
Microwave-Assisted Synthesis: Can Transition Metal Complexes Take Advantage of This “Green” Method?



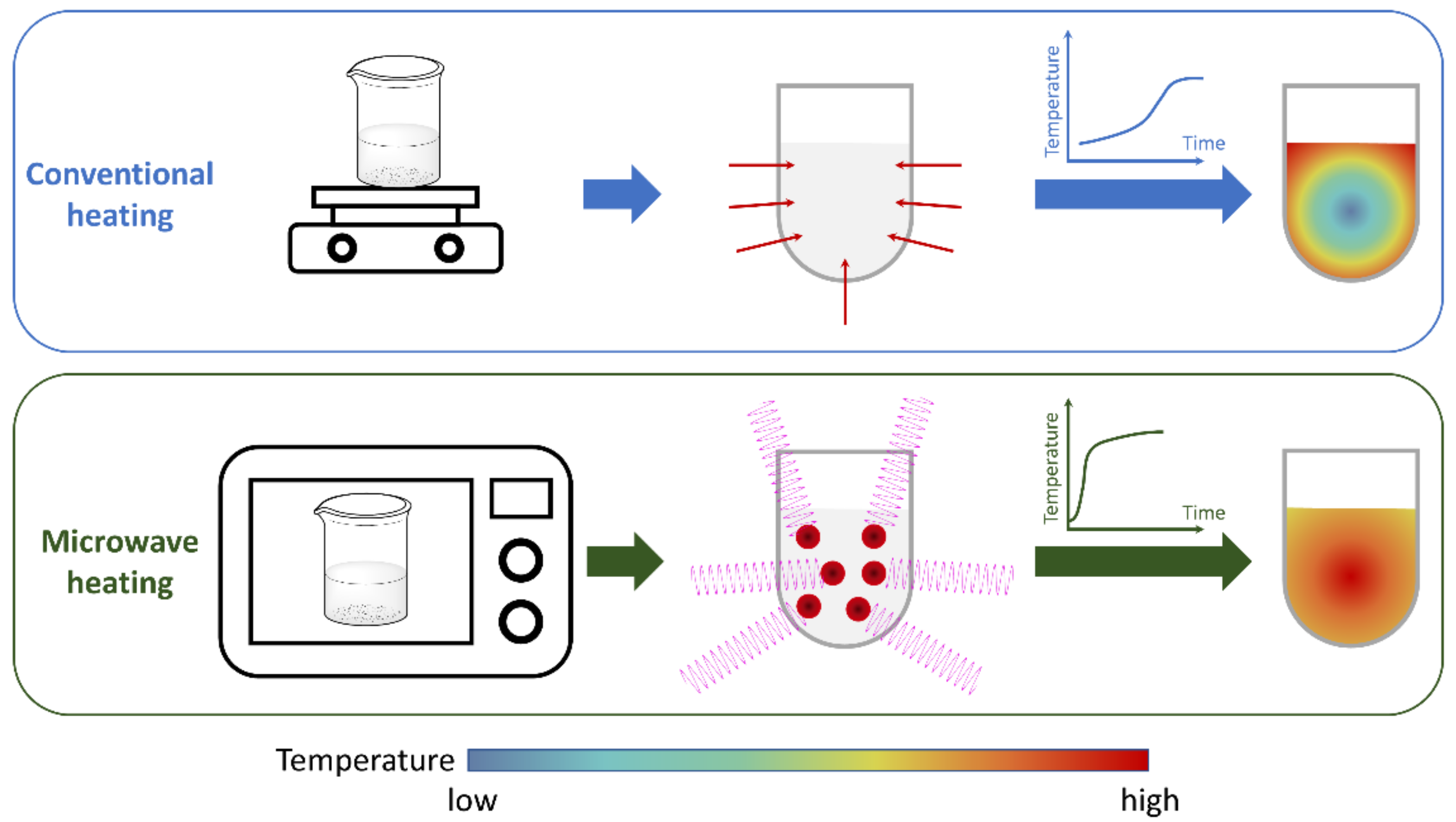{kind=link}
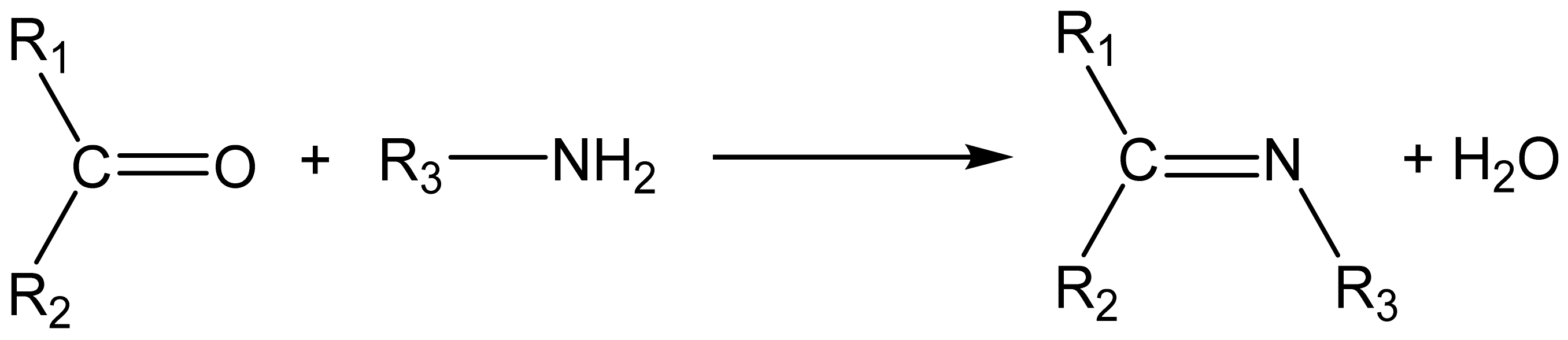{kind=link}
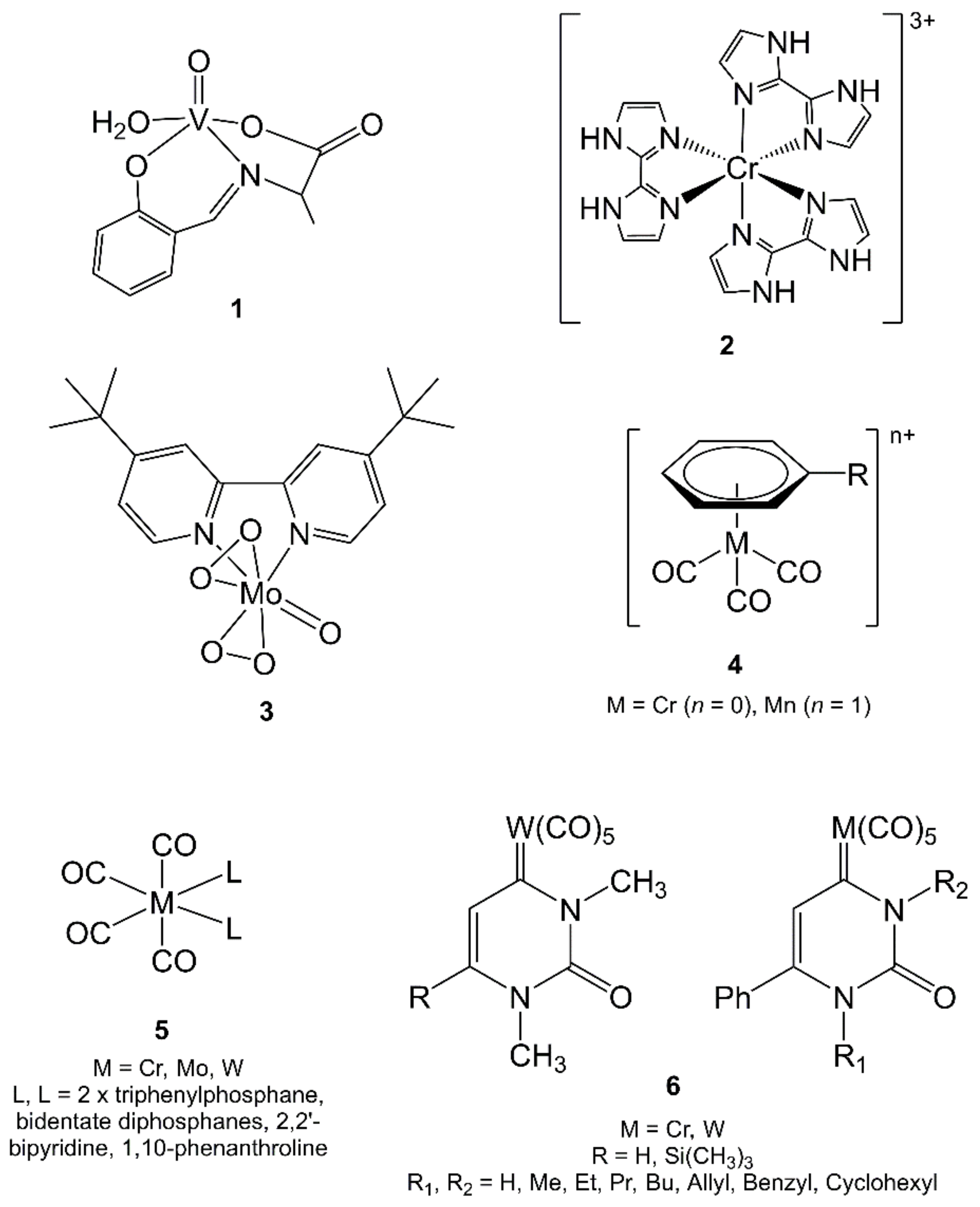{kind=link}
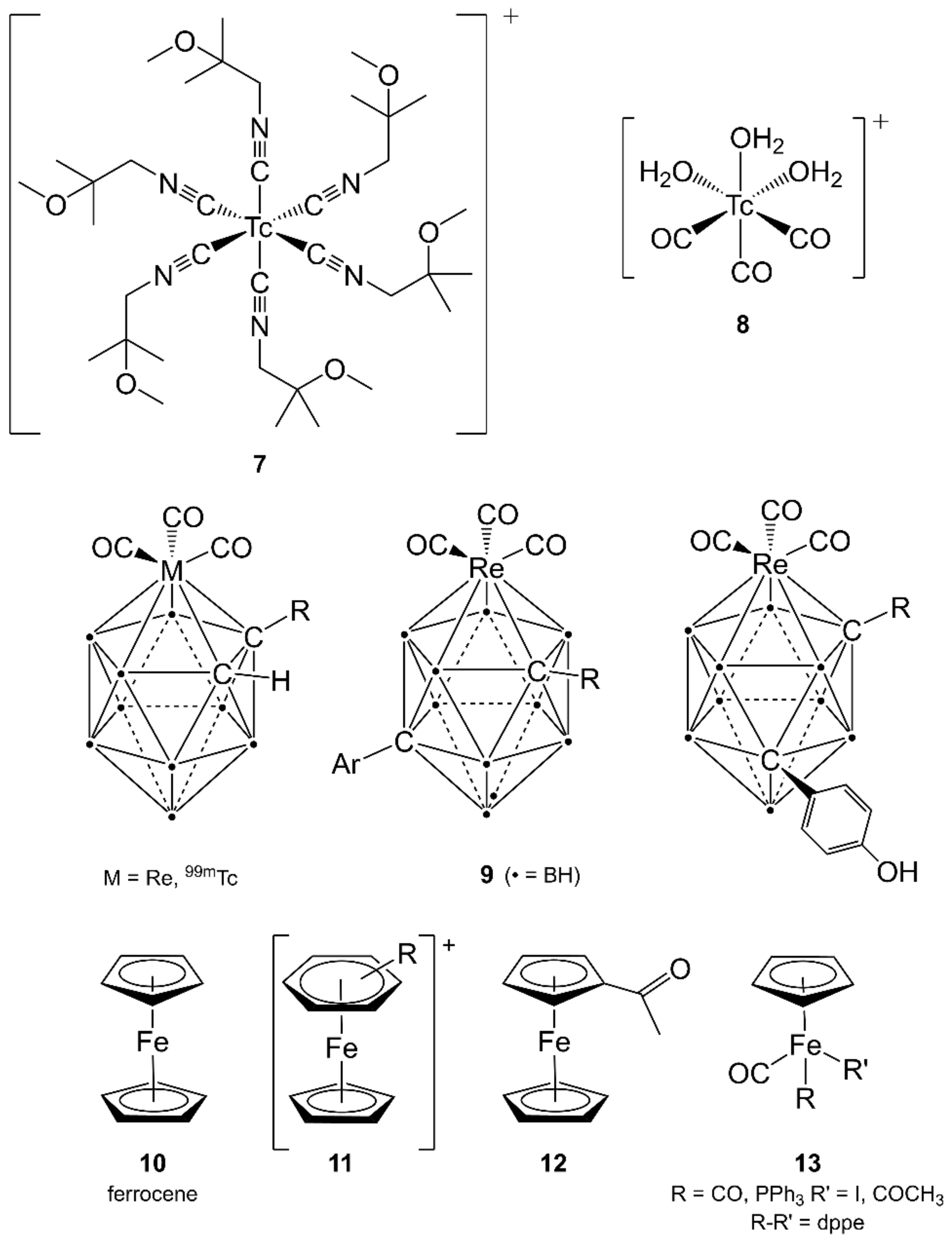{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Microwave and Chemistry: Background Information
3. Early Transition Elements of Groups 5–7
3.1. Vanadium
3.2. Chromium, Molybdenum and Tungsten
3.3. Manganese
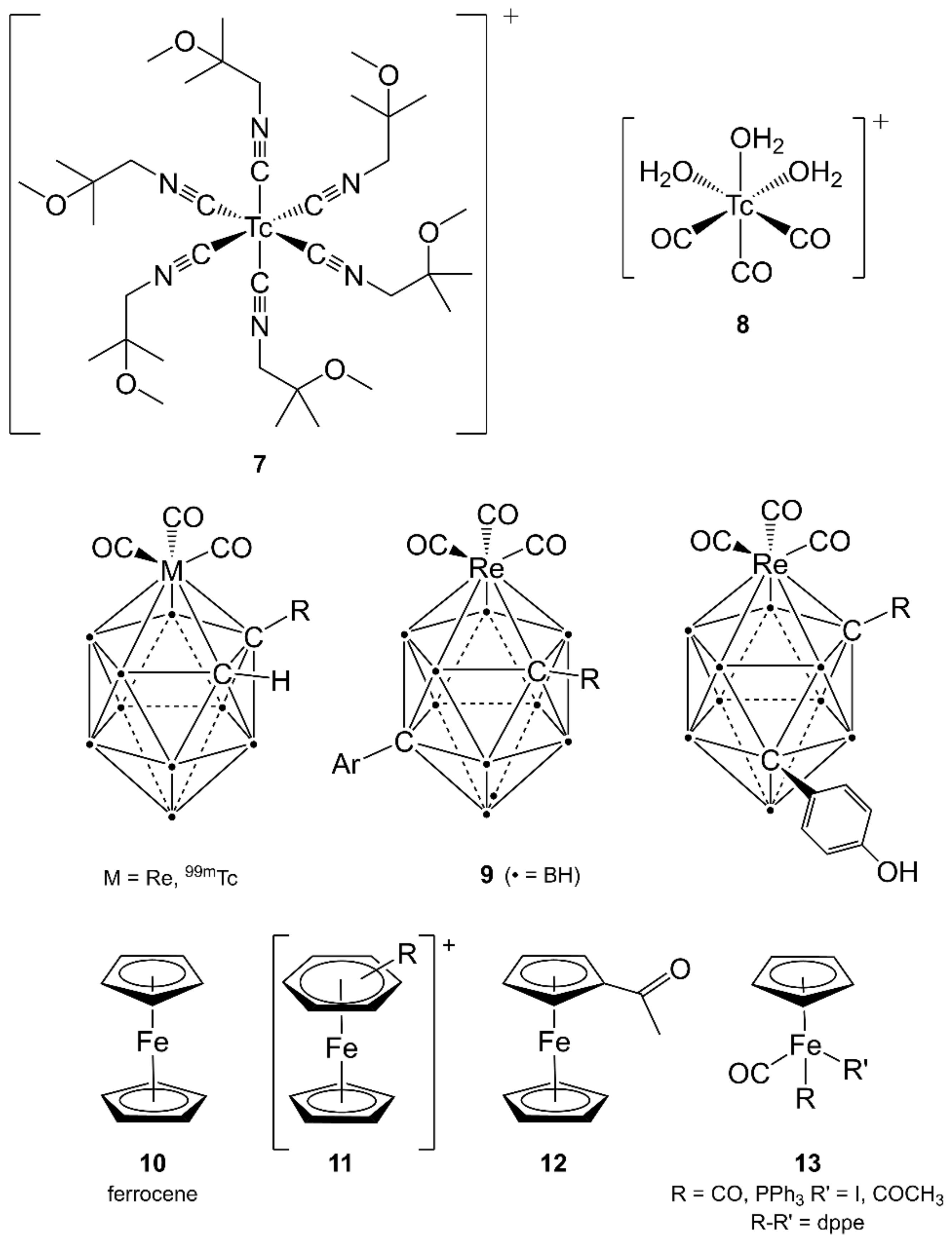
3.4. Technetium-99m and Rhenium
4. Late Transition Elements of Groups 8–12
4.1. Iron
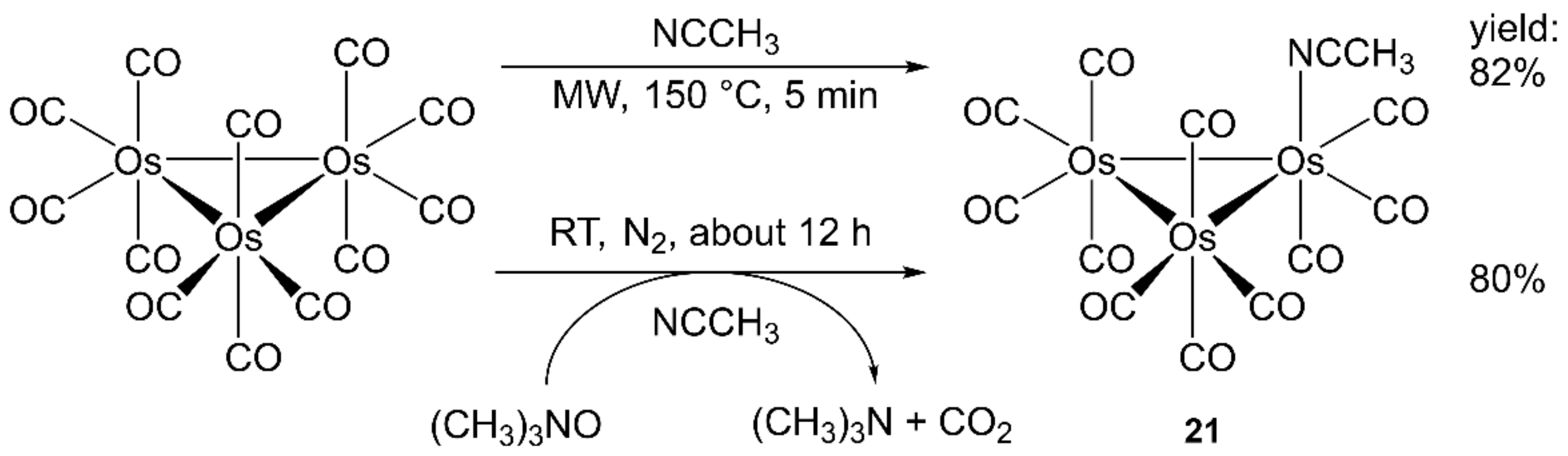
4.2. Ruthenium and Osmium
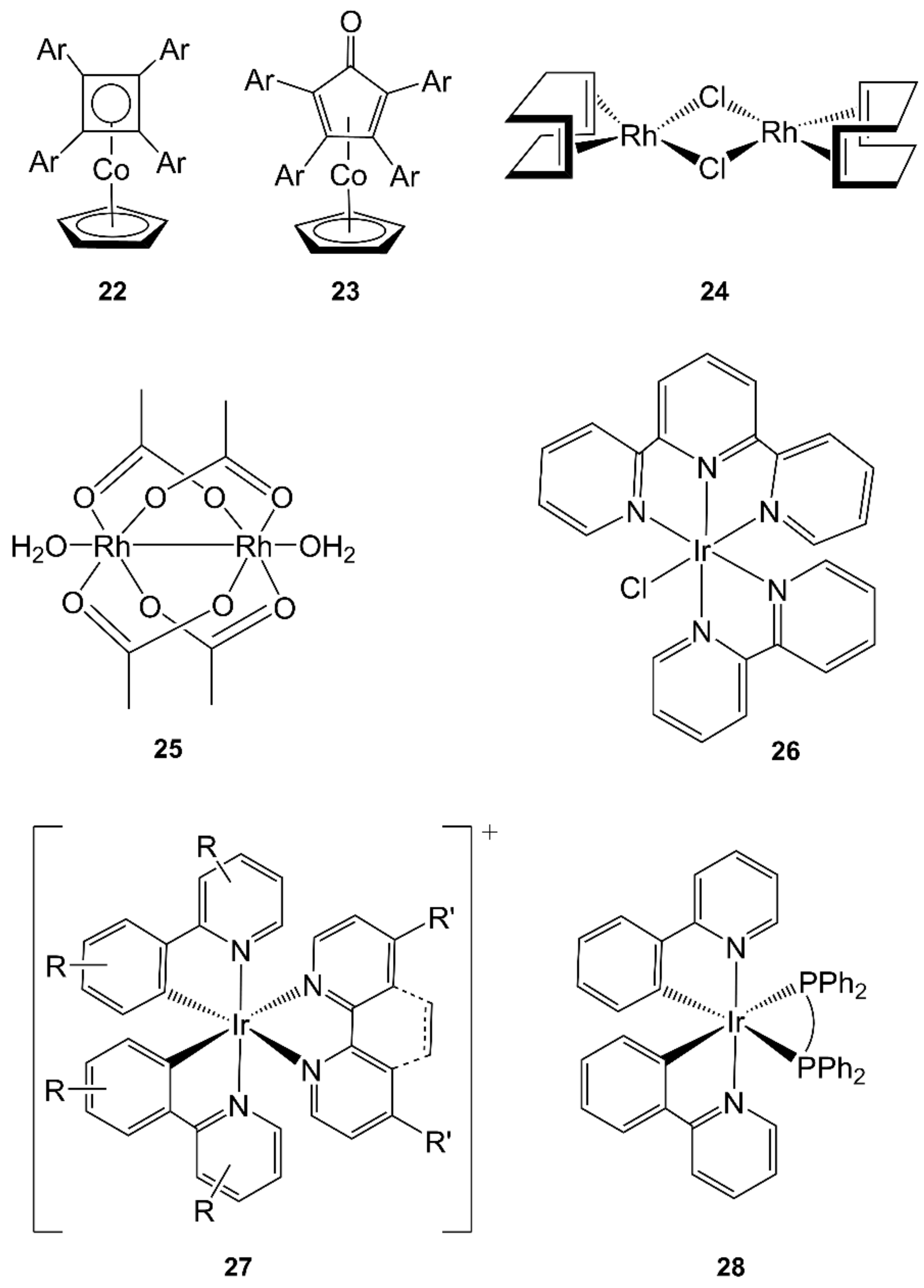
4.3. Cobalt
4.4. Rhodium
4.5. Iridium
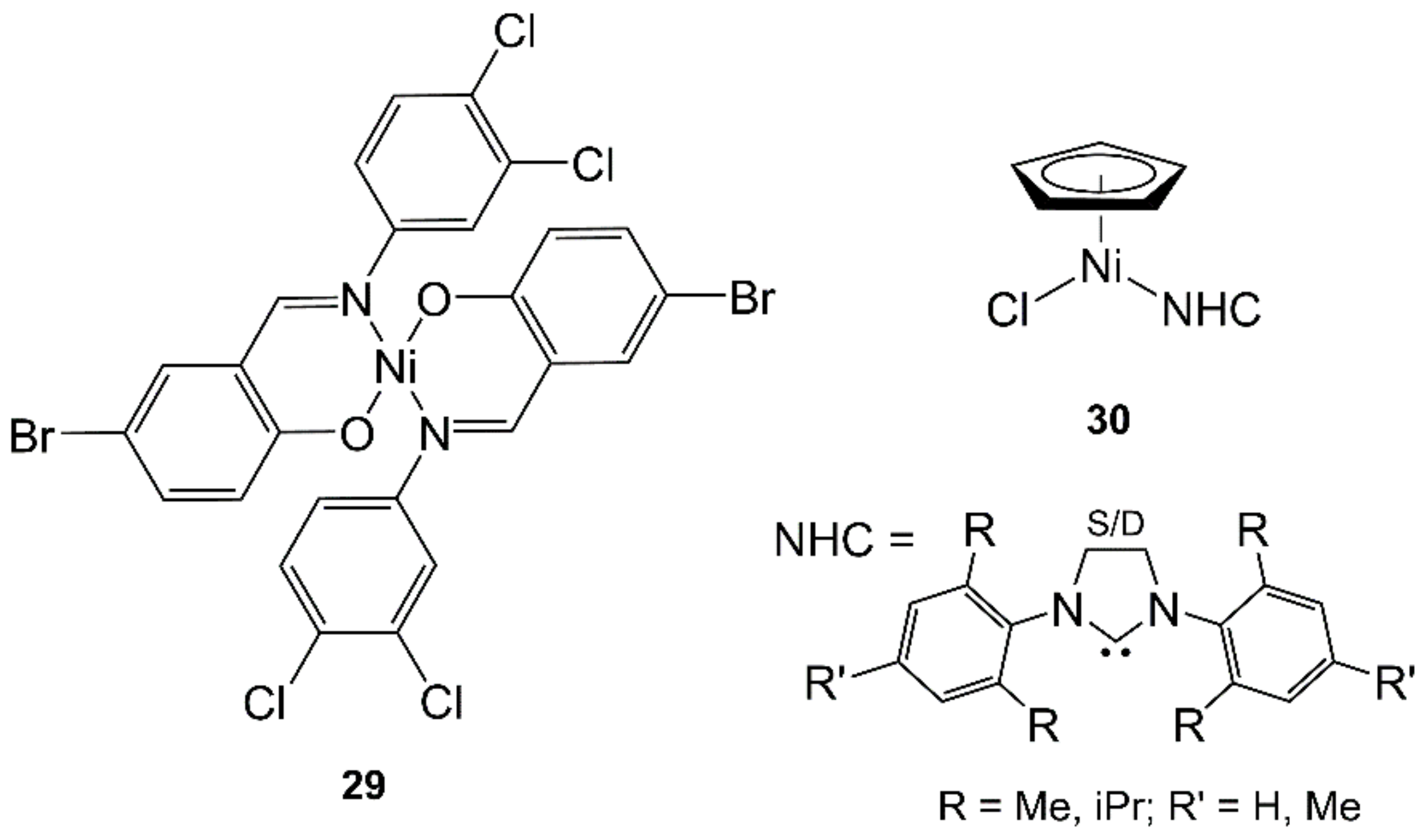
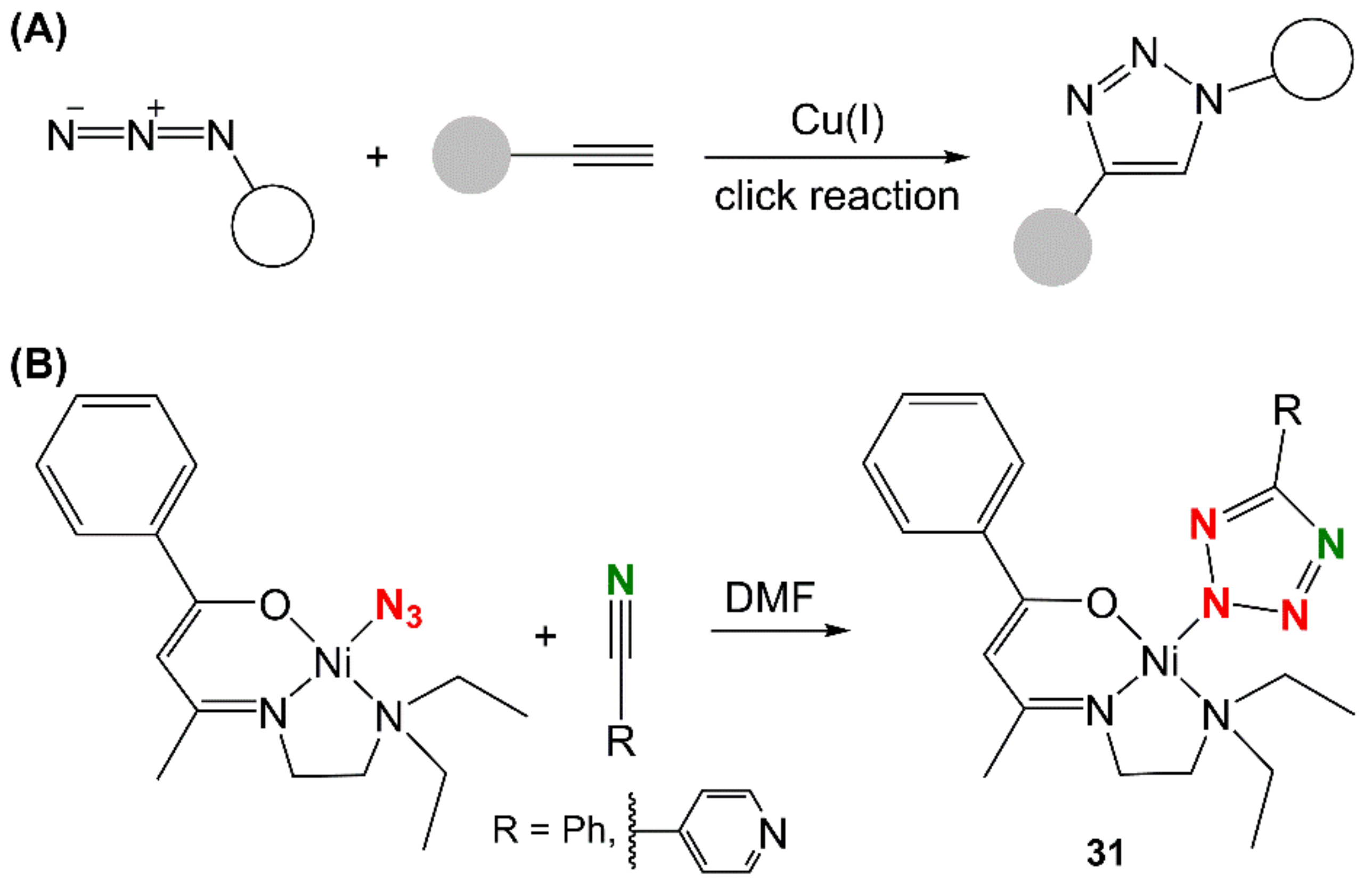
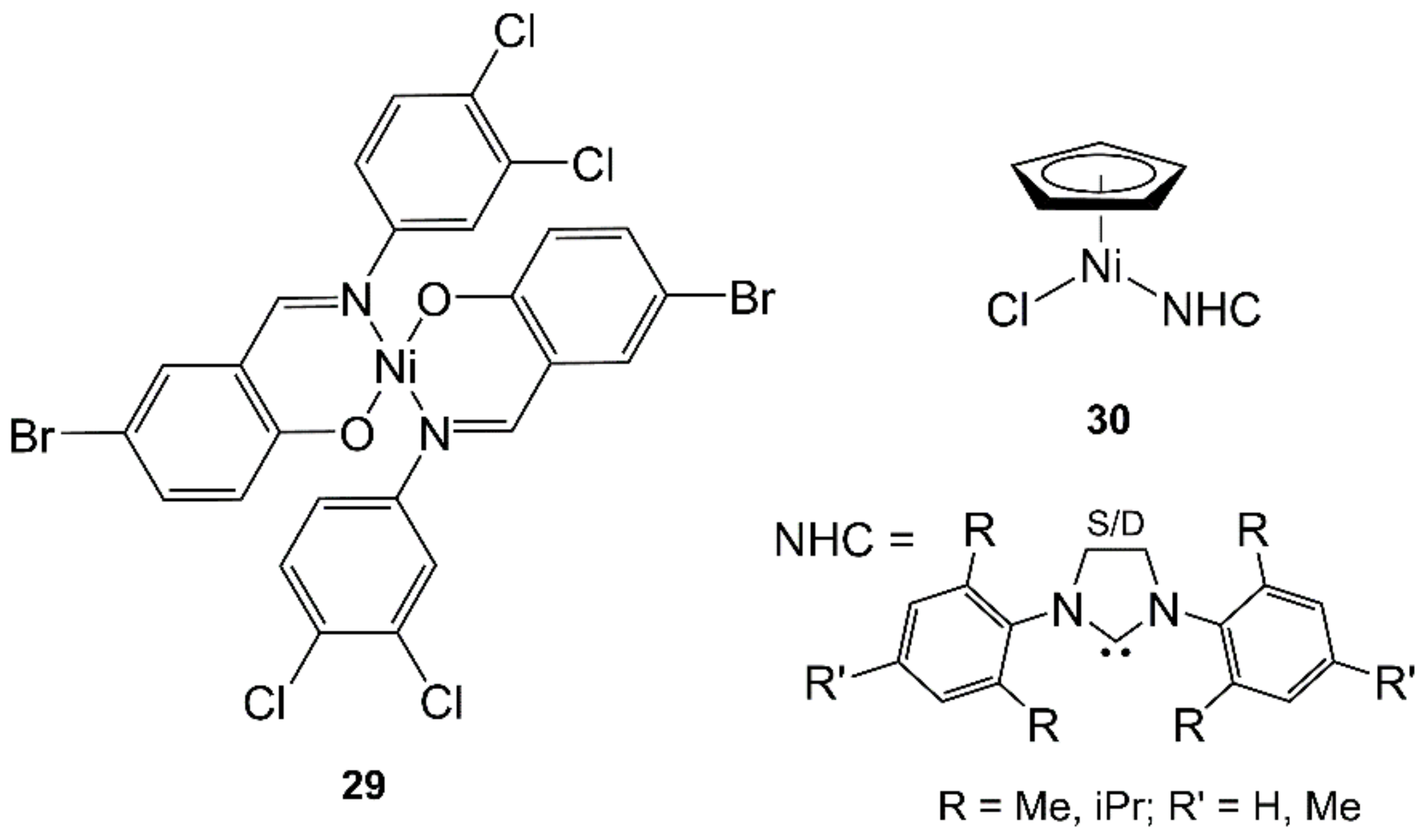
4.6. Nickel
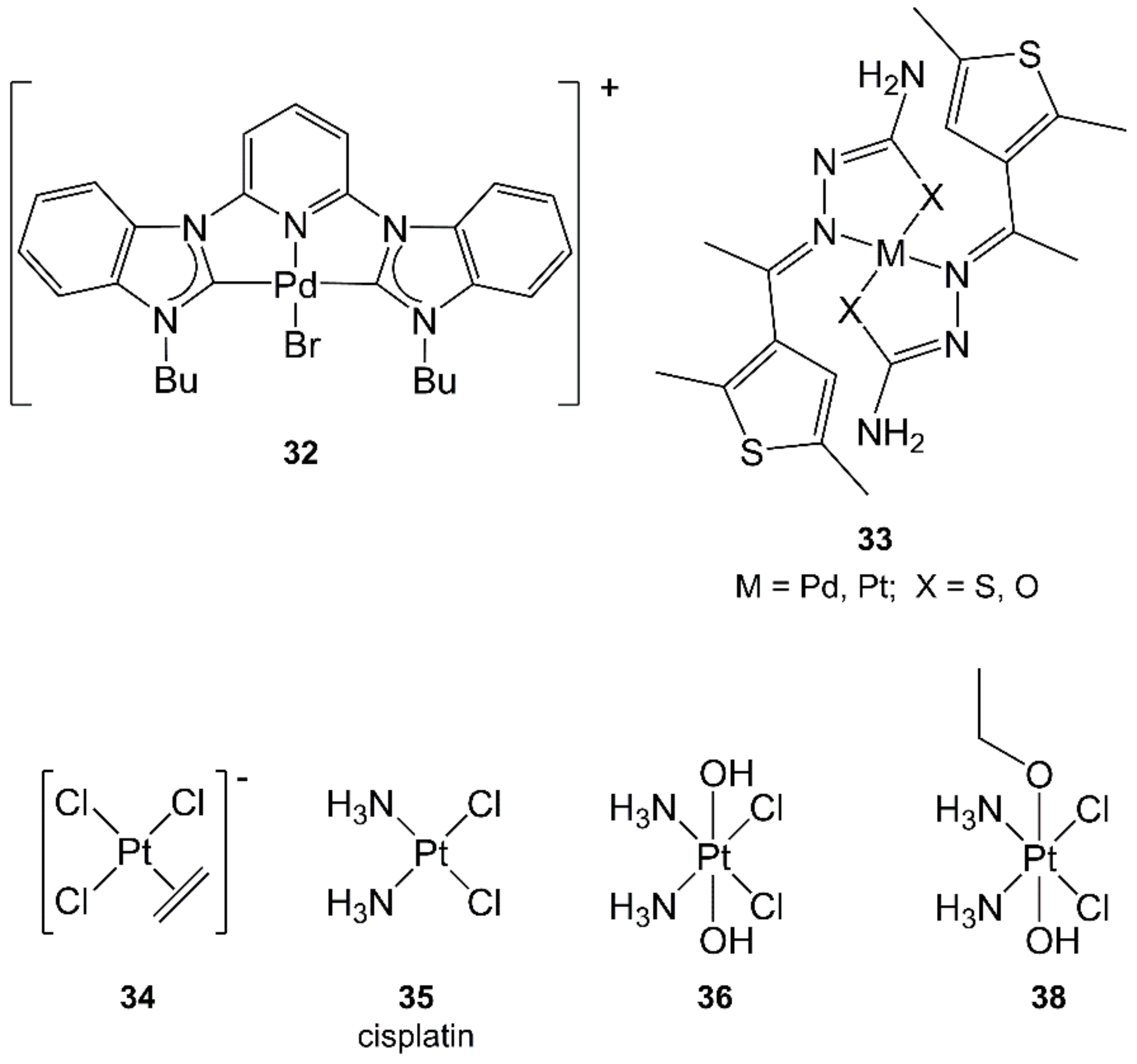
4.7. Palladium
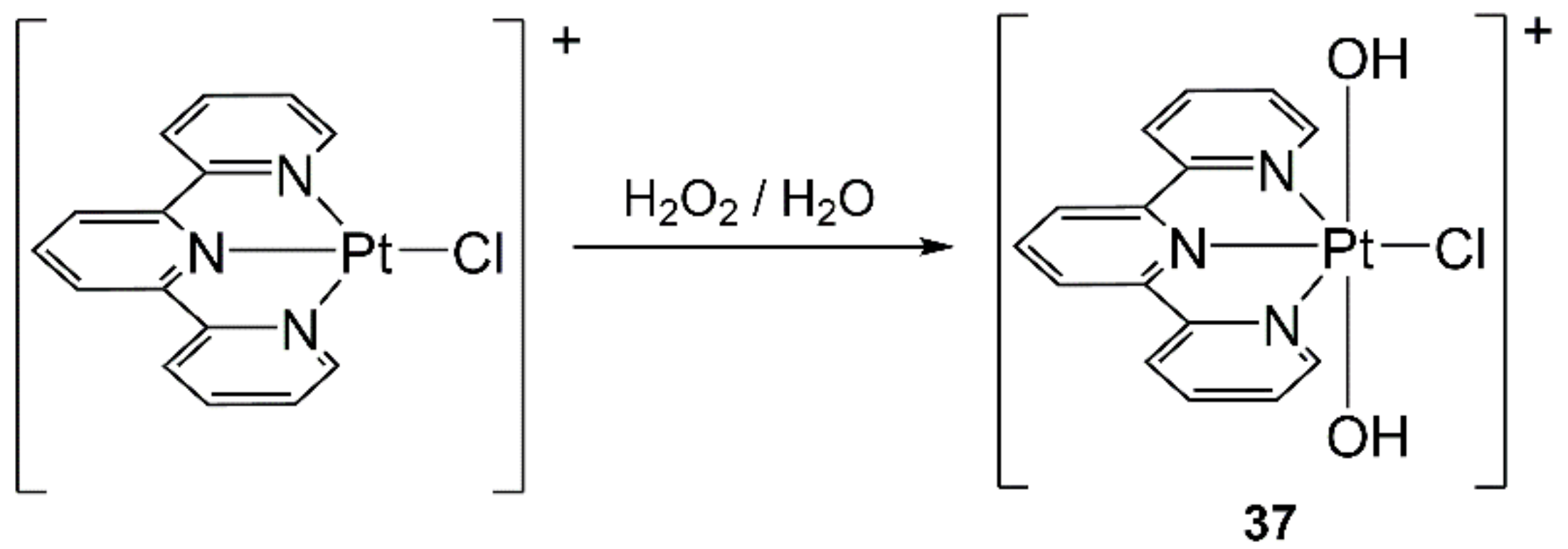
4.8. Platinum
4.9. Synthesis of a Pt(IV) Complex: An Unpublished (and Not Completely Satisfactory) Case Study
4.10. Coinage Metals
4.11. Zinc and Mercury
4.12. Lanthanides
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
Experimental Section
References
- Giguere, R.J.; Bray, T.L.; Duncan, S.M.; Majetich, G. Application of commercial microwave ovens to organic synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- De la Hoz, A.; Diaz-Ortiz, A.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Schmink, J.R.; Leadbeater, N.E. Microwave Heating as a Tool for Sustainable Chemistry: An Introduction. In Microwave Heating as a Tool for Sustainable Chemistry; CRC Press: Boca Raton, FL, USA, 2011; pp. 1–24. [Google Scholar]
- Dallinger, D.; Kappe, C.O. Microwave-assisted synthesis in water as solvent. Chem. Rev. 2007, 107, 2563–2591. [Google Scholar] [CrossRef] [PubMed]
- Mahato, A.K.; Sahoo, B.M.; Banik, B.K.; Mohanta, B.C. Microwave-assisted synthesis: Paradigm of green chemistry. J. Indian Chem. Soc. 2018, 95, 1327–1339. [Google Scholar]
- Díaz-Ortiz, Á.; Carrillo, J.R. Microwaves in green and sustainable chemistry. In Microwave Chemistry; Cravotto, G., Carnaroglio, D., Eds.; De Gruyter: Berlin, Germany, 2017; pp. 167–183. [Google Scholar]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Kappe, C.O. My Twenty Years in Microwave Chemistry: From Kitchen Ovens to Microwaves that aren’t Microwaves. Chem. Rec. 2019, 19, 15–39. [Google Scholar] [CrossRef]
- Leadbeater, N.E. Microwave-Assisted Synthesis: General Concepts. In Microwave-Assisted Polymer Synthesis; Hoogenboom, R., Schubert, U.S., Wiesbrock, F., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 274, pp. 1–44. [Google Scholar]
- Man, A.K.; Shahidan, R. Microwave-assisted chemical reactions. J. Macromol. Sci. Part A—Pure Appl. Chem. 2007, 44, 651–657. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef]
- Robinson, J.; Kingman, S.; Irvine, D.; Licence, P.; Smith, A.; Dimitrakis, G.; Obermayer, D.; Kappe, C.O. Understanding microwave heating effects in single mode type cavities—Theory and experiment. Phys. Chem. Chem. Phys. 2010, 12, 4750–4758. [Google Scholar] [CrossRef]
- Hayes, B.L. Microwave Synthesis: Chemistry at the Speed of Light; CEM Publishing: Matthews, NC, USA, 2002. [Google Scholar]
- Lei, Z.G.; Chen, B.H.; Koo, Y.M.; MacFarlane, D.R. Introduction: Ionic Liquids. Chem. Rev. 2017, 117, 6633–6635. [Google Scholar] [CrossRef] [Green Version]
- Kappe, C.O.; Pieber, B.; Dallinger, D. Microwave Effects in Organic Synthesis: Myth or Reality? Angew. Chem. Int. Ed. 2013, 52, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- De la Hoz, A.; Díaz-Ortiz, A.; Moreno, A. Review on non-thermal effects of microwave irradiation in organic synthesis. J. Microw. Power Electromagn. Energy 2007, 41, 44–64. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Li, Z.; Wu, L. Experimental demonstration of a microwave non-thermal effect in DMSO-NaCl aqueous solution. Chem. Phys. 2020, 528, 110523. [Google Scholar] [CrossRef]
- Brodie, G.; Gupta, D.; Khan, J.; Foletta, S.; Bootes, N. 5 A Brief Review of Microwave Heating. In Microwave Based Weed Control and Soil Treatment; De Gruyter Open Poland: Warsaw, Poland, 2018; pp. 43–54. [Google Scholar] [CrossRef]
- Kappe, C.O.; Dallinger, D.; Murphree, S.S. Practical Microwave Synthesis for Organic Chemists: Strategies, Instruments, and Protocols; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Ondruschka, B.; Bonrath, W.; Stuerga, D. Development and Design of Reactors in Microwave-Assisted Chemistry. In Microwaves in Organic Synthesis, 2nd ed.; Loupy, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; Volume 1, pp. 62–107. [Google Scholar]
- Caddick, S. Microwave-assisted organic-reactions. Tetrahedron 1995, 51, 10403–10432. [Google Scholar] [CrossRef]
- Lidstrom, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis—A review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Wilson, N.S.; Roth, G.P. Recent trends in microwave-assisted synthesis. Curr. Opin. Drug Discov. Dev. 2002, 5, 620–629. [Google Scholar] [CrossRef]
- Hayes, B.L. Recent advances in microwave-assisted synthesis. Aldrichimica Acta 2004, 37, 66–77. [Google Scholar]
- Suna, E.; Mutule, I. Microwave-assisted heterocyclic chemistry. In Microwave Methods in Organic Synthesis; Larhed, M., Olofsson, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 266, pp. 49–101. [Google Scholar]
- Appukkuttan, P.; Van der Eycken, E. Microwave-assisted natural product chemistry. In Microwave Methods in Organic Synthesis; Larhed, M., Olofsson, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 266, pp. 1–47. [Google Scholar]
- Kappe, C.O.; Dallinger, D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Discov. 2006, 5, 51–63. [Google Scholar] [CrossRef]
- Alcazar, J.; Diels, G.; Schoentjes, B. Microwave assisted medicinal chemistry. Mini-Rev. Med. Chem. 2007, 7, 345–369. [Google Scholar] [CrossRef]
- Brooks, W.L.A.; Sumerlin, B.S. Microwave-Assisted RAFT Polymerization. Isr. J. Chem. 2012, 52, 256–263. [Google Scholar] [CrossRef]
- Motasemi, F.; Ani, F.N. A review on microwave-assisted production of biodiesel. Renew. Sustain. Energy Rev. 2012, 16, 4719–4733. [Google Scholar] [CrossRef]
- Kaur, N. Six-membered n-heterocycles: Microwave-assisted synthesis. Synth. Commun. 2015, 45, 1–34. [Google Scholar] [CrossRef]
- Fang, L.J.; Han, G.; Zhang, H.Q. Microwave-Assisted Free Radical Polymerizations. In Microwave-Assisted Polymer Synthesis; Hoogenboom, R., Schubert, U.S., Wiesbrock, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 274, pp. 87–129. [Google Scholar]
