
Data-Driven and Multiscale Modeling of DNA-Templated Dye Aggregates

 , ,
, , 
Abstract
:1. Introduction
2. Methods
2.1. Machine Learning
2.2. Density Functional Theory
2.3. Molecular Dynamics
3. Results
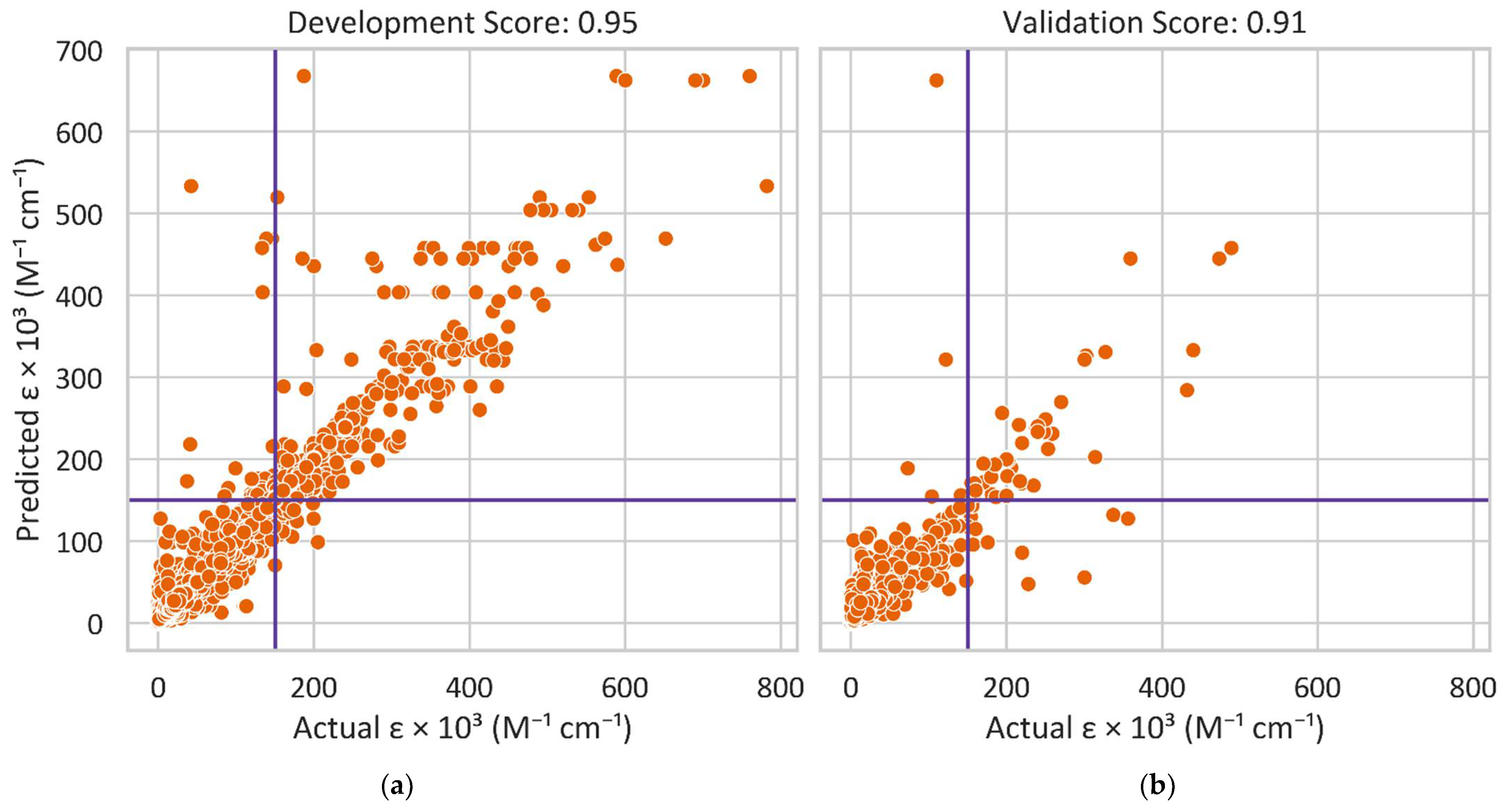
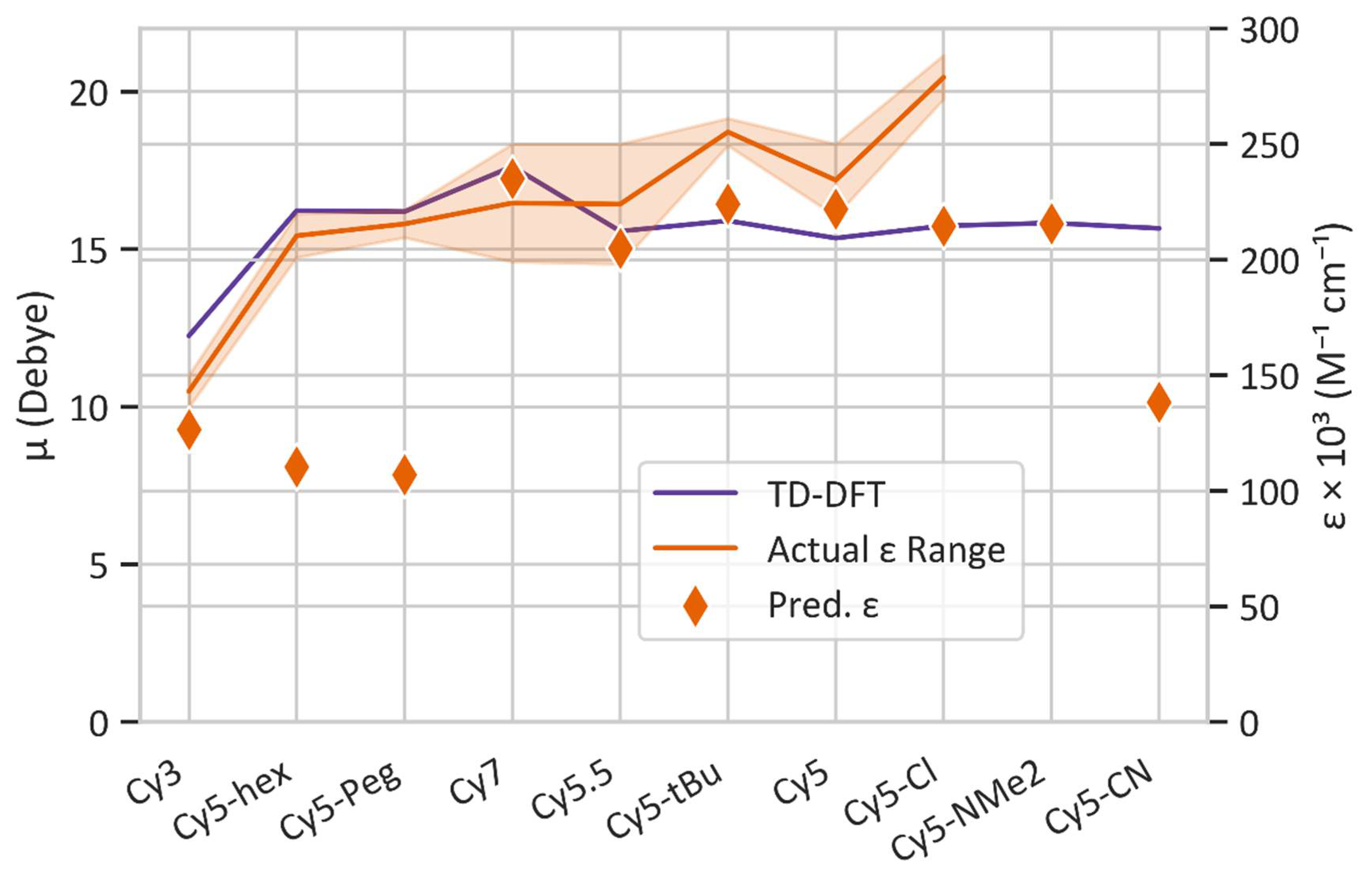
3.1. Dye Screening Using Machine Learning and Density Functional Theory
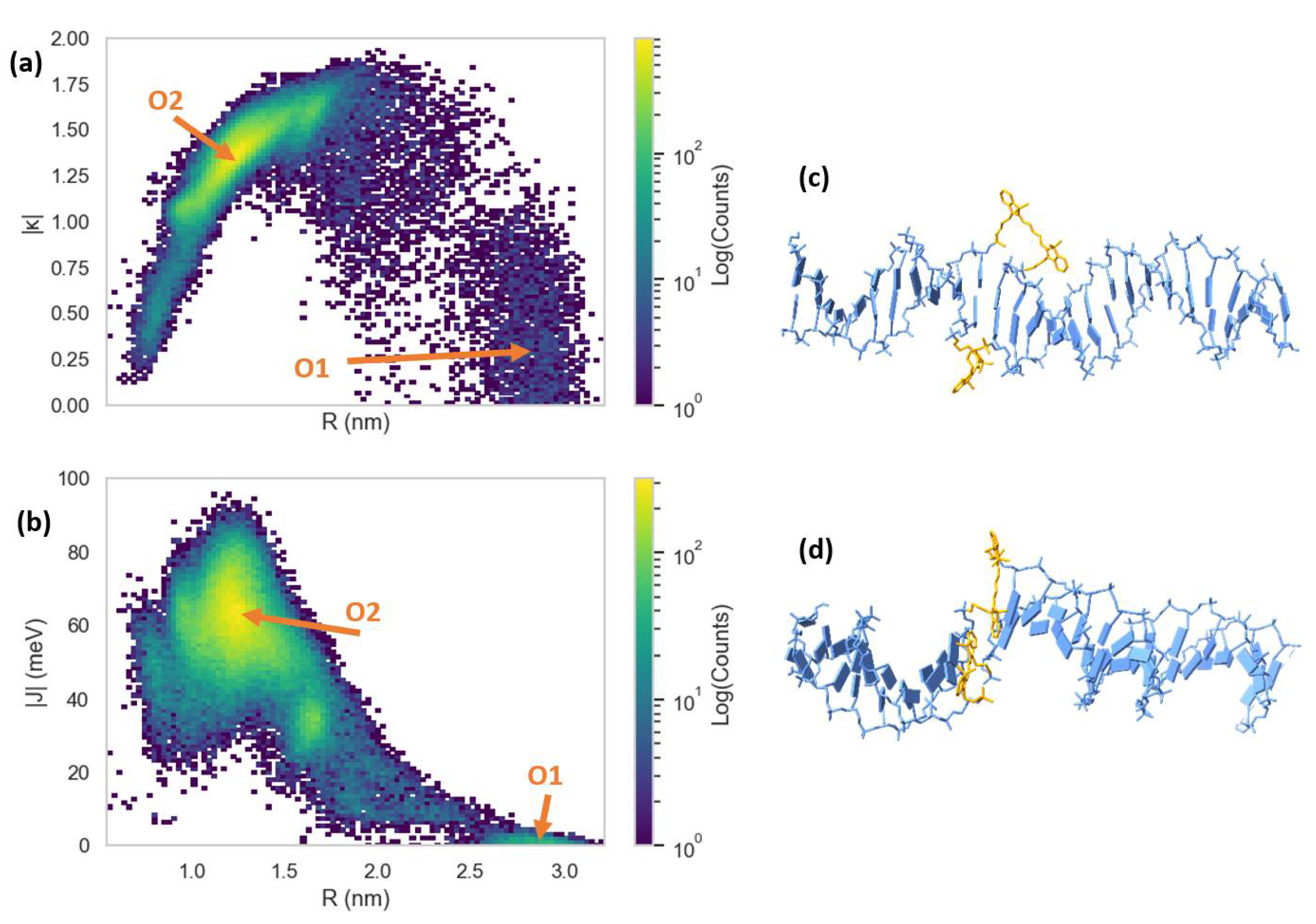
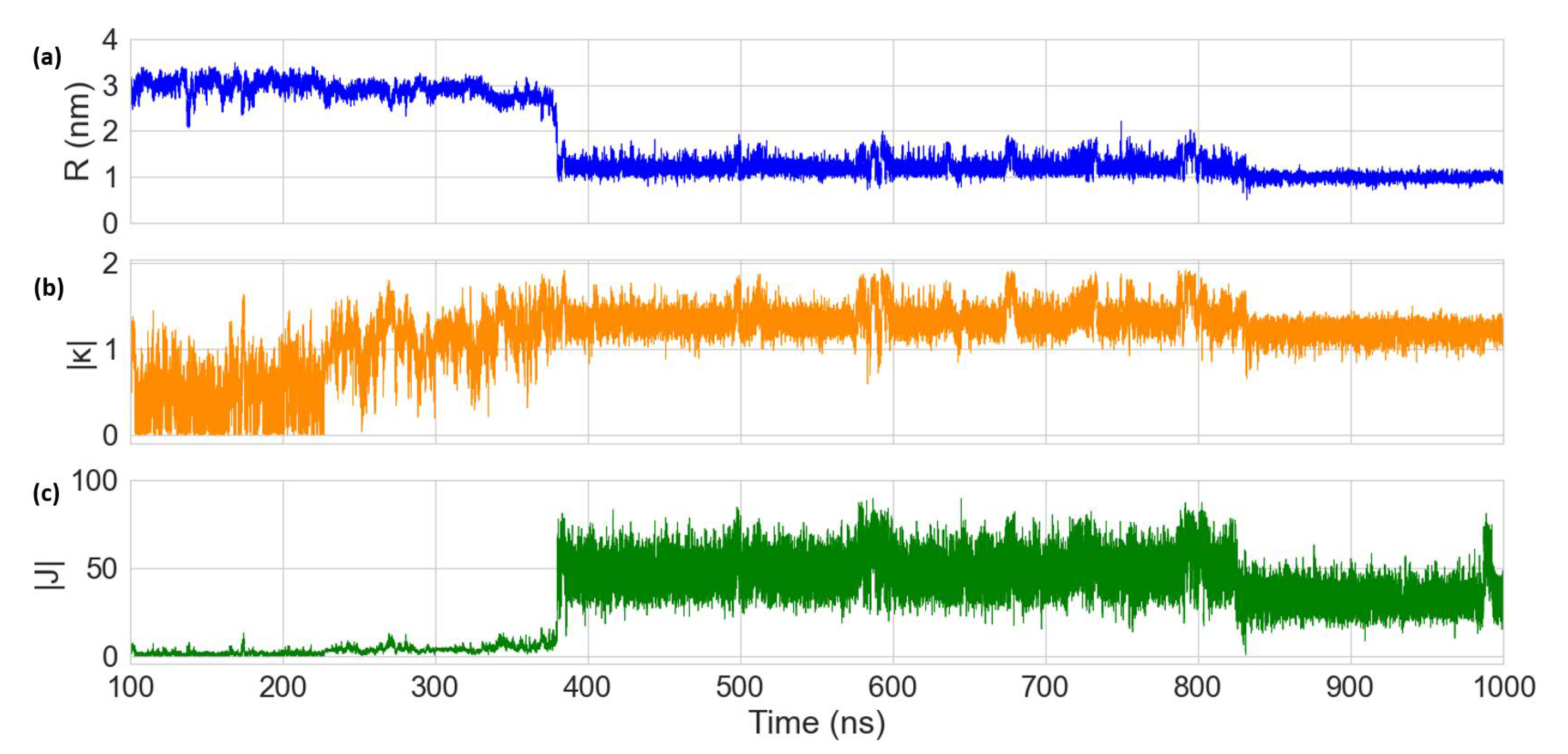
3.2. Molecular Dynamics Simulations of Dye Aggregate–DNA Duplex Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ilina, K.; MacCuaig, W.M.; Laramie, M.; Jeouty, J.N.; McNally, L.R.; Henary, M. Squaraine Dyes: Molecular Design for Different Applications and Remaining Challenges. Bioconjug. Chem. 2020, 31, 194–213. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, K.; Citterio, D.; Suzuki, K. New Trends in Near-Infrared Fluorophores for Bioimaging. Anal. Sci. 2014, 30, 327–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholes, G.D.; Fleming, G.R.; Olaya-Castro, A.; Van Grondelle, R. Lessons from nature about solar light harvesting. Nat. Chem. 2011, 3, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Collado-Fregoso, E.; Boufflet, P.; Fei, Z.; Gann, E.; Ashraf, S.; Li, Z.; Mcneill, C.R.; Durrant, J.R.; Heeney, M. Increased Exciton Dipole Moment Translates into Charge-Transfer Excitons in Thiophene-Fluorinated Low-Bandgap Polymers for Organic Photovoltaic Applications. Chem. Mater. 2015, 27, 7934–7944. [Google Scholar] [CrossRef] [Green Version]
- Markov, R.V.; Plekhanov, A.I.; Shelkovnikov, V.V.; Knoester, J. Giant Nonlinear Optical Response of Interacting One-Dimensional Frenkel Excitons in Molecular Aggregates. Phys. Status Solidi 2000, 221, 529–533. [Google Scholar] [CrossRef]
- Kellis, D.L.; Sarter, C.; Cannon, B.L.; Davis, P.H.; Graugnard, E.; Lee, J.; Pensack, R.D.; Kolmar, T.; Jäschke, A.; Yurke, B.; et al. An All-Optical Excitonic Switch Operated in the Liquid and Solid Phases. ACS Nano 2019, 13, 2986–2994. [Google Scholar] [CrossRef]
- Cannon, B.L.; Kellis, D.L.; Davis, P.H.; Lee, J.; Kuang, W.; Hughes, W.L.; Graugnard, E.; Yurke, B.; Knowlton, W.B. Excitonic AND Logic Gates on DNA Brick Nanobreadboards. ACS Photonics 2015, 2, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Outeiral, C.; Strahm, M.; Shi, J.; Morris, G.M.; Benjamin, S.C.; Deane, C.M. The prospects of quantum computing in computational molecular biology. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1481. [Google Scholar] [CrossRef]
