
Anti-Inflammatory Effects of the Iron Chelator, DIBI, in Experimental Acute Lung Injury

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
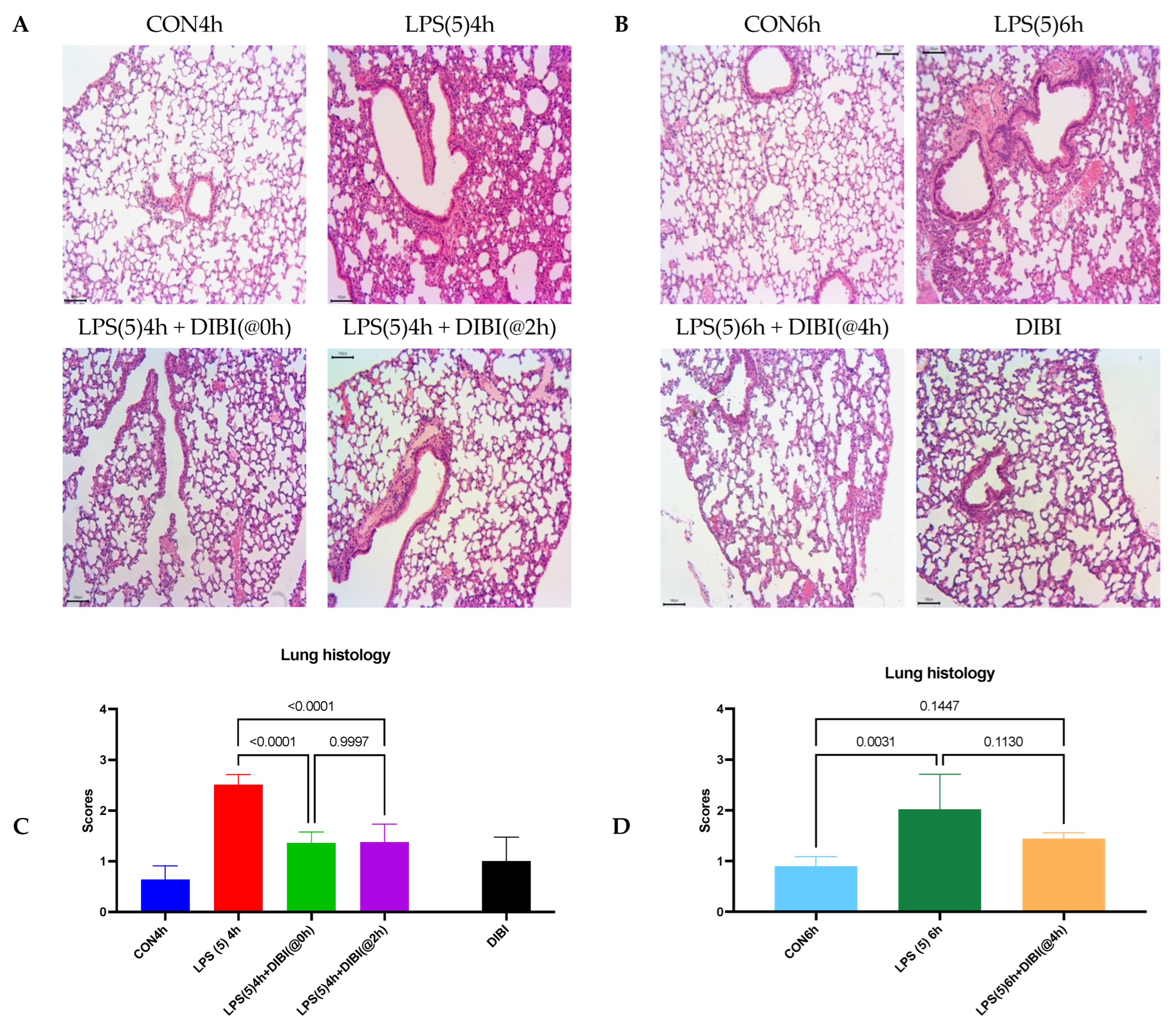
2.1. Tissue Damage by LPS
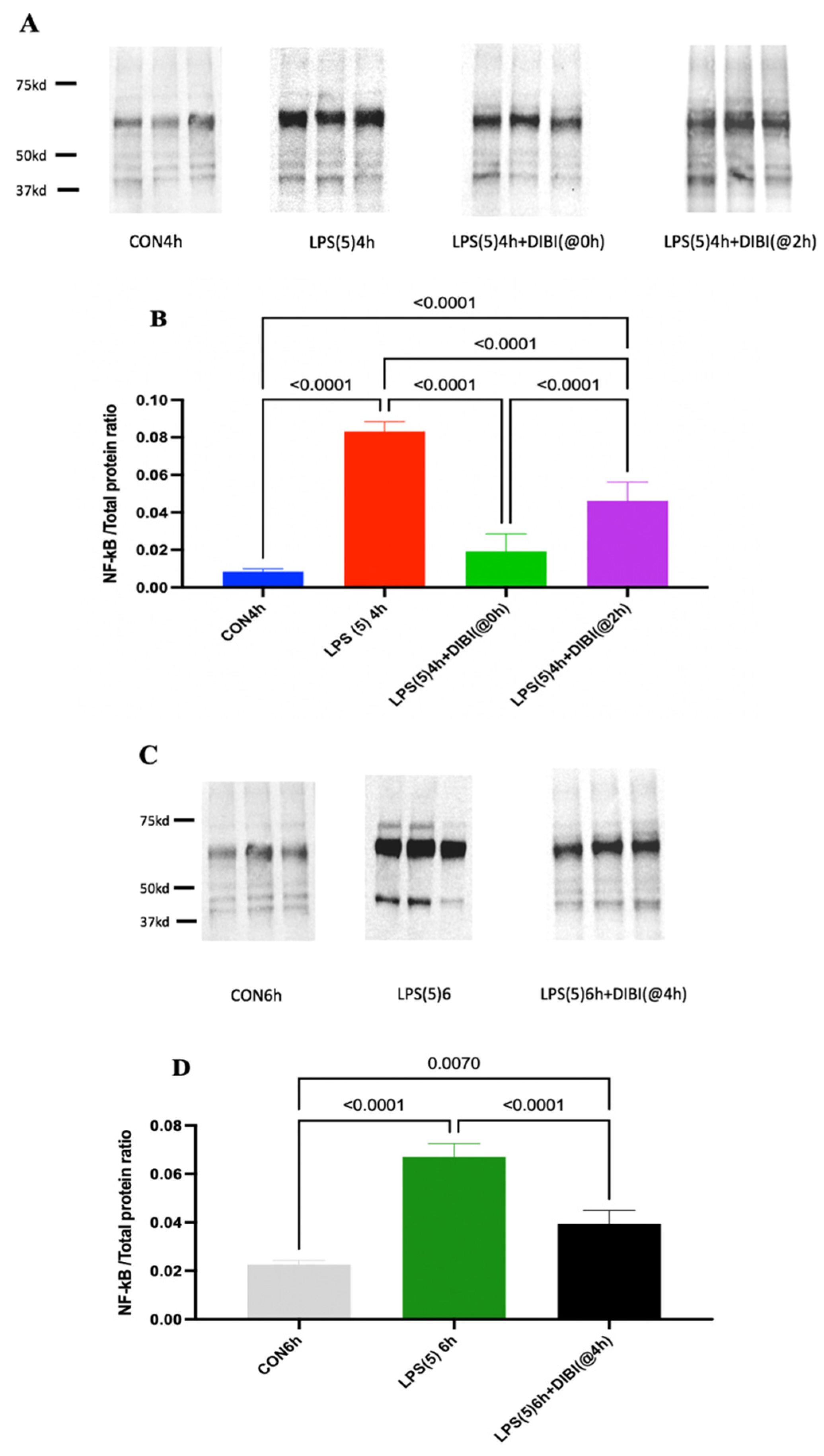
2.2. NF-B Activation
2.3. Cytokine Levels
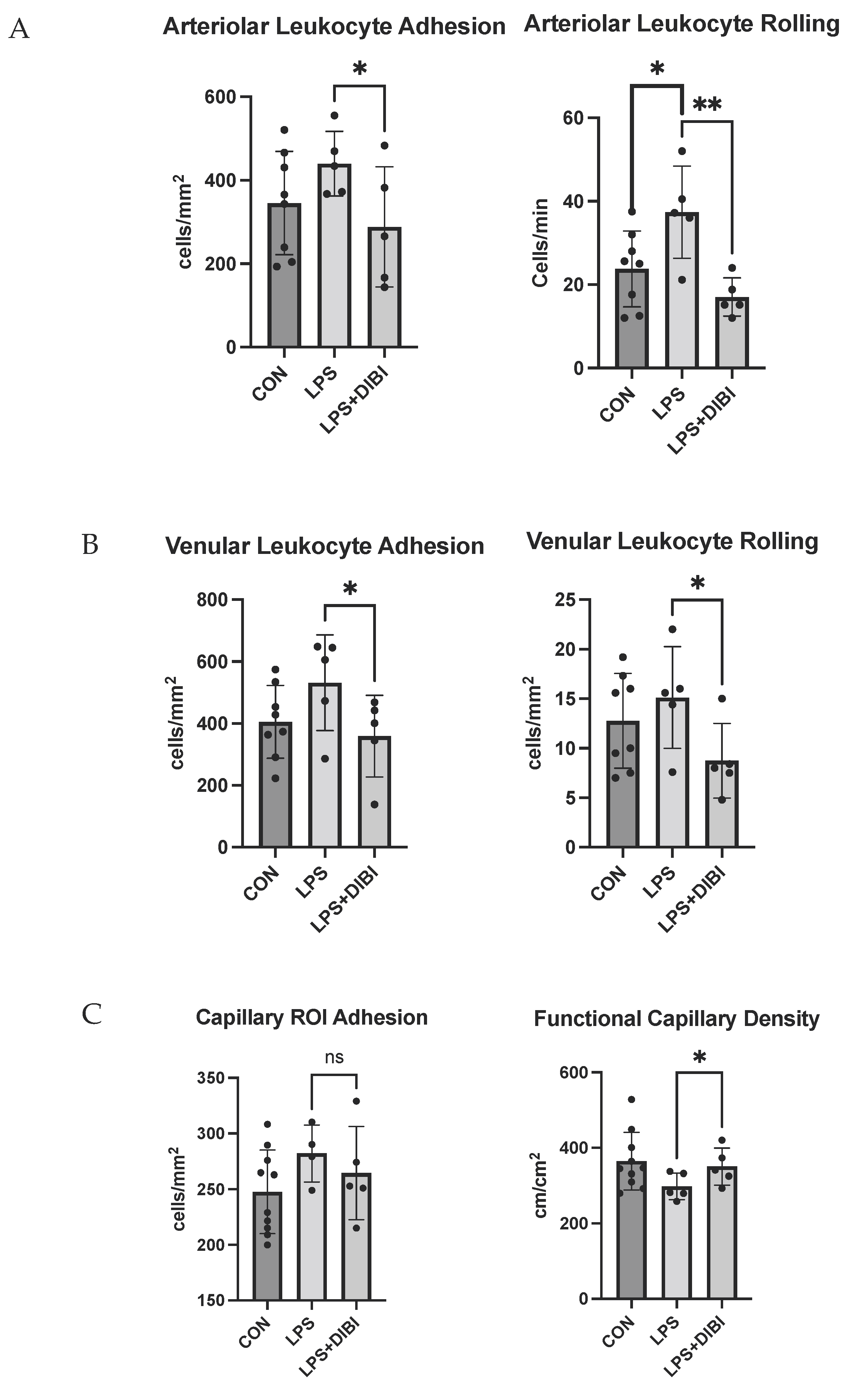
2.4. Intravital Microscopy
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Model
Experimental Groups
4.3. Histology
4.4. Western Blotting
4.5. Lung Tissue Inflammatory Mediators
4.6. IVM
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- McCormack, V.; Tolhurst-Cleaver, S. Acute Respiratory Distress Syndrome. BJA Educ. 2017, 17, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Millar, F.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Zhu, Y.-H.; Liu, D.-X.; Li, J.; Zhao, P.-C.; Zhong, Y.-P.; Chen, Y.-Q.; Xu, W.; Zhu, Z.-Q. Intranasal Application of Budesonide Attenuates Lipopolysaccharide-Induced Acute Lung Injury by Suppressing Nucleotide-Binding Oligomerization Domain-Like Receptor Family, Pyrin Domain-Containing 3 Inflammasome Activation in Mice. J. Immunol. Res. 2019, 2019, 7264383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colafrancesco, S.; Alessandri, C.; Conti, F.; Priori, R. COVID-19 Gone Bad: A New Character in the Spectrum of the Hyperferritinemic Syndrome? Autoimmun. Rev. 2020, 19, 102573. [Google Scholar] [CrossRef]
- Habib, H.M.; Ibrahim, S.; Zaim, A.; Ibrahim, W.H. The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators. Biomed. Pharmacother. 2021, 136, 111228. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal Models of Acute Lung Injury. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef] [Green Version]