- Radhika, S.; Neetha, M.; Aneeja, T.; Anilkumar, G. Microwave-assisted Amination Reactions: An Overview. Curr. Org. Chem. 2020, 24, 2235–2255. [Google Scholar] [CrossRef]
- Geetanjali; Singh, R. Microwave-assisted Organic Synthesis in Water. Curr. Microw. Chem. 2021, 8, 117–127. [Google Scholar] [CrossRef]
- Martina, K.; Cravotto, G.; Varma, R.S. Impact of Microwaves on Organic Synthesis and Strategies toward Flow Processes and Scaling Up. J. Org. Chem. 2021, 86, 13857–13872. [Google Scholar] [CrossRef]
- Dandia, A.; Bansal, S.; Indora, A.; Mahawar, D.K.; Parewa, V. Microwave-assisted stereoselective organic synthesis. In Green Sustainable Process for Chemical and Environmental Engineering and Science; Inamuddin, Boddula, R., Asiri, A.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 331–357. [Google Scholar] [CrossRef]
- Tber, Z.; Biteau, N.G.; Agrofoglio, L.; Cros, J.; Goffinont, S.; Castaing, B.; Nicolas, C.; Roy, V. Microwave-Assisted Suzuki-Miyaura and Sonogashira Coupling of 4-Chloro-2-(trifluoromethyl)pyrido 1,2-e purine Derivatives. Eur. J. Org. Chem. 2019, 2019, 5756–5767. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.C.; Wang, J.B.; Cheng, X.Y.; Liu, K.; Wu, T.Z.; Qiu, X.Q.; Kuang, Z.J.; Li, Z.Y.; Bian, J.L. Microwave-assisted unprotected Sonogashira reaction in water for the synthesis of polysubstituted aromatic acetylene compounds. Green Chem. 2020, 22, 1338–1344. [Google Scholar] [CrossRef]
- Erdelyi, M.; Gogoll, A. Rapid homogeneous-phase Sonogashira coupling reactions using controlled microwave heating. J. Org. Chem. 2001, 66, 4165–4169. [Google Scholar] [CrossRef]
- Nilsson, P.; Ofsson, K.; Larhed, M. Microwave-assisted and metal-catalyzed coupling reactions. In Microwave Methods in Organic Synthesis; Larhed, M., Olofsson, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 266, pp. 103–144. [Google Scholar]
- Panda, B. Microwave-Assisted Homogeneous Gold Catalyzed Organic Transformations. Curr. Microw. Chem. 2020, 7, 166–182. [Google Scholar] [CrossRef]
- Declerck, V.; Martinez, J.; Lamaty, F. Microwave-assisted copper-catalyzed Heck reaction in PEG solvent. Synlett 2006, 3029–3032. [Google Scholar] [CrossRef]
- Wali, A.; Pillai, S.M.; Satish, S. Heterogeneous Pd catalysts and microwave irradiation in Heck arylation. React. Kinet. Catal. Lett. 1997, 60, 189–194. [Google Scholar] [CrossRef]
- Glasnov, T.N.; Findenig, S.; Kappe, C.O. Heterogeneous Versus Homogeneous Palladium Catalysts for Ligandless Mizoroki-Heck Reactions: A Comparison of Batch/Microwave and Continuous-Flow Processing. Chem.-A Eur. J. 2009, 15, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Kaval, N.; Tomar, S.; Van der Eycken, E.; Parmar, V.S. Transition metal-catalyzed carbon-carbon bond formation Suzuki, Heck, and Sonogashira reactions using microwave and microtechnology. Org. Process Res. Dev. 2008, 12, 468–474. [Google Scholar] [CrossRef]
- Barge, A.; Tagliapietra, S.; Tei, L.; Cintas, P.; Cravotto, G. Pd-catalyzed Reactions Promoted by Ultrasound and/or Microwave Irradiation. Curr. Org. Chem. 2008, 12, 1588–1612. [Google Scholar] [CrossRef]
- Li, Q.H.; Ding, Y.; Zhang, G.; Zhang, Z.; Mo, S. Suzuki-Miyaura Cross-Coupling Reaction Catalyzed by Supported Palladium Under Microwave Irradiation. Curr. Org. Synth. 2017, 14, 462–476. [Google Scholar] [CrossRef]
- Petricci, E.; Cini, E.; Taddei, M. Metal Catalysis with Microwaves in Organic Synthesis: A Personal Account. Eur. J. Org. Chem. 2020, 2020, 4435–4446. [Google Scholar] [CrossRef]
- Barge, A.; Tagliapietra, S.; Binello, A.; Cravotto, G. Click Chemistry Under Microwave or Ultrasound Irradiation. Curr. Org. Chem. 2011, 15, 189–203. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.Q.; Cai, L.; Tang, Z.K. Microwave-Assisted Click Chemistry. Chin. J. Org. Chem. 2012, 32, 1410–1428. [Google Scholar] [CrossRef] [Green Version]
- Rathi, A.K.; Gawande, M.B.; Zboril, R.; Varma, R.S. Microwave-assisted synthesis—Catalytic applications in aqueous media. Coord. Chem. Rev. 2015, 291, 68–94. [Google Scholar] [CrossRef]
- Baqi, Y. Recent Advances in Microwave-Assisted Copper-Catalyzed Cross-Coupling Reactions. Catalysts 2021, 11, 46. [Google Scholar] [CrossRef]
- Lill, J.R.; Ingle, E.S.; Liu, P.S.; Pham, V.; Sandoval, W.N. Microwave-assisted proteomics. Mass Spectrom. Rev. 2007, 26, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.J.J. Future trends in microwave synthesis. Future Med. Chem. 2010, 2, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, H.J.; Vallance, S.K.; Kennedy, J.L.; Tapia-Ruiz, N.; Carassiti, L.; Harrison, A.; Whittaker, A.G.; Drysdale, T.D.; Kingman, S.W.; Gregory, D.H. Modern Microwave Methods in Solid-State Inorganic Materials Chemistry: From Fundamentals to Manufacturing. Chem. Rev. 2014, 114, 1170–1206. [Google Scholar] [CrossRef]
- Thomas-Hillman, I.; Laybourn, A.; Dodds, C.; Kingman, S.W. Realising the environmental benefits of metal-organic frameworks: Recent advances in microwave synthesis. J. Mater. Chem. A 2018, 6, 11564–11581. [Google Scholar] [CrossRef] [Green Version]
- Dabrowska, S.; Chudoba, T.; Wojnarowicz, J.; Lojkowski, W. Current Trends in the Development of Microwave Reactors for the Synthesis of Nanomaterials in Laboratories and Industries: A Review. Crystals 2018, 8, 379. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Das, J. Small molecules derived carbon dots: Synthesis and applications in sensing, catalysis, imaging, and biomedicine. J. Nanobiotechnol. 2019, 17, 24. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Kumar, R.; Singh, D.P.; Savu, R.; Moshkalev, S.A. Progress in microwave-assisted synthesis of quantum dots (graphene/carbon/semiconducting) for bioapplications: A review. Mater. Today Chem. 2019, 12, 282–314. [Google Scholar] [CrossRef]
- Kumar, A.; Kuang, Y.; Liang, Z.; Sun, X.M. Microwave chemistry, recent advancements, and eco-friendly microwave-assisted synthesis of nanoarchitectures and their applications: A review. Mater. Today Nano 2020, 11, 20. [Google Scholar] [CrossRef]
- Baghurst, D.R.; Michael, D.; Mingos, P.; Watson, M.J. Application of microwave dielectric loss heating effects for the rapid and convenient synthesis of organometallic compounds. J. Organomet. Chem. 1989, 368, C43–C45. [Google Scholar] [CrossRef]
- Baghurst, D.R.; Cooper, S.R.; Greene, D.L.; Mingos, D.M.P.; Reynolds, S.M. Application of microwave dielectric loss heating effects for the rapid and convenient synthesis of coordination compounds. Polyhedron 1990, 9, 893–895. [Google Scholar] [CrossRef]
