
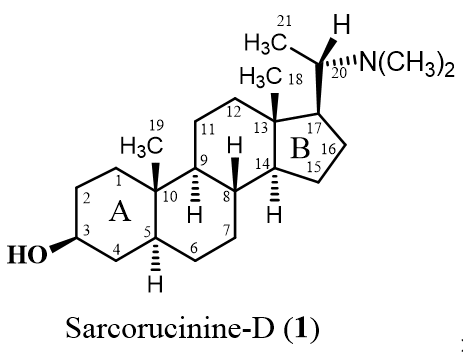
Sarcorucinine-D Inhibits Cholinesterases and Calcium Channels: Molecular Dynamics Simulation and In Vitro Mechanistic Investigations


 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
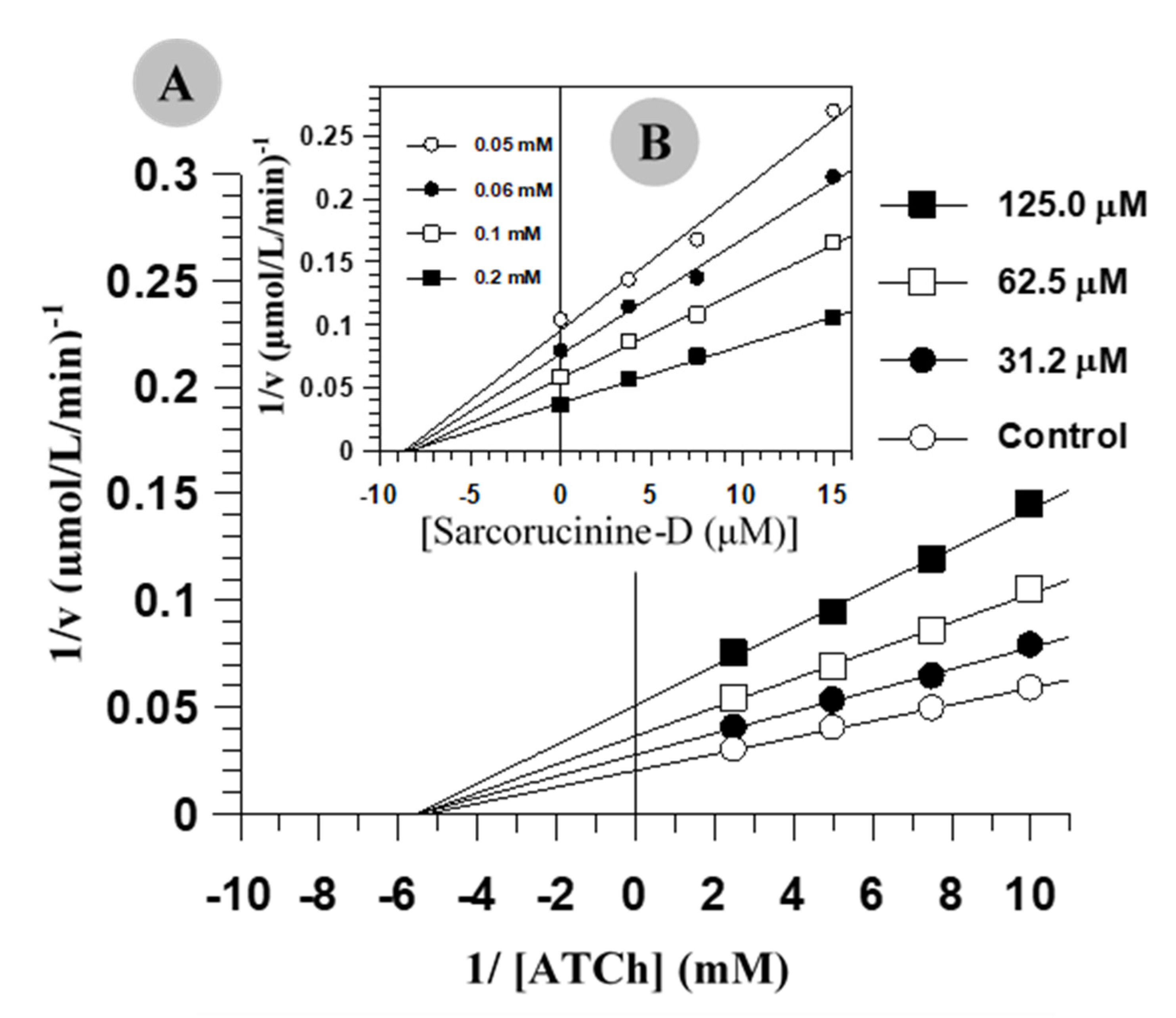
2.1. Inhibition Kinetics of AChE and BChE
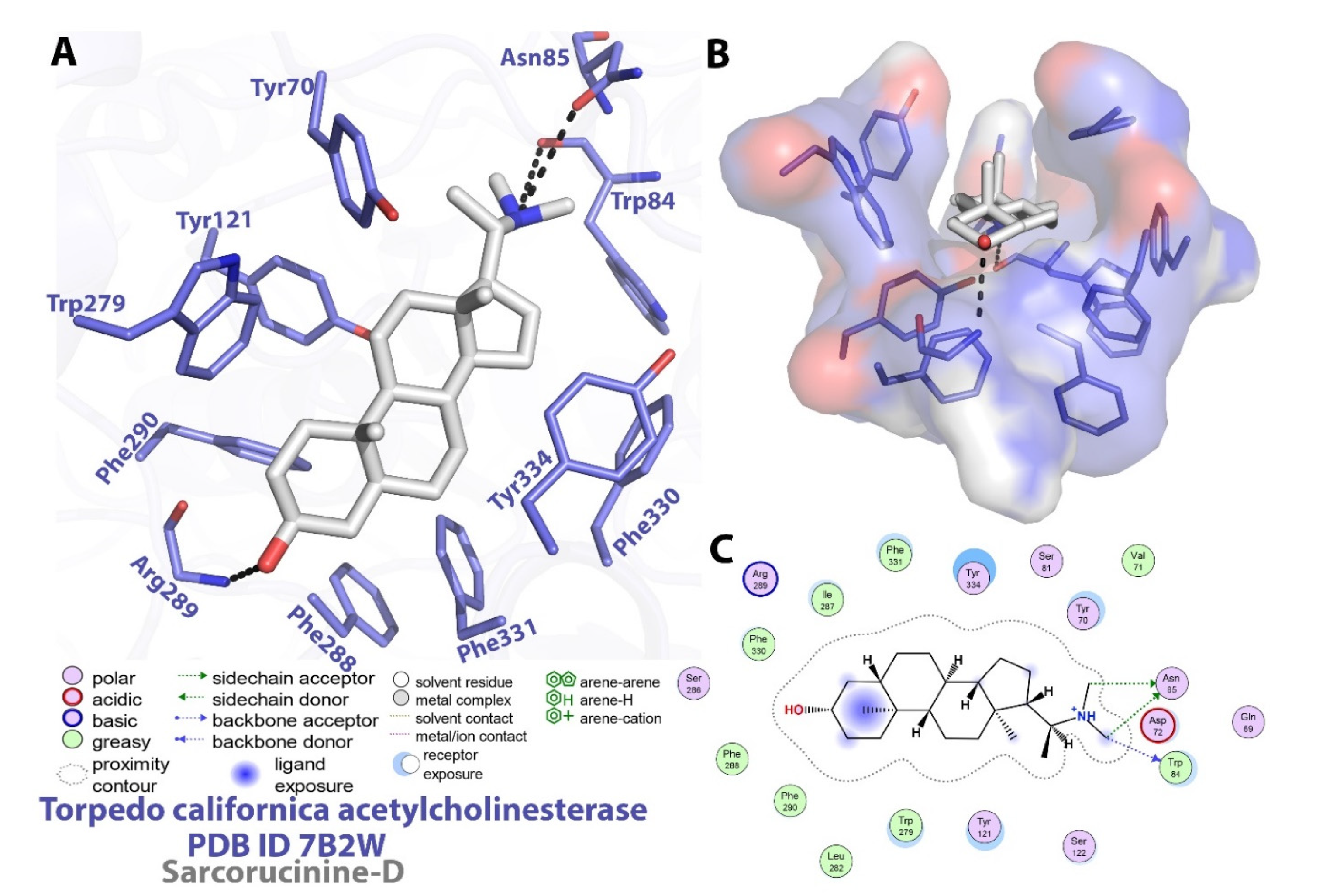
2.2. Molecular Docking Investigations against AChE
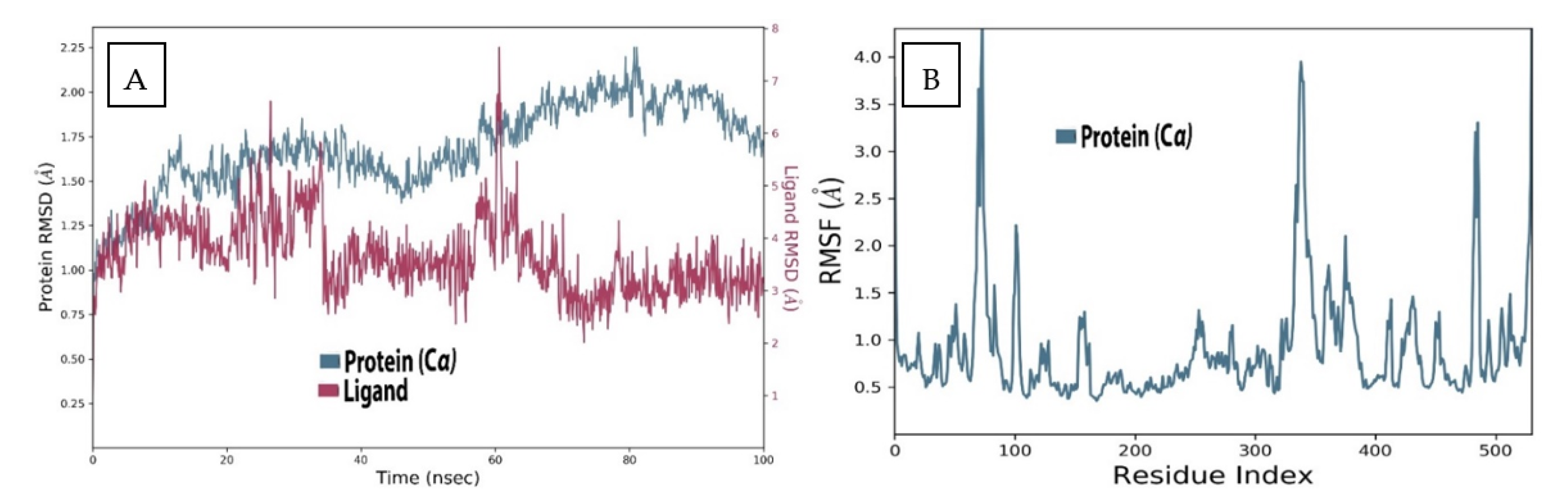
2.3. MD Analysis of Sarcorucinine-D in the Vicinity of AChE
2.4. AChE Deviation and Fluctuation in the Context of Sarcorucinine-D
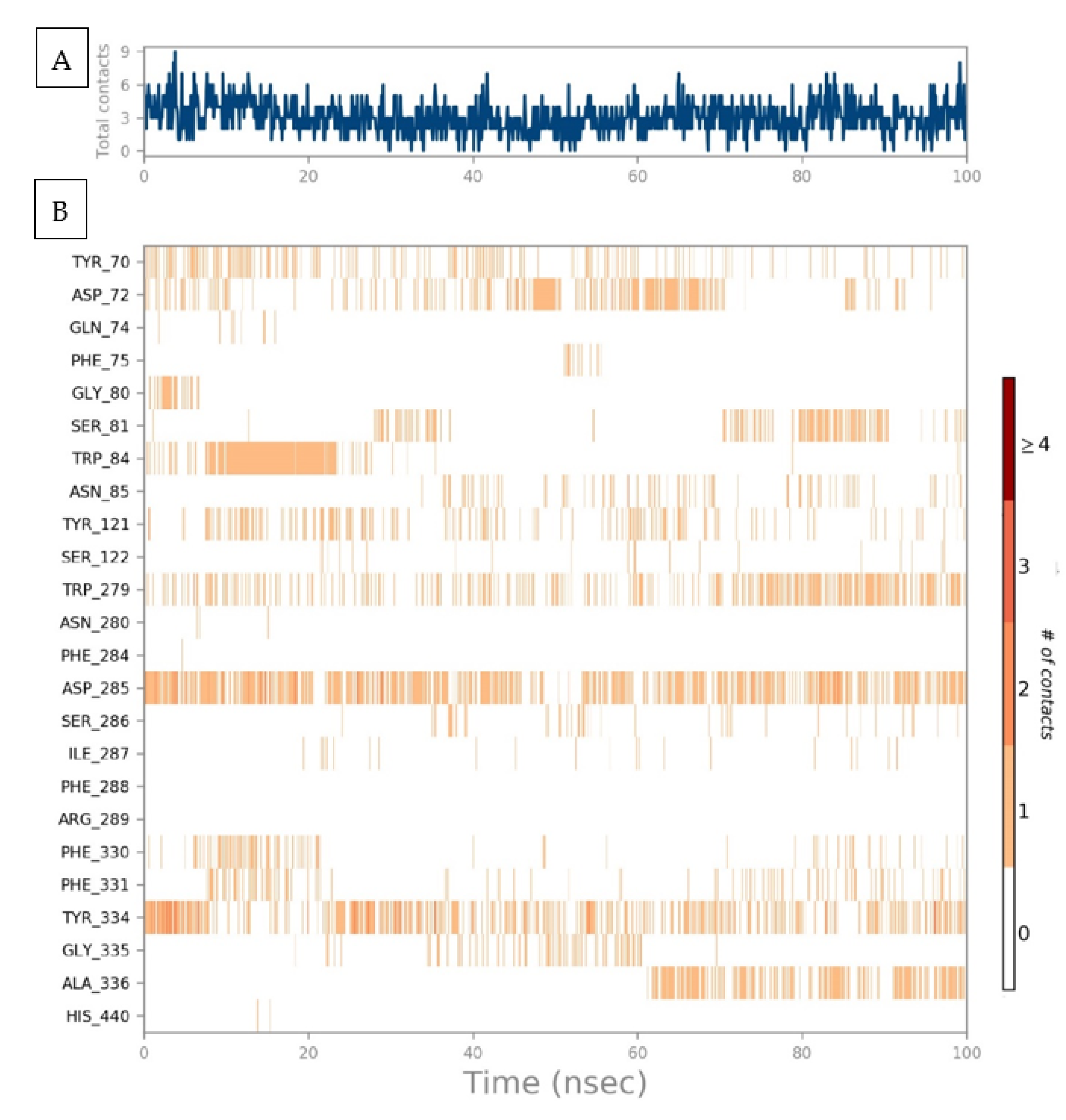
2.5. Protein–Ligand Interaction Contact Analyses
2.6. Molecular Investigation of Sarcorucinine-D’s Properties as an AChE Inhibitor
2.7. Pre- and Post-MD Results
2.8. Antispasmodic and Ca2+ Channel-Blocking (CCB) Properties
2.9. Cytotoxicity Evaluation
3. Material and Methods
3.1. Enzyme Inhibition Assays
3.2. Estimation of Inhibition and Kinetic Parameters
3.3. Molecular Ligand–Protein Docking
3.4. Molecular Dynamic Simulation Set-Up
3.5. Antispasmodic Activity in Isolated Rabbit Jejunum
3.6. Determination of Calcium Antagonist Activity
3.7. Cytotoxicity Evaluation
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Selkoe, D.J. Alzheimer’s disease: Genotypes, phenotypes, and treatments. Science 1997, 275, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.V.; Haeberlein, S.L.B.; Avil, J.A. Glycogen synthase kinase 3: A drug target for CNS therapies. J. Neurochem. 2004, 89, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, T. Therapy of Alzheimer disease. Neuro Psycho Pharm. Hung. 2009, 11, 27–33. [Google Scholar]