- Opal, S.M. Endotoxins and Other Sepsis Triggers. In Endotoxemia and Endotoxin Shock; Karger Publishers: Basel, Switzerland, 2010; Volume 167, pp. 14–24. [Google Scholar]
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [Green Version]
- Domscheit, H.; Hegeman, M.A.; Carvalho, N.; Spieth, P.M. Molecular Dynamics of Lipopolysaccharide-Induced Lung Injury in Rodents. Front. Physiol. 2020, 11, 36. [Google Scholar] [CrossRef]
- Aderem, A.; Ulevitch, R.J. Toll-like receptors in the induction of the innate immune response. Nature 2000, 406, 782–787. [Google Scholar] [CrossRef]
- Thorburn, T.; Aali, M.; Kostek, L.; LeTourneau-Paci, C.; Colp, P.; Zhou, J.; Holbein, B.; Hoskin, D.; Lehmann, C. Anti-inflammatory effects of a novel iron chelator, DIBI, in experimental sepsis. Clin. Hemorheol. Microcirc. 2017, 67, 241–250. [Google Scholar] [CrossRef]
- Arora, N.; Caldwell, A.; Wafa, K.; Szczesniak, A.; Caldwell, M.; Al-Banna, N.; Sharawy, N.; Islam, S.; Zhou, J.; Holbein, B.; et al. DIBI, a polymeric hydroxypyridinone iron chelator, reduces ocular inflammation in local and systemic endotoxin-induced uveitis. Clin. Hemorheol. Microcirc. 2018, 69, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Hagn, G.; Holbein, B.; Zhou, J.; Lehmann, C. Anti-inflammatory iron chelator, DIBI, reduces leukocyte-endothelial adhesion and clinical symptoms of LPS-induced interstitial cystitis in mice. Clin. Hemorheol. Microcirc. 2021, 79, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Islam, S.; Jarosch, S.; Zhou, J.; Hoskin, D.; Greenshields, A.; Al-Banna, N.; Sharawy, N.; Sczcesniak, A.; Kelly, M.; et al. The Utility of Iron Chelators in the Management of Inflammatory Disorders. Mediat. Inflamm. 