
Pore Formation Mechanism of A-Beta Peptide on the Fluid Membrane: A Combined Coarse-Grained and All-Atomic Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Details
2.1. Coarse Grained Simulation
2.2. All-Atom Molecular Dynamics in an Explicit Solvent
3. Results and Discussion
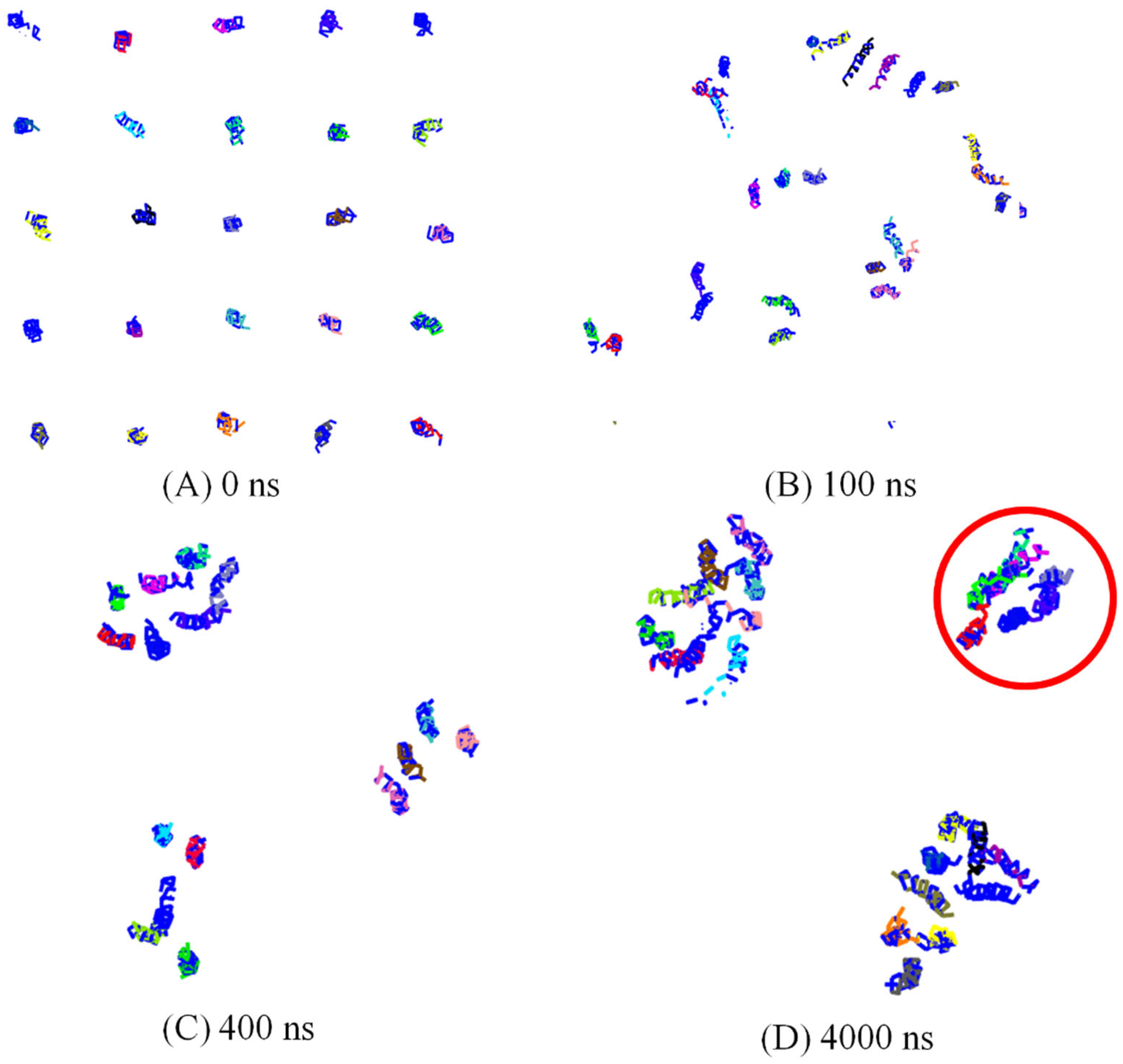
3.1. Spontaneously Aggregation of Aβ Peptides
3.2. Effect of Lipid Membrane
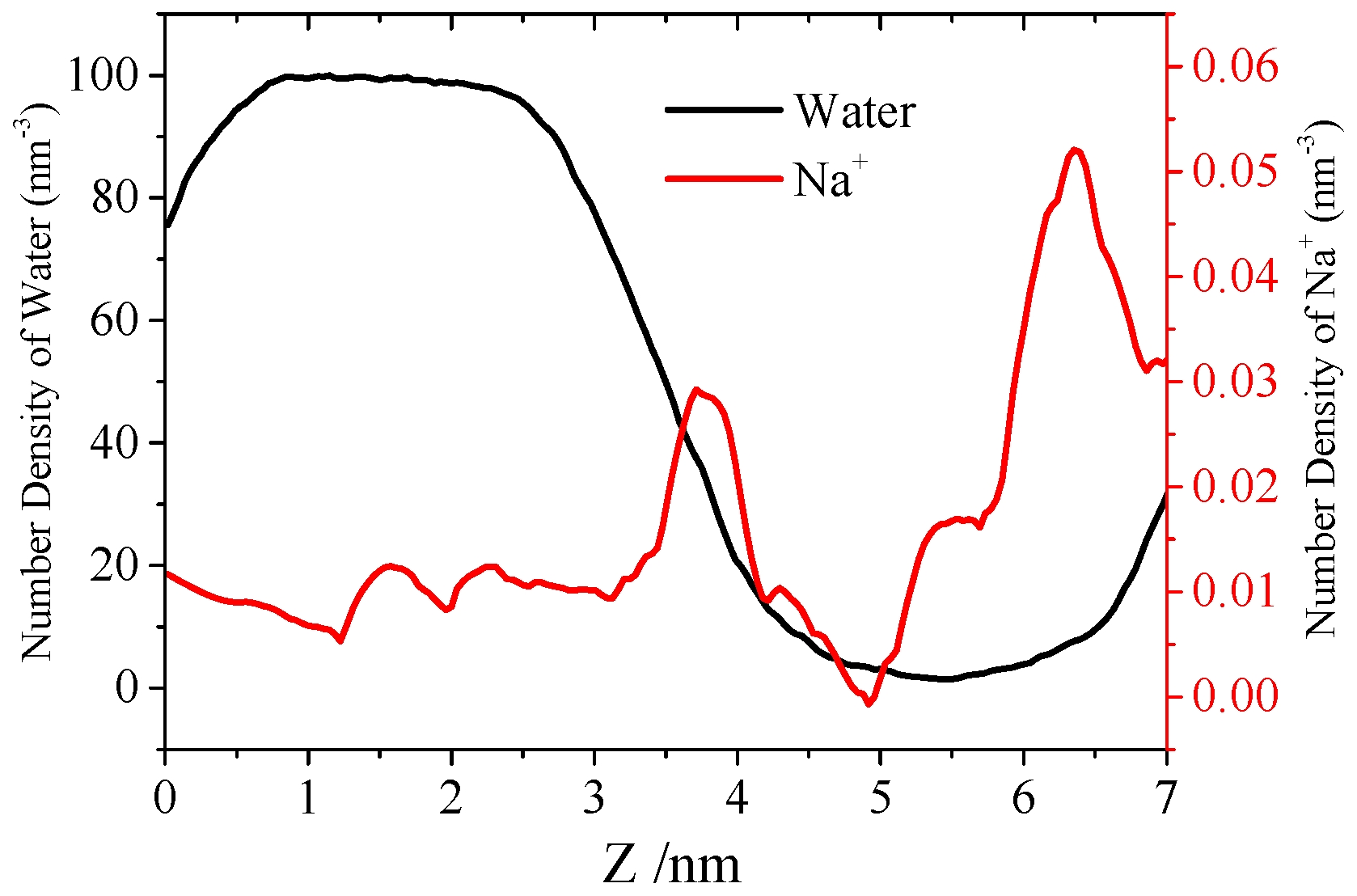
3.3. Water Permeation of the Pore

4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Strittmatter, W.J.; Roses, A.D. Apolipoprotein E and Alzheimer’s disease. Annu. Rev. Neurosci. 1996, 19, 53–77. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, I.; Soto, C. Natural animal models of neurodegenerative protein misfolding diseases. Curr. Pharm. Des. 2012, 18, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Salvadores, N.; Shahnawaz, M.; Scarpini, E.; Tagliavini, F.; Soto, C. Detection of misfolded Aβ oligomers for sensitive biochemical diagnosis of Alzheimer’s disease. Cell Rep. 2014, 7, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ (1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Benilova, I.; Karran, E.; de Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Laganowsky, A.; Liu, C.; Sawaya, M.R.; Whitelegge, J.P.; Park, J.; Zhao, M.; Pensalfini, A.; Soriaga, A.B.; Landau, M.; Teng, P.K. Atomic view of a toxic amyloid small oligomer. Science 2012, 335, 1228–1231. [Google Scholar] [CrossRef] [Green Version]
- Stroud, J.C.; Liu, C.; Teng, P.K.; Eisenberg, D.P. Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. USA 2012, 109, 7717–7722. [Google Scholar] [CrossRef] [Green Version]
- Penke, B.; Mária, S.; Ferenc, B. Oligomerization and conformational change turn monomeric β-amyloid and tau proteins toxic: Their role in Alzheimer’s pathogenesis. Molecules 2020, 25, 1659. [Google Scholar] [CrossRef] [Green Version]
- Kotler, S.A.; Walsh, P.; Brender, J.R.; Ramamoorthy, A. Differences between amyloid-β aggregation in solution and on the membrane: Insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6692–6700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacca, M.F.M.; Kotler, S.A.; Brender, J.R.; Chen, J.; Lee, D.-k.; Ramamoorthy, A. Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 2012, 103, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacca, M.F.M.; Milardi, D.; Messina, G.M.L.; Marletta, G.; Brender, J.R.; Ramamoorthy, A.; la Rosa, C. Cations as switches of amyloid-mediated membrane disruption mechanisms: Calcium and IAPP. Biophys. J. 2013, 104, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, Y.Q.; Ng, K.Y.; Chye, S.M.; Ling, A.P.K.; Koh, R.Y. Mechanisms of action of amyloid-beta and its precursor protein in neuronal cell death. Metab. Brain Dis. 2020, 35, 11–30. [Google Scholar] [CrossRef]
- Zaretsky, D.V.; Zaretskaia, M.V.; Molkov, Y.I. Membrane channel hypothesis of lysosomal permeabilization by beta-amyloid. Neurosci. Lett. 2022, 770, 136338. [Google Scholar] [CrossRef]
- di Scala, C.; Yahi, N.; Flores, A.; Boutemeur, S.; Kourdougli, N.; Chahinian, H.; Fantini, J. Comparison of the amyloid pore forming properties of rat and human Alzheimer’s beta-amyloid peptide 1–42: Calcium imaging data. Data Brief 2016, 6, 640–643. [Google Scholar] [CrossRef] [Green Version]
- Roche, J.; Shen, Y.; Lee, J.H.; Ying, J.; Bax, A. Monomeric Aβ1–40 and Aβ1–42 Peptides in Solution Adopt Very Similar Ramachandran Map Distributions That Closely Resemble Random Coil. Biochemistry 2016, 55, 762–775. [Google Scholar] [CrossRef] [Green Version]
- Brookes, D.H.; Head-Gordon, T.J. Experimental inferential structure determination of ensembles for intrinsically disordered proteins. Am. Chem. Soc. 2016, 138, 4530–4538. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Shao, H.; Zhang, Y.; Li, H.; Menon, N.K.; Neuhaus, E.B.; Brewer, J.M.; Byeon, I.-J.L.; Ray, D.G.; Vitek, M.P. Solution NMR studies of the Aβ (1–40) and Aβ (1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004, 126, 1992–2005. [Google Scholar] [CrossRef]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.-M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s ß-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R.P. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciudad, S.; Puig, E.; Botzanowski, T.; Meigooni, M.; Arango, A.S.; Do, J.; Maxim, M.; Mariam, B.; Stephane, C.; Giovanni, M.; et al. Aβ (1–42) tetramer and octamer structures reveal edge conductivity pores as a mechanism for membrane damage. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Sonar, K.; Bharadwaj, P.; Deplazes, E.; Mancera, R.L. Characterisation of the structure and oligomerisation of islet amyloid polypeptides (IAPP): A review of molecular dynamics simulation studies. Molecules 2018, 23, 2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, S.G.; Okumura, H. All-atom molecular dynamics simulation methods for the aggregation of protein and peptides: Replica exchange/permutation and nonequilibrium simulations. Comput. Simul. Aggreg. Proteins Peptides 2022, 197–220. [Google Scholar]
