
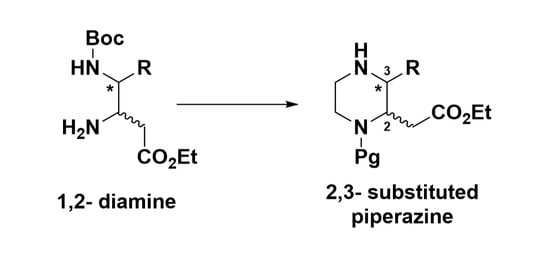
A Concise Synthetic Method for Constructing 3-Substituted Piperazine-2-Acetic Acid Esters from 1,2-Diamines

 , , and
, , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
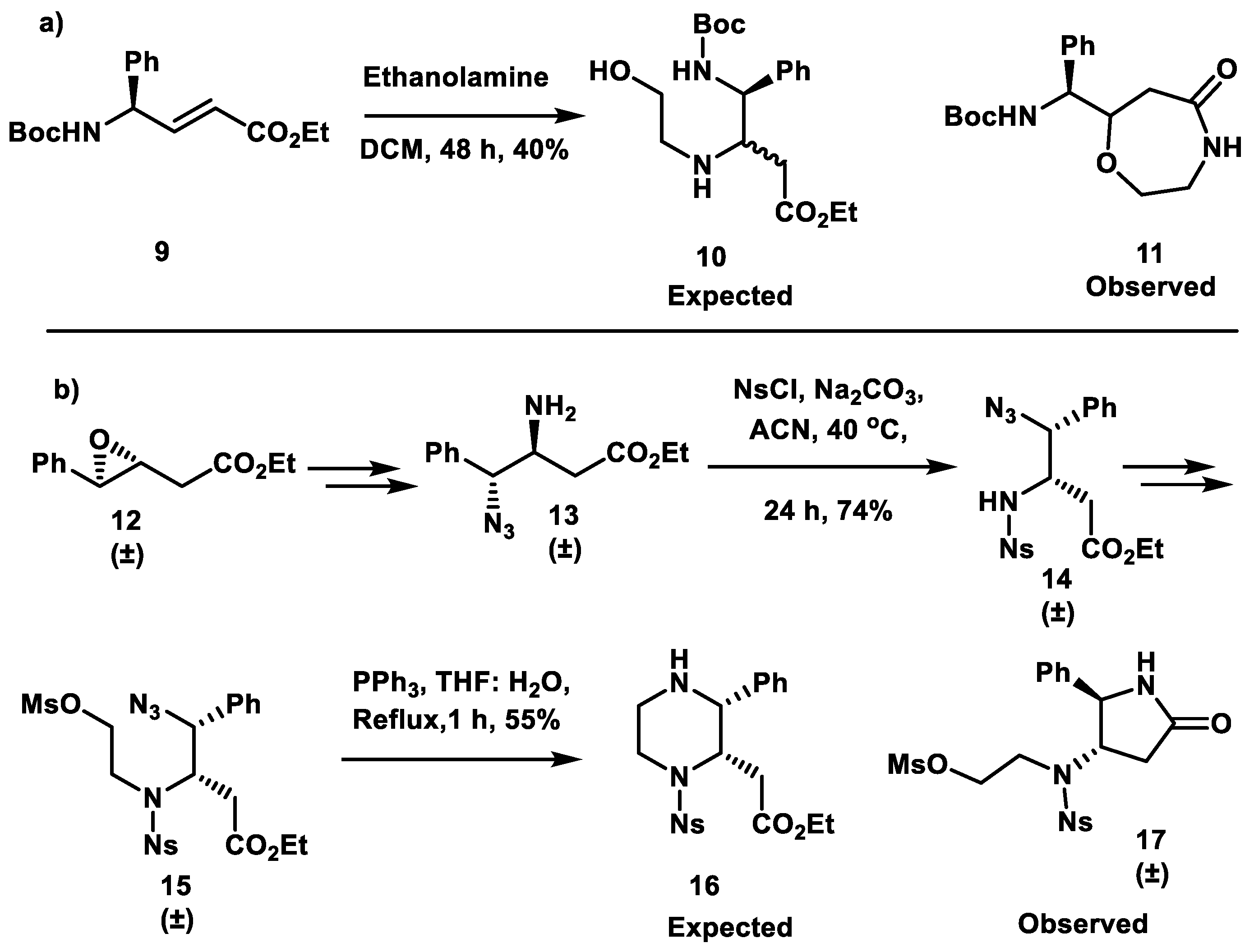
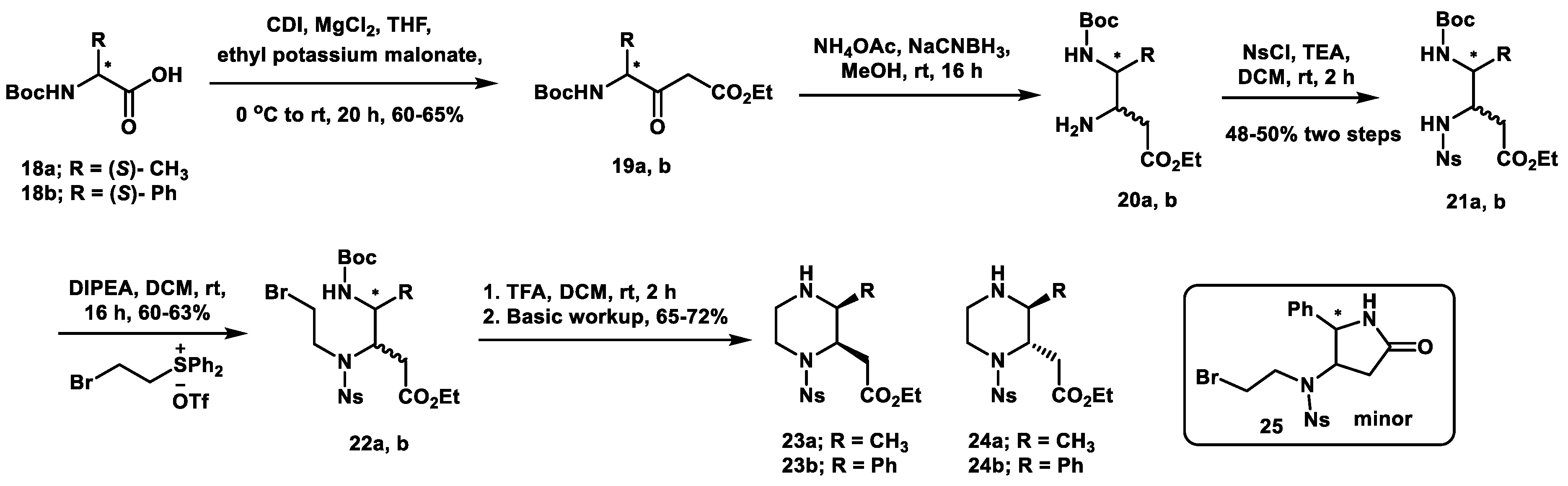
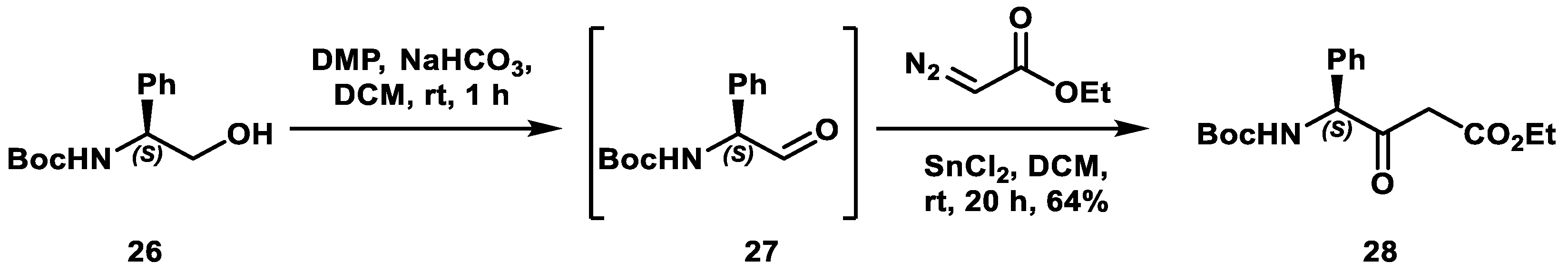
2. Results and Discussion
3. Experimental
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghorbani, M.; Bushra, B.A.; Mamatha, S.; Khanum, S.A. Piperazine and morpholine: Synthetic preview and pharmaceutical applications. Res. J. Pharm. Techol. 2015, 8, 611–628. [Google Scholar] [CrossRef]
- Rathi, A.K.; Syed, R.; Shin, H.-S.; Patel, R.V. Piperazine derivatives for therapeutic use: A patent review (2010–present). Expert Opin. Ther. Pat. 2016, 26, 777–797. [Google Scholar] [CrossRef] [PubMed]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef]
- Brito, A.F.; Moreira, L.K.; Menegatti, R.; Costa, E.A. Piperazine derivatives with central pharmacological activity used as therapeutic tools. Fundam. Clinic. Pharm. 2019, 33, 13–24. [Google Scholar] [CrossRef]
- Welsch, M.E.; Snyder, S.A.; Stockwell, B.R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Magriotis, P.A. Recent progress toward the asymmetric synthesis of carbon-substituted piperazine pharmacophores and oxidative related heterocycles. RSC Med. Chem. 2020, 11, 745–759. [Google Scholar] [CrossRef]
- Gettys, K.E.; Ye, Z.; Dai, M. Recent advances in piperazine synthesis. Synthesis 2017, 49, 2589–2604. [Google Scholar] [CrossRef]
- Mordini, A.; Reginato, G.; Calamante, M.; Zani, L. Stereoselective synthesis of polysubstituted piperazines and oxopiperazines. Useful building blocks in medicinal chemistry. Curr. Top. Med. Chem. 2014, 14, 1308–1316. [Google Scholar] [CrossRef]