2015, 2015, 516740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokam, D.; Aali, M.; Dickson, K.; Scott, C.; Holbein, B.E.; Zhou, J.; Lehmann, C. The novel iron chelator, DIBI, attenuates inflammation and improves outcome in colon ascendens stent peritonitis-induced experimental sepsis. Clin. Hemorheol. Microcirc. 2020, 76, 241–261. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; She, H.; Sung, C.K.; Tsukamoto, H. Iron-dependent activation of NF-κB in Kupffer cells: A priming mechanism for alcoholic liver disease. Alcohol 2003, 30, 107–113. [Google Scholar] [CrossRef]
- Gumbau-Brisa, R.; Ang, M.T.C.; Holbein, B.E.; Bierenstiel, M. Enhanced Fe3+ binding through cooperativity of 3-hydroxypyridin-4-one groups within a linear co-polymer: Wrapping effect leading to superior antimicrobial activity. BioMetals 2020, 33, 339–351. [Google Scholar] [CrossRef]
- Lin, M.; Rippe, R.A.; Niemela, O.; Brittenham, G.; Tsukamoto, H. Role of iron in NF-kappa B activation and cytokine gene expression by rat hepatic macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 1997, 272, G1355–G1364. [Google Scholar] [CrossRef]
- Aali, M.; Caldwell, A.; Li, A.; Holbein, B.; Chappe, V.; Lehmann, C. DIBI, a novel polymeric iron chelator modulates IL-6 and IL-8 secretion from Cystic Fibrosis airway epithelial cells in response to endotoxin induction. J. Cell. Biotechnol. 2020, 6, 161–170. [Google Scholar] [CrossRef]
- Li, L.; Frei, B. Iron Chelation Inhibits NF-ΚB–Mediated Adhesion Molecule Expression by Inhibiting P22phox Protein Expression and NADPH Oxidase Activity. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2638–2643. [Google Scholar] [CrossRef] [Green Version]