- Wasielewski, M.R.; Forbes, M.D.E.; Frank, N.L.; Kowalski, K.; Scholes, G.D.; Yuen-Zhou, J.; Baldo, M.A.; Freedman, D.E.; Goldsmith, R.H.; Goodson, T.; et al. Exploiting chemistry and molecular systems for quantum information science. Nat. Rev. Chem. 2020, 4, 490–504. [Google Scholar] [CrossRef]
- Lewis, J.E.; Maroncelli, M. On the (uninteresting) dependence of the absorption and emission transition moments of coumarin 153 on solvent. Chem. Phys. Lett. 1998, 282, 197–203. [Google Scholar] [CrossRef]
- Chako, N.Q. Absorption of light in organic compounds. J. Chem. Phys. 1934, 2, 644–653. [Google Scholar] [CrossRef]
- Marciniak, H.; Auerhammer, N.; Ricker, S.; Schmiedel, A.; Holzapfel, M.; Lambert, C. Reduction of the Fluorescence Transition Dipole Moment by Excitation Localization in a Vibronically Coupled Squaraine Dimer. J. Phys. Chem. C 2019, 123, 3426–3432. [Google Scholar] [CrossRef]
- Namuangruk, S.; Fukuda, R.; Ehara, M.; Meeprasert, J.; Khanasa, T.; Morada, S.; Kaewin, T.; Jungsuttiwong, S.; Sudyoadsuk, T.; Promarak, V. D−D−π−A-Type Organic Dyes for Dye-Sensitized Solar Cells with a Potential for Direct Electron Injection and a High Extinction Coefficient: Synthesis, Characterization, and Theoretical Investigation. J. Phys. Chem. C 2012, 116, 25653–25663. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, Y.; Zhu, H.; Chai, Q.; Liu, J.; Li, H.; Song, X.; Zhu, W.-H. Rational Molecular Engineering of Indoline-Based D-A-π-A Organic Sensitizers for Long-Wavelength-Responsive Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Yang, X.; Wang, H.; An, J.; Yu, Z.; Wang, X.; Hagfeldt, A.; Sun, L. Improving energy transfer efficiency of dye-sensitized solar cell by fine tuning of dye planarity. Sol. Energy 2019, 187, 274–280. [Google Scholar] [CrossRef]
- Sik Yoon, W.; Won Kim, D.; Park, J.-M.; Cho, I.; Kyu Kwon, O.; Ryeol Whang, D.; Hong Kim, J.; Park, J.-H.; Young Park, S. A Novel Bis-Lactam Acceptor with Outstanding Molar Extinction Coefficient and Structural Planarity for Donor−Acceptor Type Conjugated Polymer. Macromolecules 2016, 49, 8489–8497. [Google Scholar] [CrossRef]
