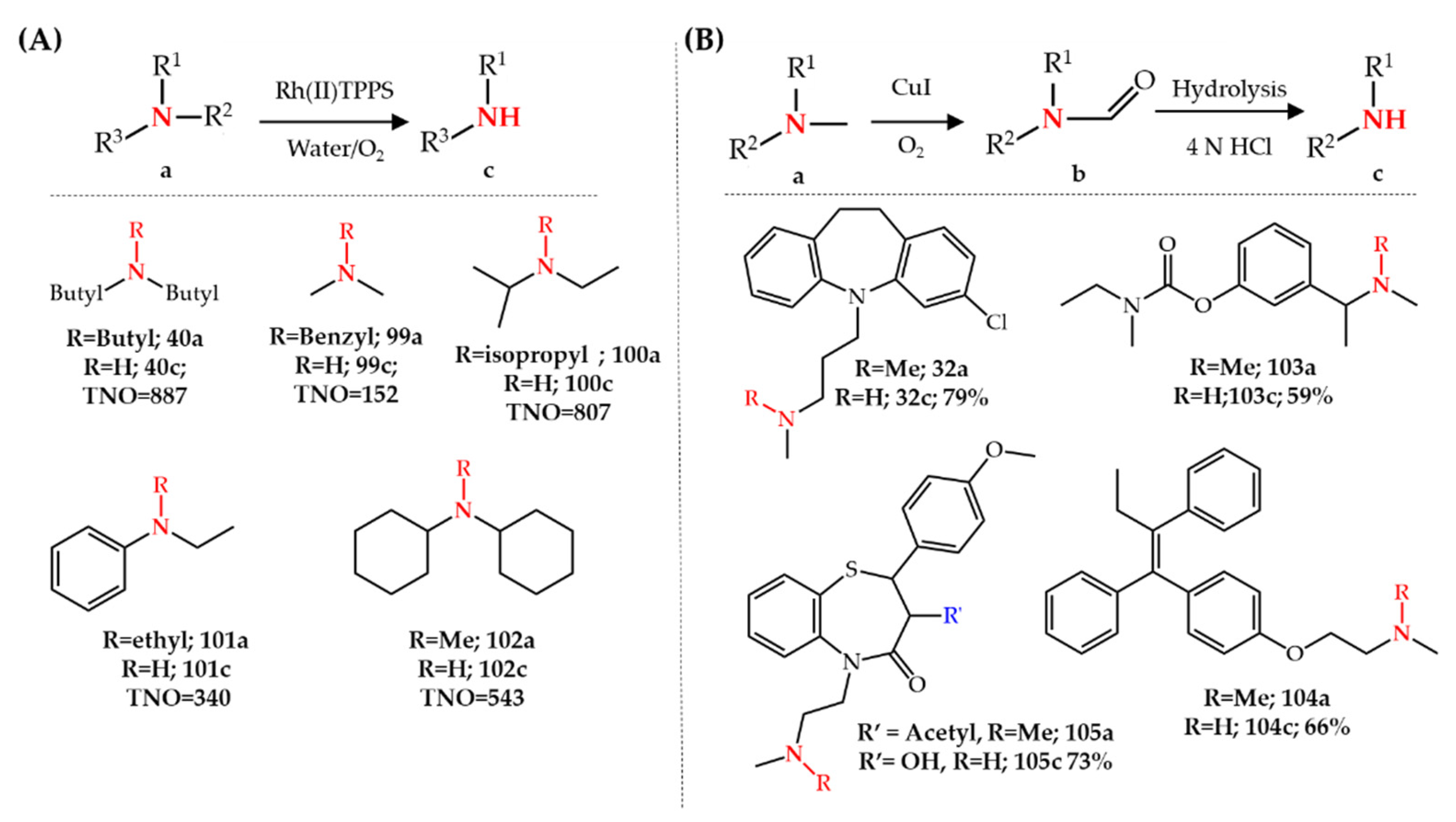
3.4. Ruthenium-, Rhodium- and Copper-Catalyzed
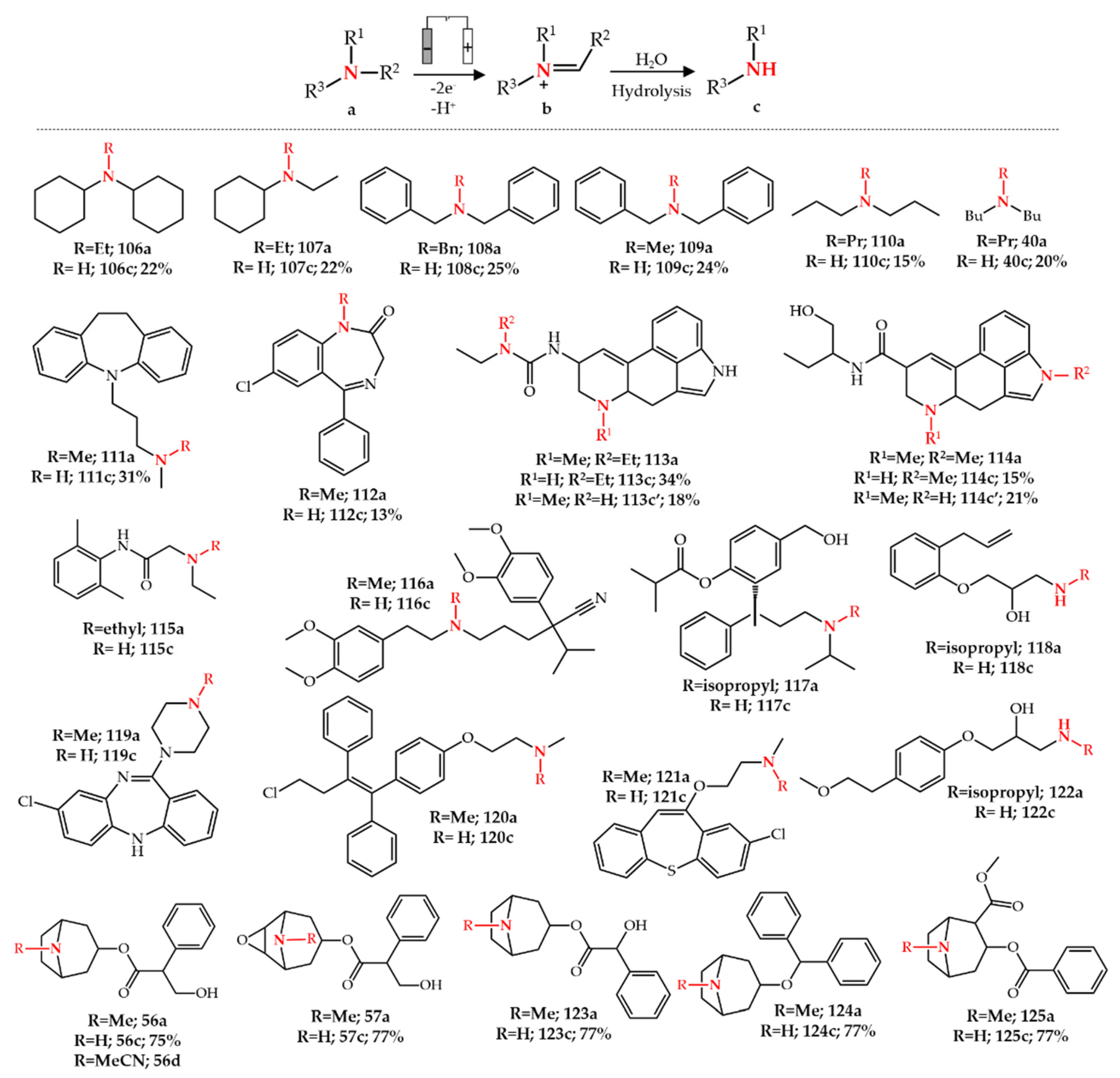
4. Electrochemical N-Dealkylation
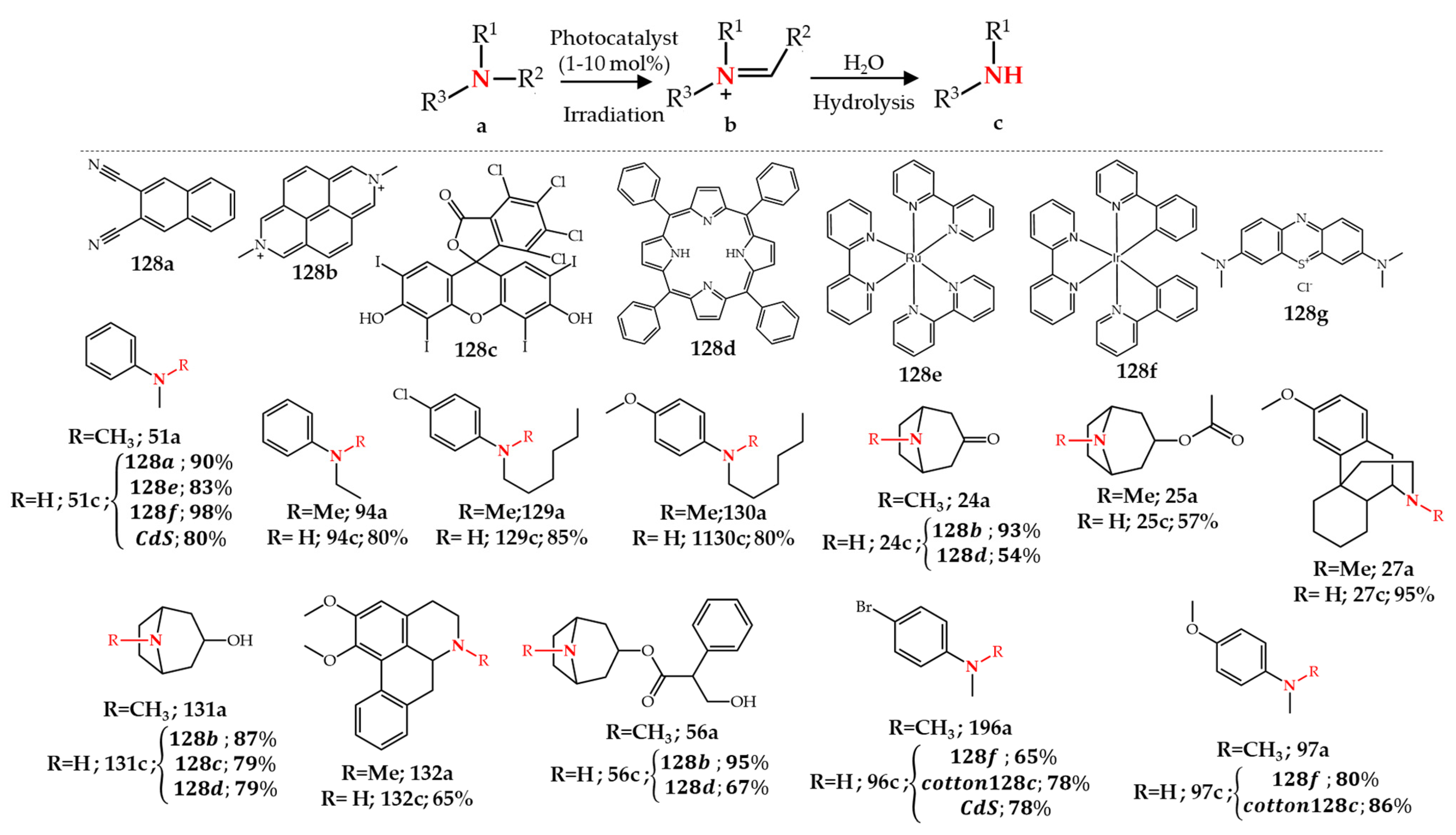
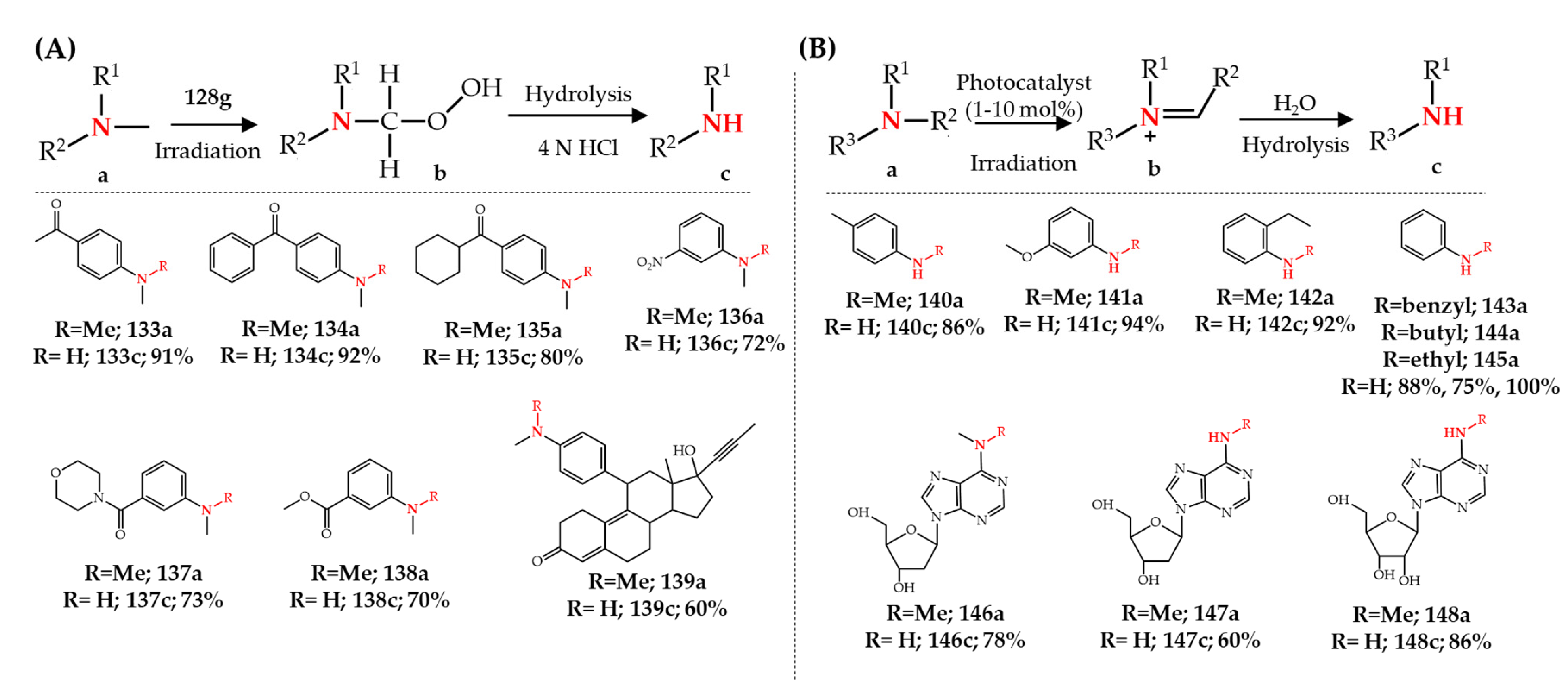
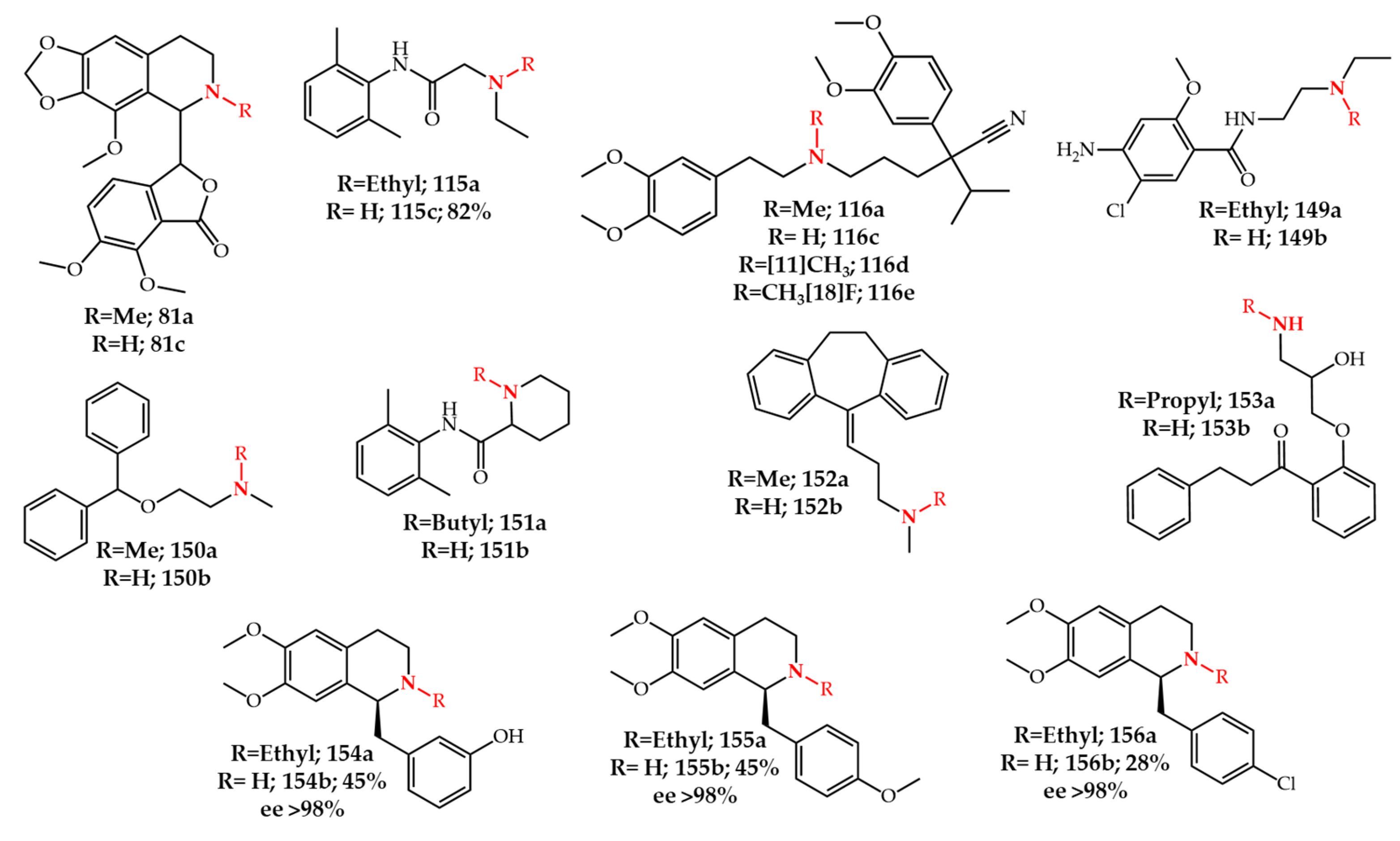
5. Photochemical N-Dealkylation
6. Enzymatic N-Dealkylation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Knaus, T.; Böhmer, W.; Mutti, F.G. Amine dehydrogenases: Efficient biocatalysts for the reductive amination of carbonyl compounds. Green Chem. 2017, 19, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, K.; Hao, W.; Zhang, W.X.; Xi, Z. Transition-metal-catalyzed cleavage of C-N single bonds. Chem. Rev. 2015, 115, 12045–12090. [Google Scholar] [CrossRef] [PubMed]
- McGrath, N.A.; Brichacek, M.; Njardarson, J.T. A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar] [CrossRef]
- Walker, D.K.; Jones, R.M.; Nedderman, A.N.R.; Wright, P.A. Primary, secondary and tertiary amines and their Isosteres. In Metabolism, Pharmacokinetics and Toxicity of Functional Groups; Royal Society of Chemistry: London, UK, 2010; pp. 168–209. [Google Scholar]
- Kärkäs, M.D. Electrochemical strategies for C-H functionalization and C-N bond formation. Chem. Soc. Rev. 2018, 47, 5786–5865. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Li, B.; Rickertsen, D.R.L.; Valles, D.A.; Seidel, D. C-H Bond functionalization of amines: A graphical overview of diverse methods. SynOpen 2021, 5, 173–228. [Google Scholar] [CrossRef]
- Rose, J.; Castagnoli, N. The metabolism of tertiary amines. Med. Res. Rev. 1983, 3, 73–88. [Google Scholar] [CrossRef]
- Nay, S.L.; O’Connor, T.R. Direct repair in mammalian cells. In New Research Directions in DNA Repair; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef] [Green Version]
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, J.L.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; et al. Key green chemistry research areas—A perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar] [CrossRef]
- Wang, Q.; Su, Y.; Li, L.; Huang, H. Transition-metal catalysed C-N bond activation. Chem. Soc. Rev. 2016, 45, 1257–1272. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Lu, J.; Shu, C.; Chen, Y.; Guo, T.; Wu, Q.; Yao, S.; Yin, P. A comparision of nalbuphine with morphine for analgesic effects and safety: Meta-analysis of randomized controlled trials. Sci. Rep. 2015, 5, 10927. [Google Scholar] [CrossRef] [Green Version]
- Najmi, A.A.; Bhat, F.; Bischoff, R.; Poelarends, G.; Permentier, H. TEMPO-mediated electrochemical N-demethylation of opiate alkaloids. ChemElectroChem 2021, 8, 2590–2596. [Google Scholar] [CrossRef]
- Gutmann, B.; Cantillo, D.; Weigl, U.; Cox, D.P.; Kappe, C.O. Design and development of Pd-catalyzed aerobic N-demethylation strategies for the synthesis of noroxymorphone in continuous flow mode. Eur. J. Org. Chem. 2017, 2017, 914–927. [Google Scholar] [CrossRef]
- Mata, A.; Cantillo, D.; Kappe, C.O. An integrated continuous-flow synthesis of a key oxazolidine intermediate to noroxymorphone from naturally occurring opioids. Eur. J. Org. Chem. 2017, 2017, 6505–6510. [Google Scholar] [CrossRef] [Green Version]
- Braun, J.V. Die einwirkung von bromcyan auf tertiäre amine. Ber. Dtsch. Chem. Ges. 1900, 33, 1438–1452. [Google Scholar] [CrossRef]
- Braun, J.V. Die Aufspaltung cyclischer Basen durch Bromcyan. Ber. Dtsch. Chem. Ges. 1907, 40, 3914–3933. [Google Scholar] [CrossRef] [Green Version]
- Braun, J.V.; Nugroho, M.B. Die Aufspaltung cyclischer Basen durch Bromcyan. Ber. Dtsch. Chem. Ges. 1909, 42, 2035–2057. [Google Scholar] [CrossRef]
- Li, J.J. Von Braun Reaction. In Name Reactions: A Collection of Detailed Reaction Mechanisms; Springer: Berlin/Heidelberg, Germany, 2003; p. 421. ISBN 978-3-662-05336-2. [Google Scholar]
- Hageman, H.A. The von Braun Cyanogen Bromide reaction. Org. React. 2004, 7, 198–262. [Google Scholar]
- Thavaneswaran, S.; Mccamley, K.; Scammells, P.J. N-Demethylation of alkaloids. Nat. Prod. Commun. 2006, 1, 2006. [Google Scholar] [CrossRef] [Green Version]
- Machara, A.; Hudlicky, T. Advances in N- and O-demethylation of opiates. Targets Heterocycl. Syst. 2016, 20, 113–138. [Google Scholar] [CrossRef]
- Braun, J.V. Untersuchungen über Morphium-Alkaloide. Ber. Dtsch. Chem. Ges. 1914, 47, 2312–2330. [Google Scholar] [CrossRef] [Green Version]
