
A Prospective Viewpoint on Neurological Diseases and Their Biomarkers


 , , , ,
, , , , 
Abstract
:1. Introduction
2. Neurodegeneration, Inflammation, and Tumorigenesis in the Central Nervous System
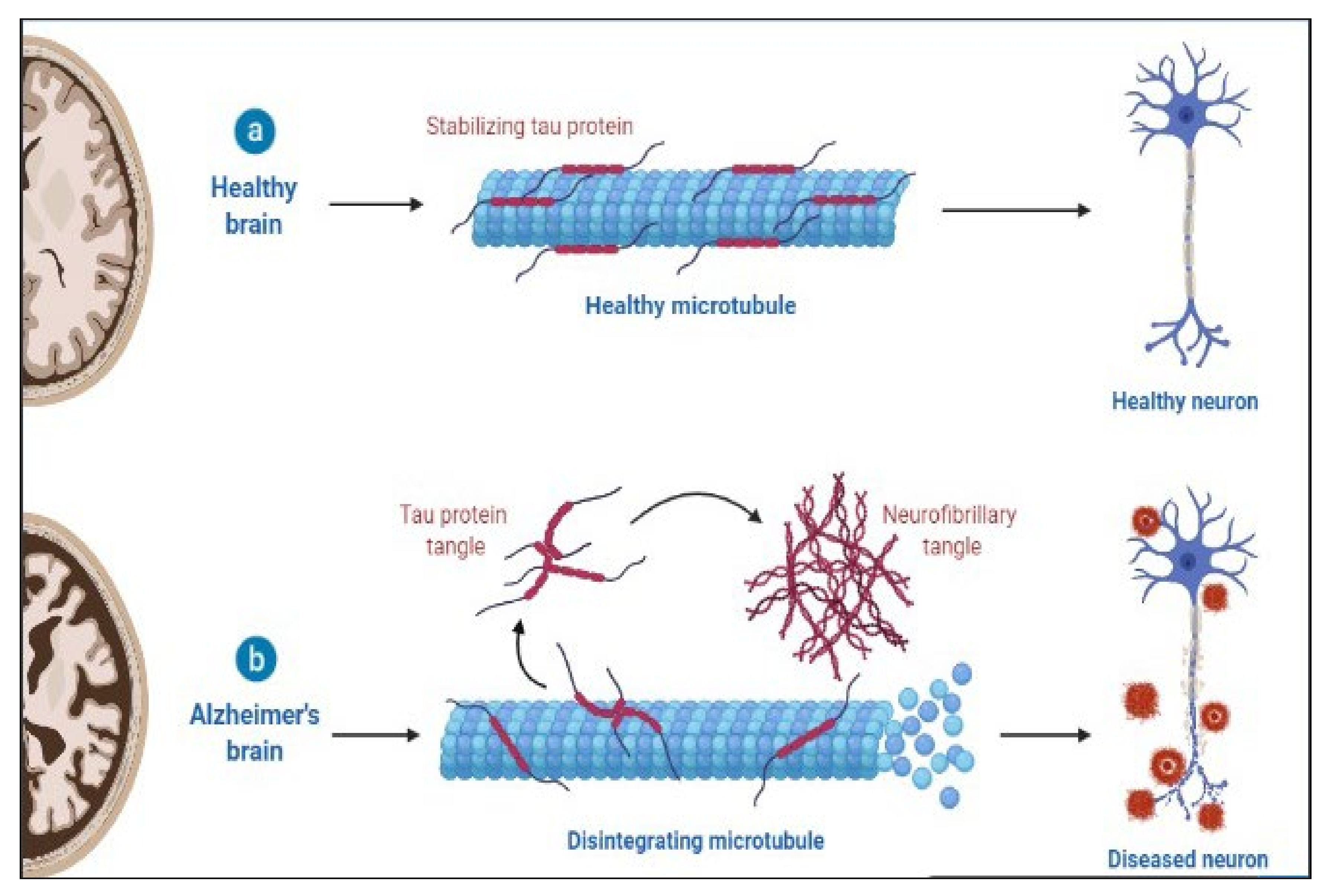
3. Neurodegenerative Diseases as Proteinopathies
4. Relationship between Neurodegeneration and Inflammation
5. Alzheimer’s and Neuroinflammation
6. Depression and Neuroinflammation
7. Infections and Neuroinflammation
8. New Potential Biomarkers
8.1. Alzheimer’s Disease
8.2. Aβ Pathology Biomarkers
8.3. Tau Pathology Biomarkers
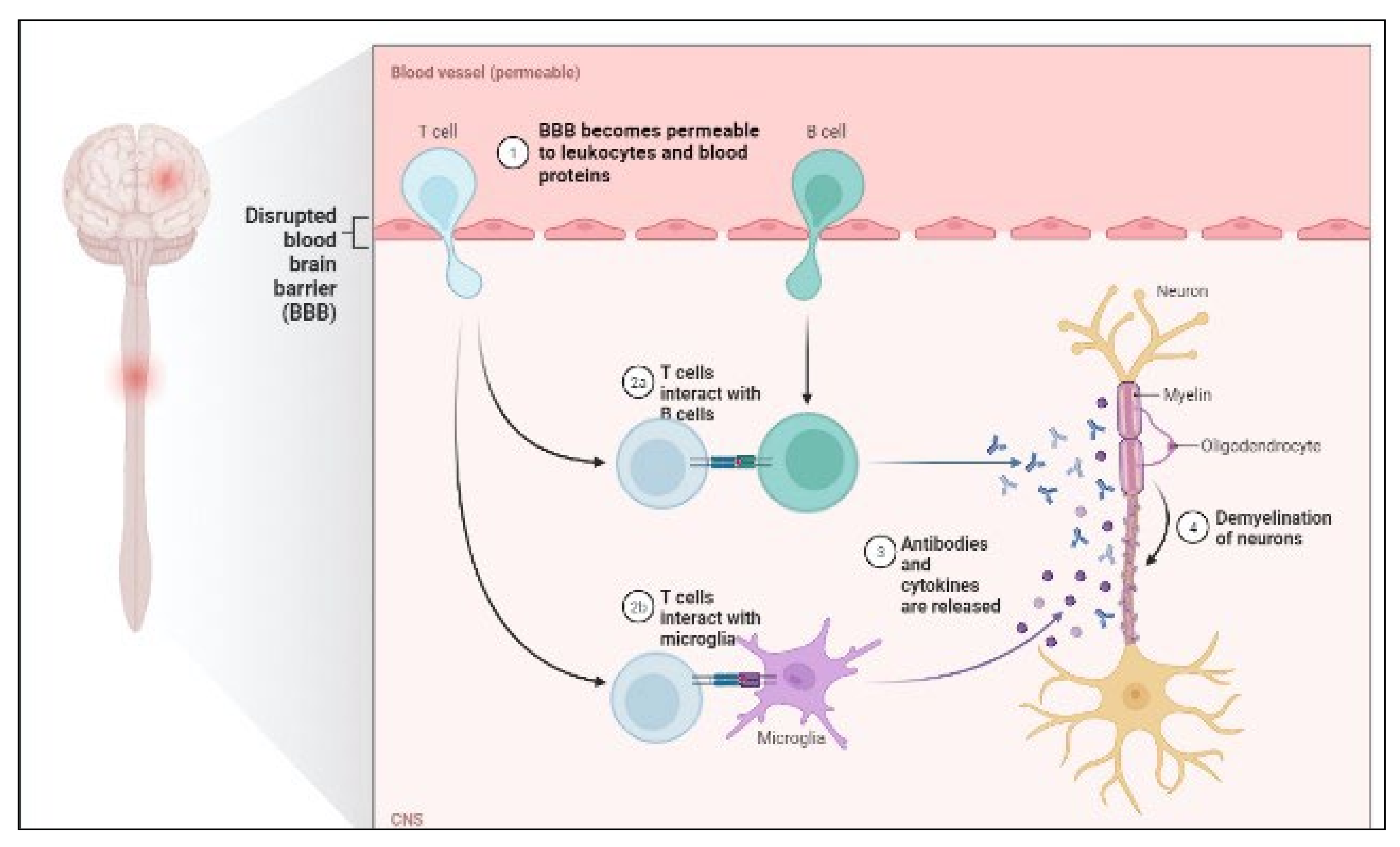
8.4. Multiple Sclerosis
8.4.1. IgG Index
8.4.2. Antinuclear antibodies
8.4.3. Anti-MOG antibodies
8.4.4. Anti-aquaporin-4 antibodies
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sochocka, M.; Zwolińska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.H.; Bajgai, J.; Fadriquela, A.; Sharma, S.; Trinh Thi, T.; Akter, R.; Goh, S.H.; Kim, C.-S.; Lee, K.-J. Redox Effects of Molecular Hydrogen and Its Therapeutic Efficacy in the Treatment of Neurodegenerative Diseases. Processes 2021, 9, 308. [Google Scholar] [CrossRef]
- Perrachione, T.K.; Perrachione, J.R. Brains and brands: Developing mutually informative research in neuroscience and marketing. J. Consum. Behav. 2008, 7, 303–318. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M.-Y. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Akter, R.; Rahman, H.; Behl, T.; Chowdhury, M.A.R.; Manirujjaman, M.; Bulbul, I.J.; Elshenaw, S.E.; Tit, D.M.; Bungau, S. Prospective Role of Polyphenolic Compounds in the Treatment of Neurodegenerative Diseases. CNS Neurol. Disord. Drug Targets 2021, 20, 430–450. [Google Scholar] [CrossRef]
- Rahman, M.H.; Akter, R.; Kamal, M.A. Prospective Function of Different Antioxidant Containing Natural Products in the Treatment of Neurodegenerative Disease. CNS Neurol. Disord. Drug Targets 2021, 20, 694–703. [Google Scholar] [CrossRef]
- Bhattacharya, T.; Dey, P.S.; Akter, R.; Kabir, T.; Rahman, H.; Rauf, A. Effect of natural leaf extracts as phytomedicine in curing geriatrics. Exp. Gerontol. 2021, 150, 111352. [Google Scholar] [CrossRef]
- Akter, R.; Chowdhury, M.A.R.; Habib Ur Rahman, M. Flavonoids and Polyphenolic Compounds as Potential Talented Agents for the Treatment of Alzheimer’s Disease with their Antioxidant Activities. Curr. Pharm. Des. 2020, 27, 345–356. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Lankatillake, C.; Dias, D.A.; Docea, A.O.; Mahomoodally, M.F.; Lobine, D.; Chazot, P.L.; Kurt, B.; Tumer, T.B.; Moreira, A.C.; et al. Impact of Natural Compounds on Neurodegenerative Disorders: From Preclinical to Pharmacotherapeutics. J. Clin. Med. 2020, 9, 1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walia, V.; Kaushik, D.; Mittal, V.; Kumar, K.; Verma, R.; Parashar, J.; Akter, R.; Rahman, H.; Bhatia, S.; Al-Harrasi, A.; et al. Delineation of Neuroprotective Effects and Possible Benefits of AntioxidantsTherapy for the Treatment of Alzheimer’s Diseases by Targeting Mitochondrial-Derived Reactive Oxygen Species: Bench to Bedside. Mol. Neurobiol. 2021, 59, 657–680. [Google Scholar] [CrossRef]
- Rahman, M.H.; Akter, R.; Bhattacharya, T.; Abdel-Daim, M.M.; Alkahtani, S.; Arafah, M.W.; Al-Johani, N.S.; Alhoshani, N.M.; Alkeraishan, N.; Alhenaky, A.; et al. Resveratrol and Neuroprotection: Impact and Its Therapeutic Potential in Alzheimer’s Disease. Front. Pharmacol. 2020, 11, 619024. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S. The 200-year journey of Parkinson disease: Reflecting on the past and looking towards the future. Park. Relat. Disord. 2018, 46, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Kobeissi, Z.A.; Zanotti-Cavazzoni, S.L. Biomarkers of sepsis. Yearb. Crit. Care Med. 2010, 2010, 227–228. [Google Scholar] [CrossRef]