- Che, Y.; Perepichka, D.F. Quantifying Planarity in the Design of Organic Electronic Materials. Angew. Chem.-Int. Ed. 2021, 60, 1364–1373. [Google Scholar] [CrossRef]
- Engel, G.S.; Calhoun, T.R.; Read, E.L.; Ahn, T.K.; Mančal, T.; Cheng, Y.C.; Blankenship, R.E.; Fleming, G.R. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 2007, 446, 782–786. [Google Scholar] [CrossRef]
- Mirkovic, T.; Ostroumov, E.E.; Anna, J.M.; Van Grondelle, R.; Govindjee; Scholes, G.D. Light absorption and energy transfer in the antenna complexes of photosynthetic organisms. Chem. Rev. 2017, 117, 249–293. [Google Scholar] [CrossRef]
- Lim, J.M.; Kim, P.; Yoon, M.C.; Sung, J.; Dehm, V.; Chen, Z.; Würthner, F.; Kim, D. Exciton delocalization and dynamics in helical π-stacks of self-assembled perylene bisimides. Chem. Sci. 2013, 4, 388–397. [Google Scholar] [CrossRef]
- Bialas, D.; Zitzler-Kunkel, A.; Kirchner, E.; Schmidt, D.; Würthner, F. Structural and quantum chemical analysis of exciton coupling in homo-and heteroaggregate stacks of merocyanines. Nat. Commun. 2016, 7, 12949. [Google Scholar] [CrossRef] [PubMed]
- Kasha, M. Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates. Radiat. Res. 1963, 20, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Abramavicius, D.; Palmieri, B.; Mukamel, S. Extracting single and two-exciton couplings in photosynthetic complexes by coherent two-dimensional electronic spectra. Chem. Phys. 2009, 357, 79–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasha, M.; Rawls, H.R.; Ashraf El-Bayoumi, M. The Exciton Model in Molecular Spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. [Google Scholar] [CrossRef] [Green Version]
- Davydov, A.S. Theory of Absorption Spectra of Molecular Crystals. Transl. Repr. Zh. Eksp. Teor. Fiz. 1948, 18, 210–218. [Google Scholar]
- Davydov, A.S. The Theory of Molecular Excitons. Sov. Phys. Uspekhi 1964, 7, 393–448. [Google Scholar] [CrossRef]
- Cannon, B.L.; Kellis, D.L.; Patten, L.K.; Davis, P.H.; Lee, J.; Graugnard, E.; Yurke, B.; Knowlton, W.B. Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121, 6905–6916. [Google Scholar] [CrossRef]
- Cannon, B.L.; Patten, L.K.; Kellis, D.L.; Davis, P.H.; Lee, J.; Graugnard, E.; Yurke, B.; Knowlton, W.B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122, 2086–2095. [Google Scholar] [CrossRef]