- Weijlard, J.; Erickson, A.E. N-Allylnormorphine. J. Am. Chem. Soc. 1942, 64, 869–870. [Google Scholar] [CrossRef]
- Selfridge, B.R.; Wang, X.; Zhang, Y.; Yin, H.; Grace, P.M.; Watkins, L.R.; Jacobson, A.E.; Rice, K.C. Structure-activity relationships of (+)-naltrexone-inspired toll-like receptor 4 (TLR4) antagonists. J. Med. Chem. 2015, 58, 5038–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentley, K.W.; Hardy, D.G. Novel analgesics and molecular rearrangements in the morphine-thebaine group. III. Alcohols of the 6, 14-endo-ethenotetrahydrooripavine series and derived analogs of N-allylnormorphine and -norcodeine. J. Am. Chem. Soc. 1967, 89, 3281–3292. [Google Scholar] [CrossRef] [PubMed]
- Mohacsi, E. Synthesis of metabolites of 3-O-t-butylmorphine. J. Heterocycl. Chem. 1987, 24, 471–472. [Google Scholar] [CrossRef]
- Rapoport, H.; Lovell, C.H.; Reist, H.R.; Warren, M.E. The synthesis of thebaine and northebaine from codeinone dimethyl ketal. J. Am. Chem. Soc. 1967, 89, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Elderfield, R.C.; Hageman, H.A. The von Braun cyanogen bromide reaction I. Application to pyrrolidines and ethylenimines. J. Org. Chem. 1949, 14, 605–637. [Google Scholar] [CrossRef]
- Iijima, I.; Minamikawa, J.; Jacobson, A.E.; Brossi, A.; Rice, K.C.; Klee, W.A. Studies in the (+)-morphinan series. 5. Synthesis and biological properties of (+)-naloxone. J. Med. Chem. 1978, 21, 398–400. [Google Scholar] [CrossRef]
- Marton, J.; Szabó, Z.; Garadnay, S.; Miklós, S.; Makleit, S. Studies on the synthesis of β-thevinone derivatives. Tetrahedron 1998, 54, 9143–9152. [Google Scholar] [CrossRef]
- Saucier, M.; Daris, J.P.; Lambert, Y.; Monkovic, I.; Pircio, A.W. 5-Allyl-9-oxobenzomorphans. 3. Potent narcotic antagonists and analgesics-antagonists in the series of substituted 2′,9beta-dihydroxy-6,7-benzomorphans. J. Med. Chem. 1977, 20, 676–682. [Google Scholar] [CrossRef]
- Machara, A.; Endoma-Arias, M.A.A.; Císařová, I.; Cox, D.P.; Hudlicky, T. Direct synthesis of noroxymorphone from thebaine: Unusual CeIV oxidation of a methoxydiene-iron complex to an enone-γ-nitrate. Eur. J. Org. Chem. 2016, 2016, 1500–1503. [Google Scholar] [CrossRef]
- Saxena, N.S.; Saxena, K.; Sata, B. Industrial Process for the Preparation of Buprenorphine and Its Intermediates. U.S. Patent B2,8,981,097, 17 March 2015. [Google Scholar]
- Lee, S.-S.; Liu, Y.-C.; Chang, S.-H.; Chen, C.-H. N-Demethylation studies of pavine alkaloids. Heterocycles 1993, 36, 1971–1974. [Google Scholar] [CrossRef]
- Molloy, B.B.; Schmiegel, K.K. Arloxyphenylpropylamines. U.S. Patent 4,314,081, 2 February 1982. [Google Scholar]
- Lawrence, A. Substitution on the Amine Nitrogen. In Category 5, Compounds with One Saturated Carbon Heteroatom Bond; Schaumann, E., Ed.; Science of Synthesis; Georg Thieme Verlag: Stuttgart, Germany, 2009; Volume 40a, ISBN 9783131839510. [Google Scholar]
- Cooley, J.H.; Evain, E.J. Amine dealkylations with acyl chlorides. Synthesis 1989, 1, 1–7. [Google Scholar] [CrossRef]
- Abdel-Monem, M.M.; Portoghese, P.S. N-Demethylation of morphine and structurally related compounds with chloroformate esters. J. Med. Chem. 1972, 15, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.K. An improved procedure for the N-Demethylation of 6,7-benzomorphans, morphine, and codeine. J. Org. Chem. 1975, 40, 1850–1851. [Google Scholar] [CrossRef] [PubMed]
- Rice, K.C.; May, E.L. Procedural refinements in the N-demethylation of morphine and codeine using phenyl chloroformate and hydrazine. J. Heterocycl. Chem. 1977, 14, 665–666. [Google Scholar] [CrossRef]
- Brine, G.A.; Boldt, K.G.; Hart, C.K.; Carroll, F.I. The N-demethylation of morphine and codeine using methyl chloroformate. Org. Prep. Proced. Int. 1976, 8, 103–106. [Google Scholar] [CrossRef]
- Dhar, N.C.; Maehr, R.B.; Masterson, L.A.; Midgley, J.M.; Stenlake, J.B.; Wastila, W.B. Approaches to short-acting neuromuscular blocking agents: Nonsymmetrical bis-tetrahydroisoquinolinium mono- and diesters. J. Med. Chem. 1996, 39, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Csutoras, C.; Zhang, A.; Bidlack, J.M.; Neumeyer, J.L. An investigation of the N-demethylation of 3-deoxymorphine and the affinity of the alkylation products to μ, δ, and κ receptors. Bioorgan. Med. Chem. 2004, 12, 2687–2690. [Google Scholar] [CrossRef]