- Messa, E.; Carturan, S.; Maffè, C.; Pautasso, M.; Bracco, E.; Roetto, A.; Messa, F.; Arruga, F.; Defilippi, I.; Rosso, V. Deferasirox Is a Powerful NF-ΚB Inhibitor in Myelodysplastic Cells and in Leukemia Cell Lines Acting Independently from Cell Iron Deprivation by Chelation and Reactive Oxygen Species Scavenging. Haematologica 2010, 95, 1308. [Google Scholar] [CrossRef] [Green Version]
- Kono, M.; Matsuhiroya, S.; Obuchi, A.; Takahashi, T.; Imoto, S.; Kawano, S.; Saigo, K. Deferasirox, an iron-chelating agent, alleviates acute lung inflammation by inhibiting neutrophil activation and extracellular trap formation. J. Int. Med Res. 2020, 48, 300060520951015. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ichinose, T.; Yoshida, S.; Nishikawa, M.; Sun, G.; Shibamoto, T. Role of iron and oxidative stress in the exacerbation of allergic inflammation in murine lungs caused by urban particulate matter <2.5 µm and desert dust. J. Appl. Toxicol. 2019, 39, 855–867. [Google Scholar] [CrossRef] [PubMed]
- O’Brien-Ladner, A.R.; Blumer, B.M.; Wesselius, L.J. Differential regulation of human alveolar macrophage-derived interleukin-1β and tumor necrosis factor-α by iron. J. Lab. Clin. Med. 1998, 132, 497–506. [Google Scholar] [CrossRef]
- Islam, S.; Jarosch, S.; Zhou, J.; Parquet, M.D.C.; Toguri, J.T.; Colp, P.; Holbein, B.E.; Lehmann, C. Anti-inflammatory and anti-bacterial effects of iron chelation in experimental sepsis. J. Surg. Res. 2016, 200, 266–273. [Google Scholar] [CrossRef]
- Lehmann, C.; Aali, M.; Zhou, J.; Holbein, B. Comparison of Treatment Effects of Different Iron Chelators in Experimental Models of Sepsis. Life 2021, 11, 57. [Google Scholar] [CrossRef]