- Huff, J.S.; Turner, D.B.; Mass, O.A.; Patten, L.K.; Wilson, C.K.; Roy, S.K.; Barclay, M.S.; Yurke, B.; Knowlton, W.B.; Davis, P.H.; et al. Excited-State Lifetimes of DNA-Templated Cyanine Dimer, Trimer, and Tetramer Aggregates: The Role of Exciton Delocalization, Dye Separation, and DNA Heterogeneity. J. Phys. Chem. B 2021, 125, 10240–10259. [Google Scholar] [CrossRef]
- Hart, S.M.; Chen, W.J.; Banal, J.L.; Bricker, W.P.; Dodin, A.; Markova, L.; Vyborna, Y.; Willard, A.P.; Häner, R.; Bathe, M.; et al. Engineering couplings for exciton transport using synthetic DNA scaffolds. Chem 2021, 7, 752–773. [Google Scholar] [CrossRef]
- Mass, O.A.; Wilson, C.K.; Roy, S.K.; Barclay, M.S.; Patten, L.K.; Terpetschnig, E.A.; Lee, J.; Pensack, R.D.; Yurke, B.; Knowlton, W.B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124, 9636–9647. [Google Scholar] [CrossRef] [PubMed]
- Barclay, M.S.; Roy, S.K.; Huff, J.S.; Mass, O.A.; Turner, D.B.; Wilson, C.K.; Kellis, D.L.; Terpetschnig, E.A.; Lee, J.; Davis, P.H.; et al. Rotaxane rings promote oblique packing and extended lifetimes in DNA-templated molecular dye aggregates. Commun. Chem. 2021, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Banal, J.L.; Kondo, T.; Veneziano, R.; Bathe, M.; Schlau-Cohen, G.S. Photophysics of J-Aggregate-Mediated Energy Transfer on DNA. J. Phys. Chem. Lett. 2017, 8, 5827–5833. [Google Scholar] [CrossRef] [PubMed]
- Markova, L.I.; Malinovskii, V.L.; Patsenker, L.D.; Häner, R. J- vs. H-type assembly: Pentamethine cyanine (Cy5) as a near-IR chiroptical reporter. Chem. Commun. 2013, 49, 5298–5300. [Google Scholar] [CrossRef] [Green Version]
- Kringle, L.; Sawaya, N.P.D.; Widom, J.; Adams, C.; Raymer, M.G.; Aspuru-Guzik, A.; Marcus, A.H. Temperature-dependent conformations of exciton-coupled Cy3 dimers in double-stranded DNA. J. Chem. Phys. 2018, 148, 085101. [Google Scholar] [CrossRef]
- Seifert, J.L.; Connor, R.E.; Kushon, S.A.; Wang, M.; Armitage, B.A. Spontaneous Assembly of Helical Cyanine Dye Aggregates on DNA Nanotemplates. J. Am. Chem. Soc. 1999, 121, 2987–2995. [Google Scholar] [CrossRef]
- Garoff, R.A.; Litzinger, E.A.; Connor, R.E.; Fishman, I.; Armitage, B.A. Helical Aggregation of Cyanine Dyes on DNA Templates: Effect of Dye Structure on Formation of Homo-and Heteroaggregates. Langmuir 2002, 18, 6330–6337. [Google Scholar] [CrossRef]