- Montzka, T.A.; Matiskella, J.D.; Partyka, R.A. 2,2,2-trichloroethyl chloroformate: A general reagent for demethylation of tertiary methylamines. Tetrahedron Lett. 1974, 15, 1325–1327. [Google Scholar] [CrossRef]
- Jozwiak, K.; Targowska-Duda, K.M.; Kaczor, A.A.; Kozak, J.; Ligeza, A.; Szacon, E.; Wrobel, T.M.; Budzynska, B.; Biala, G.; Fornal, E.; et al. Synthesis, in vitro and in vivo studies, and molecular modeling of N-alkylated dextromethorphan derivatives as non-competitive inhibitors of α3β4 nicotinic acetylcholine receptor. Bioorg. Med. Chem. 2014, 22, 6846–6856. [Google Scholar] [CrossRef]
- Peet, N.P. N-demethylation of dextromethorphan. J. Pharm. Sci. 1980, 69, 1447–1448. [Google Scholar] [CrossRef]
- Olofson, R.A.; Schnur, R.C.; Bunes, L.; Pepe, J.P. Selective N-dealkylation of tertiary amines with vinyl chloroformate: An improved synthesis of naloxone. Tetrahedron Lett. 1977, 18, 1567–1570. [Google Scholar] [CrossRef]
- Sándor, H.; Simon, C.; Sándor, M. Synthesis of N-demethyl-N-substituted-14-hydroxycodeine and morphine derivatives. Synth. Commun. 1992, 22, 2527–2541. [Google Scholar] [CrossRef]
- Kobylecki, R.J.; Carling, R.W.; Lord, J.A.H.; Smith, C.F.C.; Lane, A.C. Common anionic receptor site hypothesis: Its relevance to the antagonist action of naloxone. J. Med. Chem. 1982, 25, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Olofson, A.R.; Martz, T.J.; Senet, J.-P.; Piteau, M.; Malfroot, T. A New reagent for the selective, high-yield N-dealkylation of tertiary amines: Improved syntheses of naltrexone and nalbuphine. J. Org. Chem. 1984, 49, 2081–2082. [Google Scholar] [CrossRef]
- Jin, C.; Boldt, K.G.; Rehder, K.S.; Brine, G.A. Improved syntheses of N-desmethylcitalopram and N,N-didesmethylcitalopram. Synth. Commun. 2007, 37, 901–908. [Google Scholar] [CrossRef]
- Pelander, A.; Ojanperä, I.; Hase, T.A. Preparation of N-demethylated drug metabolites for analytical purposes using 1-chloroethyl chloroformate. Forensic Sci. Int. 1997, 85, 193–198. [Google Scholar] [CrossRef]
- Hengeveld, J.E.; Gupta, A.K.; Kemp, A.H.; Thomas, A.V. Facile N-demethylation of erythromycins. Tetrahedron Lett. 1999, 40, 2497–2500. [Google Scholar] [CrossRef]
- Flynn, E.H.; Murphy, H.W.; McMahon, R.E. Erythromycin. II. Des-N-methylerythromycin and N-methyl-C14-erythromycin. J. Am. Chem. Soc. 1955, 77, 3104–3106. [Google Scholar] [CrossRef]
- Kim, J.C. A high yield conversion of N-norapomorphine from apomorphine. Arch. Pharm. Res. 1983, 6, 137–140. [Google Scholar] [CrossRef]
- Michael Crider, A.; Grubb, R.; Bachmann, K.A.; Rawat, A.K. Convenient synthesis of 6-nor-9,10-dihydrolysergic acid methyl ester. J. Pharm. Sci. 1981, 70, 1319–1321. [Google Scholar] [CrossRef]
- Časar, Z.; Mesar, T. A DMAP-catalyzed approach to the industrial-scale preparation of N-6-demethylated 9,10-dihydrolysergic acid methyl ester: A key cabergoline and pergolide precursor. Org. Process Res. Dev. 2015, 19, 378–385. [Google Scholar] [CrossRef]
- Suzuki, Y.; Iwata, M.; Yazaki, R.; Kumagai, N.; Shibasaki, M. Concise enantioselective synthesis of duloxetine via direct catalytic asymmetric aldol reaction of thioamide. J. Org. Chem. 2012, 77, 4496–4500. [Google Scholar] [CrossRef] [PubMed]
- Deeter, J.; Frazier, J.; Staten, G.; Staszak, M.; Weigel, L. Asymmetric synthesis and absolute stereochemistry of LY248686. Tetrahedron Lett. 1990, 31, 7101–7104. [Google Scholar] [CrossRef]
- Reiter, J.; Budai, Z.; Simig, G.; Balkso, G.; Imre, J.; Nagy, K.; Tompe, P. Process for the Preparation of Fluoxetine, World Intellectual Property Organization. CH Patent Application WO 98/11054, 19 March 1998. [Google Scholar]
- International Narcotics Control Board. Narcotics Drugs 2019: Estimated World Requirements for 2020-Statistics for 2018; United Nations: Washington, DC, USA, 2020; ISBN 9789211483345. [Google Scholar]
- International Narcotics Control Board. Psychotropic Substances 2020: Statistics for 2019-Assessments of Annual Medical and Scientific Requirements for 2021; United Nations: Washington, DC, USA, 2020; ISBN 9789210481342. [Google Scholar]
- Office of the Federal Register. National Archives and Records Administration Federal Register; Office of the Federal Register: Washington, DC, USA, 2020; Volume 85, pp. 76604–76613.
