
Advances in Mixer Design and Detection Methods for Kinetics Studies of Macromolecular Folding and Binding on the Microsecond Time Scale


Abstract
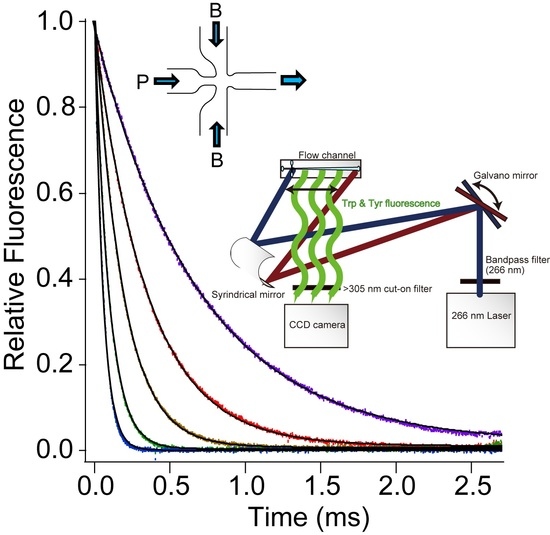
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods and Results
2.1. Design Criteria for Turbulent Mixers
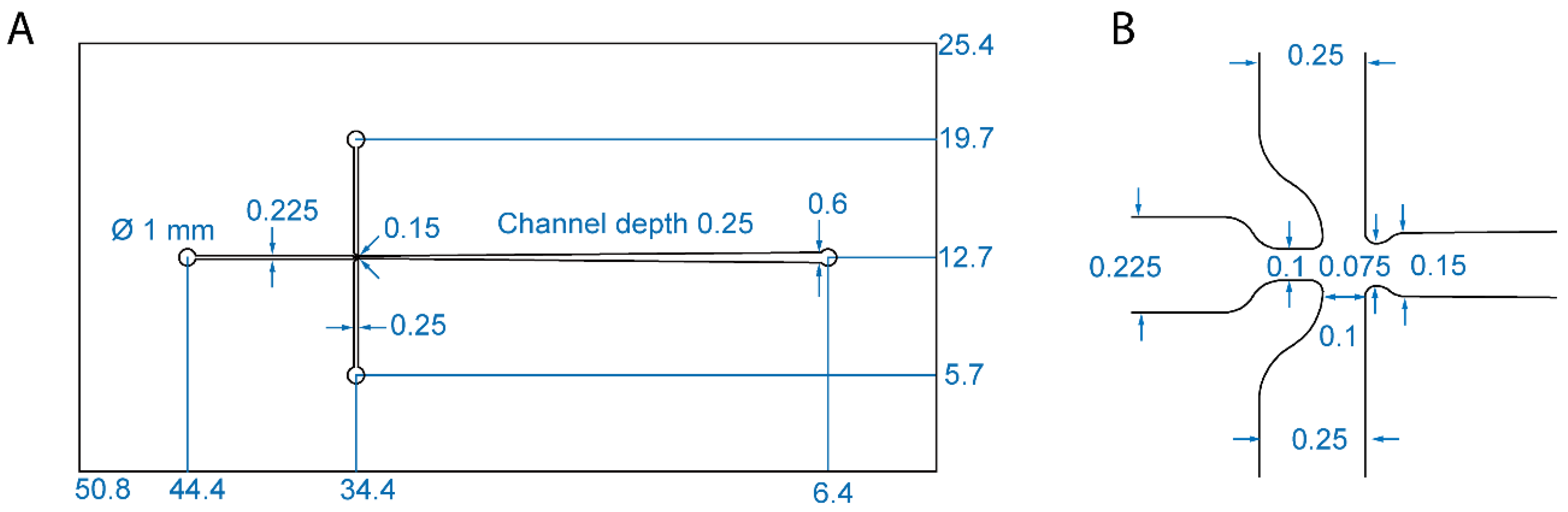
2.2. Design of a Microfabricated Mixer
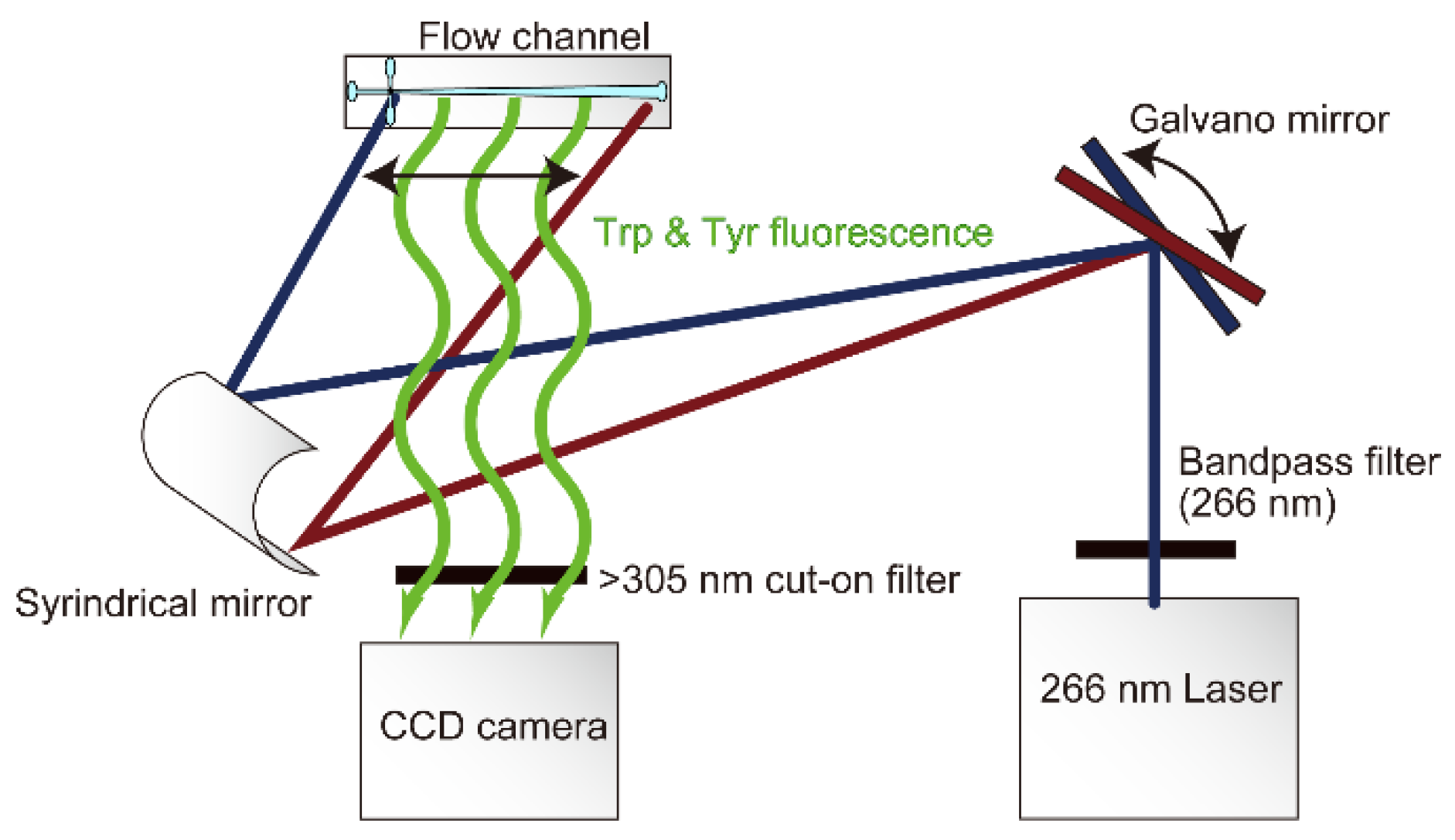
2.3. Solution Delivery and Fluorescence Detection
2.4. Data Collection and Analysis
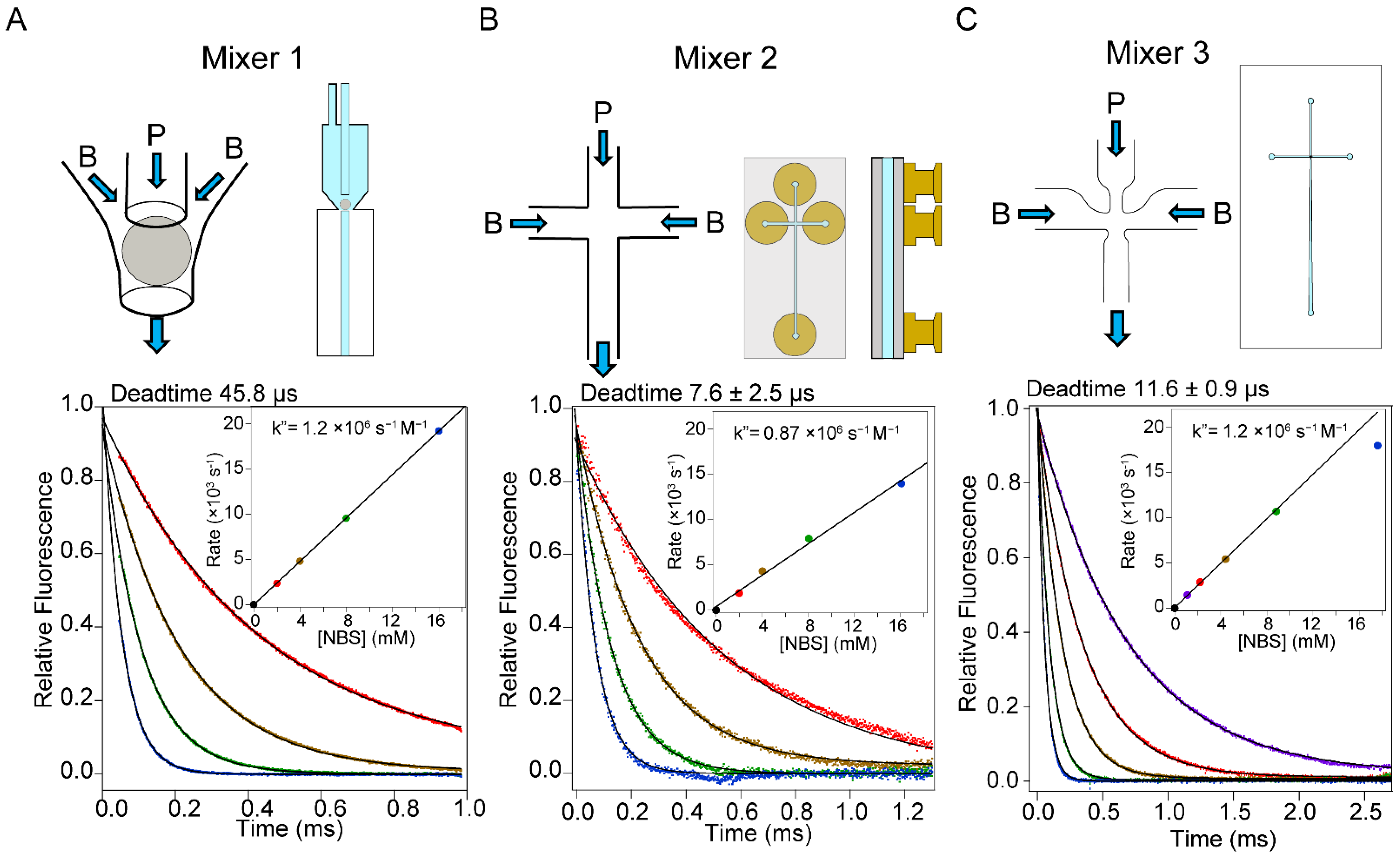
2.5. Optimization of Mixing Conditions
2.6. Mixing Efficiency
2.7. Accuracy
3. Applications
3.1. Folding of Cytochrome c
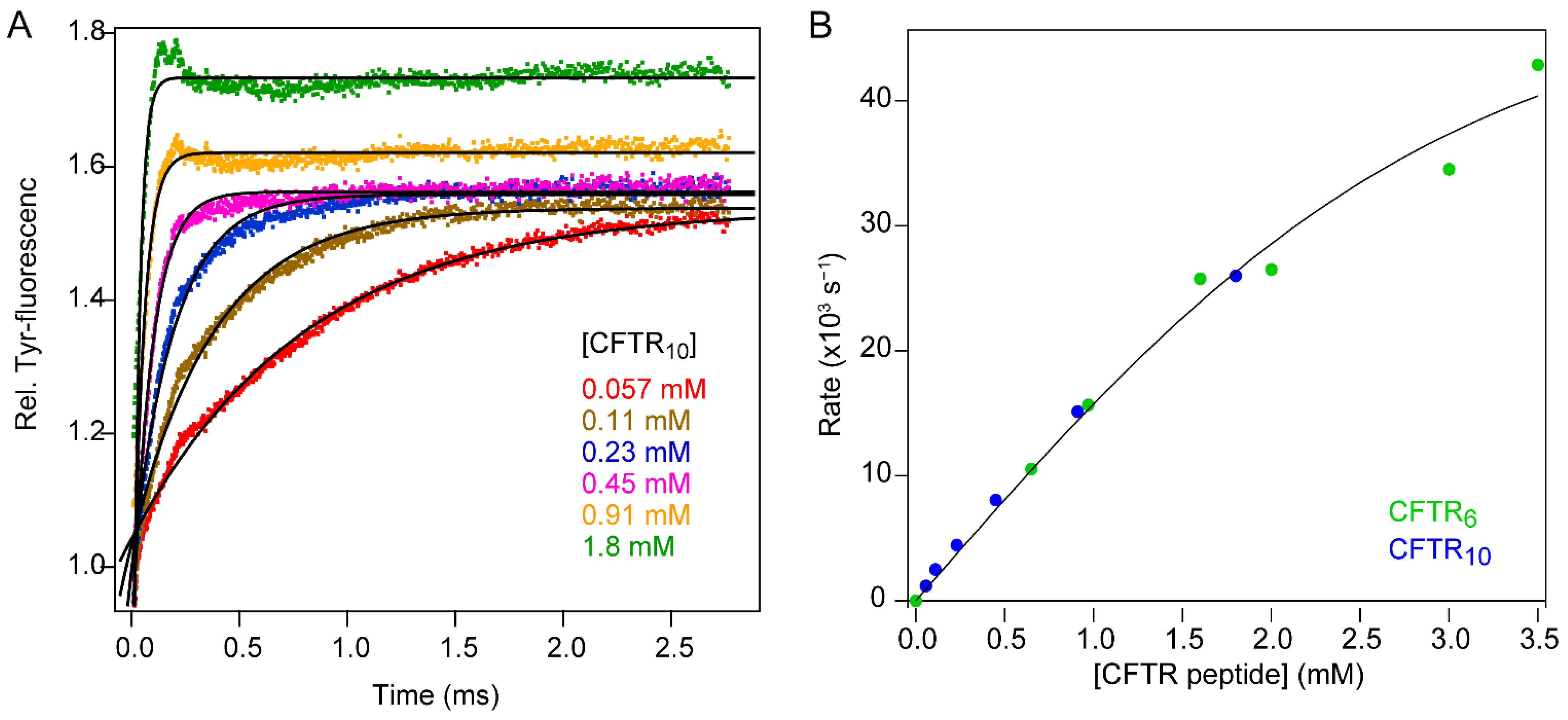
3.2. Binding of a Peptide Ligand to a PDZ Domain
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Gruebele, M.; Sabelko, J.; Ballew, R.; Ervin, J. Laser temperature jump induced protein folding. Acc. Chem. Res. 1998, 31, 699–707. [Google Scholar] [CrossRef]
- Dyer, R.B.; Brauns, E.B. Laser-induced temperature jump infrared measurements of RNA folding. Methods Enzymol. 2009, 469, 353–372. [Google Scholar] [PubMed] [Green Version]
- Prigozhin, M.B.; Liu, Y.; Wirth, A.J.; Kapoor, S.; Winter, R.; Schulten, K.; Gruebele, M. Misplaced helix slows down ultrafast pressure-jump protein folding. Proc. Natl. Acad. Sci. USA 2013, 110, 8087–8092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.M.; Henry, E.R.; Hu, Y.; Chan, C.-K.; Luck, S.D.; Bhuyan, A.; Roder, H.; Hofrichter, J.; Eaton, W.A. Fast events in protein folding initiated by nanosecond laser photolysis. Proc. Natl. Acad. Sci. USA 1993, 90, 11860–11864. [Google Scholar] [CrossRef] [Green Version]
- Pascher, T.; Chesick, J.P.; Winkler, J.R.; Gray, H.B. Protein folding triggered by electron transfer. Science 1996, 271, 1558–1560. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.B.; Vishwanath, A.; Brody, J.P.; Austin, R.H. Hydrodynamic focusing on a silicon chip: Mixing nanoliters in microseconds. Phys. Rev. Lett. 1998, 80, 3863–3866. [Google Scholar] [CrossRef]