- Chowdhury, A.U.; Díaz, S.A.; Huff, J.S.; Barclay, M.S.; Chiriboga, M.; Ellis, G.A.; Mathur, D.; Patten, L.K.; Sup, A.; Hallstrom, N.; et al. Tuning between Quenching and Energy Transfer in DNA-Templated Heterodimer Aggregates. J. Phys. Chem. Lett. 2022, 13, 2782–2791. [Google Scholar] [CrossRef]
- Roy, S.K.; Mass, O.A.; Kellis, D.L.; Wilson, C.K.; Hall, J.A.; Yurke, B.; Knowlton, W.B. Exciton Delocalization and Scaffold Stability in Bridged Nucleotide-Substituted, DNA Duplex-Templated Cyanine Aggregates. J. Phys. Chem. B 2021, 125, 13670–13684. [Google Scholar] [CrossRef]
- Jelley, E.E. Spectral absorption and fluorescence of dyes in the molecular state. Nature 1936, 138, 1009–1010. [Google Scholar] [CrossRef]
- Abou-Hatab, S.; Spata, V.A.; Matsika, S. Substituent Effects on the Absorption and Fluorescence Properties of Anthracene. J. Phys. Chem. A 2017, 121, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Navarro, F.; Glossman-Mitnik, D. DFT study of the effect of substituents on the absorption and emission spectra of Indigo. Chem. Cent. J. 2012, 6, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, C.K.; Chen, Y.J.; Chang, H.W.; Yeh, P.L.; Wang, B.C. DFT and TD-DFT investigations of metal-free dye sensitizers for solar cells: Effects of electron donors and π-conjugated linker. Comput. Theor. Chem. 2011, 971, 42–50. [Google Scholar] [CrossRef]
- Inostroza, N.; Mendizabal, F.; Arratia-Pérez, R.; Orellana, C.; Linares-Flores, C. Improvement of photovoltaic performance by substituent effect of donor and acceptor structure of TPA-based dye-sensitized solar cells. J. Mol. Model. 2016, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Barcenas, G.; Biaggne, A.; Mass, O.A.; Wilson, C.K.; Obukhova, O.M.; Kolosova, O.S.; Tatarets, A.L.; Terpetschnig, E.; Pensack, R.D.; Lee, J.; et al. First-principles studies of substituent effects on squaraine dyes. RSC Adv. 2021, 11, 19029–19040. [Google Scholar] [CrossRef]
- Biaggne, A.; Knowlton, W.B.; Yurke, B.; Lee, J.; Li, L. Substituent Effects on the Solubility and Electronic Properties of the Cyanine Dye Cy5: Density Functional and Time-Dependent Density Functional Theory Calculations. Molecules 2021, 26, 524. [Google Scholar] [CrossRef]