- Murahashi, S.I.; Watanabe, T. Palladium catalyzed hydrolysis of tertiary amines with water. J. Am. Chem. Soc. 1979, 101, 7429–7430. [Google Scholar] [CrossRef]
- Carroll, R.J.; Leisch, H.; Scocchera, E.; Hudlicky, T.; Cox, D.P. Palladium-Catalyzed N-Demethylation/N-acylation of some morphine and tropane alkaloids. Adv. Synth. Catal. 2008, 350, 2984–2992. [Google Scholar] [CrossRef]
- Machara, A.; Werner, L.; Endoma-Arias, M.A.; Cox, D.P.; Hudlicky, T. Improved synthesis of buprenorphine from thebaine and/or oripavine via palladium-catalyzed N-demethylation/acylation and/or concomitant O-demethylation. Adv. Synth. Catal. 2012, 354, 613–626. [Google Scholar] [CrossRef]
- Machara, A.; Cox, D.P.; Hudlicky, T. Direct synthesis of naltrexone by palladium-catalyzed N-demethylation/ acylation of oxymorphone: The benefit of C-H activation and the intramolecular acyl transfer from C-14 hydroxy. Adv. Synth. Catal. 2012, 354, 2713–2718. [Google Scholar] [CrossRef]
- Gutmann, B.; Elsner, P.; Cox, D.P.; Weigl, U.; Roberge, D.M.; Kappe, C.O. Toward the synthesis of noroxymorphone via aerobic palladium-catalyzed continuous flow N-demethylation strategies. ACS Sustain. Chem. Eng. 2016, 4, 6048–6061. [Google Scholar] [CrossRef]
- Gutmann, B.; Weigl, U.; Cox, D.P.; Kappe, C.O. Batch- and continuous-flow aerobic oxidation of 14-hydroxy opioids to 1,3-oxazolidines—A concise synthesis of noroxymorphone. Chem. Eur. J. 2016, 22, 10393–10398. [Google Scholar] [CrossRef]
- Murata, S.; Miura, M.; Nomura, M. Oxidation of 3- or 4-substituted N,N-dimethylanilines with molecular oxygen in the presence of either ferric chloride or [Fe(salen)]OAc. J. Org. Chem. 1989, 54, 4700–4702. [Google Scholar] [CrossRef]
- Murata, S.; Miura, M.; Nomura, M. Iron-catalysed oxidation of N,N-dimethylaniline with molecular oxygen. J. Chem. Soc. Chem. Commun. 1989, 24, 116–118. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Spencer, L.; Sugimoto, H. [FeII(MeCN)4]2+(ClO4−)2 and [FeIIICl33] as Mimics for the catalytic centers of peroxidase, catalase and cvtochrome P-450. Isr. J. Chem. 1987, 28, 3–12. [Google Scholar] [CrossRef]
- Santa, T.; Miyata, N.; Hirobe, M. Oxidation of tertiary amines and sulfides by an iron(III) porphyrin-O2-Na2S2O4 system as a model of cytochrome P-450. Chem. Pharm. Bull. 1984, 32, 1252–1255. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.R.L.; Mortimer, D.N. Oxidative N-dealkylation of N,N-dimethylbenzylamines by metalloporphyrin-catalysed model systems for cytochrome P450 mono-oxygenases. J. Chem. Soc. Chem. Commun. 1985, 2, 64–65. [Google Scholar] [CrossRef]
- Naróg, D.; Lechowicz, U.; Pietryga, T.; Sobkowiak, A. Iron(II, III)-catalyzed oxidative N-dealkylation of amines with dioxygen. J. Mol. Catal. A Chem. 2004, 212, 25–33. [Google Scholar] [CrossRef]
- Do Pham, D.D.; Kelso, G.F.; Yang, Y.; Hearn, M.T.W.W. One-pot oxidative N-demethylation of tropane alkaloids with hydrogen peroxide and a FeIII-TAML catalyst. Green Chem. 2012, 14, 1189–1195. [Google Scholar] [CrossRef]
- Do Pham, D.D.; Kelso, G.F.; Yang, Y.; Hearn, M.T.W.W. Studies on the oxidative N-demethylation of atropine, thebaine and oxycodone using a FeIII-TAML catalyst. Green Chem. 2014, 16, 1399–1409. [Google Scholar] [CrossRef]
- Grierson, D. The Polonovski reaction. Org. React. 2004, 39, 85–295. [Google Scholar]
- Fish, M.S.; Johnson, N.M.; Lawrence, E.P.; Horning, E.C. Oxidative N-dealkylation. Biochim. Biophys. Acta 1955, 18, 564–565. [Google Scholar] [CrossRef]
- Fish, M.S.; Johnson, N.M.; Horning, E.C. t-Amine oxide rearrangements. N,N-Dimethyltryptamine oxide. J. Am. Chem. Soc. 1956, 78, 3668–3671. [Google Scholar] [CrossRef]
- Sweeley, C.C.; Horning, E.C. Rearrangement and decarboxylation reactions of N,N-dimethylglycine oxide. J. Am. Chem. Soc. 1957, 79, 2620–2625. [Google Scholar] [CrossRef]
- Fish, M.S.; Sweeley, C.C.; Johnson, N.M.; Lawrence, E.P.; Horning, E.C. Chemical and enzymic rearrangements of N,N-dimethyl amino acid oxides. Biochim. Biophys. Acta 1956, 21, 196–197. [Google Scholar] [CrossRef]
- Monković, I.; Wong, H.; Bachand, C. Secondary amines from the iron(II) ion-catalyzed reaction of amine oxides: A general method for the dealkylation of tertiary amines. Synthesis 1985, 1985, 770–773. [Google Scholar] [CrossRef]
- Mary, A.; Renko, D.Z.; Guillou, C.; Thal, C. Selective N-demethylation of galanthamine to norgalanthamine via a non classical Polonovski reaction. Tetrahedron Lett. 1997, 38, 5151–5152. [Google Scholar] [CrossRef]
- McCamley, K.; Ripper, J.A.; Singer, R.D.; Scammells, P.J. Efficient N-demethylation of opiate alkaloids using a modified nonclassical Polonovski reaction. J. Org. Chem. 2003, 68, 9847–9850. [Google Scholar] [CrossRef]
- Thavaneswaran, S.; Scammells, P.J. Further investigation of the N-demethylation of tertiary amine alkaloids using the non-classical Polonovski reaction. Bioorg. Med. Chem. Lett. 2006, 16, 2868–2871. [Google Scholar] [CrossRef]
- Dong, Z.; Scammells, P.J. New methodology for the N-demethylation of opiate alkaloids. J. Org. Chem. 2007, 72, 9881–9885. [Google Scholar] [CrossRef]
- Kok, G.; Ashton, T.D.; Scammells, P.J. An improved process for the N-demethylation of opiate alkaloids using an iron(II) catalyst in acetate buffer. Adv. Synth. Catal. 2009, 351, 283–286. [Google Scholar] [CrossRef]
- Kok, G.B.; Scammells, P.J. N-Demethylation of N-methyl alkaloids with ferrocene. Bioorg. Med. Chem. Lett. 2010, 20, 4499–4502. [Google Scholar] [CrossRef]
- Kok, G.B.; Scammells, P.J. Polonovski-type N-demethylation of N-methyl alkaloids using substituted ferrocene redox catalysts. Synthesis 2012, 44, 2587–2594. [Google Scholar] [CrossRef]
- Kok, G.B.; Pye, C.C.; Singer, R.D.; Scammells, P.J. Two-step iron(0)-mediated N-demethylation of N-methyl alkaloids. J. Org. Chem. 2010, 75, 4806–4811. [Google Scholar] [CrossRef] [PubMed]
- Kok, G.B.; Scammells, P.J. Efficient iron-catalyzed N-demethylation of tertiary amine-N-oxides under oxidative conditions. Aust. J. Chem. 2011, 64, 1515–1521. [Google Scholar] [CrossRef]