- Lapidus, L.J.; Yao, S.; McGarrity, K.S.; Hertzog, D.E.; Tubman, E.; Bakajin, O. Protein hydrophobic collapse and early folding steps observed in a microfluidic mixer. Biophys. J. 2007, 93, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Shastry, M.C.R.; Luck, S.D.; Roder, H. A continuous-flow capillary mixer to monitor reactions on the microsecond time scale. Biophys. J. 1998, 74, 2714–2721. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.-K.; Hu, Y.; Takahashi, S.; Rousseau, D.L.; Eaton, W.A.; Hofrichter, J. Submillisecond protein folding kinetics studied by ultrarapid mixing. Proc. Natl. Acad. Sci. USA 1997, 94, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Bilsel, O.; Kayatekin, C.; Wallace, L.A.; Matthews, C.R. A microchannel solution mixer for studying microsecond protein folding reactions. Rev. Sci. Instr. 2005, 76, 014302. [Google Scholar] [CrossRef]
- Mitic, S.; Strampraad, M.J.F.; Hagen, W.R.; de Vries, S. Microsecond time-scale kinetics of transient biochemical reactions. PLoS ONE 2017, 12, e0185888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCamp, S.J.; Naganathan, A.N.; Waldauer, S.A.; Bakajin, O.; Lapidus, L.J. Direct observation of downhill folding of lambda-repressor in a microfluidic mixer. Biophys. J. 2009, 97, 1772–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roder, H.; Maki, K.; Cheng, H.; Shastry, M.C. Rapid mixing methods for exploring the kinetics of protein folding. Methods 2004, 34, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Roder, H.; Maki, K.; Cheng, H. Early events in protein folding explored by rapid mixing methods. Chem. Rev. 2006, 106, 1836–1861. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, S.V.; Guo, L.; Graceffa, R.; Barrea, R.; Nobrega, R.P.; Matthews, C.R.; Irving, T.C.; Bilsel, O. Minireview: Structural insights into early folding events using continuous-flow time-resolved small-angle X-ray scattering. Biopolymers 2011, 95, 550–558. [Google Scholar] [CrossRef]