- Baghurst, D.R.; Mingos, D.M.P. Design and application of a reflux modification for the synthesis of organometallic compounds using microwave dielectric loss heating effects. J. Organomet. Chem. 1990, 384, C57–C60. [Google Scholar] [CrossRef]
- Mingos, D.M.P.; Baghurst, D.R. Applications of Microwave Dielectric Heating Effects to Synthetic Problems in Chemistry. Chem. Soc. Rev. 1991, 20, 1–47. [Google Scholar] [CrossRef]
- Baghurst, D.R.; Mingos, D.M.P. A new reaction vessel for accelerated syntheses using microwave dielectric super-heating effects. J. Chem. Soc.-Dalton Trans. 1992, 1151–1155. [Google Scholar] [CrossRef]
- Mingos, D.M.P. The applications of microwaves in chemical syntheses. Res. Chem. Intermed. 1994, 20, 85–91. [Google Scholar] [CrossRef]
- Abe, T.; Miyazawa, A.; Kawanishi, Y.; Konno, H. Microwave-Assisted Synthesis of Metal Complexes. Mini-Rev. Org. Chem. 2011, 8, 315–333. [Google Scholar] [CrossRef]
- Wazalwar, S.S.; Bhave, N.S. Microwave Assisted Synthesis and Antioxidant Activity of Vanadium(IV) Complexes of Amino Acid Schiff Bases. Synth. React. Inorg. Met.Org. Nano-Met. Chem. 2012, 42, 1098–1104. [Google Scholar] [CrossRef]
- Ajbani, J.C.; Revankar, D.S.; Revanasiddappa, M.; Swamy, V.; Shankar, S. Microwave synthesis, spectroscopic, thermal and biological studies of some transition metal complexes containing heterocyclic ligand. Int. J. Chem. Sci. 2015, 13, 1673–1692. [Google Scholar]
- Mishra, A.P.; Jain, R.K. Microwave synthesis, spectroscopic, thermal and biological significance of some transition metal complexes containing heterocyclic ligands. J. Chem. Pharm. Res. 2010, 2, 51–61. [Google Scholar]
- Jain, R.K.; Mishra, A.P.; Mishra, D.K.; Gupta, S.K. Microwave Synthesis, Spectral, Thermal and Electrical Properties of Some Metal Complexes Involving 5-Bromosalicylaldehyde. E-J. Chem. 2012, 9, 1721–1727. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Fahmi, N.; Singh, R.V. Studies on chromium(III) complexes with active nitrogen, oxygen and sulfur donor ketimines synthesized under microwave conditions. J. Sulfur Chem. 2010, 31, 515–524. [Google Scholar] [CrossRef]
- Silvero, M.J.; Pelaez, W.J.; Garcia, P.F.; Arguello, G.A. Fast synthesis of a tris(N,N-diimine) chromium(III) complex by a microwave-assisted approach. RSC Adv. 2014, 4, 15507–15510. [Google Scholar] [CrossRef] [Green Version]
- Amarante, T.R.; Paz, F.A.A.; Gago, S.; Goncalves, I.S.; Pillinger, M.; Rodrigues, A.E.; Abrantes, M. Microwave-Assisted Synthesis and Crystal Structure of Oxo(diperoxo)(4,4′-di-tert-butyl-2,2′-bipyridine)-molybdenum(VI). Molecules 2009, 14, 3610–3620. [Google Scholar] [CrossRef] [PubMed]
- Adil, K.; Marrot, J.; Leblanc, M.; Maisonneuve, V. Bis tris(2-ammonioethyl)amine bis(pentafluoridooxidomolybdate) difluoride monohydrate. Acta Crystallogr. Sect. E-Struct. Rep. Online 2007, 63, M1511–M1513. [Google Scholar] [CrossRef]
- Ardon, M.; Hayes, P.D.; Hogarth, G. Microwave-assisted reflux in organometallic chemistry: Synthesis and structural determination of molybdenum carbonyl complexes—An intermediate-level organometallic-inorganic experiment. J. Chem. Educ. 2002, 79, 1249–1251. [Google Scholar] [CrossRef]
- Lee, Y.T.; Choi, S.Y.; Lee, S.I.; Chung, Y.K.; Kang, T.J. Microwave-assisted synthesis of (eta(6)-arene)tricarbonylchromium complexes. Tetrahedron Lett. 2006, 47, 6569–6572. [Google Scholar] [CrossRef]
- Ardon, M.; Hogarth, G.; Oscroft, D.T.W. Organometallic chemistry in a conventional microwave oven: The facile synthesis of group 6 carbonyl complexes. J. Organomet. Chem. 2004, 689, 2429–2435. [Google Scholar] [CrossRef]
- Birdwhistell, K.R.; Schulz, B.E.; Dizon, P.M. Rapid synthesis of Group VI carbonyl complexes by coupling borohydride catalysis and microwave heating. Inorg. Chem. Commun. 2012, 26, 69–71. [Google Scholar] [CrossRef]
- Coelho, A.C.; Paz, F.A.A.; Klinowski, J.; Pillinger, M.; Goncalves, I.S. Microwave assisted synthesis of molybdenum and tungsten tetracarbonyl complexes with a pyrazolylpyridine ligand. Crystal structure of cis- Mo(CO)4{ethyl 3-(2-pyridyl)-1-pyrazolyl acetate}. Molecules 2006, 11, 940–952. [Google Scholar] [CrossRef] [Green Version]
- Artillo, A.; Della Sala, G.; De Santis, M.; Llordes, A.; Ricart, S.; Spinella, A. Preparation of organometallic uracil-analogue Fischer carbene complexes: Comparative study of conventional heating vs microwave irradiation. J. Organomet. Chem. 2007, 692, 1277–1284. [Google Scholar] [CrossRef]
- Whittaker, A.G.; Mingos, D.M.P. Synthetic reactions using metal powders under microwave irradiation. J. Chem. Soc.-Dalton Trans. 2002, 3967–3970. [Google Scholar] [CrossRef]
- Barnard, T.M.; Leadbeater, N.E. Real-time monitoring of microwave-promoted organometallic ligand-substitutionreactions using in situ Raman spectroscopy. Chem. Commun. 2006, 3615–3616. [Google Scholar] [CrossRef] [PubMed]
- Cravotto, G.; Cintas, P. Microwave chemistry: History, development and legacy. In Microwave Chemistry; Cravotto, G., Carnaroglio, D., Eds.; De Gruyter: Berlin, Germany, 2017; pp. 1–17. [Google Scholar] [CrossRef]
- Bhojak, N.; Gudasaria, D.D.; Khiwani, N.; Jain, R. Microwave Assisted Synthesis Spectral and Antibacterial Investigations on Complexes of Mn(II) With Amide Containing Ligands. E-J. Chem. 2007, 4, 785626. [Google Scholar] [CrossRef] [Green Version]
- Ali, P.; Ramakanth, P.; Meshram, J. Exploring microwave synthesis for co-ordination: Synthesis, spectral characterization and comparative study of transition metal complexes with binuclear core derived from 4-amino-2,3-dimethyl-1-phenyl-3-pyrazolin-5-one. J. Coord. Chem. 2010, 63, 323–329. [Google Scholar] [CrossRef]
- Zhang, S.H.; Feng, C. Microwave-assisted synthesis, crystal structure and fluorescence of novel coordination complexes with Schiff base ligands. J. Mol. Struct. 2010, 977, 62–66. [Google Scholar] [CrossRef]
- Milios, C.J.; Vinslava, A.; Whittaker, A.G.; Parsons, S.; Wernsdorfer, W.; Christou, G.; Perlepes, S.P.; Brechin, E.K. Microwave-assisted synthesis of a hexanuclear Mn-III single-molecule magnet. Inorg. Chem. 2006, 45, 5272–5274. [Google Scholar] [CrossRef]
- Ledezma-Gairaud, M.; Pineda, L.W.; Aromi, G.; Sanudo, E.C. Microwave assisted synthesis: A Mn/Ni reaction system affording Mn5Ni4, Mn2Ni2 and Mn7 complexes. Polyhedron 2013, 64, 45–51. [Google Scholar] [CrossRef]