- Hansson, O. Biomarkers for neurodegenerative diseases. Nat. Med. 2021, 27, 954–963. [Google Scholar] [CrossRef]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Singleton, A.; Hardy, J. The Evolution of Genetics: Alzheimer’s and Parkinson’s Diseases. Neuron 2016, 90, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Sierksma, A.; Escott-Price, V.; De Strooper, B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 2020, 370, 61–66. [Google Scholar] [CrossRef]
- Rahman, M.H.; Bajgai, J.; Fadriquela, A.; Sharma, S.; Trinh, T.T.; Akter, R.; Jeong, Y.J.; Goh, S.H.; Kim, C.-S.; Lee, K.-J. Therapeutic Potential of Natural Products in Treating Neurodegenerative Disorders and Their Future Prospects and Challenges. Molecules 2021, 26, 5327. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Dolgacheva, L.P. Interaction of misfolded proteins and mitochondria in neurodegenerative disorders. Biochem. Soc. Trans. 2017, 45, 1025–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachurin, S.O.; Gavrilova, S.I.; Samsonova, A.; Barreto, G.E.; Aliev, G. Mild cognitive impairment due to Alzheimer disease: Contemporary approaches to diagnostics and pharmacological intervention. Pharmacol. Res. 2018, 129, 216–226. [Google Scholar] [CrossRef]
- Schneider, A.; Sari, A.T.; Alhaddad, H.; Sari, Y. Overview of Therapeutic Drugs and Methods for the Treatment of Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2020, 19, 195–206. [Google Scholar] [CrossRef]
- Al-Obaidi, M.M.J.; Desa, M.N.M. Mechanisms of Blood Brain Barrier Disruption by Different Types of Bacteria, and Bacterial–Host Interactions Facilitate the Bacterial Pathogen Invading the Brain. Cell. Mol. Neurobiol. 2018, 38, 1349–1368. [Google Scholar] [CrossRef]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Oré, M.-V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Sävman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain. Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Wu, S.-C.; Cao, Z.-S.; Chang, K.-M.; Juang, J.-L. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 2017, 8, 24. [Google Scholar] [CrossRef]
- Jellinger, K.A. Recent advances in our understanding of neurodegeneration. J. Neural Transm. 2009, 116, 1111–1162. [Google Scholar] [CrossRef]
- Skovronsky, D.M.; Lee, V.M.-Y.; Trojanowski, J.Q. Neurodegenerative Diseases: New Concepts of Pathogenesis and Their Therapeutic Implications. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Miller, V.M. Proteinopathy-induced neuronal senescence: A hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimer’s Res. Ther. 2009, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vladimir, N.U. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. 2009, 14, 5188–5238. [Google Scholar] [CrossRef]
- Tagde, P.; Tagde, P.; Tagde, S.; Bhattacharya, T.; Garg, V.; Akter, R.; Rahman, H.; Najda, A.; Albadrani, G.M.; Sayed, A.A.; et al. Natural bioactive molecules: An alternative approach to the treatment and control of glioblastoma multiforme. Biomed. Pharmacother. 2021, 141, 111928. [Google Scholar] [CrossRef] [PubMed]
- Karthika, C.; Hari, B.; Rahman, M.H.; Akter, R.; Najda, A.; Albadrani, G.M.; Sayed, A.A.; Akhtar, M.F.; Abdel-Daim, M.M. Multiple strategies with the synergistic approach for addressing colorectal cancer. Biomed. Pharmacother. 2021, 140, 111704. [Google Scholar] [CrossRef]
- Adeola, H.A.; Bano, A.; Vats, R.; Vashishtha, A.; Verma, D.; Kaushik, D.; Mittal, V.; Rahman, H.; Najda, A.; Albadrani, G.M.; et al. Bioactive compounds and their libraries: An insight into prospective phytotherapeutics approach for oral mucocutaneous cancers. Biomed. Pharmacother. 2021, 141, 111809. [Google Scholar] [CrossRef]
- Cavalu, S.; Simon, V. Proteins adsorption to orthopaedic biomaterials: Vibrational spectroscopy evidence. J. Optoelectron. Adv. Mater. 2007, 9, 3297–3302. [Google Scholar]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [Green Version]