- Tonomura, B.; Nakatani, H.; Ohnishi, M.; Yamaguchi-Ito, J.; Hiromi, K. Test reactions for a stopped-flow apparatus. Reduction of 2,6-dichlorophenolindophenol and potassium ferricyanide by L-ascorbic acid. Anal. Biochem. 1978, 84, 370–383. [Google Scholar] [CrossRef]
- Brissette, P.; Ballou, D.P.; Massey, V. Determination of the dead time of a stopped-flow fluorometer. Anal. Biochem. 1989, 181, 234–238. [Google Scholar] [CrossRef] [Green Version]
- Chance, B. Accelerated and stopped-flow apparatus. II. J. Franklin Inst. 1940, 220, 455–613. [Google Scholar] [CrossRef]
- Gibson, Q.H.; Milnes, L. Apparatus for rapid and sensitive spectrophotometry. Biochem. J. 1964, 91, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Berger, R.L.; Balko, B.; Borcherdt, W.; Friauf, W. High speed optical stopped-flow apparatus. Rev. Sci. Instrum. 1968, 39, 486–493. [Google Scholar] [CrossRef]
- Chance, B. Rapid Mixing and Sampling Techniques in Biochemistry; Academic Press: New York, NY, USA, 1964. [Google Scholar]
- Moskowitz, G.W.; Bowman, R.L. Muliticaplillary mixer of solutions. Science 1966, 153, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Regenfuss, P.; Clegg, R.M.; Fulwyler, M.J.; Barrantes, F.J.; Jovin, T.M. Mixing liquids in microseconds. Rev. Sci. Instrum. 1985, 56, 283–290. [Google Scholar] [CrossRef]
- Paeng, K.; Paeng, I.; Kincaid, J. Time-resolved resonance raman spectroscopy using a fast mixing device. Anal. Sci. 1994, 10, 157–159. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Ching, Y.-c.; Wang, J.; Rousseau, D.L. Microsecond generation of oxygen-bound cytochrome c oxidase by rapid solution mixing. J. Biol. Chem. 1995, 270, 8405–8407. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, S.; Takahashi, S.; Ishimori, K.; Morishima, I. Stepwise formation of alpha-helices during cytochrome c folding. Nat. Struct Biol. 2000, 7, 514–520. [Google Scholar] [CrossRef]
- Matsumoto, S.; Yane, A.; Nakashima, S.; Hashida, M.; Fujita, M.; Goto, Y.; Takahashi, S. A rapid flow mixer with 11-mus mixing time microfabricated by a pulsed-laser ablation technique: Observation of a barrier-limited collapse in cytochrome c folding. J. Am. Chem. Soc. 2007, 129, 3840–3841. [Google Scholar] [CrossRef]