- Sun, A.W.; Hess, S.N.; Stoltz, B.M. Enantioselective synthesis of gem-disubstituted N-Boc diazaheterocycles via decarboxylative asymmetric allylic alkylation. Chem. Sci. 2019, 10, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Lovering, F. Escape from Flatland 2: Complexity and promiscuity. MedChemComm 2013, 4, 515–519. [Google Scholar] [CrossRef]
- Zhong, C.; Wang, Y.; Hung, A.W.; Schreiber, S.L.; Young, D.W. Diastereoselective control of intramolecular aza-Michael reactions using achiral catalysts. Org. Lett. 2011, 13, 5556–5559. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Wang, Y.; O’Herin, C.; Young, D.W. Synthesis of Substituted Morpholines Using Stereodivergent Aza-Michael Reactions Catalyzed by Brønsted Acids. ACS Catal. 2013, 3, 643–646. [Google Scholar] [CrossRef]
- Durand, C.; Szostak, M. Recent Advances in the Synthesis of Piperazines: Focus on C–H Functionalization. Organics 2021, 2, 337–347. [Google Scholar] [CrossRef]
- Chamakuri, S.; Jain, P.; Reddy Guduru, S.K.; Arney, J.W.; MacKenzie, K.R.; Santini, C.; Young, D.W. Synthesis of Enantiomerically Pure 6-Substituted-Piperazine-2-Acetic Acid Esters as Intermediates for Library Production. J. Org. Chem. 2018, 83, 6541–6555. [Google Scholar] [CrossRef]
- Reddy Guduru, S.K.; Chamakuri, S.; Raji, I.O.; MacKenzie, K.R.; Santini, C.; Young, D.W. Synthesis of Enantiomerically Pure 3-Substituted Piperazine-2-acetic Acid Esters as Intermediates for Library Production. J. Org. Chem. 2018, 83, 11777–11793. [Google Scholar] [CrossRef]
- Jain, P.; Raji, I.O.; Chamakuri, S.; MacKenzie, K.R.; Ebright, B.T.; Santini, C.; Young, D.W. Synthesis of Enantiomerically Pure 5-Substituted Piperazine-2-Acetic Acid Esters as Intermediates for Library Production. J. Org. Chem. 2019, 84, 6040–6064. [Google Scholar] [CrossRef]
- Chamakuri, S.; Shah, M.M.; Yang, D.C.H.; Santini, C.; Young, D.W. Practical and scalable synthesis of orthogonally protected-2-substituted chiral piperazines. Org. Biomol. Chem. 2020, 18, 8844–8849. [Google Scholar] [CrossRef]