- Mathur, D.; Kim, Y.C.; Díaz, S.A.; Cunningham, P.D.; Rolczynski, B.S.; Ancona, M.G.; Medintz, I.L.; Melinger, J.S. Can a DNA Origami Structure Constrain the Position and Orientation of an Attached Dye Molecule? J. Phys. Chem. C 2021, 125, 1509–1522. [Google Scholar] [CrossRef]
- Cunningham, P.D.; Kim, Y.C.; Díaz, S.A.; Buckhout-White, S.; Mathur, D.; Medintz, I.L.; Melinger, J.S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. [Google Scholar] [CrossRef]
- Stennett, E.M.S.; Ma, N.; van der Vaart, A.; Levitus, M. Photophysical and Dynamical Properties of Doubly Linked Cy3–DNA Constructs. J. Phys. Chem. B 2014, 118, 152–163. [Google Scholar] [CrossRef]
- Nicoli, F.; Roos, M.K.; Hemmig, E.A.; Di Antonio, M.; de Vivie-Riedle, R.; Liedl, T. Proximity-Induced H-Aggregation of Cyanine Dyes on DNA-Duplexes. J. Phys. Chem. A 2016, 120, 9941–9947. [Google Scholar] [CrossRef]
- Kang, B.; Seok, C.; Lee, J. Prediction of Molecular Electronic Transitions Using Random Forests. J. Chem. Inf. Model. 2020, 60, 5984–5994. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.F.; Han, M.; Hwang, J.; Jeong, M.; Choi, D.H.; Park, S. Deep Learning Optical Spectroscopy Based on Experimental Database: Potential Applications to Molecular Design. JACS Au 2021, 1, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Beard, E.J.; Sivaraman, G.; Vázquez-Mayagoitia, Á.; Vishwanath, V.; Cole, J.M. Comparative dataset of experimental and computational attributes of UV/vis absorption spectra. Sci. Data 2019, 6, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, M.; Du, H.; Lindsey, J.S. PhotochemCAD 3: Diverse Modules for Photophysical Calculations with Multiple Spectral Databases. Photochem. Photobiol. 2018, 94, 277–289. [Google Scholar] [CrossRef]
- Nagasawa, S.; Al-Naamani, E.; Saeki, A. Computer-Aided Screening of Conjugated Polymers for Organic Solar Cell: Classification by Random Forest. J. Phys. Chem. Lett. 2018, 9, 2639–2646. [Google Scholar] [CrossRef]
- Cai, J.; Chu, X.; Xu, K.; Li, H.; Wei, J. Machine learning-driven new material discovery. Nanoscale Adv. 2020, 2, 3115–3130. [Google Scholar] [CrossRef]
- Joung, J.F.; Han, M.; Jeong, M.; Park, S. Experimental database of optical properties of organic compounds. Sci. Data 2020, 7, 295. [Google Scholar] [CrossRef]
- Dyomics GmbH. Available online: https://dyomics.com/en/ (accessed on 22 May 2022).
- RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org/ (accessed on 22 May 2022).
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, S.; Antonov, L.; Okuno, Y. Prediction of the color of dyes by using time-dependent density functional theory (TD-DFT). Bulg. Chem. Commun. 2014, 46, 228–237. [Google Scholar]
- Charaf-Eddin, A.; Planchat, A.; Mennucci, B.; Adamo, C.; Jacquemin, D. Choosing a functional for computing absorption and fluorescence band shapes with TD-DFT. J. Chem. Theory Comput. 2013, 9, 2749–2760. [Google Scholar] [CrossRef]
- Jacquemin, D.; Zhao, Y.; Valero, R.; Adamo, C.; Ciofini, I.; Truhlar, D.G. Verdict: Time-dependent density functional theory “not guilty” of large errors for cyanines. J. Chem. Theory Comput. 2012, 8, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to Isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Selvam, K.; Gandhi, S.; Krishnamurty, S.; Gopalakrishnan, G. Effect of substitution on the excited state photophysical and spectral properties of boron difluoride curcumin complex dye and their derivatives: A time dependent-DFT study. J. Photochem. Photobiol. B Biol. 2019, 199, 111595. [Google Scholar] [CrossRef]
- Heid, E.; Hunt, P.A.; Schröder, C. Evaluating excited state atomic polarizabilities of chromophores. Phys. Chem. Chem. Phys. 2018, 20, 8554–8563. [Google Scholar] [CrossRef]
- Fothergill, J.W.; Hernandez, A.C.; Knowlton, W.B.; Yurke, B.; Li, L. Ab Initio Studies of Exciton Interactions of Cy5 Dyes. J. Phys. Chem. A 2018, 122, 8989–8997. [Google Scholar] [CrossRef]
- Garrido, N.M.; Economou, I.G.; Queimada, A.J.; Jorge, M.; Macedo, E.A. Prediction of the n-Hexane/Water and 1-Octanol/Water Partition Coefficients for Environmentally Relevant Compounds using Molecular Simulation. AIChE J. 2012, 58, 1929–1938. [Google Scholar] [CrossRef]
- Mass, O.A.; Wilson, C.K.; Barcenas, G.; Terpetschnig, E.A.; Obukhova, O.M.; Kolosova, O.S.; Tatarets, A.L.; Li, L.; Yurke, B.; Knowlton, W.B.; et al. Influence of Hydrophobicity on Excitonic Coupling in DNA-Templated Indolenine Squaraine Dye Aggregates. J. Phys. Chem. C 2022, 126, 3475–3488. [Google Scholar] [CrossRef]
- Mananghaya, M.R.; Santos, G.N.; Yu, D.N. Solubility of amide functionalized single wall carbon nanotubes: A quantum mechanical study. J. Mol. Liq. 2017, 242, 1208–1214. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Wu, T.; Wang, Q.; Van Der Spoel, D. Comparison of Implicit and Explicit Solvent Models for the Calculation of Solvation Free Energy in Organic Solvents. J. Chem. Theory Comput. 2017, 13, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Aksimentiev, A. Improved parametrization of Li+, Na+, K+, and Mg2+ ions for all-atom molecular dynamics simulations of nucleic acid systems. J. Phys. Chem. Lett. 2012, 3, 45–50. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Czikklely, V.; Forsterling, H.D.; Kuhn, H. Extended dipole model for aggregates of dye molecules. Chem. Phys. Lett. 1970, 6, 207–210. [Google Scholar] [CrossRef]
- Mujumdar, R.B.; Ernst, L.A.; Mujumdar, S.R.; Lewis, C.J.; Waggoner, A.S. Cyanine Dye Labeling Reagents: Sulfoindocyanine Succinimidyl Esters. Bioconjug. Chem. 1993, 4, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Meares, A.; Susumu, K.; Mathur, D.; Lee, S.H.; Mass, O.A.; Lee, J.; Pensack, R.D.; Yurke, B.; Knowlton, W.B.; Melinger, J.S.; et al. Synthesis of Substituted Cy5 Phosphoramidite Derivatives and Their Incorporation into Oligonucleotides Using Automated DNA Synthesis. ACS Omega 2022, 7, 11002–11016. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Levitus, M.; Ranjit, S. Cyanine dyes in biophysical research: The photophysics of polymethine fluorescent dyes in biomolecular environments. Q. Rev. Biophys. 2011, 44, 123–151. [Google Scholar] [CrossRef]
- Zeyada, H.M.; Makhlouf, M.M.; Behairy, A.S.; Nasher, M.A. Fabrication, electrical transport mechanisms and photovoltaic properties of methyl violet 2B/n-Si hybrid organic/inorganic solar cell. Microelectron. Eng. 2016, 163, 134–139. [Google Scholar] [CrossRef]
- Chen, J.; Gao, Y.; Xu, Y.; Xu, F.; Zhang, Q.; Lu, X. Theoretical study of novel porphyrin D-π-A conjugated organic dye sensitizer in solar cells. Mater. Chem. Phys. 2019, 225, 417–425. [Google Scholar] [CrossRef]
- Li, L.L.; Diau, E.W.G. Porphyrin-sensitized solar cells. Chem. Soc. Rev. 2013, 42, 291–304. [Google Scholar] [CrossRef]
- Sameiro, M.; Gonçalves, T. Fluorescent labeling of biomolecules with organic probes. Chem. Rev. 2009, 109, 190–212. [Google Scholar] [CrossRef]
- Pan, X.; Huang, S.; Zhu, B.; Xia, R.; Peng, X. All-porphyrin organic solar cells. Dye Pigment. 2020, 180, 108503. [Google Scholar] [CrossRef]
- Wan, Y.; Stradomska, A.; Knoester, J.; Huang, L. Direct Imaging of Exciton Transport in Tubular Porphyrin Aggregates by Ultrafast Microscopy. J. Am. Chem. Soc. 2017, 139, 7287–7293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bricks, J.L.; Slominskii, Y.L.; Panas, I.D.; Demchenko, A.P. Fluorescent J-aggregates of cyanine dyes: Basic research and applications review. Methods Appl. Fluoresc. 2018, 6, 12001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AAT Bioquest. Available online: https://www.aatbio.com/ (accessed on 22 May 2022).
