
Deep Learning-Based Method for Compound Identification in NMR Spectra of Mixtures


Abstract
:1. Introduction
2. Method
2.1. Data Augmentation
2.2. Convolutional Neural Network
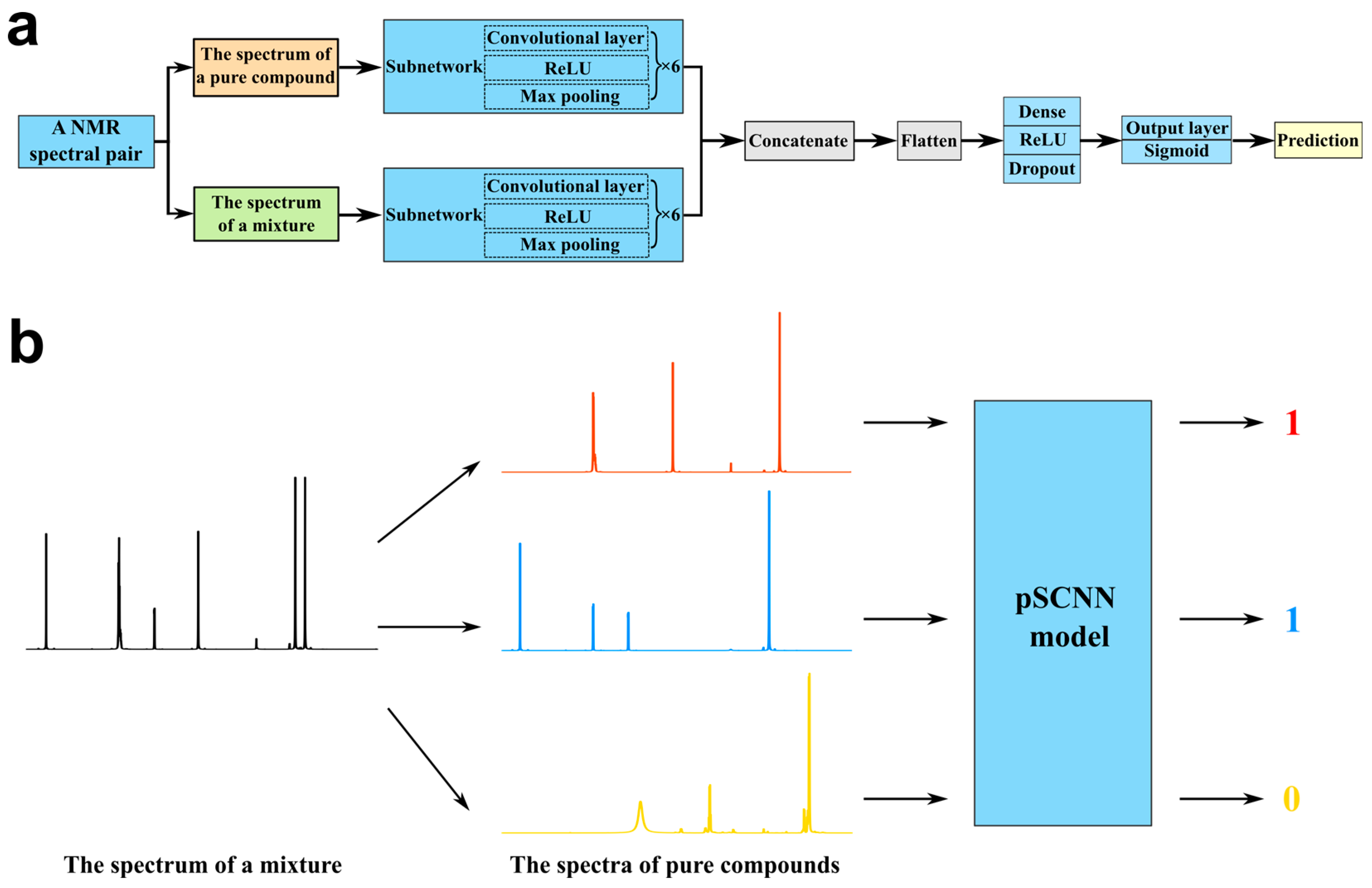
2.3. Pseudo-Siamese Convolutional Neural Network
2.4. Architecture of pSCNN for NMR
2.5. Compound Identification with pSCNN
2.6. Evaluation Metrics
3. Experiments
3.1. Flavor Standards
3.2. Known Flavor Mixtures
3.3. Additional Flavor Mixture
4. Results and Discussion
4.1. Implementation and Computing Resources
4.2. Validation of Data Augmentation
4.3. Hyperparameters Optimization and Training
4.4. Performance Evaluation
4.5. Results of Mixture Analysis
4.6. Translation Invariance for NMR Peaks
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Akash, M.S.H.; Rehman, K. Essentials of Pharmaceutical Analysis; Springer: Singapore, 2020. [Google Scholar] [CrossRef]
- Tsedilin, A.; Fakhrutdinov, A.N.; Eremin, D.; Zalesskiy, S.S.; Chizhov, A.O.; Kolotyrkina, N.G.; Ananikov, V. How sensitive and accurate are routine NMR and MS measurements? Mendeleev Commun. 2015, 25, 454–456. [Google Scholar] [CrossRef]
- Kovacs, H.; Moskau, D.; Spraul, M. Cryogenically cooled probes—A leap in NMR technology. Prog. Nucl. Magn. Reson. Spectrosc. 2005, 46, 131–155. [Google Scholar] [CrossRef]
- Elyashberg, M. Identification and structure elucidation by NMR spectroscopy. TrAC Trends Anal. Chem. 2015, 69, 88–97. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Claridge, T.D.W. Chapter 2—Introducing High-Resolution NMR. In High-Resolution NMR Techniques in Organic Chemistry, 3rd ed.; Claridge, T.D.W., Ed.; Elsevier: Boston, MA, USA, 2016; pp. 11–59. [Google Scholar]
- Edison, A.S.; Colonna, M.; Gouveia, G.J.; Holderman, N.R.; Judge, M.T.; Shen, X.; Zhang, S. NMR: Unique Strengths That Enhance Modern Metabolomics Research. Anal. Chem. 2021, 93, 478–499. [Google Scholar] [CrossRef]
- Emwas, A.-H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S. Quantitative metabolomics using NMR. TrAC Trends Anal. Chem. 2008, 27, 228–237. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, N. Applications of Solution NMR in Drug Discovery. Molecules 2021, 26, 576. [Google Scholar] [CrossRef]
- Softley, C.A.; Bostock, M.J.; Popowicz, G.M.; Sattler, M. Paramagnetic NMR in drug discovery. J. Biomol. NMR 2020, 74, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Bertini, I.; Cowburn, D.; Dalvit, C.; Giralt, E.; Jahnke, W.; James, T.L.; Homans, S.W.; Kessler, H.; Luchinat, C.; et al. Perspectives on NMR in drug discovery: A technique comes of age. Nat. Rev. Drug Discov. 2008, 7, 738–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Liu, X.; Liu, Y.; Zhai, X.; Cao, T.; Wang, A.; Qiu, J. Applications of nuclear magnetic resonance spectroscopy to the evaluation of complex food constituents. Food Chem. 2021, 342, 128258. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Fonseca, F.; Lião, L.; Alcantara, G.; Barison, A. High-resolution magic angle spinning nuclear magnetic resonance in foodstuff analysis. TrAC Trends Anal. Chem. 2015, 73, 10–18. [Google Scholar] [CrossRef]