- Jiang, L.; Zeng, Y.; Sun, Q.; Sun, Y.; Guo, Z.; Qu, J.Y.; Yao, S. Microsecond protein folding events revealed by time-resolved fluorescence resonance energy transfer in a microfluidic mixer. Anal. Chem. 2015, 87, 5589–5595. [Google Scholar] [CrossRef]
- Inguva, V.; Kathuria, S.V.; Bilsel, O.; Perot, B.J. Computer design of microfluidic mixers for protein/RNA folding studies. PLoS ONE 2018, 13, e0198534. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Li, Y.; Liu, B.F. Micromixers and their applications in kinetic analysis of biochemical reactions. Talanta 2019, 205, 120136. [Google Scholar] [CrossRef]
- Takahashi, S.; Yeh, S.-R.; Das, T.K.; Chan, C.-K.; Gottfried, D.S.; Rousseau, D.L. Folding of cytochrome c initiated by submillisecond mixing. Nat. Struct. Biol. 1997, 4, 44–50. [Google Scholar] [CrossRef]
- Shastry, M.C.R.; Roder, H. Evidence for barrier-limited protein folding kinetics on the microsecond time scale. Nat. Struct. Biol. 1998, 5, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Shastry, M.C.R.; Sauder, J.M.; Roder, H. Kinetic and structural analysis of submillisecond folding events in cytochrome c. Acc. Chem. Res. 1998, 31, 717–725. [Google Scholar] [CrossRef]
- Park, S.-H.; Shastry, M.C.R.; Roder, H. Folding dynamics of the B1 domain of protein G explored by ultrarapid mixing. Nat. Struct. Biol. 1999, 6, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Capaldi, A.P.; Shastry, R.M.C.; Kleanthous, C.; Roder, H.; Radford, S.E. Ultra-rapid mixing experiments reveal that Im7 folds via an on-pathway intermediate. Nat. Struct. Biol. 2001, 8, 68–72. [Google Scholar] [PubMed]