- Dabirmanesh, Q.; Roberts, R.M.G. The application of microwave dielectric heating to the synthesis of arene-metal complexes. Synthesis of [(eta-arene)(CO)3Mn]PF6 complexes and [(eta-arene)(eta-cyclopentadienyl)Fe]PF6 complexes with triphenylphosphine, tert-butylbenzenes and a sterically hindered phenol as arene ligands. J. Organomet. Chem. 1997, 542, 99–103. [Google Scholar] [CrossRef]
- Gagnon, A.; Taillefer, R.; Bavaria, G.; Léveillé, J. Fast Labeling of Technetium-99m-Sestamibi with Microwave Oven Heating. J. Nucl. Med. Technol. 1991, 19, 90–93. [Google Scholar]
- Hung, J.C.; Wilson, M.E.; Brown, M.L.; Gibbons, R.J. Rapid preparation and quality-control method for technetium-99m-2-methoxy isobutyl isonitrile (technetium-99m-sestamibi). J. Nucl. Med. 1991, 32, 2162–2168. [Google Scholar]
- Hung, J.C.; Gibbons, R.J. Breakage of technetium-99m-sestamibi vial with the use of a microwave-oven. J. Nucl. Med. 1992, 33, 176–178. [Google Scholar]
- Lima, M.d.J.d.C.; Yoshie Okamoto, M.R.; Tales Garcez, A.; Tatit Sapienza, M.; Alberto, B.C. Preparation and evaluation of modified composition for lyophilized kits of [Cu(MIBI)4]BF4 for [99mTc] technetium labeling. Braz. Arch. Biol. Technol. 2005, 48, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.C.; Chowdhury, S.; Redfern, M.G.; Mahoney, D.W. Rapid preparation method for technetium-99m bicisate. Eur. J. Nucl. Med. 1997, 24, 655–659. [Google Scholar] [PubMed]
- Causey, P.W.; Besanger, T.R.; Schaffer, P.; Valliant, J.F. Expedient multi-step synthesis of organometallic complexes of Tc and Re in high effective specific activity. A new platform for the production of molecular imaging and therapy agents. Inorg. Chem. 2008, 47, 8213–8221. [Google Scholar] [CrossRef]
- Simms, R.W.; Causey, P.W.; Weaver, D.M.; Sundararajan, C.; Stephenson, K.A.; Valliant, J.F. Preparation of technetium-99m bifunctional chelate complexes using a microfluidic reactor: A comparative study with conventional and microwave labeling methods. J. Label. Compd. Radiopharm. 2012, 55, 18–22. [Google Scholar] [CrossRef]
- Shah, S.Q. Formulation of technetium-99m labeled antimony trisulfide colloid, intended for sentinel lymph node imaging, using new techniques. Russ. J. Gen. Chem. 2005, 75, 1346–1350. [Google Scholar] [CrossRef]
- Green, A.E.C.; Causey, P.W.; Louie, A.S.; Armstrong, A.F.; Harrington, L.E.; Valliant, J.F. Microwave-Assisted Synthesis of 3,1,2- and 2,1,8-Re(I) and 99mTc(I)−Metallocarborane Complexes. Inorg. Chem. 2006, 45, 5727–5729. [Google Scholar] [CrossRef]
- Armstrong, A.F.; Valliant, J.F. Microwave-assisted synthesis of tricarbonyl rhenacarboranes: Steric and electronic effects on the 1,2-> 1,7 carborane cage isomerization. Inorg. Chem. 2007, 46, 2148–2158. [Google Scholar] [CrossRef]
- Causey, P.W.; Besanger, T.R.; Valliant, J.F. Synthesis and screening of mono- and di-aryl technetium and rhenium metallocarboranes. A new class of probes for the estrogen receptor. J. Med. Chem. 2008, 51, 2833–2844. [Google Scholar] [CrossRef]
- Reed, C.R.; Feeney, C.; Merritt, M.A. Microwave synthesis of dirhenium paddlewheel complexes. J. Coord. Chem. 2015, 68, 3449–3456. [Google Scholar] [CrossRef]
- Kunz, P.C.; Berghahn, M.; Bruckmann, N.E.; Dickmeis, M.; Kettel, M.; Spingler, B. Functionalised Tris(pyrazolyl)methane Ligands and Re(CO)3 Complexes Thereof. Z. Anorg. Allg. Chem. 2009, 635, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Merillas, B.; Cuellar, E.; Diez-Varga, A.; Asensio-Bartolome, M.; Garcia-Herbosa, G.; Torroba, T.; Martin-Alvarez, J.M.; Miguel, D.; Villafane, F. Whole microwave syntheses of pyridylpyrazole and of Re and Ru luminescent pyridylpyrazole complexes. Inorg. Chim. Acta 2019, 484, 1–7. [Google Scholar] [CrossRef]
- Winstead, A.J.; Alabrash, K.; Powell, B.V.; Parnell, S.J.; Hinton, T.V.; Odebode, T.; Peng, J.N.; Krause, J.A.; Zavalij, P.Y.; Mandal, S.K. Microwave-assisted synthesis of organometallic rhenium(I) pentylcarbonato complexes: New synthon for carboxylato, sulfonato and chlorido complexes. J. Organomet. Chem. 2021, 936, 121718. [Google Scholar] [CrossRef] [PubMed]
- Dabirmanesh, Q.; Roberts, R.M.G. The synthesis of iron sandwich complexes by microwave dielectric heating using a simple solid CO2-cooled apparatus in an unmodified commercial microwave oven. J. Organomet. Chem. 1993, 460, C28–C29. [Google Scholar] [CrossRef]
- Dabirmanesh, Q.; Fernando, S.I.S.; Roberts, R.M.G. Synthesis and decomplexation of (η-arene)(η-cyclopentadienyl)-iron(II) hexafluorophosphates using microwave dielectric heating. J. Chem. Soc. Perkin Trans. 1995, 1, 743–749. [Google Scholar] [CrossRef]
- Roberts, R.M.G. Synthesis of (η6-arene)(η5-cyclopentadienyl)iron(II) complexes with heteroatom and carbonyl substituents. Part I: Oxygen and carbonyl substituents. J. Organomet. Chem. 2006, 691, 2641–2647. [Google Scholar] [CrossRef]
- Roberts, R.M.G. Synthesis of (eta(6)-arene)(mu(5)-cyclopentadienyl)iron(II) complexes with heteroatom and carbonyl substituents—Part II, amino substituents. J. Organomet. Chem. 2006, 691, 4926–4930. [Google Scholar] [CrossRef]
- Puciová, M.; Ertl, P.; Toma, Š. Synthesis of Ferrocenyl-Substituted Heterocycles: The Beneficial Effect of the Microwave Irradiation. Collect. Czech. Chem. Commun. 1994, 59, 175–185. [Google Scholar] [CrossRef]
- Janková, Š.; Císařová, I.; Uhlík, F.; Štepnička, P.; Kotora, M. Synthesis and characterisation of Dewar benzene–ferrocene conjugates. Dalton Trans. 2009, 3137–3139. [Google Scholar] [CrossRef]
- Villemin, D.; Martin, B.; Puciova, M.; Toma, S. Dry synthesis under microwave irradiation: Synthesis of ferrocenylenones. J. Organomet. Chem. 1994, 484, 27–31. [Google Scholar] [CrossRef]
- Garringer, S.M.; Hesse, A.J.; Magers, J.R.; Pugh, K.R.; O’Reilly, S.A.; Wilson, A.M. Microwave Synthesis of Benchmark Organo-Iron Complexes. Organometallics 2009, 28, 6841–6844. [Google Scholar] [CrossRef]
- Pagnoux-Ozherelyeva, A.; Bolien, D.; Gaillard, S.; Peudru, F.; Lohier, J.F.; Whitby, R.J.; Renaud, J.L. Microwave irradiation and flow chemistry for a straightforward synthesis of piano-stool iron complexes. J. Organomet. Chem. 2014, 774, 35–42. [Google Scholar] [CrossRef]
- Naik, A.; Zhou, J.; Gao, C.; Wang, L. Microwave Synthesis of LiFePO4 from Iron Carbonyl Complex. Electrochim. Acta 2014, 142, 215–222. [Google Scholar] [CrossRef]
- Greene, D.L.; Mingos, D.M.P. Application of microwave dielectric loss heating effects for the rapid and convenient synthesis of ruthenium(II) polypyridine complexeS. Transit. Met. Chem. 1991, 16, 71–72. [Google Scholar] [CrossRef]
- Matsumura-Inoue, T.; Tanabe, M.; Minami, T.; Ohashi, T. A Remarkably Rapid Synthesis of Ruthenium(II) Polypyridine Complexes by Microwave Irradiation. Chem. Lett. 1994, 23, 2443–2446. [Google Scholar] [CrossRef]