- Vachharajani, T.J.; Work, J.; Issekutz, A.C.; Granger, D.N. Heme oxygenase modulates selectin expression in different regional vascular beds. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1613–H1617. [Google Scholar] [CrossRef]
- Sarelius, I.H.; Kuebel, J.M.; Wang, J.; Huxley, V.H. Macromolecule permeability of in situ and excised rodent skeletal muscle arterioles and venules. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H474–H480. [Google Scholar] [CrossRef] [Green Version]
- Perlman, R.L. Mouse Models of Human Disease: An Evolutionary Perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Card, J.W.; Carey, M.A.; Bradbury, J.A.; DeGraff, L.M.; Morgan, D.L.; Moorman, M.P.; Flake, G.P.; Zeldin, D.C. Gender Differences in Murine Airway Responsiveness and Lipopolysaccharide-Induced Inflammation. J. Immunol. 2006, 177, 621–630. [Google Scholar] [CrossRef]
- Gahima, I.; Twizeyimana, E.; LuckGonzales, E.; Remonde, C.G.; Jeon, S.J.; Shin, C.Y. Strain, Age, and Gender Differences in Response to Lipopolysaccharide (LPS) Animal Model of Sepsis in Mice. Yakhak Hoeji 2021, 65, 17–22. [Google Scholar] [CrossRef]
- Vlahakos, V.D.; Marathias, K.P.; Arkadopoulos, N.; Vlahakos, D.V. Hyperferritinemia in Patients with COVID-19: An Opportunity for Iron Chelation? Artif. Organs 2021, 45, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Perricone, C.; Bartoloni, E.; Bursi, R.; Cafaro, G.; Guidelli, G.M.; Shoenfeld, Y.; Gerli, R. COVID-19 as part of the hyperferritinemic syndromes: The role of iron depletion therapy. Immunol. Res. 2020, 68, 213–224. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CON4h | LPS(5)4h | LPS(5)4h + DIBI(@0h) | LPS(5)4h + DIBI(@2h) | CON6h | LPS(5)6h | LPS(5)6h + DIBI(@4h) | |