- Greig, N.H.; Utsuki, T.; Yu, Q.; Zhu, X.; Holloway, H.W.; Perry, T.; Lee, B.; Ingram, D.K.; Lahiri, D.K. A new therapeutic target in Alzheimer's disease treatment: Attention to butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Holloway, H.W.; Utsuki, T.; Brossi, A.; Greig, N.H. Synthesis of Novel Phenserine-based-selective Inhibitors of Butyrylcholinesterase for Alzheimer's Disease. J. Med. Chem. 1999, 42, 1855–1861. [Google Scholar] [CrossRef]
- Quinn, D.M. Acetylcholinesterase: Enzyme Structure, Reaction Dynamics, and Virtual Transition States. Chem. Rev. 1987, 87, 955–979. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [Green Version]
- Eichler, J.; Anselmet, A.; Sussman, J.L.; Massoulie, J.; Silman, I. Differential Effects of Peripheral Site Ligands on Torpedo and Chicken Acetylcholinesterase. Mol. Pharmacol. 1994, 45, 335–340. [Google Scholar]
- Kiamuddin, M.; Hye, H. Activity of an Alkaloids form Sarcococca saligna. Pak. J. Sci. Ind. Res. 1970, 13, 59–62. [Google Scholar]
- Khalid, A.; Choudhary, M.I.; Zaheer-ul-Haq; Anjum, S.; Khan, M.R.; Atta-ur-Rahma. Kinetics and Structure-Activity Relationship studies on Steroidal Alkaloids That Inhibit Cholinesterases. Bioorg. Med. Chem. 2004, 12, 1995–2003. [Google Scholar] [CrossRef] [PubMed]
- Zaheer-ul-Haq; Wellenzohn, B.; Tonmunphean, S.; Khalid, A.; Choudhary, M.I.; Rode, B.M. 3D-QSAR Studies on Natural Acetylcholinesterase Inhibitors of Sarcococca saligna by Comparative Molecular FIeld Analysis (CoMFA). Bioorg. Med. Chem. Lett. 2003, 13, 4375–4380. [Google Scholar] [CrossRef] [PubMed]
- Atta-ur-Rahman; Zaheer-ul-Haq; Feroz, F.; Khalid, A.; Nawaz, S.A.; Khan, M.R.; Choudhary, M.I. New Cholinesterase-Inhibiting Steroidal Alkaloids from Sarcococca saligna. Helv. Chim. Acta 2004, 87, 439–448. [Google Scholar] [CrossRef]