- Wang, Z.-F.; You, Y.-L.; Li, F.-F.; Kong, W.-R.; Wang, S.-Q. Research Progress of NMR in Natural Product Quantification. Molecules 2021, 26, 6308. [Google Scholar] [CrossRef] [PubMed]
- Robinette, S.L.; Brüschweiler, R.; Schroeder, F.C.; Edison, A.S. NMR in Metabolomics and Natural Products Research: Two Sides of the Same Coin. Acc. Chem. Res. 2012, 45, 288–297. [Google Scholar] [CrossRef]
- Martin, G.J.; Martin, M.L. Thirty Years of Flavor NMR. In Flavor Chemistry: Thirty Years of Progress; Teranishi, R., Wick, E.L., Hornstein, I., Eds.; Springer: Boston, MA, USA, 1999; pp. 19–30. [Google Scholar]
- Singh, P.; Singh, M.K.; Beg, Y.R.; Nishad, G.R. A review on spectroscopic methods for determination of nitrite and nitrate in environmental samples. Talanta 2019, 191, 364–381. [Google Scholar] [CrossRef]
- Santos, A.; Dutra, L.; Menezes, L.; Santos, M.; Barison, A. Forensic NMR spectroscopy: Just a beginning of a promising partnership. TrAC Trends Anal. Chem. 2018, 107, 31–42. [Google Scholar] [CrossRef]
- Proietti, N.; Capitani, D.; Di Tullio, V. Nuclear Magnetic Resonance, a Powerful Tool in Cultural Heritage. Magnetochemistry 2018, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, P.; Viereck, N.; Bro, R.; Engelsen, S.B. Chemometric Analysis of NMR Spectra. In Modern Magnetic Resonance; Webb, G.A., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–20. [Google Scholar]
- Kwon, Y.; Lee, D.; Choi, Y.-S.; Kang, S. Molecular search by NMR spectrum based on evaluation of matching between spectrum and molecule. Sci. Rep. 2021, 11, 20998. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C.; Krause, S.; Kuhn, S. NMRShiftDBConstructing a Free Chemical Information System with Open-Source Components. J. Chem. Inf. Comput. Sci. 2003, 43, 1733–1739. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Lewis, I.A.; Hegeman, A.D.; Anderson, M.E.; Li, J.; Schulte, C.F.; Westler, W.M.; Eghbalnia, H.R.; Sussman, M.R.; Markley, J.L. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 2008, 26, 162–164. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Ballabio, D.; Consonni, V. Distances and Similarity Measures in Chemometrics and Chemoinformatics. In Encyclopedia of Analytical Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2020; pp. 1–40. [Google Scholar] [CrossRef]
- Schaller, R.B.; Pretsch, E. A computer program for the automatic estimation of 1H NMR chemical shifts. Anal. Chim. Acta 1994, 290, 295–302. [Google Scholar] [CrossRef]
- De Meyer, T.; Sinnaeve, D.; Van Gasse, B.; Tsiporkova, E.; Rietzschel, E.R.; De Buyzere, M.L.; Gillebert, T.C.; Bekaert, S.; Martins, J.C.; Van Criekinge, W. NMR-Based Characterization of Metabolic Alterations in Hypertension Using an Adaptive, Intelligent Binning Algorithm. Anal. Chem. 2008, 80, 3783–3790. [Google Scholar] [CrossRef]