- Lumiprobe. Available online: https://www.lumiprobe.com/ (accessed on 22 May 2022).
- Glen Research. Available online: https://www.glenresearch.com/ (accessed on 22 May 2022).
- Interchim. Available online: https://www.interchim.com/ (accessed on 22 May 2022).
- Markova, L.I.; Terpetschnig, E.A.; Patsenker, L.D. Comparison of a series of hydrophilic squaraine and cyanine dyes for use as biological labels. Dye Pigment. 2013, 99, 561–570. [Google Scholar] [CrossRef]
- Murashima, T.; Hayata, K.; Saiki, Y.; Matsui, J.; Miyoshi, D.; Yamada, T.; Miyazawa, T.; Sugimoto, N. Synthesis, structure and thermal stability of fully hydrophobic porphyrin-DNA conjugates. Tetrahedron Lett. 2007, 48, 8514–8517. [Google Scholar] [CrossRef]
- Ben-Dror, S.; Bronshtein, I.; Wiehe, A.; Röder, B.; Senge, M.O.; Ehrenberg, B. On the Correlation Between Hydrophobicity, Liposome Binding and Cellular Uptake of Porphyrin Sensitizers. Photochem. Photobiol. 2006, 82, 695. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | ML-Predicted | TD-DFT , Debye |
|---|---|---|
| Cy3 | 126 | 12.25 |
| Cy5 | 222 | 15.35 |
| Cy5-CN | 138 | 15.66 |
| Cy5-NMe2 | 216 | 15.83 |
| Cy5-Cl | 214 | 15.74 |
| Cy5-hex | 110 | 16.22 |
| Cy5-Peg | 107 | 16.19 |
| Cy5-tBu | 224 | 15.90 |
| Cy5.5 | 205 | 15.57 |
| Cy7 | 235 | 17.62 |
| 1 | 307 | 9.08 |
| 2 | 288 | 7.84 |
| 3 | 265 | 20.25 |
| 4 | 240 | 7.99 |
| 5 | 235 | 20.17 |
| 6 | 229 | 14.00 |
| 7 | 227 | 10.66 |
| 8 | 227 | 15.49 |
| 9 | 226 | 14.68 |
| 10 | 223 | 16.19 |
| 11 | 222 | 14.00 |
| 12 | 218 | 11.26 |
| 13 | 216 | 9.52 |
| 14 | 212 | 9.34 |
| 15 | 210 | 10.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biaggne, A.; Spear, L.; Barcenas, G.; Ketteridge, M.; Kim, Y.C.; Melinger, J.S.; Knowlton, W.B.; Yurke, B.; Li, L. Data-Driven and Multiscale Modeling of DNA-Templated Dye Aggregates. Molecules 2022, 27, 3456. https://doi.org/10.3390/molecules27113456
Biaggne A, Spear L, Barcenas G, Ketteridge M, Kim YC, Melinger JS, Knowlton WB, Yurke B, Li L. Data-Driven and Multiscale Modeling of DNA-Templated Dye Aggregates. Molecules. 2022; 27(11):3456. https://doi.org/10.3390/molecules27113456
Chicago/Turabian StyleBiaggne, Austin, Lawrence Spear, German Barcenas, Maia Ketteridge, Young C. Kim, Joseph S. Melinger, William B. Knowlton, Bernard Yurke, and Lan Li. 2022. "Data-Driven and Multiscale Modeling of DNA-Templated Dye Aggregates" Molecules 27, no. 11: 3456. https://doi.org/10.3390/molecules27113456
APA StyleBiaggne, A., Spear, L., Barcenas, G., Ketteridge, M., Kim, Y. C., Melinger, J. S., Knowlton, W. B., Yurke, B., & Li, L. (2022). Data-Driven and Multiscale Modeling of DNA-Templated Dye Aggregates. Molecules, 27(11), 3456. https://doi.org/10.3390/molecules27113456