- Xiao, X.M.; Sakamoto, J.; Tanabe, M.; Yamazaki, S.; Yamabe, S.; Matsumura-Inoue, T. Microwave synthesis and electrospectrochemical study on ruthenium(II) polypyridine complexes. J. Electroanal. Chem. 2002, 527, 33–40. [Google Scholar] [CrossRef]
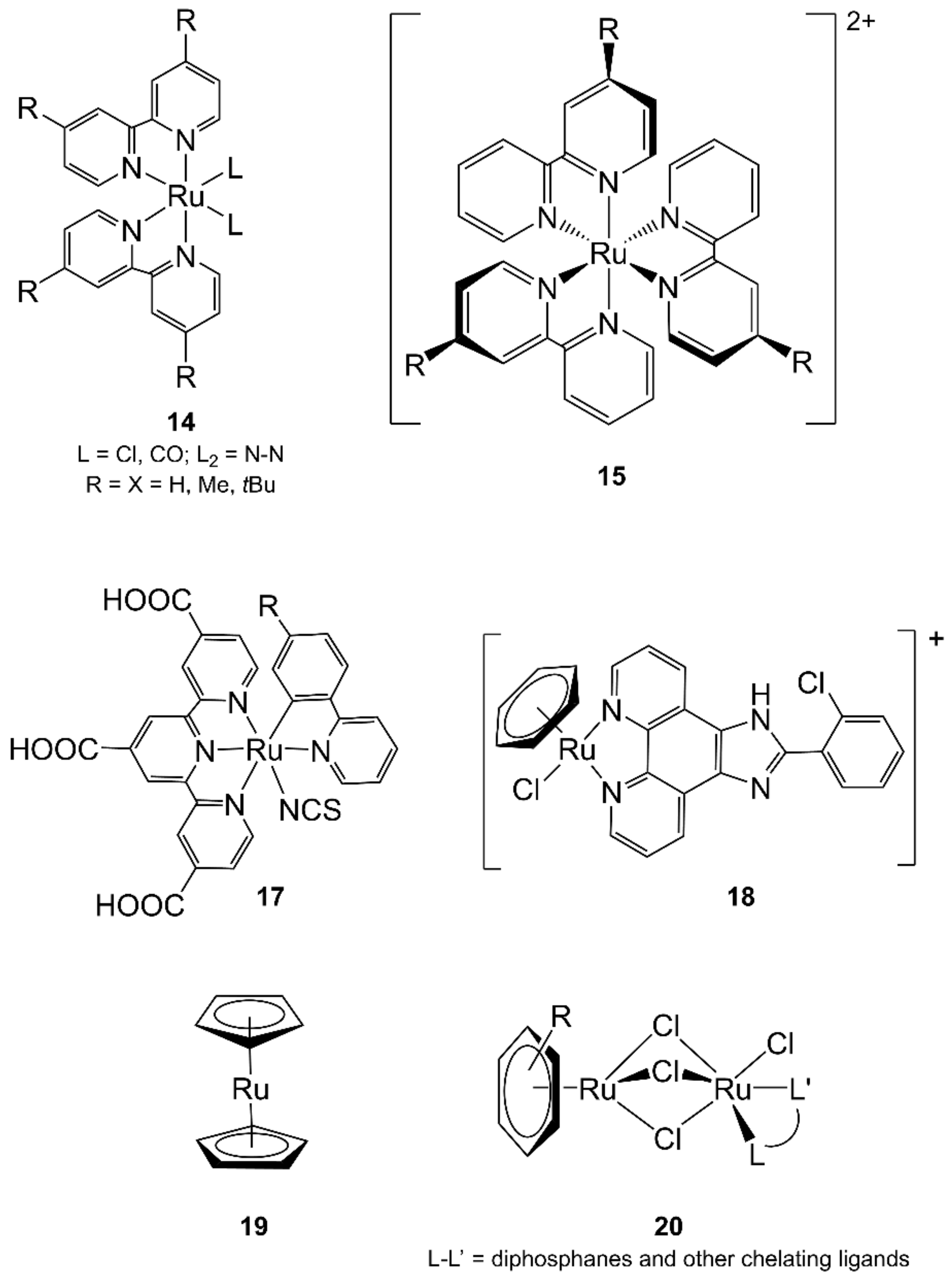
- Rau, S.; Shafer, B.; Grussing, A.; Schebesta, S.; Lamm, K.; Vieth, J.; Gorls, H.; Walther, D.; Rudolph, M.; Grummt, U.W.; et al. Efficient synthesis of ruthenium complexes of the type (R-bpy)2RuCl2 and [(R-bpy)2Ru(L-L)]Cl2 by microwave-activated reactions (R: H, Me, tert-But) (L-L: Substituted bibenzimidazoles, bipyrimidine, and phenanthroline). Inorg. Chim. Acta 2004, 357, 4496–4503. [Google Scholar] [CrossRef]
- Ziegler, M.; Monney, V.; Stoeckli-Evans, H.; Von Zelewsky, A.; Sasaki, I.; Dupic, G.; Daran, J.C.; Balavoine, G.G.A. Complexes of new chiral terpyridyl ligands. Synthesis and characterization of their ruthenium(II) and rhodium(III) complexes. J. Chem. Soc. Dalton Trans. 1999, 667–675. [Google Scholar] [CrossRef]
- Beves, J.E.; Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Schaffner, S.; Zampese, J.A. 4′-Chloro-2,2′: 6′,2″-terpyridine (L): Ethyl sulfate salts of [H2L]2+ and the single crystal structures of [H2L][EtOSO3]Cl∙H2O and [ML2][PF6]2 with M = Fe and Ru. Inorg. Chem. Commun. 2008, 11, 1006–1008. [Google Scholar] [CrossRef]
- Glasson, C.R.K.; Meehan, G.V.; Clegg, J.K.; Lindoy, L.F.; Smith, J.A.; Keene, F.R.; Motti, C. Microwave Synthesis of a Rare Ru2L34+ Triple Helicate and Its Interaction with DNA. Chem.-A Eur. J. 2008, 14, 10535–10538. [Google Scholar] [CrossRef]
- Martineau, D.; Beley, M.; Gros, P.C.; Cazzanti, S.; Caramori, S.; Bignozzi, C.A. Tuning of ruthenium complex properties using pyrrole- and pyrrolidine-containing polypyridine ligands. Inorg. Chem. 2007, 46, 2272–2277. [Google Scholar] [CrossRef]
- Grabulosa, A.; Beley, M.; Gros, P.C. Remarkable Effect of 4-Substituted 2,2′-Bipyridine Ligands on the Stereochemistry of Ruthenium(II) Complexes. Eur. J. Inorg. Chem. 2008, 2008, 1747–1751. [Google Scholar] [CrossRef]
- Schwalbe, M.; Schafer, B.; Gorls, H.; Rau, S.; Tschierlei, S.; Schmitt, M.; Popp, J.; Vaughan, G.; Henry, W.; Vos, J.G. Synthesis and characterisation of poly(bipyridine)ruthenium complexes as building blocks for heterosupramolecular arrays. Eur. J. Inorg. Chem. 2008, 2008, 3310–3319. [Google Scholar] [CrossRef]
- Wu, F.Y.; Thummel, R.P. Ru(II) complexes of crowded delocalized diimine ligands. Inorg. Chim. Acta 2002, 327, 26–30. [Google Scholar] [CrossRef]
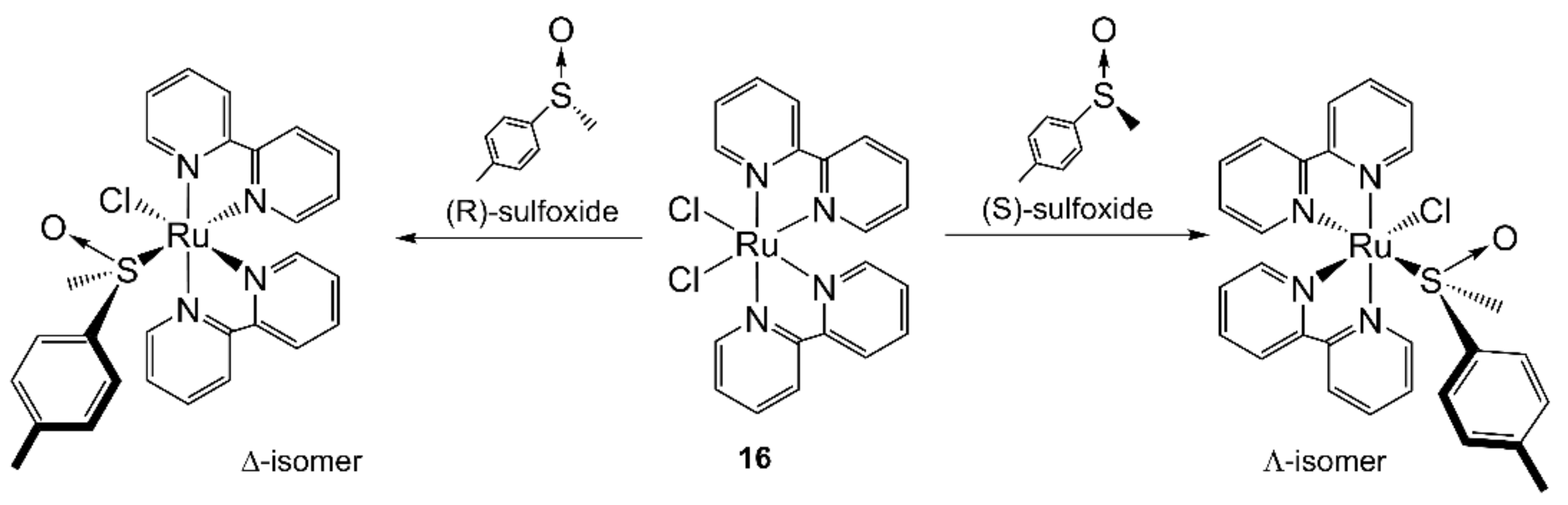
- Pezet, F.; Daran, J.C.; Sasaki, I.; Ait-Haddou, H.; Balavoine, G.G.A. Highly diastereoselective preparation of ruthenium bis(diimine) sulfoxide complexes: New concept in the preparation of optically active octahedral ruthenium complexes. Organometallics 2000, 19, 4008–4015. [Google Scholar] [CrossRef]
- Funaki, T.; Yanagida, M.; Onozawa-Komatsuzaki, N.; Kasuga, K.; Kawanishi, Y.; Kurashige, M.; Sayama, K.; Sugihara, H. Synthesis of a new class of cyclometallated ruthenium(II) complexes and their application in dye-sensitized solar cells. Inorg. Chem. Commun. 2009, 12, 842–845. [Google Scholar] [CrossRef]
- Jager, M.; Kumar, R.J.; Gorls, H.; Bergquist, J.; Johansson, O. Facile Synthesis of Bistridentate Ru-II Complexes Based on 2,6-Di(quinolin-8-yl)pyridyl Ligands: Sensitizers with Microsecond (MLCT)-M-3 Excited State Lifetimes. Inorg. Chem. 2009, 48, 3228–3238. [Google Scholar] [CrossRef] [PubMed]
- Jasimuddin, S.; Byabartta, P.; Mostafa, G.; Lu, T.H.; Sinha, C. Synthesis, spectral studies, crystal structure and redox properties of homoleptic tris-chelated ruthenium(II)-arylazoimidazoles. Polyhedron 2004, 23, 727–733. [Google Scholar] [CrossRef]
- Cortijo, M.; Delgado-Martinez, P.; Gonzalez-Prieto, R.; Herrero, S.; Jimenez-Aparicio, R.; Perles, J.; Priego, J.L.; Torres, M.R. Microwave and solvothermal methods for the synthesis of nickel and ruthenium complexes with 9-anthracene carboxylate ligand. Inorg. Chim. Acta 2015, 424, 176–185. [Google Scholar] [CrossRef]
- Herrero, S.; Jimenez-Aparicio, R.; Perles, J.; Priego, J.L.; Urbanos, F.A. First microwave synthesis of multiple metal-metal bond paddlewheel compounds. Green Chem. 2010, 12, 965–967. [Google Scholar] [CrossRef]
- Bashilov, A.V.; Fedorova, A.A.; Runov, V.K. Reduction of ruthenium(IV) to ruthenium(III) in aqueous alcohol solutions of hydrochloric acid under microwave radiation. J. Anal. Chem. 2000, 55, 1123–1127. [Google Scholar] [CrossRef]
- Tardiff, B.J.; Decken, A.; McGrady, G.S. Microwave-assisted synthesis of [Os2Cl3(PEt2Ph)6]Cl, featuring the first reported X-ray crystal structure. Inorg. Chem. Commun. 2008, 11, 44–46. [Google Scholar] [CrossRef]
- Kuhnert, N.; Danks, T.N. Microwave accelerated synthesis of cyclopentadienyl bis-phosphine ruthenium(II) thiolato complexes using focused microwave irradiation. J. Chem. Res. 2002, 2002, 66–68. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, J.; Mei, W.J.; Wang, Q.; Zhang, Z.; Wu, X.H.; Sun, F.Y.; Wu, W.L.; Chen, Y.H.; Hu, X.Y.; et al. Microwave-Assisted Synthesis of Arene Ruthenium(II) Complex as Apoptosis Inducer of A549 Cells. Aust. J. Chem. 2013, 66, 1422–1427. [Google Scholar] [CrossRef]
- Bocekova-Gajdosikova, E.; Epik, B.; Chou, J.Y.; Akiyama, K.; Fukui, N.; Guenee, L.; Kundig, E.P. Microwave-Assisted Synthesis and Transformations of Cationic CpRu(II)(naphthalene) and CpRu(II)(naphthoquinone) Complexes. Helv. Chim. Acta 2019, 102, e1900076. [Google Scholar] [CrossRef]
- Albrecht, C.; Gauthier, S.; Wolf, J.; Scopelliti, R.; Severin, K. Microwave-Assisted Organometallic Syntheses: Formation of Dinuclear [(Arene)Ru(μ-Cl)3RuCl(L-L′)] Complexes (L-L′: Chelate Ligands with P-, N-, or S-Donor Atoms) by Displacement of Arene pi Ligands. Eur. J. Inorg. Chem. 2009, 2009, 1003–1010. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Shoemaker, K.A. Preparation of ruthenium and osmium carbonyl complexes using microwave heating: Demonstrating the use of a gas-loading accessory and real-time reaction monitoring by means of a digital camera. Organometallics 2008, 27, 1254–1258. [Google Scholar] [CrossRef]
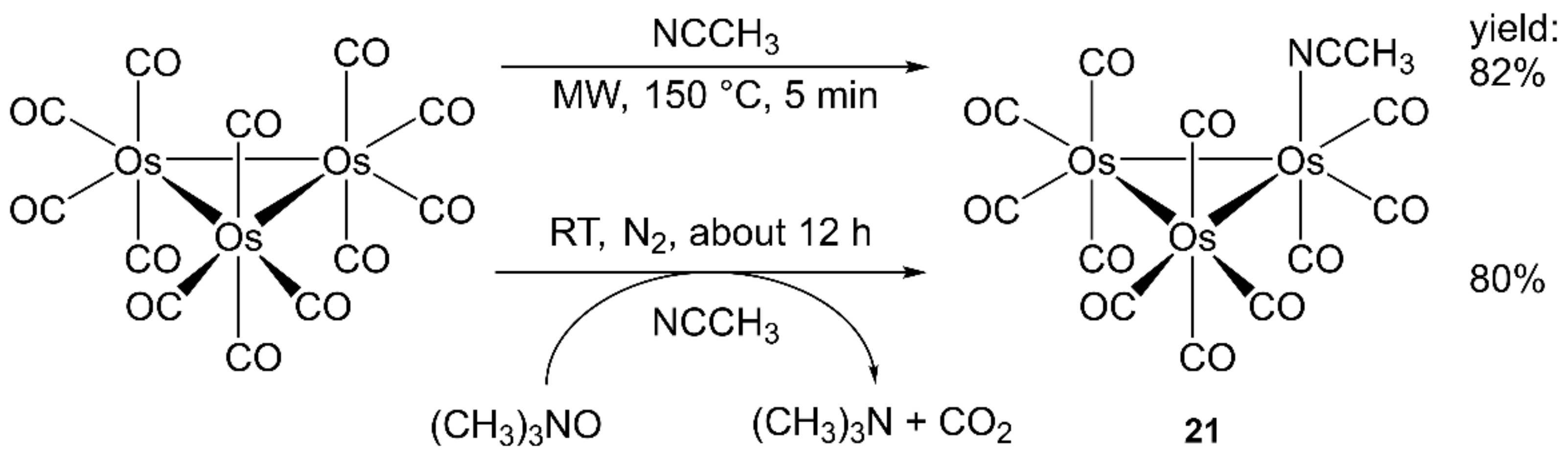
- Jung, J.Y.; Newton, B.S.; Tonkin, M.L.; Powell, C.B.; Powell, G.L. Efficient microwave syntheses of the compounds Os-3(CO)11L, L = NCMe, py, PPh3. J. Organomet. Chem. 2009, 694, 3526–3528. [Google Scholar] [CrossRef]