- Teilum, K.; Maki, K.; Kragelund, B.B.; Poulsen, F.M.; Roder, H. Early kinetic intermediate in the folding of acyl-CoA binding protein detected by fluorescence labeling and ultrarapid mixing. Proc. Natl. Acad. Sci. USA 2002, 99, 9807–9812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, K.; Cheng, H.; Dolgikh, D.A.; Shastry, M.C.; Roder, H. Early Events During Folding of Wild-type Staphylococcal Nuclease and a Single-tryptophan Variant Studied by Ultrarapid Mixing. J. Mol. Biol. 2004, 338, 383–400. [Google Scholar] [CrossRef]
- Uzawa, T.; Akiyama, S.; Kimura, T.; Takahashi, S.; Ishimori, K.; Morishima, I.; Fujisawa, T. Collapse and search dynamics of apomyoglobin folding revealed by submillisecond observations of α-helical content and compactness. Proc. Natl. Acad. Sci. USA 2004, 101, 1171–1176. [Google Scholar] [CrossRef] [Green Version]
- Gianni, S.; Walma, T.; Arcovito, A.; Calosci, N.; Bellelli, A.; Engstrom, A.; Travaglini-Allocatelli, C.; Brunori, M.; Jemth, P.; Vuister, G.W. Demonstration of long-range interactions in a PDZ domain by NMR, kinetics, and protein engineering. Structure 2006, 14, 1801–1809. [Google Scholar] [CrossRef] [Green Version]
- Maki, K.; Cheng, H.; Dolgikh, D.A.; Roder, H. Folding kinetics of staphylococcal nuclease studied by tryptophan engineering and rapid mixing methods. J. Mol. Biol. 2007, 368, 244–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apetri, A.C.; Maki, K.; Roder, H.; Surewicz, W.K. Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. J. Am. Chem. Soc. 2006, 128, 11673–11678. [Google Scholar] [CrossRef] [Green Version]
- Arai, M.; Kondrashkina, E.; Kayatekin, C.; Matthews, C.R.; Iwakura, M.; Bilsel, O. Microsecond hydrophobic collapse in the folding of Escherichia coli dihydrofolate reductase, an alpha/beta-type protein. J. Mol. Biol. 2007, 368, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Xu, M.; Wedemeyer, W.J.; Roder, H. Microsecond unfolding kinetics of sheep prion protein reveals an intermediate that correlates with susceptibility to classical scrapie. Biophys. J. 2011, 101, 1221–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Beresneva, O.; Rosario, R.; Roder, H. Microsecond folding dynamics of apomyoglobin at acidic pH. J. Phys. Chem. B 2012, 116, 