- Åberg, K.M.; Alm, E.; Torgrip, R.J.O. The correspondence problem for metabonomics datasets. Anal. Bioanal. Chem. 2009, 394, 151–162. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. Generalized adaptive intelligent binning of multiway data. Chemom. Intell. Lab. Syst. 2015, 146, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, T.N.; Laukens, K. Getting Your Peaks in Line: A Review of Alignment Methods for NMR Spectral Data. Metabolites 2013, 3, 259–276. [Google Scholar] [CrossRef]
- Savorani, F.; Tomasi, G.; Engelsen, S.B. icoshift: A versatile tool for the rapid alignment of 1D NMR spectra. J. Magn. Reson. 2010, 202, 190–202. [Google Scholar] [CrossRef]
- Veselkov, K.A.; Lindon, J.C.; Ebbels, T.M.D.; Crockford, D.; Volynkin, V.V.; Holmes, E.; Davies, D.B.; Nicholson, J.K. Recursive Segment-Wise Peak Alignment of Biological (1)H NMR Spectra for Improved Metabolic Biomarker Recovery. Anal. Chem. 2009, 81, 56–66. [Google Scholar] [CrossRef]
- Castillo, A.M.; Uribe, L.; Patiny, L.; Wist, J. Fast and shift-insensitive similarity comparisons of NMR using a tree-representation of spectra. Chemom. Intell. Lab. Syst. 2013, 127, 1–6. [Google Scholar] [CrossRef]
- Bodis, L.; Ross, A.; Pretsch, E. A novel spectra similarity measure. Chemom. Intell. Lab. Syst. 2007, 85, 1–8. [Google Scholar] [CrossRef]
- Mishra, R.; Marchand, A.; Jacquemmoz, C.; Dumez, J.-N. Ultrafast diffusion-based unmixing of 1H NMR spectra. Chem. Commun. 2021, 57, 2384–2387. [Google Scholar] [CrossRef]
- Lin, M.; Shapiro, M.J. Mixture Analysis by NMR Spectroscopy. Anal. Chem. 1997, 69, 4731–4733. [Google Scholar] [CrossRef]
- Zhang, F.; Brüschweiler, R. Robust Deconvolution of Complex Mixtures by Covariance TOCSY Spectroscopy. Angew. Chem. Int. Ed. 2007, 46, 2639–2642. [Google Scholar] [CrossRef]
- Castellanos, E.R.R.; Wist, J. Decomposition of mixtures’ spectra by multivariate curve resolution of rapidly acquired TOCSY experiments. Magn. Reson. Chem. 2010, 48, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Brüschweiler, R. Deconvolution of Chemical Mixtures with High Complexity by NMR Consensus Trace Clustering. Anal. Chem. 2011, 83, 7412–7417. [Google Scholar] [CrossRef] [Green Version]
- Toumi, I.; Caldarelli, S.; Torrésani, B. A review of blind source separation in NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 81, 37–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poggetto, G.D.; Castañar, L.; Adams, R.W.; Morris, G.A.; Nilsson, M. Dissect and Divide: Putting NMR Spectra of Mixtures under the Knife. J. Am. Chem. Soc. 2019, 141, 5766–5771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, J.S.; Donarski, J.A.; Wilson, J.C.; Charlton, A.J. Analysis of complex mixtures using high-resolution nuclear magnetic resonance spectroscopy and chemometrics. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 336–359. [Google Scholar] [CrossRef]