- Mishra, A.P.; Tiwari, A.; Jain, R.K. Microwave Induced Synthesis and Characterization of Semiconducting 2-Thiophenecarboxaldehyde Metal Complexes. Adv. Mater. Lett. 2012, 3, 213–219. [Google Scholar] [CrossRef]
- Jain, R.K.; Mishra, A.P. Microwave synthesis, spectral, thermal, and antimicrobial activities of some transition metal complexes involving 5-bromosalicylaldehyde moiety. Curr. Chem. Lett. 2012, 1, 163–174. [Google Scholar] [CrossRef]
- Jain, R.K.; Mishra, A.P. Microwave synthesis and spectral, thermal and antimicrobial activities of some novel transition metal complexes with tridentate Schiff base ligands. J. Serb. Chem. Soc. 2012, 77, 1013–1029. [Google Scholar] [CrossRef]
- Mishra, A.P.; Jain, R.K. Microwave Synthesis, Spectral, Thermal and Antimicrobial Activities of Some Transition Metal Complexes Involving 2-Amino-6-nitrobenzothiazole Moiety. Proc. Natl. Acad. Sci. India Sect. A-Phys. Sci. 2013, 83, 213–223. [Google Scholar] [CrossRef]
- Mishra, A.P.; Jain, R.K. Conventional and microwave synthesis, spectral, thermal and antimicrobial studies of some transition metal complexes containing 2-amino-5-methylthiazole moiety. J. Saudi Chem. Soc. 2014, 18, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Zayed, M.E.M.; Asiri, A.M.; Khan, S.A. Microwave Assisted Synthesis, Spectrofluorometric Characterization of Azomethine as Intermediate for Transition Metal Complexes with Biological Application. J. Fluoresc. 2016, 26, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Aswathy, R.; Mohanan, K. Microwave Assisted Synthesis, Characterisation and Fluorescence Studies of some Transition Metal Complexes with a Luminol Derivative. J. Fluoresc. 2017, 27, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Devi, P.R.S.; David, S.T.; Joel, C.; Bennie, R.B.; Abraham, S.D. Microwave synthesis, characterization and biological activities of transition metal complexes with novel SNSN donor Schiff base ligand. Indian J. Chem. Sect. A (IJCA) 2021, 60, 1416–1426. [Google Scholar]
- Murugavel, R.; Davis, P.; Walawalkar, M.G. First examples of metal cyclohexylphosphonates: Influence of the choice of synthetic route on the product. Z. Anorg. Allg. Chem. 2005, 631, 2806–2811. [Google Scholar] [CrossRef]
- Collet, A.; Wilson, C.; Murrie, M. Microwave-assisted synthesis: From a mononuclear {CoII} complex to {CoII9} solvomorphs. Dalton Trans. 2019, 48, 854–858. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Katano, S.; Ohta, K. Microwave-assisted Synthesis of Phthalocyanine Metal Complexes: Relationship between Yield and Maximum Temperature Reached by Microwave Irradiation. J. Jpn. Pet. Inst. 2018, 61, 140–149. [Google Scholar] [CrossRef] [Green Version]
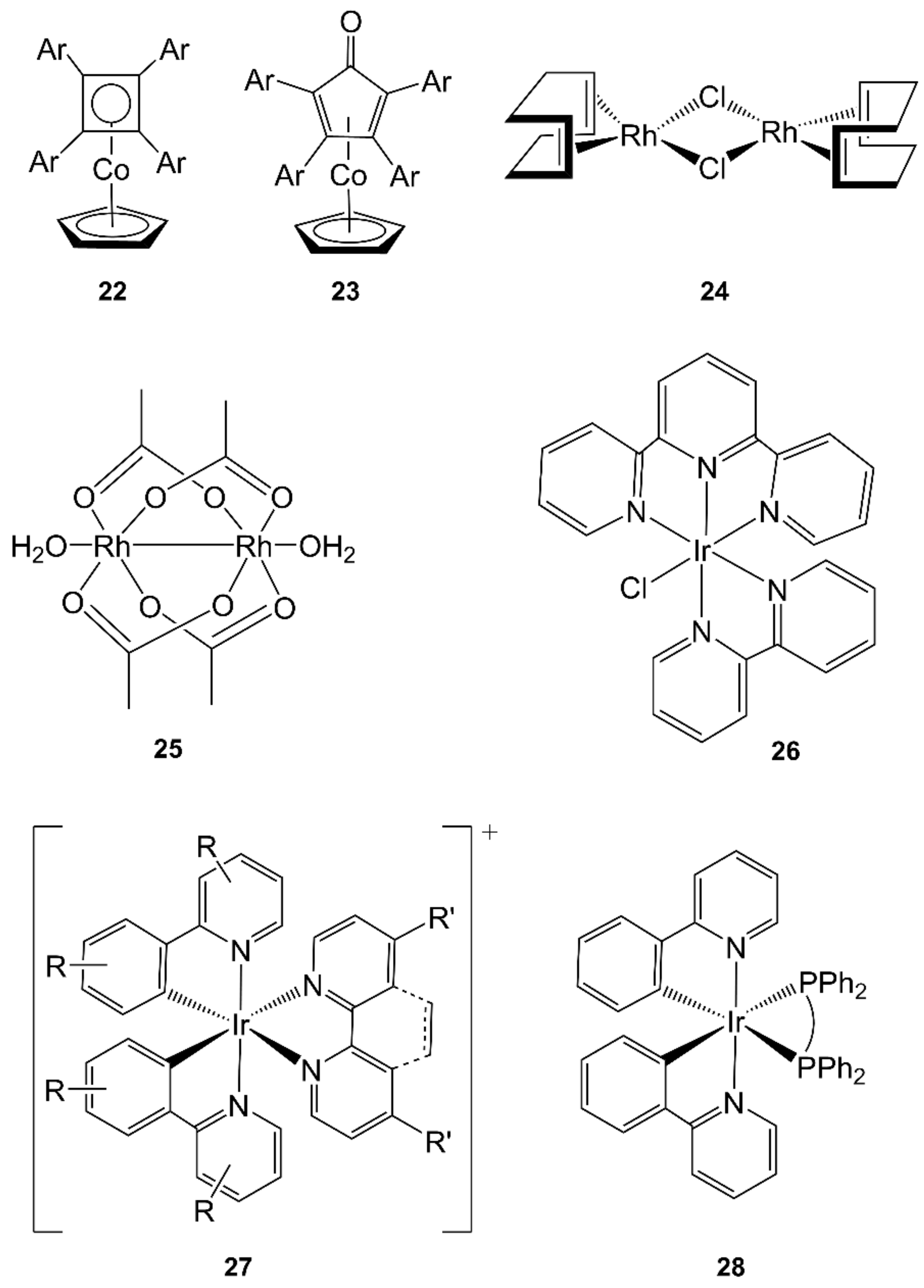
- Harcourt, E.M.; Yonis, S.R.; Lynch, D.E.; Hamilton, D.G. Microwave-assisted synthesis of cyclopentadienyl-cobalt sandwich complexes from diaryl acetylenes. Organometallics 2008, 27, 1653–1656. [Google Scholar] [CrossRef]
- Amarante, D.; Cherian, C.; Ernmel, C.; Chen, H.Y.; Dayal, S.; Koshy, M.; Megehee, E.G. Improved synthetic routes to rhodium bipyridine complexes: Comparison of microwave vs. conventional synthesis. Inorg. Chim. Acta 2005, 358, 2231–2238. [Google Scholar] [CrossRef]
- Ezerskaya, N.A.; Toropchenova, E.S.; Kubrakova, I.V.; Krasheninnikova, S.V.; Kudinova, T.F.; Fomina, T.A.; Kiseleva, I.N. Preparation of binuclear rhodium(II) tetraacetate (initial compound for the coulometric determination of rhodium) under the action of microwave radiation. J. Anal. Chem. 2000, 55, 1132–1135. [Google Scholar] [CrossRef]
- Pruszynski, M.; Bilewicz, A.; Zalutsky, M.R. Preparation of Rh[16aneS4-diol]211At and Ir[16aneS4-diol]211At complexes as potential precursors for astatine radiopharmaceuticals. Part I: Synthesis. Bioconj. Chem. 2008, 19, 958–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Matsusue, N.; Kanno, H.; Hamada, Y.; Takahashi, H.; Matsumura, T. Microwave Synthesis of Iridium(III) Complexes: Synthesis of Highly Efficient Orange Emitters in Organic Light-Emitting Devices. Jpn. J. Appl. Phys. 2004, 43, 2733–2734. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Matsumura-Inoue, T. Electrochemical and phosphorescent properties of new mixed-ligand Ir(III) complexes coordinated with both terpyridine and various bipyridine derivatives. Anal. Sci. 2003, 19, 761–765. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, N.; Sakamoto, J.; Matsumura-Inoue, T.; Takashima, H.; Tsukahara, K.; Kanehisa, N.; Kai, Y. Electrochemical and phosphorescent properties of new Ir(III) complexes coordinated by various bipyridine derivatives. Anal. Sci. 2004, 20, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, N.; Yamabe, S.; Kanehisa, N.; Kai, Y.; Takashima, H.; Tsukahara, K. Synthesis, characterization, and DFT investigation of Ir-III tolylterpyridine complexes. Eur. J. Inorg. Chem. 2007, 2007, 1911–1919. [Google Scholar] [CrossRef]
- Monos, T.M.; Sun, A.C.; McAtee, R.C.; Devery, J.J.; Stephenson, C.R.J. Microwave-Assisted Synthesis of Heteroleptic Ir(III)(+) Polypyridyl Complexes. J. Org. Chem. 2016, 81, 6988–6994. [Google Scholar] [CrossRef]
- Konno, H.; Sasaki, Y. Selective one-pot synthesis of facial tris-ortho-metalated iridium(III) complexes using microwave irradiation. Chem. Lett. 2003, 32, 252–253. [Google Scholar] [CrossRef]
- Alam, P.; Laskar, I.R.; Climent, C.; Casanova, D.; Alemany, P.; Karanam, M.; Choudhury, A.R.; Butcher, J.R. Microwave-assisted facile and expeditive syntheses of phosphorescent cyclometallated iridium(III) complexes. Polyhedron 2013, 53, 286–294. [Google Scholar] [CrossRef]
- Mishra, A.P.; Purwar, H.; Jain, R.K.; Gupta, S.K. Microwave Synthesis, Spectral, Thermal and Antimicrobial Studies of Some Co(II), Ni(II) and Cu(II) Complexes Containing 2-Aminothiazole Moiety. E-J. Chem. 2012, 9, 106460. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.H.; Tang, M.F.; Ge, C.M. Microwave Synthesis, Crystal Structure and Magnetic Behavior of a Schiff Base Trinuclear Nickel Cluster. Z. Anorg. Allg. Chem. 2009, 635, 1442–1446. [Google Scholar] [CrossRef]
- Piquer, L.R.; Sanudo, E.C. Microwave assisted synthesis of polynuclear Ni(II) complexes. Polyhedron 2019, 169, 195–201. [Google Scholar] [CrossRef]
- Dangwal, K.L.; Semwal, A.R. Microwave-assisted synthesis and characterization of oxime derivative of substituted chalcone and its nickel(II) complex. Curr. Sci. 2018, 115, 476–481. [Google Scholar] [CrossRef]
- Pawara, J.M.; Patil, S.S. Microwave-Assisted Synthesis of Some New Mixed Ligand Ni(II) Complexes Its Characterization and Its Antimicrobial Study. J. Pharm. Res. Int. 2021, 33, 143–152. [Google Scholar] [CrossRef]