7014–7025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizukami, T.; Xu, M.; Cheng, H.; Roder, H.; Maki, K. Nonuniform chain collapse during early stages of staphylococcal nuclease folding detected by fluorescence resonance energy transfer and ultrarapid mixing methods. Protein Sci. 2013, 22, 1336–1348. [Google Scholar] [CrossRef]
- Honda, R.P.; Xu, M.; Yamaguchi, K.; Roder, H.; Kuwata, K. A Native-like Intermediate Serves as a Branching Point between the Folding and Aggregation Pathways of the Mouse Prion Protein. Structure 2015, 23, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Goluguri, R.R.; Udgaonkar, J.B. Microsecond Rearrangements of Hydrophobic Clusters in an Initially Collapsed Globule Prime Structure Formation during the Folding of a Small Protein. J. Mol. Biol. 2016, 428, 3102–3117. [Google Scholar] [CrossRef]
- Sen, S.; Goluguri, R.R.; Udgaonkar, J.B. A Dry Transition State More Compact Than the Native State Is Stabilized by Non-Native Interactions during the Unfolding of a Small Protein. Biochemistry 2017, 56, 3699–3703. [Google Scholar] [CrossRef]
- Srour, B.; Strampraad, M.J.F.; Hagen, W.R.; Hagedoorn, P.L. Refolding kinetics of cytochrome c studied with microsecond timescale continuous-flow UV-vis spectroscopy and rapid freeze-quench EPR. J. Inorg. Biochem. 2018, 184, 42–49. [Google Scholar] [CrossRef]
- Mizukami, T.; Xu, M.; Fazlieva, R.; Bychkova, V.E.; Roder, H. Complex Folding Landscape of Apomyoglobin at Acidic pH Revealed by Ultrafast Kinetic Analysis of Core Mutants. J. Phys. Chem. B 2018, 122, 11228–11239. [Google Scholar] [CrossRef]
- Mizukami, T.; Bedford, J.T.; Liao, S.; Greene, L.H.; Roder, H. Effects of ionic strength on the folding and stability of SAMP1, a ubiquitin-like halophilic protein. Biophys. J. 2022, 121, 552–564. [Google Scholar] [CrossRef]