- Kok, G.B.; Scammells, P.J. Further investigations into the N-demethylation of oripavine using iron and stainless steel. Org. Biomol. Chem. 2011, 9, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Savage, G.P.; Saubern, S.; Scammells, P.J.; Polyzos, A. A multi-step continuous flow process for the N-demethylation of alkaloids. Aust. J. Chem. 2013, 66, 178–182. [Google Scholar] [CrossRef]
- Awalt, J.K.; Lam, R.; Kellam, B.; Graham, B.; Scammells, P.J.; Singer, R.D. Utility of iron nanoparticles and a solution-phase iron species for the N-demethylation of alkaloids. Green Chem. 2017, 19, 2587–2594. [Google Scholar] [CrossRef]
- Awalt, J.K.; Scammells, P.J.; Singer, R.D. liquid assisted grinding (LAG) for the N-demethylation of alkaloids. ACS Sustain. Chem. Eng. 2018, 6, 10052–10057. [Google Scholar] [CrossRef]
- Berényi, S.; Gyulai, Z.; Udvardy, A.; Sipos, A. One-pot N-dealkylation and acid-catalyzed rearrangement of morphinans into aporphines. Tetrahedron Lett. 2010, 51, 1196–1198. [Google Scholar] [CrossRef]
- Smith, C.; Purcell, S.; Waddell, L.; Hayes, N.; Ritchie, J. World Intellectual Property Organization. CH Patent Application WO 2005/028483A1, 20 January 2005. [Google Scholar]
- Werner, L.; Wernerova, M.; MacHara, A.; Endoma-Arias, M.A.; Duchek, J.; Adams, D.R.; Cox, D.P.; Hudlicky, T. Unexpected N-demethylation of oxymorphone and oxycodone N-oxides mediated by the burgess reagent: Direct synthesis of naltrexone, naloxone, and other antagonists from oxymorphone. Adv. Synth. Catal. 2012, 354, 2706–2712. [Google Scholar] [CrossRef]
- Endoma-Arias, M.A.A.; Cox, D.P.; Hudlicky, T. General method of synthesis for naloxone, naltrexone, nalbuphone, and nalbuphine by the reaction of Grignard reagents with an oxazolidine derived from oxymorphone. Adv. Synth. Catal. 2013, 355, 1869–1873. [Google Scholar] [CrossRef]
- Singh, G.; Koerner, T.B.; Godefroy, S.B.; Armand, C. N-demethylation of cyamemazine via non-classical Polonovski reaction and its conjugation to bovine serum albumin. Bioorg. Med. Chem. Lett. 2012, 22, 2160–2162. [Google Scholar] [CrossRef]
- Fődi, T.; Ignácz, G.; Decsi, B.; Béni, Z.; Túrós, G.I.; Kupai, J.; Weiser, D.B.; Greiner, I.; Huszthy, P.; Balogh, G.T. Biomimetic synthesis of drug metabolites in batch and continuous-flow reactors. Chem. Eur. J. 2018, 24, 9385–9392. [Google Scholar] [CrossRef] [PubMed]
- Antier, C.; Kudsk, P.; Reboud, X.; Ulber, L.; Baret, P.V.; Messéan, A. Glyphosate use in the European agricultural sector and a framework for its further monitoring. Sustainability 2020, 12, 5682. [Google Scholar] [CrossRef]
- Pyrjaev, P.A.; Yushchenko, D.Y.; Moroz, B.L.; Pai, Z.P.; Bukhtiyarov, V.I. Aqueous-phase oxidation of N-substituted N-phosphonomethyl glycines into glyphosate with hydrogen peroxide in the presence of carbon-supported gold catalysts. Chem. Select 2019, 4, 10756–10764. [Google Scholar] [CrossRef]
- Yushchenko, D.Y.; Khlebnikova, T.B.; Pai, Z.P.; Bukhtiyarov, V.I. Glyphosate: Methods of synthesis. Kinet. Catal. 2021, 62, 331–341. [Google Scholar] [CrossRef]
- Morgenstern, D. Process for the Preparation of N-(Phosphonomethyl)glycine by Oxidizing N-Substituted N-(Phosphonomethyl)glycine. U.S. Patent 7,297,657 B2, 20 November 2007. [Google Scholar]
- Yushchenko, D.Y.; Simonov, P.A.; Khlebnikova, T.B.; Pai, Z.P.; Bukhtiyarov, V.I. Oxidation of N-isopropyl phosphonomethyl glycine with hydrogen peroxide catalyzed by carbon-supported gold nanoparticles. Catal. Commun. 2019, 121, 57–61. [Google Scholar] [CrossRef]
- Parry, D.R.; Tomlin, C.D.S. Preparation of N-Phosphonomethylglycine. U.S. Patent 3,956,370, 11 May 1976. [Google Scholar]
- Gaertner, R.V. Process for Producing N-Phosphonomethyl Glycine. U.S. Patent 3,927,080, 16 December 1975. [Google Scholar]
- Miller, H.W.; Balthazor, M.T. Thermal Dealkylation of N-Alkyl N-Phosphonomethylglycine. U.S. Patent 5,068,404, 26 November 1991. [Google Scholar]
- Murahashi, S.S.; Naota, T.; Yonemura, K. Ruthenium-catalyzed cytochrome P-450 type oxidation of tertiary amines with alkyl hydroperoxides. J. Am. Chem. Soc. 1988, 110, 8256–8258. [Google Scholar] [CrossRef]
- Murahashi, S.I.; Naota, T.; Miyaguchi, N.; Nakato, T. Ruthenium-catalyzed oxidation of tertiary amines with hydrogen peroxide in the presence of methanol. Tetrahedron Lett. 1992, 33, 6991–6994. [Google Scholar] [CrossRef]
- Hollmann, D.; Bähn, S.; Tillack, A.; Beller, M. N-Dealkylation of aliphatic amines and selective synthesis of monoalkylated aryl amines. Chem. Commun. 2008, 27, 3199–3201. [Google Scholar] [CrossRef]
- Ling, Z.; Yun, L.; Liu, L.; Wu, B.; Fu, X. Aerobic oxidative N-dealkylation of tertiary amines in aqueous solution catalyzed by rhodium porphyrins. Chem. Commun. 2013, 49, 4214–4216. [Google Scholar] [CrossRef]
- Yun, L.; Zhen, L.; Wang, Z.; Fu, X. Aerobic oxidative N-dealkylation of secondary amines in aqueous solution catalyzed by rhodium porphyrins. J. Porphyr. Phthalocyanines 2014, 18, 937–943. [Google Scholar] [CrossRef]
- Maiti, D.; Narducci Sarjeant, A.A.; Karlin, K.D. Copper(II)-hydroperoxo complex induced oxidative N-dealkylation chemistry. J. Am. Chem. Soc. 2007, 129, 6720–6721. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ginsbach, J.W.; Lee, J.Y.; Peterson, R.L.; Liu, J.J.; Siegler, M.A.; Sarjeant, A.A.; Solomon, E.I.; Karlin, K.D. Amine oxidative N-dealkylation via cupric hydroperoxide Cu-OOH homolytic cleavage followed by site-specific fenton chemistry. J. Am. Chem. Soc. 2015, 137, 2867–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovino, J.; Lütz, S.; Sames, D.; Touré, B.B. Complementation of biotransformations with chemical C-H oxidation: Copper-catalyzed oxidation of tertiary amines in complex pharmaceuticals. J. Am. Chem. Soc. 2013, 135, 12346–12352. [Google Scholar] [CrossRef] [PubMed]
- Genovino, J.; Sames, D.; Touré, B.B. Access to drug metabolites via C-H functionalization: Copper-catalyzed aerobic oxidation of N,N-dimethylalkylamines in complex pharmaceuticals. Tetrahedron Lett. 2015, 56, 3066–3069. [Google Scholar] [CrossRef]