- Re, D.B.; Le Verche, V.; Yu, C.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis Drives Motor Neuron Death in Models of Both Sporadic and Familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of Necroptosis in Multiple Sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; de Vries, R.L.A.; Tocilescu, M.; Przedborski, S. PINK1/Parkin direct mitochondria to autophagy. Autophagy 2010, 6, 315–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.G.; Robinson, J.L.; Xie, S.X.; Lee, E.B.; Grossman, M.; Wolk, D.A.; Irwin, D.; Weintraub, D.; Kim, C.F.; Schuck, T.; et al. Evaluating the Patterns of Aging-Related Tau Astrogliopathy Unravels Novel Insights Into Brain Aging and Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2017, 76, 270–288. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Kovacs, G.G.; Vonsattel, J.P.G.; Davey, K.; Mok, K.Y.; Hardy, J.; Morris, H.R.; Warner, T.T.; Holton, J.L.; Revesz, T. Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain 2016, 139, 3237–3252. [Google Scholar] [CrossRef] [Green Version]
- Lucchinetti, C.F.; Guo, Y.; Popescu, B.F.G.; Fujihara, K.; Itoyama, Y.; Misu, T. The Pathology of an Autoimmune Astrocytopathy: Lessons Learned from Neuromyelitis Optica. Brain Pathol. 2013, 24, 83–97. [Google Scholar] [CrossRef]
- Jung, Y.J.; Tweedie, D.; Scerba, M.T.; Greig, N.H. Neuroinflammation as a Factor of Neurodegenerative Disease: Thalidomide Analogs as Treatments. Front. Cell Dev. Biol. 2019, 7, 313. [Google Scholar] [CrossRef]
- Bajgai, J.; Lee, K.-J.; Rahman, H.; Fadriquela, A.; Kim, C.-S. Role of Molecular Hydrogen in Skin Diseases and its Impact in Beauty. Curr. Pharm. Des. 2021, 27, 737–746. [Google Scholar] [CrossRef]
- Boiko, D.I.; Skrypnikov, A.M.; Shkodina, A.D.; Hasan, M.M.; Ashraf, G.M.; Rahman, H. Circadian rhythm disorder and anxiety as mental health complications in post-COVID-19. Environ. Sci. Pollut. Res. 2022, 29, 28062–28069. [Google Scholar] [CrossRef]
- Miere, F.; Fritea, L.; Cavalu, S.; Vicaș, S.I. Formulation, Characterization, and Advantages of Using Liposomes in Multiple Therapies. Pharmacophore 2020, 11, 1–12. [Google Scholar]
- Bhattacharya, T.; Chopra, H.; Rahman, M.M.; Hasan, Z.; Swain, S.S.; Cavalu, S. Applications of Phyto-Nanotechnology for the Treatment of Neurodegenerative Disorders. Materials 2022, 15, 804. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.-P.; Firlej, V.; Allory, Y.; Roméo, P.-H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.; McAleese, K.E.; Thomas, A.J.; Johnson, M.; Martin-Ruiz, C.; Parker, C.; Colloby, S.J.; Jellinger, K.; Attems, J. Neuropathologically mixed Alzheimer’s and Lewy body disease: Burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol. 2015, 129, 729–748. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, C.; Finkelstein, D.; Lawson, V.A.; Hill, A.F. Pathogenic mechanisms of prion protein, amyloid-β and α-synuclein misfolding: The prion concept and neurotoxicity of protein oligomers. J. Neurochem. 2016, 139, 162–180. [Google Scholar] [CrossRef]
- Bayer, T.A. Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur. Neuropsychopharmacol. 2015, 25, 713–724. [Google Scholar] [CrossRef]
- Bae, E.-J.; Lee, H.-J.; Rockenstein, E.; Ho, D.H.; Park, E.-B.; Yang, N.-Y.; Desplats, P.; Masliah, E.; Lee, S.-J. Antibody-Aided Clearance of Extracellular -Synuclein Prevents Cell-to-Cell Aggregate Transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef]
- Deleidi, M.; Maetzler, W. Protein Clearance Mechanisms of Alpha-Synuclein and Amyloid-Beta in Lewy Body Disorders. Int. J. Alzheimer’s Dis. 2012, 2012, 391438. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Gärtner, U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J. Neural Transm. 1998, 105, 949–960. [Google Scholar] [CrossRef]