- Kalauni, S.K.; Choudhary, M.I.; Shaheen, F.; Manandhar, M.D.; Atta-ur-Rahma; Gewali, M.B.; Khalid, A. Steroidal alkaloids from the leaves of Sarcococca coriacea of Nepalese origin. J. Nat. Prod. 2001, 64, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Kalauni, S.K.; Choudhary, M.I.; Khalid, A.; Manandhar, M.D.; Shaheen, F.; Atta-ur-Rahma; Gewali, M.B. New Cholinesterase Inhibiting Steroidal Alkaloids from the Leaves of Sarcococca Coriacea of Nepalese origin. Chem. Pharm. Bull. 2002, 50, 1423–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atta-ur-Rahman; Akhtar, M.N.; Choudhary, M.I.; Tsuda, Y.; Sener, B.; Khalid, A.; Parvez, M. New steroidal alkaloids from Fritillaria imperialis and their cholinesterase inhibiting activities. Chem. Pharm. Bull. 2002, 50, 1013–1016. [Google Scholar] [CrossRef] [Green Version]
- Atta-ur-Rahman; Parveen, S.; Khalid, A.; Farooq, A.; Ayattollahi, S.A.M.; Choudhary, M.I. Acetylcholinesterase inhibiting triterpenoidal alkaloids from Buxus hyrcana. Heterocycles 1998, 49, 481–487. [Google Scholar] [CrossRef]
- Atta-ur-Rahman; Zaheer-ul-Haq; Khalid, A.; Anjum, S.; Khan, M.R.; Choudhary, M.I. Pregnane-type Steroidal Alkaloids of Sarcococca saligna:A New Class of Cholinesterases Inhibitors. Helv. Chim. Acta 2002, 85, 678–688. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Do, P.C.; Lee, E.H.; Le, L. Steered molecular dynamics simulation in rational drug design. J. Chem. Inf. Mod. 2018, 58, 1473–1482. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.; C Rosales-Hernández, M.; Mendieta-Wejebe, J.E.; Martínez-Archundia, M.; Correa Basurto, J. Current tools and methods in molecular dynamics (MD) simulations for drug design. Curr. Med. Chem. 2016, 23, 3909–3924. [Google Scholar] [CrossRef] [PubMed]