- Murahashi, S.I.; Zhang, D. Ruthenium catalyzed biomimetic oxidation in organic synthesis inspired by cytochrome P-450. Chem. Soc. Rev. 2008, 37, 1490–1501. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, Y.; Xue, D.; Wang, Z.; Xiao, J.; Wang, C. Highly efficient binuclear copper-catalyzed oxidation of N,N-dimethylanilines with O2. ChemCatChem 2020, 12, 2221–2225. [Google Scholar] [CrossRef]
- Shatskiy, A.; Lundberg, H.; Kärkäs, M.D. Organic electrosynthesis: Applications in complex molecule synthesis. ChemElectroChem 2019, 6, 4067–4092. [Google Scholar] [CrossRef]
- Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S.R. Electrifying organic synthesis. Angew. Chem. Int. Ed. 2018, 57, 5594–5619. [Google Scholar] [CrossRef]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef]
- Shono, T. Electroorganic chemistry in organic synthesis. Tetrahedron 1984, 40, 811–850. [Google Scholar] [CrossRef]
- Bussy, U.; Boujtita, M. Advances in the electrochemical simulation of oxidation reactions mediated by cytochrome P450. Chem. Res. Toxicol. 2014, 27, 1652–1668. [Google Scholar] [CrossRef] [PubMed]
- Nouri-Nigjeh, E.; Bischoff, R.; Bruins, P.A.; Permentier, P.H. Electrochemistry in the mimicry of oxidative drug metabolism by cytochrome P450s. Curr. Drug Metab. 2011, 12, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dapo, R.F.; Mann, C.K. Anodic oxidation of triethylamine. Anal. Chem. 1963, 35, 677–680. [Google Scholar] [CrossRef]
- Mann, C.K. Cyclic stationary electrode voltammetry of some aliphatic amines. Anal. Chem. 1964, 36, 2424–2426. [Google Scholar] [CrossRef]
- Masui, M.; Sayo, H.; Tsuda, Y. Anodic oxidation of amines. Part I. Cyclic voltammetry of aliphatic amines at a stationary glassy-carbon electrode. J. Chem. Soc. B Phys. Org. 1968, 973–976. [Google Scholar] [CrossRef]
- Barnes, K.K.; Mann, C.K. Electrochemical oxidation of primary aliphatic amines. J. Org. Chem. 1967, 32, 1474–1479. [Google Scholar] [CrossRef]
- Smith, P.J.; Mann, C.K. Electrochemical dealkylation of aliphatic amines. J. Org. Chem. 1969, 34, 1821–1826. [Google Scholar] [CrossRef]
- Portis, L.C.; Bhat, V.V.; Mann, C.K. Electrochemical dealkylation of aliphatic tertiary and secondary amines. J. Org. Chem. 1970, 35, 2175–2178. [Google Scholar] [CrossRef]
- Shono, T.; Toda, T.; Oshino, N. Preparation of N-dealkylated drug metabolites by electrochemical simulation of biotransformation. Drug Metab. Dispos. 1981, 9, 481–482. [Google Scholar]
- Shono, T.; Toda, T.; Oshino, N. Electron transfer from nitrogen in microsomal oxidation of amine and amide. Simulation of microsomal oxidation by anodic oxidation. J. Am. Chem. Soc. 1982, 104, 2639–2641. [Google Scholar] [CrossRef]
- Pedersen, A.J.; Ambach, L.; König, S.; Weinmann, W. Electrochemical simulation of phase I metabolism for 21 drugs using four different working electrodes in an automated screening setup with MS detection. Bioanalysis 2014, 6, 2607–2621. [Google Scholar] [CrossRef] [PubMed]
- Jahn, S.; Baumann, A.; Roscher, J.; Hense, K.; Zazzeroni, R.; Karst, U. Investigation of the biotransformation pathway of verapamil using electrochemistry/liquid chromatography/mass spectrometry—A comparative study with liver cell microsomes. J. Chromatogr. A 2011, 1218, 9210–9220. [Google Scholar] [CrossRef] [PubMed]
- Nouri-Nigjeh, E.; Permentier, H.P.; Bischoff, R.; Bruins, A.P. Lidocaine oxidation by electrogenerated reactive oxygen species in the light of oxidative drug metabolism. Anal. Chem. 2010, 82, 7625–7633. [Google Scholar] [CrossRef] [PubMed]
- Nouri-Nigjeh, E.; Bischoff, R.; Bruins, A.P.; Permentier, H.P. Electrochemical oxidation by square-wave potential pulses in the imitation of phenacetin to acetaminophen biotransformation. Analyst 2011, 136, 5064–5067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouri-Nigjeh, E.; Bruins, A.P.; Bischoff, R.; Permentier, H.P. Electrocatalytic oxidation of hydrogen peroxide on a platinum electrode in the imitation of oxidative drug metabolism of lidocaine. Analyst 2012, 137, 4698. [Google Scholar] [CrossRef] [PubMed]
- Gul, T.; Bischoff, R.; Permentier, H.P. Optimization of reaction parameters for the electrochemical oxidation of lidocaine with a design of experiments approach. Electrochim. Acta 2015, 171, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.; Brown, R.; Szucs, R.; Hawkins, J.M.; Scrivens, G.; Pettman, A.; Kraus, D.; Taylor, M.R. Rapid synthesis of pharmaceutical oxidation products using electrochemistry: A systematic study of N-dealkylation reactions of fesoterodine using a commercially available synthesis cell. Org. Process Res. Dev. 2015, 19, 1596–1603. [Google Scholar] [CrossRef]
- Roth, H.G.; Romero, N.A.; Nicewicz, D.A. Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett 2016, 27, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Torriero, A.A.J.; Shiddiky, M.J.A.; Burgar, I.; Bond, A.M. Homogeneous electron-transfer reaction between electrochemically generated ferrocenium ions and amine-containing compounds. Organometallics 2013, 32, 5731–5739. [Google Scholar] [CrossRef]
- Torriero, A.A.J.J.; Morda, J.; Saw, J. Electrocatalytic dealkylation of amines mediated by ferrocene. Organometallics 2019, 38, 4280–4287. [Google Scholar] [CrossRef]
- Sasano, Y.; Sato, F.; Iwabuchi, Y.; Ono, T.; Kashiwagi, Y.; Dairaku, T.; Yoshida, K.; Kumano, M.; Sato, K. Electrochemical oxidation of amines using a nitroxyl radical catalyst and the electroanalysis of lidocaine. Catalysts 2018, 8, 649. [Google Scholar] [CrossRef] [Green Version]
- Bussy, U.; Delaforge, M.; El-Bekkali, C.; Ferchaud-Roucher, V.; Krempf, M.; Tea, I.; Galland, N.; Jacquemin, D.; Boujtita, M. Acebutolol and alprenolol metabolism predictions: Comparative study of electrochemical and cytochrome P450-catalyzed reactions using liquid chromatography coupled to high-resolution mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 6077–6085. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, S.M.; Blankert, B.; Kauffmann, J.M.; Karst, U. Prediction of clozapine metabolism by on-line electrochemistry/liquid chromatography/mass spectrometry. Anal. Bioanal. Chem. 2005, 382, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, W.; Karst, U. Electrochemistry meets enzymes: Instrumental on-line simulation of oxidative and conjugative metabolism reactions of toremifene. Anal. Bioanal. Chem. 2009, 394, 1341–1348. [Google Scholar] [CrossRef]