- Yang, Y.; Geldmacher, D.S.; Herrup, K. DNA Replication Precedes Neuronal Cell Death in Alzheimer’s Disease. J. Neurosci. 2001, 21, 2661–2668. [Google Scholar] [CrossRef] [PubMed]
- Mina-Osorio, P.; Rosas-Ballina, M.; Valdes-Ferrer, S.I.; Al-Abed, Y.; Tracey, K.J.; Diamond, B. Neural Signaling in the Spleen Controls B-Cell Responses to Blood-Borne Antigen. Mol. Med. 2012, 18, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Heesters, B.A.; Ghasemlou, N.; Von Hehn, C.A.; Zhao, F.; Tran, J.; Wainger, B.; Strominger, A.; Muralidharan, S.; Horswill, A.R.; et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013, 501, 52–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas-Ballina, M.; Valdés-Ferrer, S.I.; Dancho, M.E.; Ochani, M.; Katz, D.; Cheng, K.F.; Olofsson, P.S.; Chavan, S.S.; Al-Abed, Y.; Tracey, K.J.; et al. Xanomeline suppresses excessive pro-inflammatory cytokine responses through neural signal-mediated pathways and improves survival in lethal inflammation. Brain. Behav. Immun. 2014, 44, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Satapathy, S.K.; Ochani, M.; Dancho, M.; Hudson, L.K.; Rosas-Ballina, M.; Valdés-Ferrer, S.I.; Olofsson, P.S.; Harris, Y.T.; Roth, J.; Chavan, S.; et al. Galantamine Alleviates Inflammation and Other Obesity-Associated Complications in High-Fat Diet-Fed Mice. Mol. Med. 2011, 17, 599–606. [Google Scholar] [CrossRef]
- Ji, H.; Rabbi, M.F.; Labis, B.; Pavlov, V.; Tracey, K.J.; Ghia, J.-E. Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol. 2013, 7, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Deutschman, C.S.; Tracey, K.J. Sepsis: Current Dogma and New Perspectives. Immunity 2014, 40, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Mak, A.; Cheung, M.W.J.; Chiew, H.J.; Liu, Y.; Ho, R.C.-M. Global Trend of Survival and Damage of Systemic Lupus Erythematosus: Meta-Analysis and Meta-Regression of Observational Studies from the 1950s to 2000s. Semin. Arthritis Rheum. 2012, 41, 830–839. [Google Scholar] [CrossRef]
- Jeltsch-David, H.; Muller, S. Neuropsychiatric systemic lupus erythematosus: Pathogenesis and biomarkers. Nat. Rev. Neurol. 2014, 10, 579–596. [Google Scholar] [CrossRef]
- Faust, T.W.; Chang, E.H.; Kowal, C.; Berlin, R.; Gazaryan, I.G.; Bertini, E.; Zhang, J.; Sanchez-Guerrero, J.; Fragoso-Loyo, H.E.; Volpe, B.T.; et al. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc. Natl. Acad. Sci. 2010, 107, 18569–18574. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Zehnder, M.; Toledo, E.M.; Segovia-Miranda, F.; Serrano, F.G.; Benito, M.J.; Metz, C.; Retamal, C.; Álvarez, A.; Massardo, L.; Inestrosa, N.C.; et al. Anti-Ribosomal P Protein Autoantibodies From Patients With Neuropsychiatric Lupus Impair Memory in Mice. Arthritis Rheumatol. 2014, 67, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Bullmore, E.T.; Sporns, O. Complex brain networks: Graph theoretical analysis of structural and functional systems. Nat. Rev. Neurosci. 2009, 10, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Toga, A.W.; Clark, K.A.; Thompson, P.M.; Shattuck, D.W.; Van Horn, J.D. Mapping the Human Connectome. Neurosurgery 2012, 71, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Bullmore, E.; Sporns, O. The economy of brain network organization. Nat. Rev. Neurosci. 2012, 13, 336–349. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.-O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 842–857. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, V.V.; Masud, F.; Petronilho, F.; Dal-Pizzol, F.; Barichello, T. Infection-Induced Systemic Inflammation Is a Potential Driver of Alzheimer’s Disease Progression. Front. Aging Neurosci. 2019, 11, 122. [Google Scholar] [CrossRef]