- Ul-Haq, Z.; Wellenzohn, B.; Liedl, K.R.; Rode, B.M. Molecular Docking Studies of Natural Cholinesterase-inhibiting Steroidal Alkaloids from Sarcococca saligna. J. Med. Chem. 2003, 46, 5087–5090. [Google Scholar] [CrossRef] [PubMed]
- Khalid, A.; Azim, M.K.; Parveen, S.; Atta-ur-Rahman; Choudhary, M.I. Structural basis of acetylcholinesterase inhibition by triterpenoidal alkaloids. Biochem. Biophys. Res. Commun. 2005, 331, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Godfraind, T.; Miller, R.; Wibo, M. Calcium Antagonism and Calcium Entry Blockade. Pharmacol. Rev. 1986, 38, 321–416. [Google Scholar]
- Sobolev, V.; Sorokine, A.; Prilusky, J.; Abola, E.E.; Edelman, M. Automated analysis of interatomic contacts in proteins. Bioinformatics 1999, 15, 327–332. [Google Scholar] [CrossRef]
- Williamson, E.M.; Okpako, D.T.; Evans, F.J. Selection, Preperation and Pharmacological Evaluation of Plant Material; John Wiley & Sons Ltd.: Chichester, UK, 1996. [Google Scholar]
- Triggle, D.J. Drugs Affecting Calcium Regulation and Action. In TextbooK of Pharma-Cology; Smith, G.M., Reynard, A.M., Eds.; W B Saunders: Philadelphia, PA, USA, 1992; pp. 453–479. [Google Scholar]
- Brading, A.F. How Do Drugs Initiate Contraction in Smooth Muscles. Trends. Pharmacol. Sci. 1981, 2, 261–265. [Google Scholar] [CrossRef]
- Bolton, T.B. Mechanism of Action of Transmitters and other Substances on Smooth Muscles. Physiol. Rev. 1979, 59, 606–718. [Google Scholar] [CrossRef]
- Karaki, H.; Weise, G.B. Mini Reviews: Calcium Release in Smooth Muscles. Life Sci. 1988, 42, 111–122. [Google Scholar] [CrossRef]
- Silman, H.I.; Karlin, A. Effect of local pH changes caused by substrate hydrolysis on the activity of membrane-bound acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1967, 58, 1664–1668. [Google Scholar] [CrossRef] [Green Version]