- Tulpan, D.; Léger, S.; Belliveau, L.; Culf, A.; Čuperlović-Culf, M. MetaboHunter: An automatic approach for identification of metabolites from 1H-NMR spectra of complex mixtures. BMC Bioinform. 2011, 12, 400. [Google Scholar] [CrossRef]
- Wei, S.; Zhang, J.; Liu, L.; Ye, T.; Gowda, G.A.N.; Tayyari, F.; Raftery, D. Ratio Analysis Nuclear Magnetic Resonance Spectroscopy for Selective Metabolite Identification in Complex Samples. Anal. Chem. 2011, 83, 7616–7623. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, K. CRAFT (complete reduction to amplitude frequency table)—Robust and time-efficient Bayesian approach for quantitative mixture analysis by NMR. Magn. Reson. Chem. 2013, 51, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.; Nuzillard, J.-M.; Purson, S.; Hamzaoui, M.; Borie, N.; Reynaud, R.; Renault, J.-H. Identification of Natural Metabolites in Mixture: A Pattern Recognition Strategy Based on 13C NMR. Anal. Chem. 2014, 86, 2955–2962. [Google Scholar] [CrossRef]
- Kuhn, S.; Colreavy-Donnelly, S.; de Souza, J.S.; Borges, R.M. An integrated approach for mixture analysis using MS and NMR techniques. Faraday Discuss. 2019, 218, 339–353. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet Classification with Deep Convolutional Neural Networks. In Proceedings of the Advances in Neural Information Processing Systems 2012, Lake Tahoe, NV, USA, 3–6 December 2012; pp. 1097–1105. [Google Scholar]
- Graves, A.; Mohamed, A.; Hinton, G. Speech recognition with deep recurrent neural networks. In Proceedings of the IEEE International Conference on Acoustics, Speech and Signal Processing, Vancouver, BC, Canada, 26–31 May 2013; pp. 6645–6649. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.u.; Polosukhin, I. Attention is All you Need. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–6 December 2017; pp. 5998–6008. [Google Scholar]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P.S. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2020, 32, 4–24. [Google Scholar] [CrossRef] [Green Version]
- Bengio, Y.; Courville, A.; Vincent, P. Representation Learning: A Review and New Perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 2013, 35, 1798–1828. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Pu, H.; Wang, F.; Hu, Z.; Wang, L. The expressive power of neural networks: A view from the width. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–6 December 2017; pp. 6232–6240. [Google Scholar]
- Chen, D.; Wang, Z.; Guo, D.; Orekhov, V.; Qu, X. Review and prospect: Deep learning in nuclear magnetic resonance spectroscopy. Chem. Eur. J. 2020, 26, 10391–10401. [Google Scholar] [CrossRef] [Green Version]