- Landers, B.; Navarro, O. Microwave-assisted synthesis of (N-heterocyclic carbene)Ni(Cp)Cl complexes. Inorg. Chim. Acta 2012, 380, 350–353. [Google Scholar] [CrossRef]
- Kappe, C.O.; Van der Eycken, E. Click chemistry under non-classical reaction conditions. Chem. Soc. Rev. 2010, 39, 1280–1290. [Google Scholar] [CrossRef]
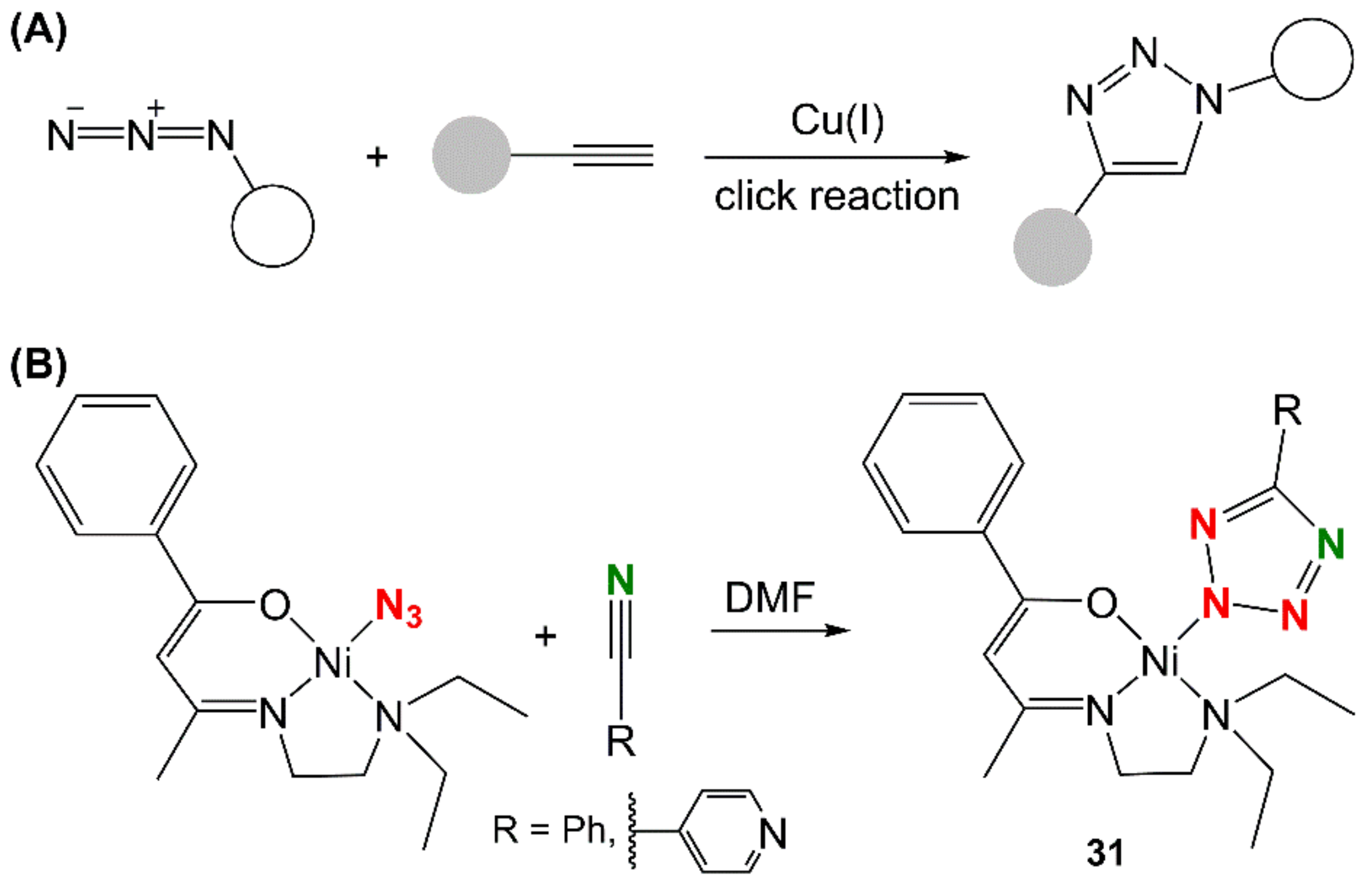
- Malviya, N.; Mandal, P.; Das, M.; Ganguly, R.; Mukhopadhyay, S. Nickel tetrazolato complexes synthesized by microwave irradiation: Catecholase like activity and interaction with biomolecules. J. Coord. Chem. 2017, 70, 261–278. [Google Scholar] [CrossRef]
- Lasri, J.; Rodriiguez, M.J.F.; da Silva, M.; Smolenski, P.; Kopylovich, M.N.; da Silva, J.; Pombeiro, A.J.L. Microwave synthesis of bis(tetrazolato)-Pd-II complexes with PPh3 and water-soluble 1,3,5-triaza-7-phosphaadamantane (PTA). The first example of C-CN bond cleavage of propionitrile by a Pd-II Centre. J. Organomet. Chem. 2011, 696, 3513–3520. [Google Scholar] [CrossRef]
- Tu, T.; Malineni, J.; Dotz, K.H. A novel pyridine-bridged bis-benzimidazolylidene pincer palladium complex: Synthesis and catalytic properties. Adv. Synth. Catal. 2008, 350, 1791–1795. [Google Scholar] [CrossRef]
- Gosiewska, S.; Herreras, S.M.; Lutz, M.; Spek, A.L.; Havenith, R.W.A.; van Klink, G.P.M.; van Koten, G.; Gebbink, R. Synthesis, structure, and catalytic performance of diastereopure five-coordinated NCN-pincer palladium(II) complexes bearing bulky amino acid substituents. Organometallics 2008, 27, 2549–2559. [Google Scholar] [CrossRef] [Green Version]
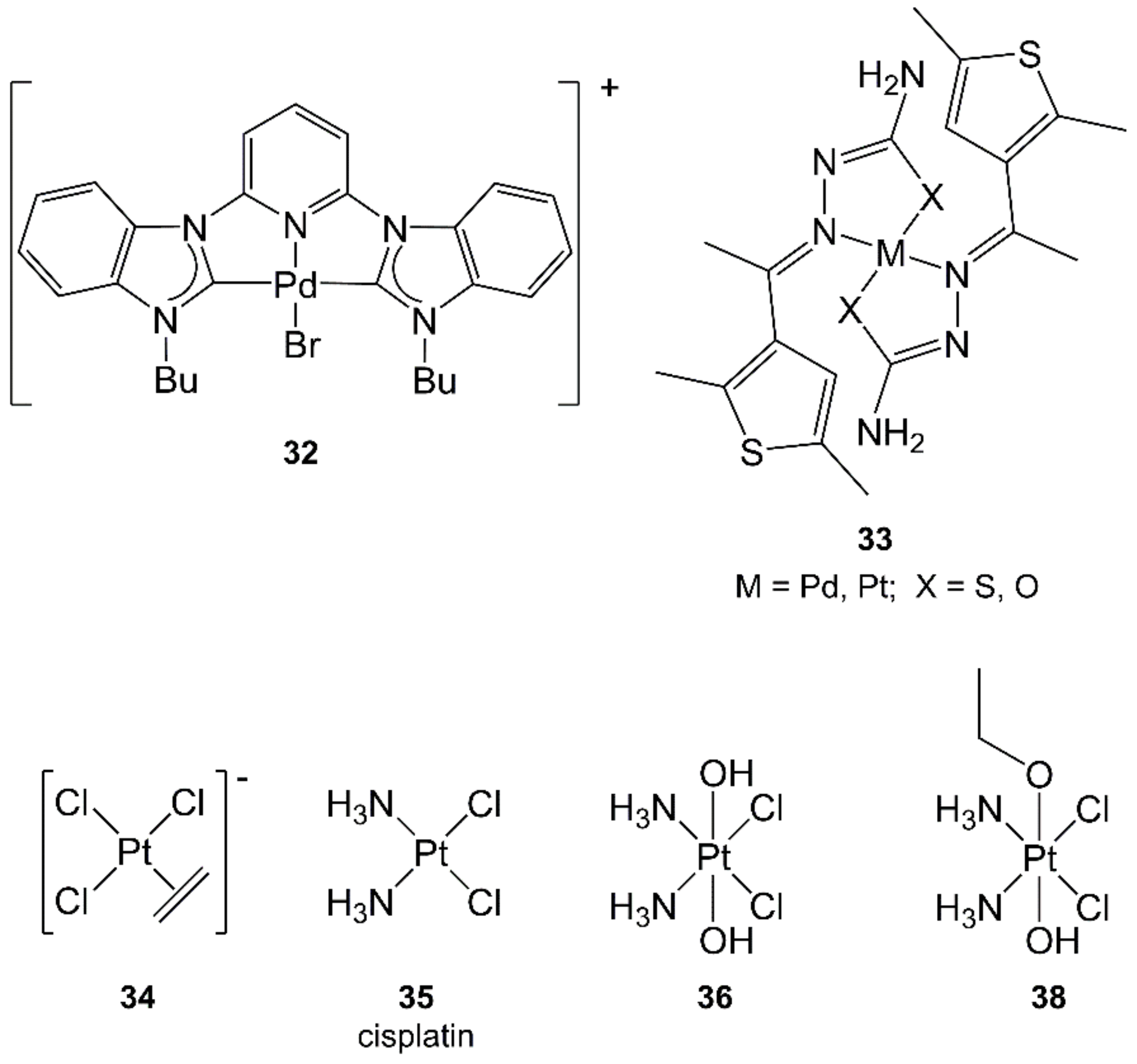
- Winkelmann, O.H.; Navarro, O. Microwave-Assisted Synthesis of N-Heterocyclic Carbene-Palladium(II) Complexes. Adv. Synth. Catal. 2010, 352, 212–214. [Google Scholar] [CrossRef]
- Castan, P.; Labiad, B.; Villemin, D.; Wimmer, F.L.; Wimmer, S. solid-state cyclometalation of the 1-methyl-2,4′-bipyridinium complexes of palladium(II) and platinum(II). J. Organomet. Chem. 1994, 479, 153–157. [Google Scholar] [CrossRef]
- Sharma, K.; Singh, R.; Fahmi, N.; Singh, R.V. Microwave assisted synthesis, characterization and biological evaluation of palladium and platinum complexes with azomethines. Spectrochim. Acta Part A-Mol. Biomol. Spectrosc. 2010, 75, 422–427. [Google Scholar] [CrossRef]
- Lopez-Vidal, E.M.; Blanco, V.; Garcia, M.D.; Peinador, C.; Quintela, J.M. Synthesis of Platinum(II) Metallocycles Using Microwave-Assisted Heating. Org. Lett. 2012, 14, 580–583. [Google Scholar] [CrossRef]
- Gabano, E.; Gama, S.; Mendes, F.; Fregonese, F.; Paulo, A.; Ravera, M. Application of microwave-assisted heating to the synthesis of Pt(II) complexes. Inorg. Chim. Acta 2015, 437, 16–19. [Google Scholar] [CrossRef]
- La Manna, S.; Florio, D.; Iacobucci, I.; Napolitano, F.; De Benedictis, I.; Malfitano, A.M.; Monti, M.; Ravera, M.; Gabano, E.; Marasco, D. A Comparative Study of the Effects of Platinum (II) Complexes on beta-Amyloid Aggregation: Potential Neurodrug Applications. Int. J. Mol. Sci. 2021, 22, 3015. [Google Scholar] [CrossRef] [PubMed]
- Godbert, N.; Pugliese, T.; Aiello, I.; Bellusci, A.; Crispini, A.; Ghedini, M. Efficient, ultrafast, microwave-assisted syntheses of cycloplatinated complexes. Eur. J. Inorg. Chem. 2007, 2007, 5105–5111. [Google Scholar] [CrossRef]
- Wang, Z.X.; Turner, E.; Mahoney, V.; Madakuni, S.; Groy, T.; Li, J.A. Facile Synthesis and Characterization of Phosphorescent Pt(N boolean AND C boolean AND N)X Complexes. Inorg. Chem. 2010, 49, 11276–11286. [Google Scholar] [CrossRef]
- Shoemaker, K.A.; Leadbeater, N.E. A fast and easy approach to the synthesis of Zeise’s salt using microwave heating. Inorg. Chem. Commun. 2009, 12, 341–342. [Google Scholar] [CrossRef]
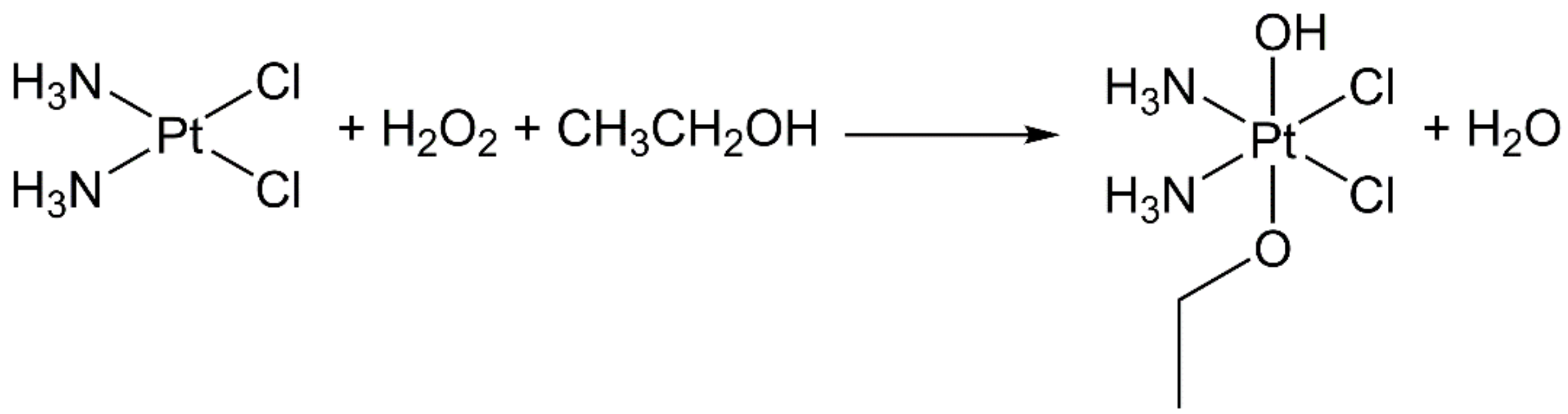
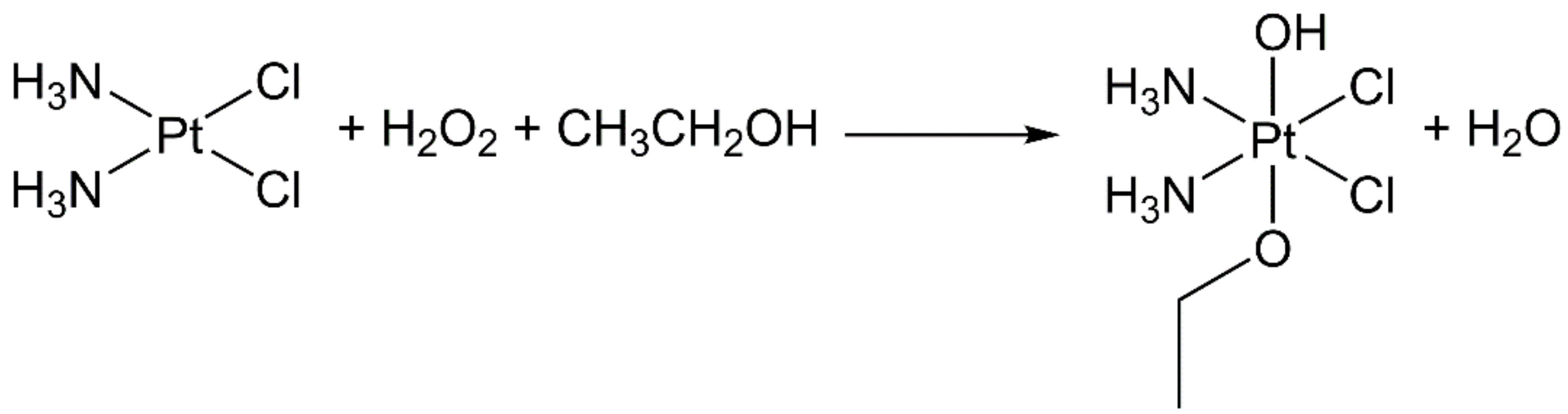
- Pedrick, E.A.; Leadbeater, N.E. Preparation of cisplatin using microwave heating and continuous-flow processing as tools. Inorg. Chem. Commun. 2011, 14, 481–483. [Google Scholar] [CrossRef]
- Petruzzella, E.; Chirosca, C.V.; Heidenga, C.S.; Hoeschele, J.D. Microwave-assisted synthesis of the anticancer drug cisplatin, cis-[Pt(NH3)2Cl2]. Dalton Trans. 2015, 44, 3384–3392. [Google Scholar] [CrossRef] [PubMed]
- Dhara, S.C. A rapid method for the synthesis of cis-[Pt(NH3)2Cl2]. Indian J. Chem. 1970, 8, 193–194. [Google Scholar]
- Lasri, J.; Januário Charmier, M.A.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Direct synthesis of (imine)platinum(II) complexes by iminoacylation of ketoximes with activated organonitrile ligands. Dalton Trans. 2006, 5062–5067. [Google Scholar] [CrossRef] [PubMed]
- Charmier, M.A.J.; Kukushkin, V.Y.; Pombeiro, A.J.L. Microwave-assisted 2+3 cycloaddition of nitrones to platinum-(II) and -(IV) bound organonitriles. Dalton Trans. 2003, 2540–2543. [Google Scholar] [CrossRef]
- Lasri, J.; Charmier, M.A.J.; Haukka, M.; Pombeiro, A.J.L. Stereospecific synthesis of polysubstituted E-olefins by reaction of acyclic nitrones with free and platinum(II) coordinated organonitriles. J. Org. Chem. 2007, 72, 750–755. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Lasri, J.; Charmier, M.A.J.; da Silva, M.; Pombeiro, A.J.L. Microwave synthesis of mono- and bis-tetrazolato complexes via 1,3-dipolar cycloaddition of organonitriles with platinum(II)-bound azides. Dalton Trans. 2007, 5297–5304. [Google Scholar] [CrossRef]
- Smolenski, P.; Mukhopadhyay, S.; Silva, M.; Charmier, M.A.J.; Pombeiro, A.J.L. New water-soluble azido- and derived tetrazolato-platinum(II) complexes with PTA. Easy metal-mediated synthesis and isolation of 5-substituted tetrazoles. Dalton Trans. 2008, 6546–6555. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Lasri, J.; da Silva, M.; Charmier, M.A.J.; Pombeiro, A.J.L. trans-Bis 5-(4-fluorophenyl)tetrazolato bis(triphenylphosphine)platinum(II). Acta Crystallogr. Sect. E-Crystallogr. Commun. 2007, E63, m2656. [Google Scholar] [CrossRef]
- Desai, B.; Danks, T.N.; Wagner, G. Ligand discrimination in the reaction of nitrones with PtCl2(PhCN)2. Selective formation of mono-oxadiazoline and mixed bis-oxadiazoline complexes under thermal and microwave conditions. Dalton Trans. 2004, 166–171. [Google Scholar] [CrossRef]
- Dopke, N.C.; Oemke, H.E. The microwave synthesis of platinum(II) phosphine complexes. Inorg. Chim. Acta 2011, 376, 638–640. [Google Scholar] [CrossRef]
- Carlsson, M.; Eliasson, B. One-pot synthesis of trans mono- or diarylalkynyl substituted platinum(II) compounds with tertiary phosphine or phosphite ligands. Organometallics 2006, 25, 5500–5502. [Google Scholar] [CrossRef]