- Chamakuri, S.; Chung, M.K.; Samuel, E.L.G.; Tran, K.A.; Chen, Y.C.; Nyshadham, P.; Santini, C.; Matzuk, M.M.; Young, D.W. Design and construction of a stereochemically diverse piperazine-based DNA-encoded chemical library. Bioorg. Med. Chem. 2021, 48, 116387. [Google Scholar] [CrossRef]
- Nordstrøm, L.U.; Madsen, R. Iridium catalysed synthesis of piperazines from diols. Chem. Comm. 2007, 47, 5034–5036. [Google Scholar] [CrossRef] [PubMed]
- Yar, M.; McGarrigle, E.M.; Aggarwal, V.K. An Annulation Reaction for the Synthesis of Morpholines, Thiomorpholines, and Piperazines from β-Heteroatom Amino Compounds and Vinyl Sulfonium Salts. Angew. Chem. 2008, 120, 3844–3846. [Google Scholar] [CrossRef]
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Yokoshima, S.; Watanabe, K.; Uehara, F.; Usui, Y.; Tanaka, H. Asymmetric synthesis of 2-arylpiperazines. Bioorg. Med. Chem. Lett. 2014, 24, 5749–5751. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rosello, M.; Delgado, O.; Mateu, N.; Trabanco, A.A.; Van Gool, M.; Fustero, S. Diastereoselective synthesis of 2-phenyl-3-(trifluoromethyl) piperazines as building blocks for drug discovery. J. Org. Chem. 2014, 79, 5887–5894. [Google Scholar] [CrossRef]
- Viso, A.; Fernandez de la Pradilla, R.; Flores, A.; García, A.; Tortosa, M.; López-Rodríguez, M.L. Synthesis of highly substituted enantiopure piperazines and ketopiperazines from vicinal N-sulfinyl diamines. J. Org. Chem. 2006, 71, 1442–1448. [Google Scholar] [CrossRef]
- Bisol, T.B.; Bortoluzzi, A.J.; Sa, M.M. Nucleophilic ring-opening of epoxide and aziridine acetates for the stereodivergent synthesis of β-hydroxy and β-amino γ-lactams. J. Org. Chem. 2011, 76, 948–962. [Google Scholar] [CrossRef]
- Theberge, C.R.; Zercher, C.K. Chain extension of amino acid skeletons: Preparation of ketomethylene isosteres. Tetrahedron 2003, 59, 1521–1527. [Google Scholar] [CrossRef]