- Grobe-Einsler, R. Clinical aspects of nimodipine. Clin. Neuropharmacol. 1993, 16 (Suppl. S1), S39–S45. [Google Scholar] [CrossRef]
- Feldman, H.H.; van Baelen, B.; Kavanagh, S.M.; Torfs, K.E. Cognition, function, and caregiving time patterns in patients with mild-to-moderate Alzheimer disease: A 12-month analysis. Alzheimer Dis. Assoc. Disord. 2005, 19, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Ahmad, V.U.; Abbasi, M.A.; Hussain, H.; Akhtar, M.N.; Farooq, U.; Fatima, N.; Choudhary, M.I. Phenolic glycosides from Symplocos racemosa: Natural inhibitors of phosphodiesterase I. Phytochemistry 2003, 63, 217–220. [Google Scholar] [CrossRef]
- Amtul, Z.; Rasheed, M.; Choudhary, M.I.; Supino, R.; Khan, K.M.; Atta-ur-Rahman. Kinetics of novel competitive inhibitors of urease enzymes by a focused library of oxadiazoles/thiadiazoles and triazoles. Biochem. Biophys. Res. Commun. 2004, 319, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Anis, E.; Anis, I.; Ahmed, S.; Mustafa, G.; Malik, A.; Afza, N.; Hai, S.M.; Shahzad-ul-hussan, S.; Choudhary, M.I. Alpha-glucosidase inhibitory constituents from Cuscuta reflexa. Chem. Pharm. Bull. 2002, 50, 112–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.M.; Shujaat, S.; Rahat, S.; Hayat, S.; Atta-ur-Rahman; Choudhary, M.I. Beta-N-cyanoethyl acyl hydrazide derivatives: A new class of beta-glucuronidase inhibitors. Chem. Pharm. Bull. 2002, 50, 1443–1446. [Google Scholar] [CrossRef] [Green Version]