- Balin, B.J.; Little, C.S.; Hammond, C.J.; Appelt, D.M.; Whittum-Hudson, J.A.; Gérard, H.C.; Hudson, A.P. Chlamydophila Pneumoniae and the Etiology of Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 13, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-Y.Y.; Mucke, L.; Nie, Q.; Du, X.X.; Geng, M.M.; Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; et al. SM Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimers disease. ACS Chem. Neurosci. 2018, 13, 340ra72. [Google Scholar]
- Golde, T.E. Host immune defence, amyloid-β peptide and Alzheimer disease. Nat. Rev. Neurol. 2016, 12, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflamm. 2012, 9, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, D.J.; Ditor, D.S. The common inflammatory etiology of depression and cognitive impairment: A therapeutic target. J. Neuroinflammation 2014, 11, 151. [Google Scholar] [CrossRef] [Green Version]
- Schiffrin, E.L. Inflammation, immunity and development of essential hypertension. J. Hypertens. 2014, 32, 228–229. [Google Scholar] [CrossRef]
- Shimizu, M.; Ishikawa, J.; Yano, Y.; Hoshide, S.; Shimada, K.; Kario, K. The relationship between the morning blood pressure surge and low-grade inflammation on silent cerebral infarct and clinical stroke events. Atherosclerosis 2011, 219, 316–321. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, N.; Androulakis, E.; Antoniades, C.; Toutouzas, K.; Stefanadis, C. Pathophysiology of Atherosclerosis: The Role of Inflammation. Curr. Pharm. Des. 2011, 17, 4089–4110. [Google Scholar] [CrossRef]
- Di Napoli, M.; Godoy, D.A.; Campi, V.; Masotti, L.; Smith, C.J.; Jones, A.R.P.; Hopkins, S.J.; Slevin, M.; Papa, F.; Mogoanta, L.; et al. C-reactive protein in intracerebral hemorrhage: Time course, tissue localization, and prognosis. Neurology 2012, 79, 690–699. [Google Scholar] [CrossRef]
- Di Napoli, M.; Parry-Jones, A.R.; Smith, C.; Hopkins, S.; Slevin, M.; Masotti, L.; Campi, V.; Singh, P.; Papa, F.; Popa-Wagner, A.; et al. C-Reactive Protein Predicts Hematoma Growth in Intracerebral Hemorrhage. Stroke 2014, 45, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Goossens, G.H.; Bizzarri, A.; Venteclef, N.; Essers, Y.; Cleutjens, J.P.; Konings, E.; Jocken, J.W.E.; Čajlaković, M.; Ribitsch, V.; Clément, K.; et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation 2011, 124, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howcroft, T.K.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The role of inflammation in age-related disease. Aging 2013, 5, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The Role of Inflammation in Age-Related Sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruunsgaard, H.; Pedersen, M.; Pedersen, B.K. Aging and proinflammatory cytokines. Curr. Opin. Hematol. 2001, 8, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Su, S.; Miller, A.H.; Bremner, J.D.; Goldberg, J.; Vogt, G.J.; Maisano, C.; Jones, L.; Murrah, N.V.; Vaccarino, V. Depressive Symptoms and Metabolic Syndrome: Is Inflammation the Underlying Link? Biol. Psychiatry 2008, 64, 896–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Capuron, L.; Poitou, C.; Machaux-Tholliez, D.; Frochot, V.; Bouillot, J.-L.; Basdevant, A.; Layé, S.; Clément, K. Relationship between adiposity, emotional status and eating behaviour in obese women: Role of inflammation. Psychol. Med. 2010, 41, 1517–1528. [Google Scholar] [CrossRef]
- McCrimmon, R.J.; Ryan, C.M.; Frier, B.M. Diabetes and cognitive dysfunction. Lancet 2012, 379, 2291–2299. [Google Scholar] [CrossRef]
- Wolkowitz, O.M.; Epel, E.S.; Reus, V.; Mellon, S.H. Depression gets old fast: Do stress and depression accelerate cell aging? Depress. Anxiety 2010, 27, 327–338. [Google Scholar] [CrossRef]
- Cavalu, S.; Antoniac, I.V.; Mohan, A.; Bodog, F.; Doicin, C.; Mates, I.; Ulmeanu, M.; Murzac, R.; Semenescu, A. Nanoparticles and Nanostructured Surface Fabrication for Innovative Cranial and Maxillofacial Surgery. Materials 2020, 13, 5391. [Google Scholar] [CrossRef]