- Nudelman, A.; Marcovici-Mizrahi, D.; Nudelman, A.; Flint, D.; Wittenbach, V. Inhibitors of biotin biosynthesis as potential herbicides. Tetrahedron 2004, 60, 1731–1748. [Google Scholar] [CrossRef]
- Ganesh Kumar, M.; Thombare, V.J.; Bhaisare, R.D.; Adak, A.; Gopi, H.N. Synthesis of Tetrasubstituted Symmetrical Pyrazines from β-Keto γ-Amino Esters: A Mild Strategy for Self-Dimerization of Peptides. Eur. J. Org. Chem. 2015, 2015, 135–141. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Agrawal, N.; Mali, S.M.; Jadhav, S.V.; Gopi, H.N. Tin (II) chloride assisted synthesis of N-protected γ-amino β-keto esters through semipinacol rearrangement. Org. Biomol. Chem. 2010, 8, 4855–4860. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamakuri, S.; Tang, S.A.; Tran, K.A.; Guduru, S.K.R.; Bolin, P.K.; MacKenzie, K.R.; Young, D.W. A Concise Synthetic Method for Constructing 3-Substituted Piperazine-2-Acetic Acid Esters from 1,2-Diamines. Molecules 2022, 27, 3419. https://doi.org/10.3390/molecules27113419
Chamakuri S, Tang SA, Tran KA, Guduru SKR, Bolin PK, MacKenzie KR, Young DW. A Concise Synthetic Method for Constructing 3-Substituted Piperazine-2-Acetic Acid Esters from 1,2-Diamines. Molecules. 2022; 27(11):3419. https://doi.org/10.3390/molecules27113419
Chicago/Turabian StyleChamakuri, Srinivas, Sunny Ann Tang, Kevin A. Tran, Shiva Krishna Reddy Guduru, Peter K. Bolin, Kevin R. MacKenzie, and Damian W. Young. 2022. "A Concise Synthetic Method for Constructing 3-Substituted Piperazine-2-Acetic Acid Esters from 1,2-Diamines" Molecules 27, no. 11: 3419. https://doi.org/10.3390/molecules27113419
APA StyleChamakuri, S., Tang, S. A., Tran, K. A., Guduru, S. K. R., Bolin, P. K., MacKenzie, K. R., & Young, D. W. (2022). A Concise Synthetic Method for Constructing 3-Substituted Piperazine-2-Acetic Acid Esters from 1,2-Diamines. Molecules, 27(11), 3419. https://doi.org/10.3390/molecules27113419