- Dingfelder, F.; Wunderlich, B.; Benke, S.; Zosel, F.; Zijlstra, N.; Nettels, D.; Schuler, B. Rapid Microfluidic Double-Jump Mixing Device for Single-Molecule Spectroscopy. J. Am. Chem. Soc. 2017, 139, 6062–6065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, R.L.; Balko, B.; Chapman, H.F. High resolution mixer for the study of the kinetics of rapid reactions in solution. Rev. Sci. Instrum. 1968, 39, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Bellouard, Y.; Said, A.; Dugan, M.; Bado, P. Fabrication of high-aspect ratio, micro-fluidic channels and tunnels using femtosecond laser pulses and chemical etching. Opt. Express 2004, 12, 2120–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roder, H.; Elöve, G.A.; Englander, S.W. Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature 1988, 335, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabit, S.A.; Roder, H.; Hagen, S.J. Internal friction controls the speed of protein folding from a compact configuration. Biochemistry 2004, 43, 12532–12538. [Google Scholar] [CrossRef]
- Tsong, T.Y. The Trp-59 fluorescence of ferricytochrome c as a sensitive measure of the over-all protein conformation. J. Biol. Chem. 1974, 249, 1988–1990. [Google Scholar] [CrossRef]
- Elöve, G.A.; Chaffotte, A.F.; Roder, H.; Goldberg, M.E. Early steps in cytochrome c folding probed by time-resolved circular dichroism and fluorescence spectroscopy. Biochemistry 1992, 31, 6876–6883. [Google Scholar] [CrossRef]
- Hagen, S.J.; Eaton, W.A. Two-state expansion and collapse of a polypeptide. J. Mol. Biol. 2000, 297, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Fazelinia, H.; Xu, M.; Cheng, H.; Roder, H. Ultrafast hydrogen exchange reveals specific structural events during the initial stages of folding of cytochrome c. J. Am. Chem. Soc. 2014, 136, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Vogt, A.D.; Di Cera, E. Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry 2012, 51, 5894–5902. [Google Scholar] [CrossRef]