- Cobas, C. NMR signal processing, prediction, and structure verification with machine learning techniques. Magn. Reson. Chem. 2020, 58, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Huang, Y.; Lu, H.; Qiu, T.; Guo, D.; Agback, T.; Orekhov, V.; Chen, Z. Accelerated Nuclear Magnetic Resonance Spectroscopy with Deep Learning. Angew. Chem. Int. Ed. 2020, 59, 10297–10300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Zeng, Q.; Wu, K.; Lin, Y. Fast reconstruction of non-uniform sampling multidimensional NMR spectroscopy via a deep neural network. J. Magn. Reson. 2020, 317, 106772. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.F. Using Deep Neural Networks to Reconstruct Non-uniformly Sampled NMR Spectra. J. Biomol. NMR 2019, 73, 577–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Luo, J.; Zeng, Q.; Dong, X.; Chen, J.; Zhan, C.; Chen, Z.; Lin, Y. Improvement in Signal-to-Noise Ratio of Liquid-State NMR Spectroscopy via a Deep Neural Network DN-Unet. Anal. Chem. 2021, 93, 1377–1382. [Google Scholar] [CrossRef]
- Klukowski, P.; Augoff, M.; Zieba, M.; Drwal, M.; Gonczarek, A.; Walczak, M.J. NMRNet: A deep learning approach to automated peak picking of protein NMR spectra. Bioinformatics 2018, 34, 2590–2597. [Google Scholar] [CrossRef]
- Li, D.-W.; Hansen, A.L.; Yuan, C.; Bruschweiler-Li, L.; Brüschweiler, R. DEEP picker is a deep neural network for accurate deconvolution of complex two-dimensional NMR spectra. Nat. Commun. 2021, 12, 5229. [Google Scholar] [CrossRef]
- Jonas, E.; Kuhn, S. Rapid prediction of NMR spectral properties with quantified uncertainty. J. Cheminformatics 2019, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Lee, D.; Choi, Y.-S.; Kang, M.; Kang, S. Neural Message Passing for NMR Chemical Shift Prediction. J. Chem. Inf. Model. 2020, 60, 2024–2030. [Google Scholar] [CrossRef]
- Gerrard, W.; Bratholm, L.A.; Packer, M.J.; Mulholland, A.J.; Glowacki, D.R.; Butts, C.P. IMPRESSION—Prediction of NMR parameters for 3-dimensional chemical structures using machine learning with near quantum chemical accuracy. Chem. Sci. 2020, 11, 508–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Shree Sowndarya, S.V.; Gallegos, L.C.; John, P.C.S.; Paton, R.S. Real-time prediction of 1H and 13C chemical shifts with DFT accuracy using a 3D graph neural network. Chem. Sci. 2021, 12, 12012–12026. [Google Scholar] [CrossRef]
- Yang, Z.; Chakraborty, M.; White, A.D. Predicting chemical shifts with graph neural networks. Chem. Sci. 2021, 12, 10802–10809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Idelbayev, Y.; Roberts, N.; Tao, Y.; Nannapaneni, Y.; Duggan, B.M.; Min, J.; Lin, E.C.; Gerwick, E.C.; Cottrell, G.W.; et al. Small Molecule Accurate Recognition Technology (SMART) to Enhance Natural Products Research. Sci. Rep. 2017, 7, 14243. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Terayama, K.; Sumita, M.; Yoshizoe, K.; Ito, K.; Kikuchi, J.; Tsuda, K. NMR-TS: De novo molecule identification from NMR spectra. Sci. Technol. Adv. Mater. 2020, 21, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, M.S.; Woroch, C.P.P.; Markland, T.E.; Kanan, M.W. A framework for automated structure elucidation from routine NMR spectra. Chem. Sci. 2021, 12, 15329–15338. [Google Scholar] [CrossRef]