- Tagde, P.; Tagde, S.; Bhattacharya, T.; Tagde, P.; Chopra, H.; Akter, R.; Kaushik, D.; Rahman, H. Blockchain and artificial intelligence technology in e-Health. Environ. Sci. Pollut. Res. 2021, 28, 52810–52831. [Google Scholar] [CrossRef] [PubMed]
- Baune, B.T.; Smith, E.; Reppermund, S.; Air, T.; Samaras, K.; Lux, O.; Brodaty, H.; Sachdev, P.; Trollor, J. Inflammatory biomarkers predict depressive, but not anxiety symptoms during aging: The prospective Sydney Memory and Aging Study. Psychoneuroendocrinology 2012, 37, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.; Pearlman, D.M.; Devinsky, O.; Najjar, A.; Zagzag, D. Neurovascular unit dysfunction with blood-brain barrier hyperpermeability contributes to major depressive disorder: A review of clinical and experimental evidence. J. Neuroinflammation 2013, 10, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunszain, P.A.; Hepgul, N.; Pariante, C.M. Inflammation and Depression. In Behavioral Neurobiology of Depression and Its Treatment; Springer: New York, NY, USA, 2012; Volume 14, pp. 135–151. [Google Scholar] [CrossRef]
- Papakostas, G.I.; Shelton, R.C.; Kinrys, G.; Henry, M.E.; Bakow, B.R.; Lipkin, S.H.; Pi, B.; Thurmond, L.; Bilello, J.A. Assessment of a multi-assay, serum-based biological diagnostic test for major depressive disorder: A Pilot and Replication Study. Mol. Psychiatry 2013, 18, 332–339. [Google Scholar] [CrossRef]
- Leonard, B.E. Inflammation and depression: A causal or coincidental link to the pathophysiology? Acta Neuropsychiatr. 2018, 30, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Schnell, G.; Joseph, S.; Spudich, S.; Price, R.W.; Swanstrom, R. HIV-1 Replication in the Central Nervous System Occurs in Two Distinct Cell Types. PLOS Pathog. 2011, 7, e1002286. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, J.; Xu, C.; Keblesh, J.; Zang, W.; Xiong, H. HIV-1gp120 Induces Neuronal Apoptosis through Enhancement of 4-Aminopyridine-Senstive Outward K+ Currents. PLoS ONE 2011, 6, e25994. [Google Scholar] [CrossRef]
- Chang, J.R.; Mukerjee, R.; Bagashev, A.; Del Valle, L.; Chabrashvili, T.; Hawkins, B.J.; He, J.J.; Sawaya, B.E. HIV-1 Tat Protein Promotes Neuronal Dysfunction through Disruption of MicroRNAs. J. Biol. Chem. 2011, 286, 41125–41134. [Google Scholar] [CrossRef] [Green Version]
- Brew, B.J.; Crowe, S.M.; Landay, A.; Cysique, L.; Guillemin, G. Neurodegeneration and Ageing in the HAART Era. J. Neuroimmune Pharmacol. 2008, 4, 163–174. [Google Scholar] [CrossRef]
- Tohidpour, A.; Morgun, A.V.; Boitsova, E.B.; Malinovskaya, N.A.; Martynova, G.P.; Khilazheva, E.D.; Kopylevich, N.V.; Gertsog, G.E.; Salmina, A.B. Neuroinflammation and Infection: Molecular Mechanisms Associated with Dysfunction of Neurovascular Unit. Front. Cell. Infect. Microbiol. 2017, 7, 276. [Google Scholar] [CrossRef] [Green Version]
- Fattakhov, N.; Torices, S.; Stangis, M.; Park, M.; Toborek, M. Synergistic Impairment of the Neurovascular Unit by HIV-1 Infection and Methamphetamine Use: Implications for HIV-1-Associated Neurocognitive Disorders. Viruses 2021, 13, 1883. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.; Cryan, J.F. The impact of gut microbiota on brain and behaviour: Implications for psychiatry. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 552–558. [Google Scholar] [CrossRef] [PubMed]
- El Aidy, S.; Dinan, T.G.; Cryan, J.F. Immune modulation of the brain-gut-microbe axis. Front. Microbiol. 2014, 5, 146. [Google Scholar] [CrossRef] [PubMed]
- United Nations Environment Programme; International Labour Organization; World Health Organization; International Program on Chemical Safety. Biomarkers in Risk Assessment: Validity and Validation; World Health Organization: Geneva, Switzerland, 2014; pp. 1–21.