- Segel, I.H. Enzyme Kinetics, Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; John Wiley and Sons Inc.: New York, NY, USA, 1975. [Google Scholar]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- WebLab. ViewerPro; Molecular Simulation Inc.: San Diego, CA, USA, 2000. [Google Scholar]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A Program to Generate Schematic Diagrams of Protein-ligand Interactions. Prot. Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Gilani, A.H.; Ghayur, M.N.; Khalid, A.; Zaheer-ul-Haq; Choudhary, M.I.; Atta-ur-Rahma. Presence of antispasmodic, antidiarrheal, antisecretory, calcium antagonist and acetylcholinesterase inhibitory steroidal alkaloids in Sarcococca saligna. Planta Med. 2005, 71, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Farre, A.J.; Colombo, M.; Fort, M.; Gutierrez, B. Differential effects of various Ca2+ antagonists. Gen. Pharmacol. 1991, 22, 177–181. [Google Scholar] [CrossRef]
- Van-Rossu, J.M. Commutative dose-response curves. II. Techniques for the making of dose-response curves in isolated organs and the evaluation of drug parameters. Arch Int. Pharmacodyn. Ther. 1963, 143, 199–230. [Google Scholar]
- Mesaik, M.A.; Rahat, S.; Khan, K.M.; Zia, U.; Choudhary, M.I.; Murad, S.; Ismail, Z.; Atta-ur-Rahman; Ahmad, A. Synthesis and immunomodulatory properties of selected oxazolone derivatives. Bioorg. Med. Chem. 2004, 12, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Leatherbarrow, R.J. GraFit; Erithacus Software Ltd.: Stains, UK, 1999. [Google Scholar]
- Cass, T. Easy data analysis. Science 2000, 289, 1158. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
| Compound | Acetylcholinesterase | Butyrylcholinesterase | ||||
| IC50 (µM)a | Ki b (µM)a | Inhibition | IC50 (µM)a | Ki (µM)a | Inhibition c | |
| Sarcorucinine-D | 42.9 ± 3.13 | 103.3 ± 6.67 | NC | 8.87 ± 0.56 | 4.66 ± 0.3 | NC |
| Tacrine | 0.021 ± 0.002 | 0.23 ± 0.02 | MT | 0.051 ± 0.005 | 0.025 ± 0.003 | MT |