- Nozaki, K.; Osaka, I.; Kawasaki, H.; Arakawa, R. Application of on-line electrochemistry/electrospray/tandem mass spectrometry to a quantification method for the antipsychotic drug zotepine in human serum. Anal. Sci. 2009, 25, 1197–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, T.; Weidolf, L.; Jurva, U. Mimicry of phase I drug metabolism–novel methods for metabolite characterization and synthesis. Rapid Commun. Mass Spectrom. 2007, 21, 2323–2331. [Google Scholar] [CrossRef]
- Alipour Najmi, A.; Xiao, Z.; Bischoff, R.; Dekker, F.J.; Permentier, H.P. Electrochemical N-demethylation of tropane alkaloids. Green Chem. 2020, 22, 6455–6463. [Google Scholar] [CrossRef]
- Glotz, G.; Kappe, C.O.; Cantillo, D. Electrochemical N-demethylation of 14-hydroxy morphinans: Sustainable access to opioid antagonists. Org. Lett. 2020, 22, 6891–6896. [Google Scholar] [CrossRef]
- Frazier, W.H.; Coeur, C.; Smith, R.L.; Wagenknecht, H.J. Electrolytic Process for Producing N-Phosphonomethyl Glycine. U.S. Patent 3,835,000, 10 September 1974. [Google Scholar]
- Xiang, J.; Peng, M.; Pan, Y.; Luo, L.-J.; Cheng, S.-C.; Jin, X.-X.; Yiu, S.-M.; Man, W.-L.; Ko, C.-C.; Lau, K.-C.; et al. Visible light-induced oxidative N-dealkylation of alkylamines by a luminescent osmium(VI) nitrido complex. Chem. Sci. 2021, 12, 14494–14498. [Google Scholar] [CrossRef]
- Görner, H.; Döpp, D. Photoinduced demethylation of 4-nitro-N,N-dimethylaniline. Photochem. Photobiol. Sci. 2002, 1, 270–277. [Google Scholar] [CrossRef]
- Fawi, M.; Latif, A. El Solar and ultraviolet N-dealkylation of N,N-dimethylaminobenzylidene malonic acid derivatives via photoexcited polycyclic nitroaromatic compounds. J. Photochem. Photobiol. A Chem. 2001, 141, 241–245. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, S.; Duan, W.; Lian, Q.; Wei, W. Catalyst-free photoinduced selective oxidative C(sp3)-C(sp3) bond cleavage in arylamines. Green Chem. 2021, 23, 3261–3267. [Google Scholar] [CrossRef]
- Tesema, T.E.; Annesley, C.; Habteyes, T.G. Plasmon-enhanced autocatalytic N-demethylation. J. Phys. Chem. C 2018, 122, 19831–19841. [Google Scholar] [CrossRef]
- Pandey, G.; Rani, K.S.; Bhalerao, U.T.; Pandey, C.; Rani, K.S. Photooxidative set initiated N-demethylation of N,N′-dimethylanlines: Mimicking the cytochrome P-450 type oxygenations. Tetrahedron Lett. 1990, 31, 1199–1202. [Google Scholar] [CrossRef]
- Santamaria, J.; Ouchabane, R.; Rigaudy, J. Electron-transfer activation. Photochemical N-demethylation of tertiary amines. Tetrahedron Lett. 1989, 30, 2927–2928. [Google Scholar] [CrossRef]
- Ripper, J.A.; Tiekink, R.T.; Scammells, P.J. Photochemical N-Demethylation of alkaloids. Bioorg. Med. Chem. Lett. 2001, 11, 443–445. [Google Scholar] [CrossRef]
- Chen, Y.; Glotz, G.; Cantillo, D.; Kappe, C.O. Organophotocatalytic N-demethylation of oxycodone using molecular oxygen. Chem. Eur. J. 2020, 26, 2973–2979. [Google Scholar] [CrossRef]
- Rueping, M.; Vila, C.; Szadkowska, A.; Koenigs, R.M.; Fronert, J. Photoredox catalysis as an efficient tool for the aerobic oxidation of amines and alcohols: Bioinspired demethylations and condensations. ACS Catal. 2012, 2, 2810–2815. [Google Scholar] [CrossRef]
- Xiao, L.; Huang, Y.; Luo, Y.; Yang, B.; Liu, Y.; Zhou, X.; Zhang, J. Organic cotton photocatalysis. ACS Sustain. Chem. Eng. 2018, 6, 14759–14766. [Google Scholar] [CrossRef]
- Firoozi, S.; Hosseini-Sarvari, M. Nanosized CdS as a reusable photocatalyst: The study of different reaction pathways between tertiary amines and aryl sulfonyl chlorides through visible-light-induced N-dealkylation and C-H activation processes. J. Org. Chem. 2021, 86, 2117–2134. [Google Scholar] [CrossRef]
- Wu, G.; Li, Y.; Yu, X.; Gao, Y.; Chen, H. Acetic acid accelerated visible-light photoredox catalyzed N-demethylation of N,N-dimethylaminophenyl derivatives. Adv. Synth. Catal. 2017, 359, 687–692. [Google Scholar] [CrossRef]
- Zhao, H.; Leonori, D. Minimization of back-electron transfer enables the elusive sp3 C–H functionalization of secondary anilines. Angew. Chem. Int. Ed. 2021, 60, 7669–7674. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.J.; Wang, R.L.; Wang, D.; Liu, L.; Cheng, L. Visible-light-mediated oxidative demethylation of: N6-methyl adenines. Chem. Commun. 2017, 53, 10734–10737. [Google Scholar] [CrossRef]
- Gul, T.; Bischoff, R.; Permentier, H.P. Electrosynthesis methods and approaches for the preparative production of metabolites from parent drugs. TrAC-Trends Anal. Chem. 2015, 70, 58–66. [Google Scholar] [CrossRef]
- Caswell, J.M.; O’Neill, M.; Taylor, S.J.C.; Moody, T.S. Engineering and application of P450 monooxygenases in pharmaceutical and metabolite synthesis. Curr. Opin. Chem. Biol. 2013, 17, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Hrynchak, I.; Sousa, E.; Pinto, M.; Costa, V.M. The importance of drug metabolites synthesis: The case-study of cardiotoxic anticancer drugs. Drug Metab. Rev. 2017, 49, 158–196. [Google Scholar] [CrossRef]
- Cusack, K.P.; Koolman, H.F.; Lange, U.E.W.; Peltier, H.M.; Piel, I.; Vasudevan, A. Emerging technologies for metabolite generation and structural diversification. Bioorg. Med. Chem. Lett. 2013, 23, 5471–5483. [Google Scholar] [CrossRef] [Green Version]
- Genovino, J.; Sames, D.; Hamann, L.G.; Touré, B.B. Accessing drug metabolites via transition-metal catalyzed C−H oxidation: The liver as synthetic inspiration. Angew. Chem. Int. Ed. 2016, 55, 14218–14238. [Google Scholar] [CrossRef]
- Bonn, B.; Masimirembwa, C.M.; Aristei, Y.; Zamora, I. The molecular basis of CYP2D6-mediated N-dealkylation: Balance between metabolic clearance routes and enzyme inhibition. Drug Metab. Dispos. 2008, 36, 2199–2210. [Google Scholar] [CrossRef] [Green Version]