- Strimbu, K.; Tavel, J.A. What Are Biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Palmqvist, S.; Insel, P.S.; Stomrud, E.; Janelidze, S.; Zetterberg, H.; Brix, B.; Eichenlaub, U.; Dage, J.; Chai, X.; Blennow, K.; et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer’s disease. EMBO Mol. Med. 2019, 11, e11170. [Google Scholar] [CrossRef]
- Jack, C.R.; Holtzman, D.M. Biomarker Modeling of Alzheimer’s Disease. Neuron 2013, 80, 1347–1358. [Google Scholar] [CrossRef] [Green Version]
- Arya, A.; Chahal, R.; Rao, R.; Rahman, H.; Kaushik, D.; Akhtar, M.; Saleem, A.; Khalifa, S.; El-Seedi, H.; Kamel, M.; et al. Acetylcholinesterase Inhibitory Potential of Various Sesquiterpene Analogues for Alzheimer’s Disease Therapy. Biomolecules 2021, 11, 350. [Google Scholar] [CrossRef]
- Pérez, V.; Sarasa, L.; Allue, J.A.; Casabona, D.; Montanes, M.; Insua, D.; Badi, H.; Monleón, I.; Lacosta, A.M.; Jose, I.S.; et al. O2-03-05: Beta-amyloid-17 is a major beta-amyloid fragment isoform in cerebrospinal fluid and blood that shows diagnostic value. Alzheimer’s Dement. 2012, 8, P240. [Google Scholar] [CrossRef]
- Portelius, E.; Dean, R.A.; Gustavsson, M.K.; Andreasson, U.; Zetterberg, H.; Siemers, E.; Blennow, K. A novel Aβ isoform pattern in CSF reflects γ-secretase inhibition in Alzheimer disease. Alzheimer’s Res. Ther. 2010, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Grijalba, V.; Pesini, P.; Monleón, I.; Boada, M.; Tárraga, L.; Ruiz-Laza, A.; Martínez-Lage, P.; San-José, I.; Sarasa, M. Several Direct and Calculated Biomarkers from the Amyloid-β Pool in Blood are Associated with an Increased Likelihood of Suffering from Mild Cognitive Impairment. J. Alzheimer’s Dis. 2013, 36, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.D.; Landau, S.M.; Snitz, B.E.; Klunk, W.E.; Blennow, K.; Zetterberg, H. Fluid and PET biomarkers for amyloid pathology in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 97, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Koeppe, R.A.; Price, J.C.; Benzinger, T.L.; Devous, M.D.; Jagust, W.J.; Johnson, K.A.; Mathis, C.A.; Minhas, D.; Pontecorvo, M.J.; et al. The Centiloid Project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimer’s Dement. 2014, 11, 1–15.e4. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.A.; Minoshima, S.; Bohnen, N.I.; Donohoe, K.J.; Foster, N.L.; Herscovitch, P.; Karlawish, J.H.; Rowe, C.C.; Hedrick, S.; Pappas, V.; et al. Update on appropriate use criteria for amyloid PET imaging: Dementia experts, mild cognitive impairment, and education. Amyloid Imaging Task Force of the Alzheimer’s Association and Society for Nuclear Medicine and Molecular Imaging. Alzheimer’s Dement. 2013, 9, e106–e109. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.J. Tauopathies as clinicopathological entities. Park. Relat. Disord. 2016, 22, S29–S33. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Skillbäck, T.; Farahmand, B.Y.; Rosén, C.; Mattsson, N.; Nägga, K.; Kilander, L.; Religa, D.; Wimo, A.; Winblad, B.; Schott, J.M.; et al. Cerebrospinal fluid tau and amyloid-β1-42 in patients with dementia. Brain 2015, 138, 2716–2731. [Google Scholar] [CrossRef] [Green Version]
- Maia, L.F.; Kaeser, S.A.; Reichwald, J.; Hruscha, M.; Martus, P.; Staufenbiel, M.; Jucker, M. Changes in Amyloid-β and Tau in the Cerebrospinal Fluid of Transgenic Mice Overexpressing Amyloid Precursor Protein. Sci. Transl. Med. 2013, 5, 194re2. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Barthélemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 98, 861–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, R.; Giovannoni, G. Multiple sclerosis–A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef] [Green Version]
- Weksler, B.B.; Subileau, E.A.; Perrière, N.; Charneau, P.; Holloway, C.J.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef] [PubMed]
- Ziemssen, T.; Kern, R.; Thomas, K. Multiple sclerosis: Clinical profiling and data collection as prerequisite for personalized medicine approach. BMC Neurol. 2016, 16, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Kooij, G.; Kroon, J.; Paul, D.; Reijerkerk, A.; Geerts, D.; van der Pol, S.M.A.; Hof, B.V.H.; Drexhage, J.A.; van Vliet, S.J.; Hekking, L.H.P.; et al. P-glycoprotein regulates trafficking of CD8+ T cells to the brain parenchyma. Acta Neuropathol. 2014, 127, 699–711. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H.G. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef]
- Pryce, G.; Baker, D. Oligoclonal bands in multiple sclerosis; Functional significance and therapeutic implications. Does the specificity matter? Mult. Scler. Relat. Disord. 2018, 25, 131–137. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Petzold, A. Applying the 2017 McDonald diagnostic criteria for multiple sclerosis. Lancet Neurol. 2018, 17, 496–497. [Google Scholar] [CrossRef]