- Kuhn, S.; Tumer, E.; Colreavy-Donnelly, S.; Borges, R.M. A Pilot Study for Fragment Identification Using 2D NMR and Deep Learning. Magn. Reson. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chicco, D. Siamese Neural Networks: An Overview. In Artificial Neural Networks; Cartwright, H., Ed.; Springer: New York, NY, USA, 2021; pp. 73–94. [Google Scholar]
- Huber, F.; van der Burg, S.; van der Hooft, J.J.J.; Ridder, L. MS2DeepScore: A novel deep learning similarity measure to compare tandem mass spectra. J. Cheminform. 2021, 13, 84. [Google Scholar] [CrossRef]
- Fan, X.; Ming, W.; Zeng, H.; Zhang, Z.; Lu, H. Deep learning-based component identification for the Raman spectra of mixtures. Analyst 2019, 144, 1789–1798. [Google Scholar] [CrossRef]
- Mater, A.C.; Coote, M.L. Deep Learning in Chemistry. J. Chem. Inf. Model. 2019, 59, 2545–2559. [Google Scholar] [CrossRef]
- Debus, B.; Parastar, H.; Harrington, P.; Kirsanov, D. Deep learning in analytical chemistry. TrAC Trends Anal. Chem. 2021, 145, 116459. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Models | Epoch | The Number of Convolutional Layers * | Learning Rate | ACC |
|---|---|---|---|---|
| M1 | 100 | 6 | 10−2 | 0.4900 |
| M2 | 100 | 6 | 10−3 | 0.4935 |
| M3 | 100 | 6 | 10−4 | 0.9990 |
| M4 | 100 | 6 | 10−5 | 0.9935 |
| M5 | 100 | 5 | 10−4 | 0.9975 |
| M6 | 100 | 7 | 10−4 | 0.9975 |
| M7 | 100 | 8 | 10−4 | 0.9935 |
| M8 | 100 | 9 | 10−4 | 0.9925 |
| M9 | 100 | 10 | 10−4 | 0.9860 |
| Datasets | ACC | TPR | FPR |
|---|---|---|---|
| flavor mixtures dataset | 97.62% | 96.44% | 2.29% |
| additional flavor mixture dataset | 91.67% | 100.00% | 10.53% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, W.; Liao, Y.; Wang, Y.; Wang, S.; Du, W.; Lu, H.; Kong, B.; Yang, H.; Zhang, Z. Deep Learning-Based Method for Compound Identification in NMR Spectra of Mixtures. Molecules 2022, 27, 3653. https://doi.org/10.3390/molecules27123653
Wei W, Liao Y, Wang Y, Wang S, Du W, Lu H, Kong B, Yang H, Zhang Z. Deep Learning-Based Method for Compound Identification in NMR Spectra of Mixtures. Molecules. 2022; 27(12):3653. https://doi.org/10.3390/molecules27123653
Chicago/Turabian StyleWei, Weiwei, Yuxuan Liao, Yufei Wang, Shaoqi Wang, Wen Du, Hongmei Lu, Bo Kong, Huawu Yang, and Zhimin Zhang. 2022. "Deep Learning-Based Method for Compound Identification in NMR Spectra of Mixtures" Molecules 27, no. 12: 3653. https://doi.org/10.3390/molecules27123653
APA StyleWei, W., Liao, Y., Wang, Y., Wang, S., Du, W., Lu, H., Kong, B., Yang, H., & Zhang, Z. (2022). Deep Learning-Based Method for Compound Identification in NMR Spectra of Mixtures. Molecules, 27(12), 3653. https://doi.org/10.3390/molecules27123653