| Galanthamine | 0.45 ± 0.02 | 0.19 ± 0.01 | MT | 39.1 ± 0.032 | 32.0 ± 0.33 | NC |
| Binding energy (kcal/mol) a | −12.73 | |
| Estimated Ki b | 4.66 × 10−10 | |
| Compl. values c | 0.53 | |
| Solv. accessible surface d | 45.1/564.4 | |
| Residues involved in specific contacts with Sarcorucinine-D e | polar contacts (Enz-Inh.) | Glu199 (O…O-H) Inh. (2.7Å) Tyr130 (O-H…O) Inh. (2.8Å) |
| non-polar contacts | Tyr70; Asp72; Trp84; Ser122; Leu127; Trp279; Phe290; Phe330; Phe331; Tyr334; His440; Ile444 | |
| destabilizing contacts | Asp72; Trp84; Gly117; Gly118; Ser122; Gly123; Tyr130; Glu199; Ser200; Phe330; Tyr334; His440; Ile444 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalid, A.; Abdalla, M.; Saeed, M.; Ghayur, M.N.; Kalauni, S.K.; Albratty, M.; Alhazmi, H.A.; Mesaik, M.A.; Gilani, A.H.; Ul-Haq, Z. Sarcorucinine-D Inhibits Cholinesterases and Calcium Channels: Molecular Dynamics Simulation and In Vitro Mechanistic Investigations. Molecules 2022, 27, 3361. https://doi.org/10.3390/molecules27113361
Khalid A, Abdalla M, Saeed M, Ghayur MN, Kalauni SK, Albratty M, Alhazmi HA, Mesaik MA, Gilani AH, Ul-Haq Z. Sarcorucinine-D Inhibits Cholinesterases and Calcium Channels: Molecular Dynamics Simulation and In Vitro Mechanistic Investigations. Molecules. 2022; 27(11):3361. https://doi.org/10.3390/molecules27113361
Chicago/Turabian StyleKhalid, Asaad, Mohnad Abdalla, Maria Saeed, Muhammad Nabeel Ghayur, Surya Kant Kalauni, Mohammed Albratty, Hassan A. Alhazmi, Mohammed Ahmed Mesaik, Anwarul Hassan Gilani, and Zaheer Ul-Haq. 2022. "Sarcorucinine-D Inhibits Cholinesterases and Calcium Channels: Molecular Dynamics Simulation and In Vitro Mechanistic Investigations" Molecules 27, no. 11: 3361. https://doi.org/10.3390/molecules27113361