- Coutts, R.T.; Su, P.; Baker, G.B. Involvement of CYP2D6, CYP3A4, and other cytochrome P-450 isozymes in N-dealkylation reactions. J. Pharmacol. Toxicol. Methods 1994, 31, 177–186. [Google Scholar] [CrossRef]
- Lundemo, M.T.; Woodley, J.M. Guidelines for development and implementation of biocatalytic P450 processes. Appl. Microbiol. Biotechnol. 2015, 99, 2465–2483. [Google Scholar] [CrossRef] [PubMed]
- Zelasko, S.; Palaria, A.; Das, A. Optimizations to achieve high-level expression of cytochrome P450 proteins using Escherichia coli expression systems. Protein Expr. Purif. 2013, 92, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Schroer, K.; Kittelmann, M.; Lütz, S. Recombinant human cytochrome P450 monooxygenases for drug metabolite synthesis. Biotechnol. Bioeng. 2010, 106, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 monooxygenases in biotechnology and synthetic biology. Trends Biotechnol. 2019, 37, 882–897. [Google Scholar] [CrossRef] [PubMed]
- Olsen, D.; Nelson, L.; Ludwidshafen, A.G.; Ger, W.; Nelson, W.L.; Olsen, L.D. Regiochemistry and enantioselectivity in the oxidative N-dealkylation of verapamil. Drug Metab. Dispos. 1988, 16, LP834–LP841. [Google Scholar]
- Shen, C.; Liu, H.; Dai, W.; Liu, X.; Liu, J.; Yu, B. Specific N-demethylation of verapamil by cytochrome P450 from Streptomyces griseus ATCC 13273. Eng. Life Sci. 2019, 19, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luurtsema, G.; Windhorst, A.D.; Mooijer, M.P.J.; Herscheid, J.D.M.; Lammertsma, A.A.; Franssen, E.J.F. Fully automated high yield synthesis of (R)- and (S)-[11C]verapamil for measuring P-glycoprotein function with positron emission tomography. J. Label. Compd. Radiopharm. 2002, 45, 1199–1207. [Google Scholar] [CrossRef]
- Elsinga, P.H.; Windhorst, A.D.; Lammertsma, A.A.; Luurtsema, G.; Schuit, R.C.; Kooijman, E.J.M.; Raaphorst, R.M. Synthesis and evaluation of new fluorine-18 labeled verapamil analogs to investigate the function of P-glycoprotein in the blood–brain barrier. ACS Chem. Neurosci. 2017, 8, 1925–1936. [Google Scholar] [CrossRef] [Green Version]
- Livezey, M.R.; Briggs, E.D.; Bolles, A.K.; Nagy, L.D.; Fujiwara, R.; Furge, L.L. Metoclopramide is metabolized by CYP2D6 and is a reversible inhibitor, but not inactivator, of CYP2D6. Xenobiotica 2014, 44, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Akutsu, T.; Kobayashi, K.; Sakurada, K.; Ikegaya, H.; Furihata, T.; Chiba, K. Identification of human cytochrome P450 isozymes involved in diphenhydramine N-demethylation. Drug Metab. Dispos. 2007, 35, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Gantenbein, M.; Attolini, L.; Bruguerolle, B.; Villard, P.H.; Puyoou, F.; Durand, A.; Lacarelle, B.; Hardwigsen, J.; Le-Treut, Y.P. Oxidative metabolism of bupivacaine into pipecolylxylidine in humans is mainly catalyzed by CYP3A. Drug Metab. Dispos. 2000, 28, 383–385. [Google Scholar] [PubMed]
- Ghahramani, P.; Ellis, S.W.; Lennard, M.S.; Ramsay, L.E.; Tucker, G.T. Cytochromes P450 mediating the N-demethylation of amitriptyline. Br. J. Clin. Pharmacol. 1997, 43, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botsch, S.; Gautier, J.C.; Beaune, P.; Eichelbaum, M.; Kroemer, H.K. Identification and characterization of the cytochrome P450 enzymes involved in N-dealkylation of propafenone: Molecular base for interaction potential and variable disposition of active metabolites. Mol. Pharmacol. 1993, 43, 120–126. [Google Scholar] [PubMed]
- Ren, X.; Yorke, J.A.; Taylor, E.; Zhang, T.; Zhou, W.; Wong, L.L. Drug oxidation by cytochrome P450BM3: Metabolite synthesis and discovering new P450 reaction types. Chem. Eur. J. 2015, 21, 15039–15047. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H.G.; Brazzale, A.; Ma, Z. Biomimetic synthesis of macrolide/ketolide metabolites through a selective N-demethylation reaction. J. Org. Chem. 2000, 65, 3875–3876. [Google Scholar] [CrossRef]
- Richards, L.; Lutz, A.; Chalmers, D.K.; Jarrold, A.; Bowser, T.; Stevens, G.W.; Gras, S.L. Production of metabolites of the anti-cancer drug noscapine using a P450BM3 mutant library. Biotechnol. Rep. 2019, 24, e00372. [Google Scholar] [CrossRef]
- Richards, L.; Jarrold, A.; Bowser, T.; Stevens, G.W.; Gras, S.L. Cytochrome P450-mediated N-demethylation of noscapine by whole-cell biotransformation: Process limitations and strategies for optimisation. J. Ind. Microbiol. Biotechnol. 2020, 47, 449–464. [Google Scholar] [CrossRef]
- Gandomkar, S.; Fischereder, E.-M.; Schrittwieser, J.H.; Wallner, S.; Habibi, Z.; Macheroux, P.; Kroutil, W. Enantioselective oxidative aerobic dealkylation of N-ethyl benzylisoquinolines by employing the berberine bridge enzyme. Angew. Chem. Int. Ed. 2015, 54, 15051–15054. [Google Scholar] [CrossRef]
- O’Reilly, E.; Iglesias, C.; Turner, N.J. Monoamine oxidase-ω-transaminase cascade for the deracemisation and dealkylation of amines. ChemCatChem 2014, 6, 992–995. [Google Scholar] [CrossRef]
- Xin, Y.; Hao, M.; Fan, G.; Zhang, Y.; Zhang, L. Soluble expression of Thermomicrobium roseum sarcosine oxidase and characterization of N-demethylation activity. Mol. Catal. 2019, 464, 48–56. [Google Scholar] [CrossRef]
- Augustin, M.M.; Augustin, J.M.; Brock, J.R.; Kutchan, T.M. Enzyme morphinan N-demethylase for more sustainable opiate processing. Nat. Sustain. 2019, 2, 465–474. [Google Scholar] [CrossRef]
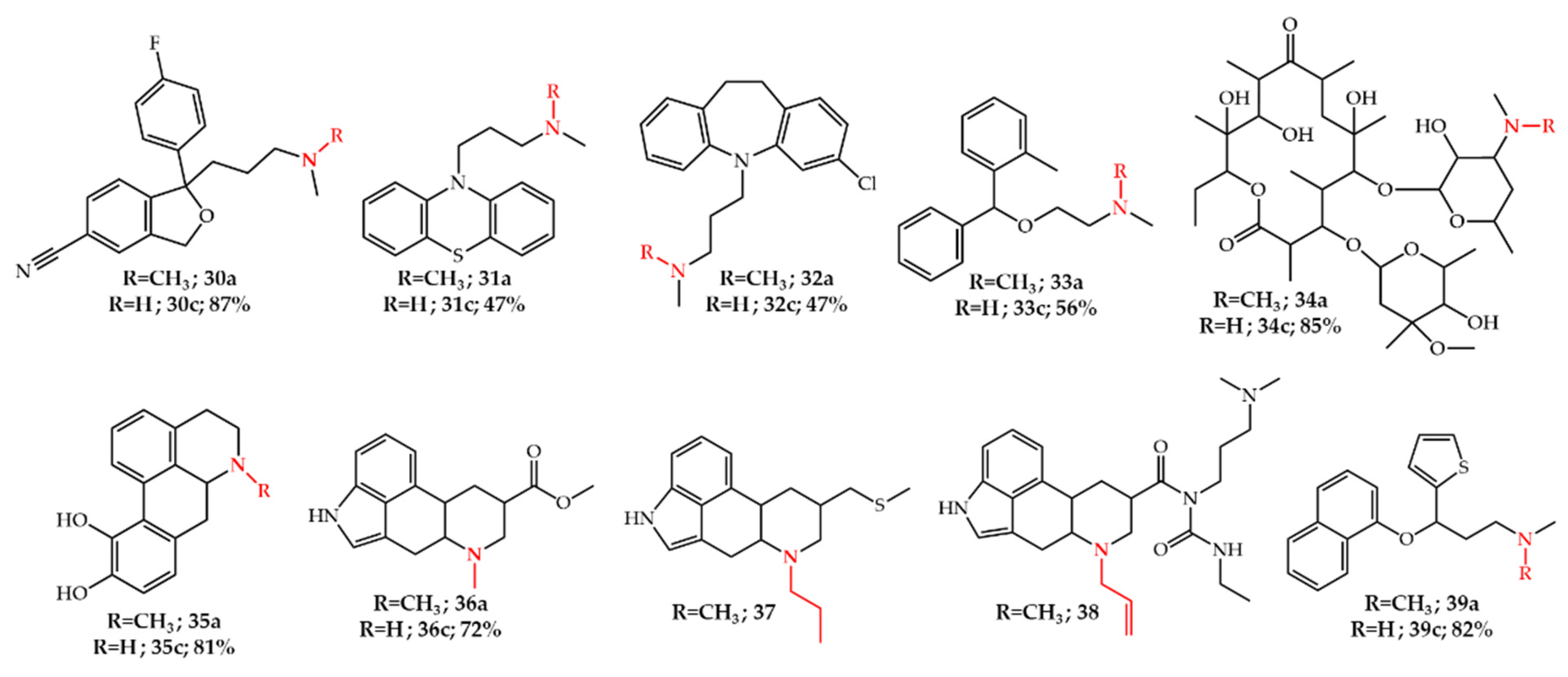
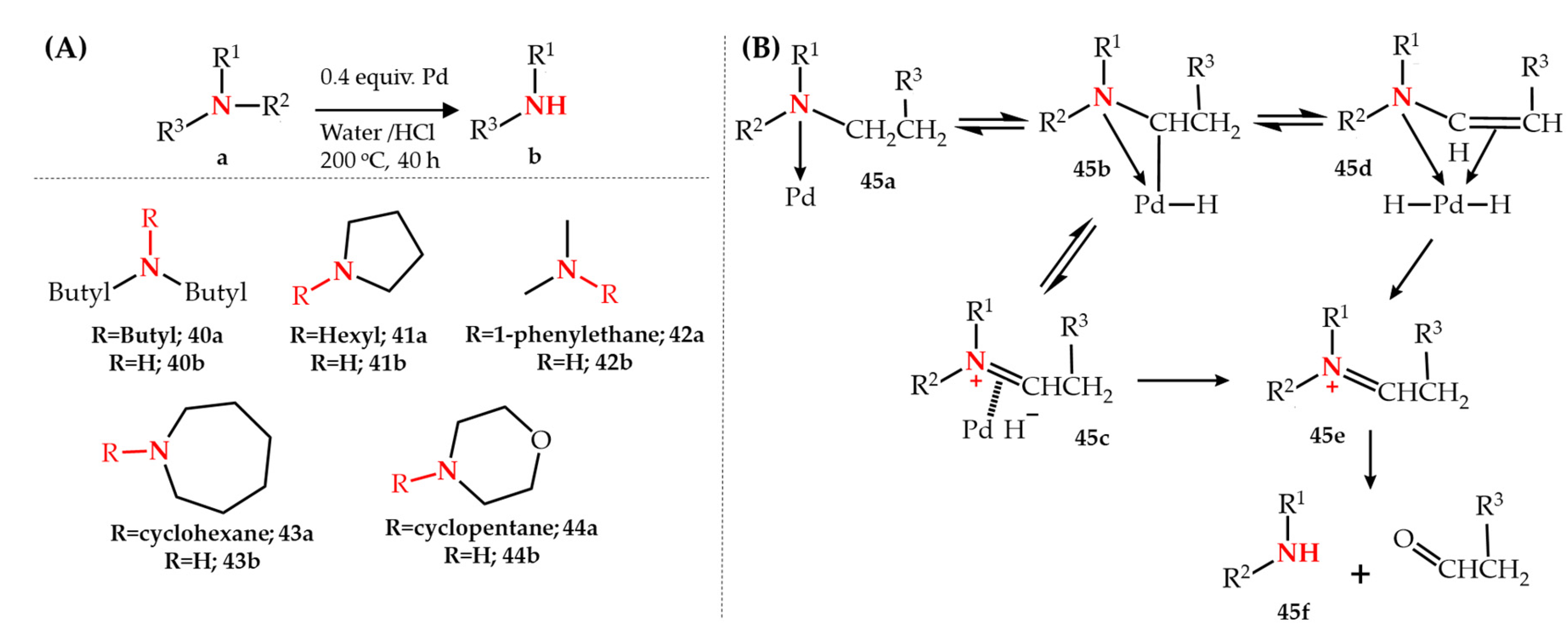



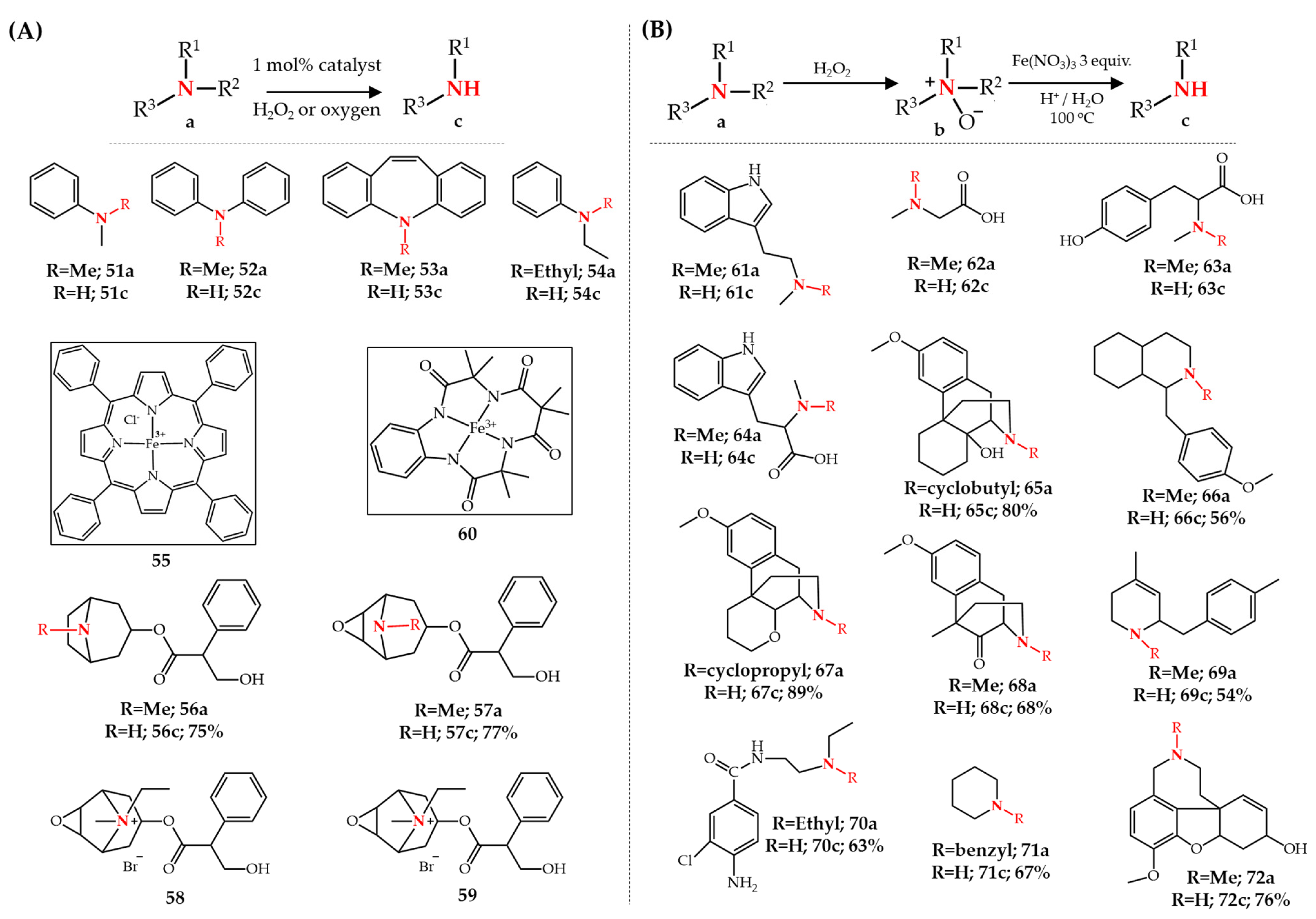
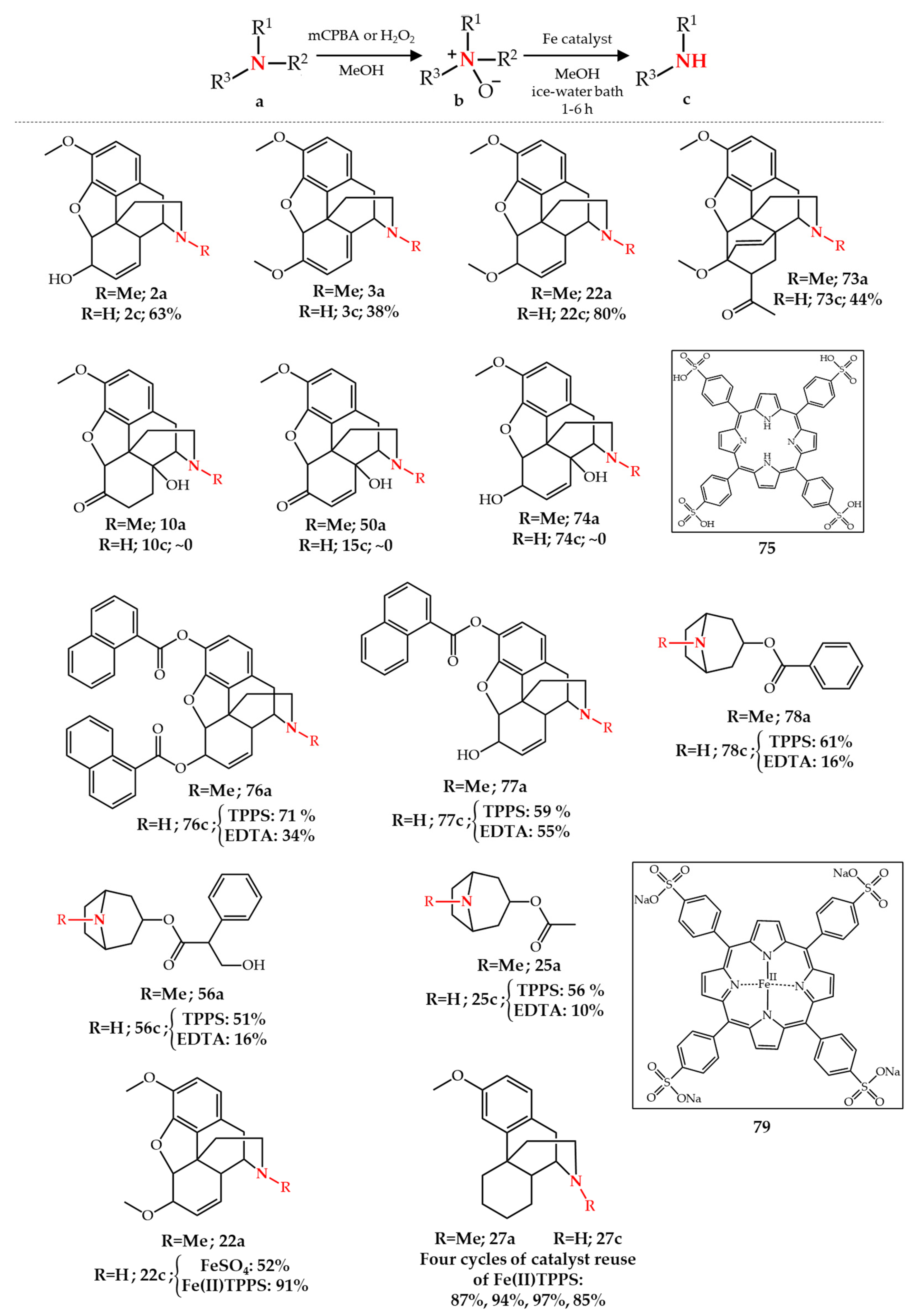


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najmi, A.A.; Bischoff, R.; Permentier, H.P. N-Dealkylation of Amines. Molecules 2022, 27, 3293. https://doi.org/10.3390/molecules27103293
Najmi AA, Bischoff R, Permentier HP. N-Dealkylation of Amines. Molecules. 2022; 27(10):3293. https://doi.org/10.3390/molecules27103293
Chicago/Turabian StyleNajmi, Ali Alipour, Rainer Bischoff, and Hjalmar P. Permentier. 2022. "N-Dealkylation of Amines" Molecules 27, no. 10: 3293. https://doi.org/10.3390/molecules27103293