- Gianni, S.; Dogan, J.; Jemth, P. Distinguishing induced fit from conformational selection. Biophys. Chem. 2014, 189, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammes, G.G.; Chang, Y.C.; Oas, T.G. Conformational selection or induced fit: A flux description of reaction mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 13737–13741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizukami, T.; Furuzawa, S.; Itoh, S.G.; Segawa, S.; Ikura, T.; Ihara, K.; Okumura, H.; Roder, H.; Maki, K. Energetics and kinetics of substrate analog-coupled staphylococcal nuclease folding revealed by a statistical mechanical approach. Proc. Natl. Acad. Sci. USA 2020, 117, 19953–19962. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, S.; Leung, T.; Birrane, G.; Webster, G.; Ladias, J.A. Crystal structure of the PDZ1 domain of human Na(+)/H(+) exchanger regulatory factor provides insights into the mechanism of carboxyl-terminal leucine recognition by class I PDZ domains. J. Mol. Biol. 2001, 308, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Cushing, P.R.; Fellows, A.; Villone, D.; Boisguerin, P.; Madden, D.R. The relative binding affinities of PDZ partners for CFTR: A biochemical basis for efficient endocytic recycling. Biochemistry 2008, 47, 10084–10098. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizukami, T.; Roder, H. Advances in Mixer Design and Detection Methods for Kinetics Studies of Macromolecular Folding and Binding on the Microsecond Time Scale. Molecules 2022, 27, 3392. https://doi.org/10.3390/molecules27113392
Mizukami T, Roder H. Advances in Mixer Design and Detection Methods for Kinetics Studies of Macromolecular Folding and Binding on the Microsecond Time Scale. Molecules. 2022; 27(11):3392. https://doi.org/10.3390/molecules27113392
Chicago/Turabian StyleMizukami, Takuya, and Heinrich Roder. 2022. "Advances in Mixer Design and Detection Methods for Kinetics Studies of Macromolecular Folding and Binding on the Microsecond Time Scale" Molecules 27, no. 11: 3392. https://doi.org/10.3390/molecules27113392
APA StyleMizukami, T., & Roder, H. (2022). Advances in Mixer Design and Detection Methods for Kinetics Studies of Macromolecular Folding and Binding on the Microsecond Time Scale. Molecules, 27(11), 3392. https://doi.org/10.3390/molecules27113392