- Arrambide, G.; Tintore, M.; Montalban, X. Oligoclonal bands do not represent dissemination in time in the 2017 revisions to the McDonald criteria. Mult. Scler. J. 2019, 25, 1690–1691. [Google Scholar] [CrossRef] [PubMed]
- Trojano, M.; Tintore, M.; Montalban, X.; Hillert, J.; Kalincik, T.; Iaffaldano, P.; Spelman, T.; Sormani, M.P.; Butzkueven, H. Treatment decisions in multiple sclerosis-insights from real-world observational studies. Nat. Rev. Neurol. 2017, 13, 105–118. [Google Scholar] [CrossRef]
- Levine, S.M. Albumin and multiple sclerosis. BMC Neurol. 2016, 16, 47. [Google Scholar] [CrossRef] [Green Version]
- Maggi, P.; Absinta, M.; Grammatico, M.; Vuolo, L.; Emmi, G.; Carlucci, G.; Spagni, G.; Barilaro, A.; Repice, A.M.; Emmi, L.; et al. Central vein sign differentiates Multiple Sclerosis from central nervous system inflammatory vasculopathies. Ann. Neurol. 2018, 83, 283–294. [Google Scholar] [CrossRef]
- Bonnan, M. Intrathecal IgG Synthesis: A Resistant and Valuable Target for Future Multiple Sclerosis Treatments. Mult. Scler. Int. 2015, 2015, 296184. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B.; Rogacka, N.; Puszczewicz, M. Antinuclear antibodies in healthy people and non-rheumatic diseases–diagnostic and clinical implications. Reumatologia 2018, 56, 243–248. [Google Scholar] [CrossRef]
- Becker, J.; Geffken, M.; Diehl, R.R.; Berlit, P.; Krämer, M. Choosing wisely? Multiple Sclerosis and Laboratory Screening for Autoimmune Differential Diagnoses. Neurol. Int. Open 2017, 01, E256–E263. [Google Scholar] [CrossRef] [Green Version]
- Narayan, R.; Simpson, A.; Fritsche, K.; Salama, S.; Pardo, S.; Mealy, M.; Paul, F.; Levy, M. MOG antibody disease: A review of MOG antibody seropositive neuromyelitis optica spectrum disorder. Mult. Scler. Relat. Disord. 2018, 25, 66–72. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Aktas, O.; Asgari, N.; Dale, R.C.; De Seze, J.; Franciotta, D.; Fujihara, K.; Jacob, A.; Kim, H.J.; et al. MOG encephalomyelitis: International recommendations on diagnosis and antibody testing. J. Neuroinflamm. 2018, 15, 134. [Google Scholar] [CrossRef] [PubMed]
- Peschl, P.; Bradl, M.; Höftberger, R.; Berger, T.; Reindl, M. Myelin Oligodendrocyte Glycoprotein: Deciphering a Target in Inflammatory Demyelinating Diseases. Front. Immunol. 2017, 8, 529. [Google Scholar] [CrossRef]
- Mitsdoerffer, M.; Kuchroo, V.; Korn, T. Immunology of neuromyelitis optica: A T cell-B cell collaboration. Ann. New York Acad. Sci. 2013, 1283, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkman, A.S. Aquaporins in Clinical Medicine. Annu. Rev. Med. 2012, 63, 303–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCreary, M.; Mealy, M.A.; Wingerchuk, D.M.; Levy, M.; DeSena, A.; Greenberg, B.M. Updated diagnostic criteria for neuromyelitis optica spectrum disorder: Similar outcomes of previously separate cohorts. Mult. Scler. J. Exp. Transl. Clin. 2018, 4, 2055217318815925. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Amyloid-Beta | Tau Protein | Phosphorylated Tau |
|---|---|---|
| Aβ plaque depositions commonly define AD. The amyloidogenic pathways produce these 42-amino-acid peptides (Aβ1-42), clumping in the brain. The amount of Aβ in AD patients’ CSF is reduced by roughly 500 pg/mL compared with healthy controls (79,420 pg/mL) [121]. | Tau inclusion intraneuronal microtubule-associated protein is another well-established AD biomarker. There is an exponential increase in tau protein levels in AD patients from 300 to 600 pg/mL, which grows with age from 21–50 years to >71 years (in patients aged 51–70 years). Therefore it is an excellent prognostic biomarker [122]. | In AD, tau protein is phosphorylated in about 39 places. Position 181 is a distinct biomarker in AD versus controls. Tau protein phosphorylation causes function loss and neuronal malfunction. There are also phosphorylated tau-199, tau-231, tau-235, tau-396 and tau-400 [123]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zehravi, M.; Kabir, J.; Akter, R.; Malik, S.; Ashraf, G.M.; Tagde, P.; Ramproshad, S.; Mondal, B.; Rahman, M.H.; Mohan, A.G.; et al. A Prospective Viewpoint on Neurological Diseases and Their Biomarkers. Molecules 2022, 27, 3516. https://doi.org/10.3390/molecules27113516
Zehravi M, Kabir J, Akter R, Malik S, Ashraf GM, Tagde P, Ramproshad S, Mondal B, Rahman MH, Mohan AG, et al. A Prospective Viewpoint on Neurological Diseases and Their Biomarkers. Molecules. 2022; 27(11):3516. https://doi.org/10.3390/molecules27113516
Chicago/Turabian StyleZehravi, Mehrukh, Janisa Kabir, Rokeya Akter, Sumira Malik, Ghulam Md. Ashraf, Priti Tagde, Sarker Ramproshad, Banani Mondal, Md. Habibur Rahman, Aurel George Mohan, and et al. 2022. "A Prospective Viewpoint on Neurological Diseases and Their Biomarkers" Molecules 27, no. 11: 3516. https://doi.org/10.3390/molecules27113516