- Strodel, B. Amyloid aggregation simulations: Challenges, advances and perspectives. Curr. Opin. Struc. Biol. 2021, 67, 145–152. [Google Scholar] [CrossRef]
- Rezaei-Ghaleh, N.; Amininasab, M.; Kumar, S.; Walter, J.; Zweckstetter, M. Phosphorylation modifies the molecular stability of β-amyloid deposits. Nat. Commun. 2016, 7, 111359. [Google Scholar] [CrossRef] [Green Version]
- Berhanu, W.M.; Alred, E.J.; Bernhardt, N.A.; Hansmann, U.H.E. All-atom simulation of amyloid aggregates. Phys. Proc. 2015, 68, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Arce, F.T.; Capone, R.; Ramachandran, S.; Lal, R.; Nussinov, R. Misfolded amyloid ion channels present mobile β-sheet subunits in contrast to conventional ion channels. Biophys. J. 2009, 97, 3029–3037. [Google Scholar] [CrossRef] [Green Version]
- Mustata, M.; Capone, R.; Jang, H.; Arce, F.T.; Ramachandran, S.; Lal, R.; Nussinov, R. K3 Fragment of Amyloidogenic β2-Microglobulin Forms Ion Channels: Implication for Dialysis Related Amyloidosis. J. Am. Chem. Soc. 2009, 131, 14938–14945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Luo, Y.; Zhou, R.; Wei, G. Probing the self-assembly mechanism of diphenylalanine-based peptide nanovesicles and nanotubes. ACS Nano 2012, 6, 3907–3918. [Google Scholar] [CrossRef]
- Bond, P.J.; Holyoake, J.; Ivetac, A.; Khalid, S.; Sansom, M.S.P. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struc. Biol. 2007, 157, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Tsanai, M.; Frederix, P.W.J.M.; Schroer, C.F.; Souza, P.C.; Marrink, S.J. Coacervate formation studied by explicit solvent coarse-grain molecular dynamics with the Martini model. Chem. Sci. 2021, 12, 8521–8530. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sampath, J.; Alamdari, S.; Shen, G.; Chen, C.L.; Mundy, C.J.; Pfaendtner, J.; Ferguson, A.L. MARTINI-compatible coarse-grained model for the mesoscale simulation of peptoids. J. Phys. Chem. B 2020, 124, 7745–7764. [Google Scholar] [CrossRef] [PubMed]