|---|---|---|---|---|---|---|---|
| LIX | 3.3 (2.7) | 43.4 (19.2) * | 21.5 (7.1) ** | 14.9 (4.7) ** | 2.1 (0.8) | 25.7 (10.2) * | 23.1 (8.7) * |
| CXCL2 | 0.6 (0.8) | 26.1 (16.6) * | 4.5 (1.4) ** | 9.1 (1.6) ** | 0.6 (0.4) | 6.1 (0.6) * | 3.4 (0.9) *,** |
| CCL5 | 3.2 (1.9) | 35.6 (23.2) * | 9.4 (1.1) | 11.2 (2.4) | 2.6 (1.6) | 26.4 (11.9) * | 14.1 (7.8) |
| CXCL10 | 4.9 (7.5) | 38.3 (28.1) * | 14.9 (5.7) | 24.0 (4.6) | 0.9 (0.2) | 18.9 (13.2) * | 22.5 (5.6) * |
| IL-1β | 3.6 (0.4) | 12.9 (9.2) | 8.7 (1.6) | 8.2 (1.8) | 4.0 (1.6) | 10.3 (2.7) * | 8.1 (0.7) * |
| IL-6 | 1.4 (0.2) | 27.9 (13.7) * | 0.8 (0.4) ** | 1.6 (0.8) ** | 0.5 (0.2) | 8.1 (1.1) * | 2.4 (2.6) ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehmann, C.; Alizadeh-Tabrizi, N.; Hall, S.; Faridi, S.; Euodia, I.; Holbein, B.; Zhou, J.; Chappe, V. Anti-Inflammatory Effects of the Iron Chelator, DIBI, in Experimental Acute Lung Injury. Molecules 2022, 27, 4036. https://doi.org/10.3390/molecules27134036
Lehmann C, Alizadeh-Tabrizi N, Hall S, Faridi S, Euodia I, Holbein B, Zhou J, Chappe V. Anti-Inflammatory Effects of the Iron Chelator, DIBI, in Experimental Acute Lung Injury. Molecules. 2022; 27(13):4036. https://doi.org/10.3390/molecules27134036
Chicago/Turabian StyleLehmann, Christian, Nazli Alizadeh-Tabrizi, Stefan Hall, Sufyan Faridi, Irene Euodia, Bruce Holbein, Juan Zhou, and Valerie Chappe. 2022. "Anti-Inflammatory Effects of the Iron Chelator, DIBI, in Experimental Acute Lung Injury" Molecules 27, no. 13: 4036. https://doi.org/10.3390/molecules27134036