- Thøgersen, L.; Schiøtt, B.; Vosegaard, T.; Nielsen, N.C.; Tajkhorshid, E. Peptide aggregation and pore formation in a lipid bilayer: A combined coarse-grained and all atom molecular dynamics study. Biophys. J. 2008, 95, 4337–4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talafous, J.; Marcinowski, K.J.; Klopman, G.; Zagorski, M.G. Solution Structure of Residues 1-28 of the Amyloid. beta.-Peptide. Biochemistry 1994, 33, 7788–7796. [Google Scholar] [CrossRef]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.-J. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; Jr, A.D.M.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Wassenaar, T.A.; Pluhackova, K.; Böckmann, R.A.; Marrink, S.J.; Tieleman, D.P. Going backward: A flexible geometric approach to reverse transformation from coarse grained to atomistic models. J. Chem. Theory Comput. 2014, 10, 676–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L.J. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Schatz, G.C. Free Energy Profile and Mechanism of Self-Assembly of Peptide Amphiphiles Based on a Collective Assembly Coordinate. J. Phys. Chem. B 2013, 117, 9004–9013. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R. Trp-cage: Folding free energy landscape in explicit water. Proc. Natl. Acad. Sci. USA 2003, 100, 13280–13285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, L.; Wang, L.W.; Shen, J.W. The self-assembly mechanism of tetra-peptides from the motif of β-amyloid peptides: A combined coarse-grained and all-atom molecular dynamics simulation. RSC Adv. 2016, 6, 100072–100078. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Y.; Xie, Z.; Liang, L. Pore Formation Mechanism of A-Beta Peptide on the Fluid Membrane: A Combined Coarse-Grained and All-Atomic Model. Molecules 2022, 27, 3924. https://doi.org/10.3390/molecules27123924
Dai Y, Xie Z, Liang L. Pore Formation Mechanism of A-Beta Peptide on the Fluid Membrane: A Combined Coarse-Grained and All-Atomic Model. Molecules. 2022; 27(12):3924. https://doi.org/10.3390/molecules27123924
Chicago/Turabian StyleDai, Yuxi, Zhexing Xie, and Lijun Liang. 2022. "Pore Formation Mechanism of A-Beta Peptide on the Fluid Membrane: A Combined Coarse-Grained and All-Atomic Model" Molecules 27, no. 12: 3924. https://doi.org/10.3390/molecules27123924
APA StyleDai, Y., Xie, Z., & Liang, L. (2022). Pore Formation Mechanism of A-Beta Peptide on the Fluid Membrane: A Combined Coarse-Grained and All-Atomic Model. Molecules, 27(12), 3924. https://doi.org/10.3390/molecules27123924