- Giandomenico, C.M.; Abrams, M.J.; Murrer, B.A.; Vollano, J.F.; Rheinheimer, M.I.; Wyer, S.B.; Bossard, G.E.; Higgins, J.D. Carboxylation of Kinetically Inert Platinum(IV) Hydroxy Complexes. An Entrée into Orally Active Platinum(IV) Antitumor Agents. Inorg. Chem. 1995, 34, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Papa, E.; Luini, M.; Monti, E.; Gariboldi, M.B.; Ravera, M.; Gabano, E.; Gaviglio, L.; Osella, D. Antiproliferative Pt(IV) complexes: Synthesis, biological activity, and quantitative structure-activity relationship modeling. J. Biol. Inorg. Chem. 2010, 15, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Gabano, E.; Ravera, M.; Trivero, F.; Tinello, S.; Gallina, A.; Zanellato, I.; Gariboldi, M.B.; Monti, E.; Osella, D. The cisplatin-based Pt(IV)-diclorofibrato multi-action anticancer prodrug exhibits excellent performances also under hypoxic conditions. Dalton Trans. 2018, 47, 8268–8282. [Google Scholar] [CrossRef] [PubMed]
- Corinti, D.; Crestoni, M.E.; Fornarini, S.; Dabbish, E.; Sicilia, E.; Gabano, E.; Perin, E.; Osella, D. A multi-methodological inquiry of the behavior of cisplatin-based Pt(IV) derivatives in the presence of bioreductants with a focus on the isolated encounter complexes. J. Biol. Inorg. Chem. 2020, 25, 655–670. [Google Scholar] [CrossRef]
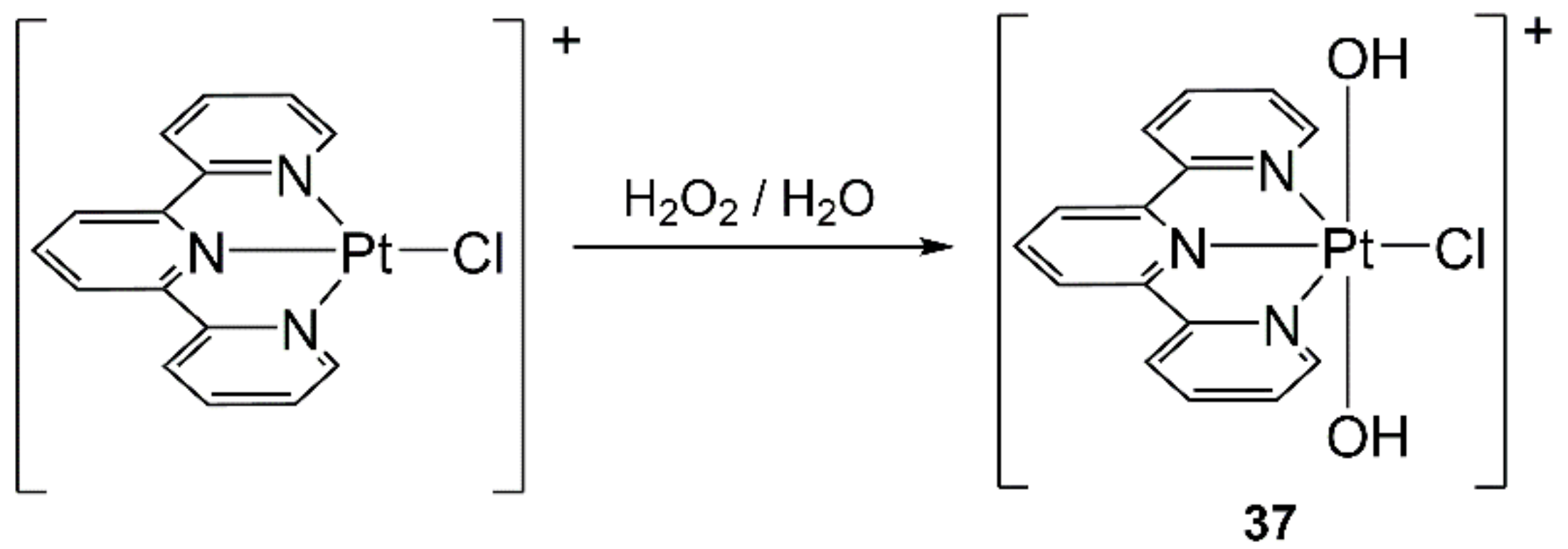
- Gabano, E.; Perin, E.; Fielden, C.; Platts, J.A.; Gallina, A.; Rangone, B.; Ravera, M. How to obtain Pt(IV) complexes suitable for conjugation to nanovectors from the oxidation of PtCl(terpyridine). Dalton Trans. 2017, 46, 10246–10254. [Google Scholar] [CrossRef] [Green Version]
- Sabbatini, M.; Zanellato, I.; Ravera, M.; Gabano, E.; Perin, E.; Rangone, B.; Osella, D. Pt(IV) Bifunctional Prodrug Containing 2-(2-Propynyl)octanoato Axial Ligand: Induction of Immunogenic Cell Death on Colon Cancer. J. Med. Chem. 2019, 62, 3395–3406. [Google Scholar] [CrossRef]
- Gabano, E.; Rangone, B.; Perin, E.; Caron, G.; Ermondi, G.; Vallaro, M.; Gandin, V.; Marzano, C.; Barbanente, A.; Margiotta, N.; et al. Pt(IV) complexes based on cyclohexanediamines and the histone deacetylase inhibitor 2-(2-propynyl)octanoic acid: Synthesis, characterization, cell penetration properties and antitumor activity. Dalton Trans. 2021, 50, 4663–4672. [Google Scholar] [CrossRef]
- Ravera, M.; Gabano, E.; Zanellato, I.; Rangone, B.; Perin, E.; Ferrari, B.; Bottone, M.G.; Osella, D. Cis,cis,trans-[PtIVCl2(NH3)2(perillato)2], a dual-action prodrug with excellent cytotoxic and antimetastatic activity. Dalton Trans. 2021, 50, 3161–3177. [Google Scholar] [CrossRef]
- Ravera, M.; Zanellato, I.; Gabano, E.; Perin, E.; Rangone, B.; Coppola, M.; Osella, D. Antiproliferative Activity of Pt(IV) Conjugates Containing the Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) Ketoprofen and Naproxen. Int. J. Mol. Sci. 2019, 20, 3074. [Google Scholar] [CrossRef] [Green Version]
- Gabano, E.; Ravera, M.; Perin, E.; Zanellato, I.; Rangone, B.; McGlinchey, M.J.; Osella, D. Synthesis and characterization of cyclohexane-1R,2R-diamine-based Pt(IV) dicarboxylato anticancer prodrugs: Their selective activity against human colon cancer cell lines. Dalton Trans. 2019, 48, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Lombard, C.K.; Myers, K.L.; Platt, Z.H.; Holland, A.W. Kinetics of Reductive Elimination from Platinum(IV) as a Probe for Nonthermal Effects in Microwave-Heated Reactions. Organometallics 2009, 28, 3303–3306. [Google Scholar] [CrossRef]
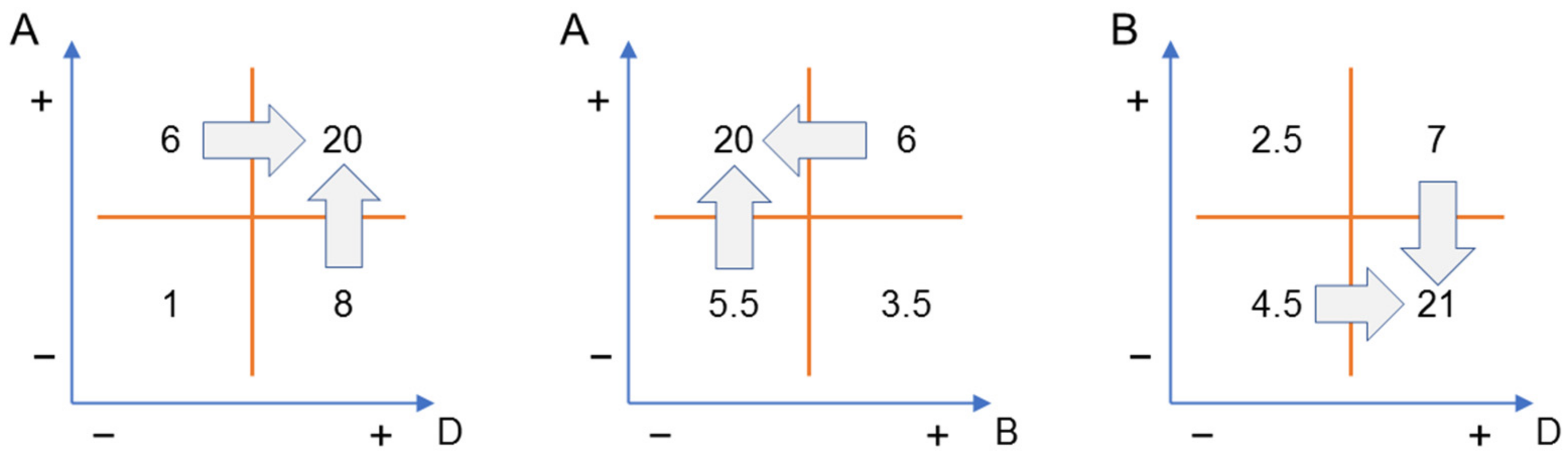
- Owen, M.R.; Luscombe, C.; Lai, L.W.; Godbert, S.; Crookes, D.L.; Emiabata-Smith, D. Efficiency by design: Optimisation in process research. Org. Process Res. Dev. 2001, 5, 308–323. [Google Scholar] [CrossRef]
- Leardi, R. Experimental design in chemistry: A tutorial. Anal. Chim. Acta 2009, 652, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Berger, P.D.; Maurer, R.E.; Celli, G.B. Experimental Design; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Riedwyl, H. Modifying and using Yates’ algorithm. Stat. Pap. 1998, 39, 41–60. [Google Scholar] [CrossRef] [Green Version]
- Ravera, M.; Perin, E.; Gabano, E.; Zanellato, I.; Panzarasa, G.; Sparnacci, K.; Laus, M.; Osella, D. Functional fluorescent nonporous silica nanoparticles as carriers for Pt(IV) anticancer prodrugs. J. Inorg. Biochem. 2015, 151, 132–142. [Google Scholar] [CrossRef]
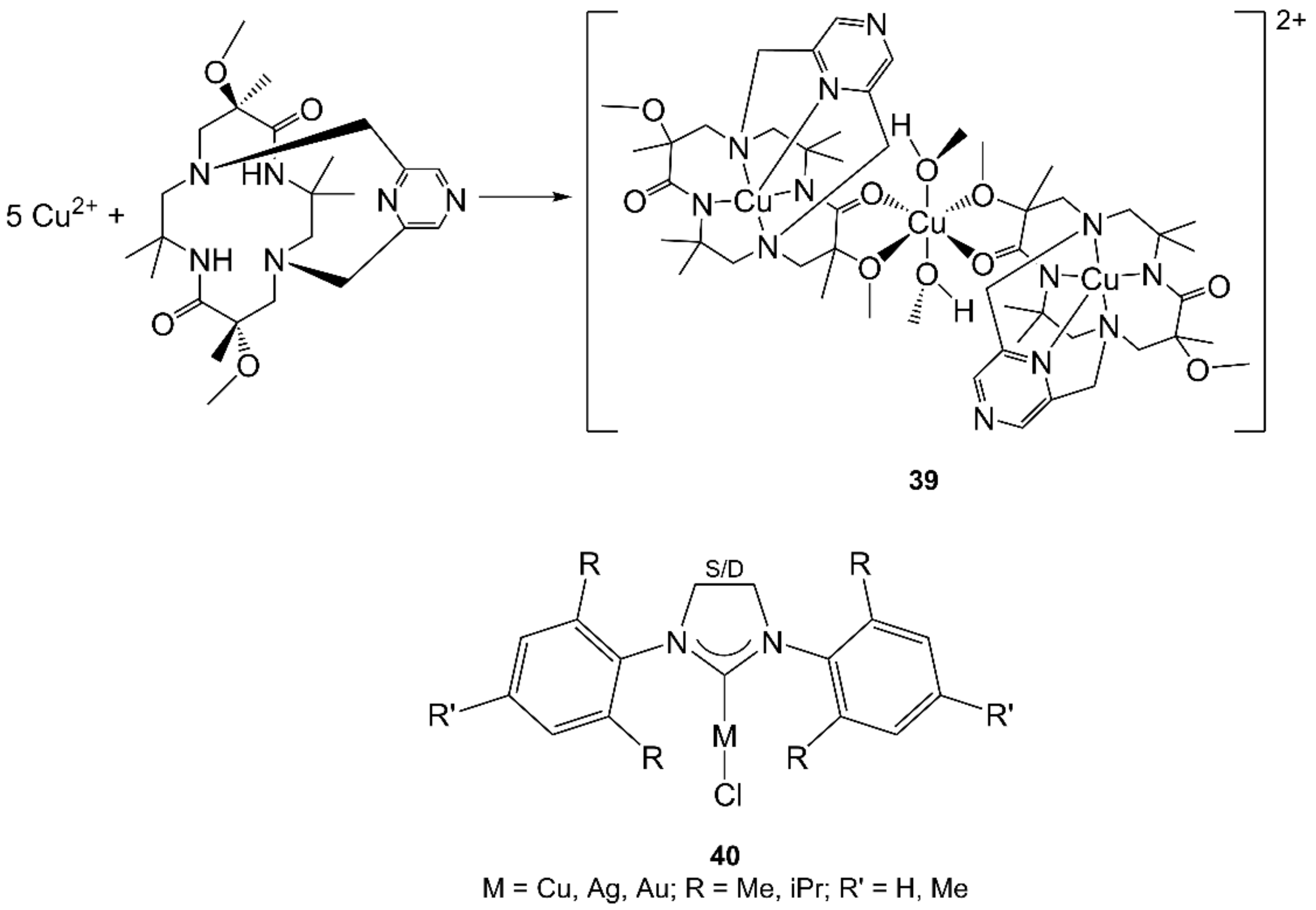
- Otani, N.; Furuya, T.; Katsuumi, N.; Haraguchi, T.; Akitsu, T. Synthesis of amino acid derivative Schiff base copper(II) complexes by microwave and wet mechanochemical methods. J. Indian Chem. Soc. 2021, 98, 100004. [Google Scholar] [CrossRef]
- Singh, A.; Chaudhary, A. Microwave-assisted synthesis, structural elucidation, antimicrobial and pesticidal activity of heterobimetallic complexes of Copper(II). J. Iran. Chem. Soc. 2020, 17, 973–983. [Google Scholar] [CrossRef]
- Hegedus, L.S.; Sundermann, M.J.; Dorhout, P.K. Synthesis, complexation, and coordination oligomerization of 1,8-pyrazine-capped 5,12-dioxocyclams. Inorg. Chem. 2003, 42, 4346–4354. [Google Scholar] [CrossRef]
- Phetmung, H.; Wongsawat, S.; Pakawatchai, C.; Harding, D.J. Microwave synthesis, spectroscopy, thermal analysis and crystal structure of an one-dimensional polymeric {[Cu(4,4′-bipy)(H2O)3(SO4)]●2H2O}n complex. Inorg. Chim. Acta 2009, 362, 2435–2439. [Google Scholar] [CrossRef]
- Carballo, R.; Covelo, B.; Fernandez-Hermida, N.; Garcia-Martinez, E.; Lago, A.B.; Vazquez-Lopez, E.M. Anion effect on the construction of copper(II) coordination polymers with the twisted ligand bis(4-pyridylthio)methane. Polyhedron 2008, 27, 3247–3254. [Google Scholar] [CrossRef]
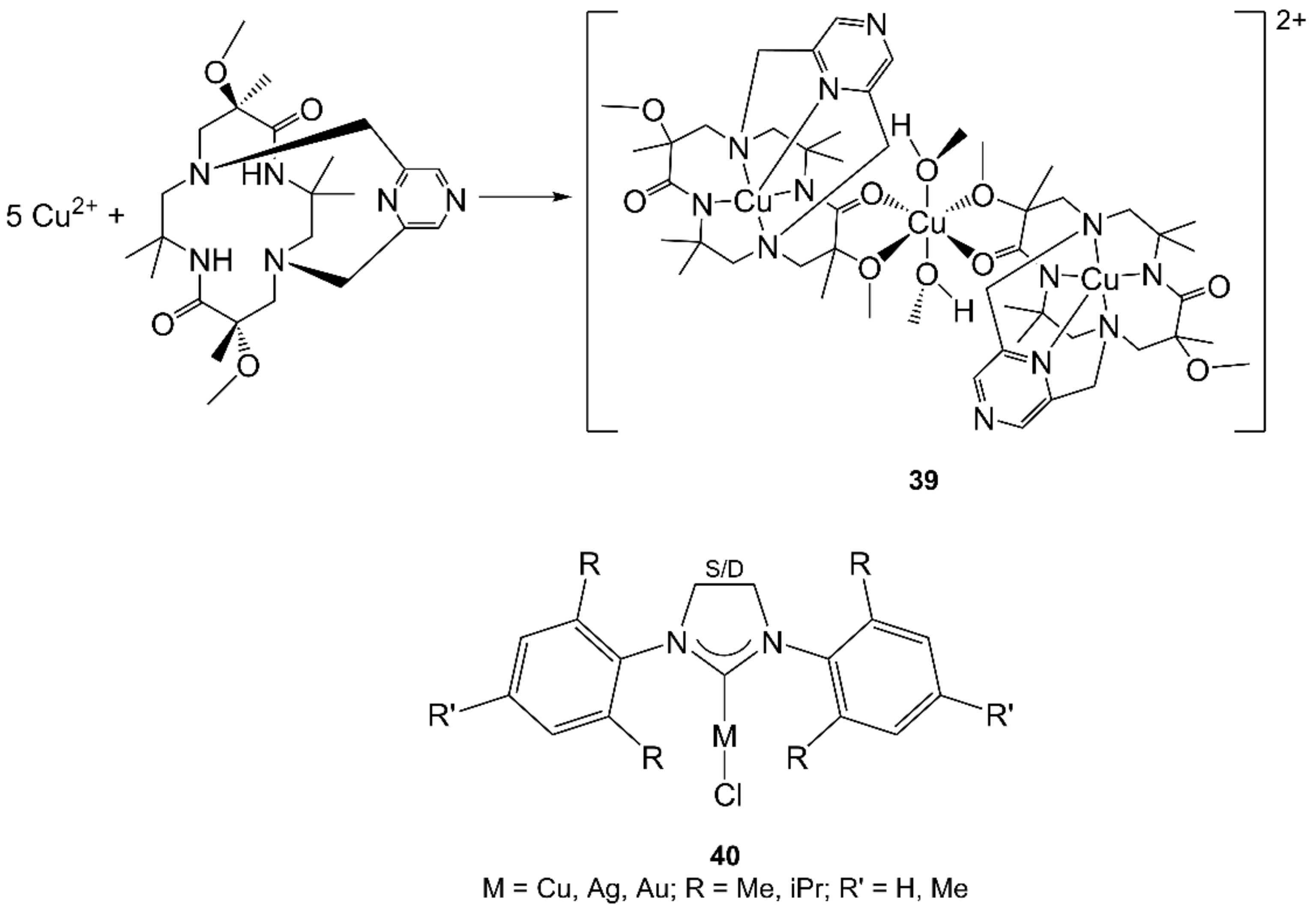
- Desbois, N.; Pacquelet, S.; Dubois, A.; Michelin, C.; Gros, C.P. Easy access to heterobimetallic complexes for medical imaging applications via microwave-enhanced cycloaddition. Beilstein J. Org. Chem. 2015, 11, 2202–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landers, B.; Navarro, O. Microwave-Assisted Synthesis of (N-Heterocyclic carbene)MCl Complexes of Group 11 Metals. Eur. J. Inorg. Chem. 2012, 2012, 2980–2982. [Google Scholar] [CrossRef]
- Shaw, A.P.; Tilset, M.; Heyn, R.H.; Jakobsen, S. Microwave methods for the synthesis of gold(III) complexes. J. Coord. Chem. 2011, 64, 38–47. [Google Scholar] [CrossRef]
- Guino-o, M.A.; Bustrom, B.; Tigaa, R.A.; de Bettencourt-Dias, A. Microwave-assisted synthesis of ternary lanthanide (2-thenoyltrifluoroacetone)(3)(triphenylphosphine oxide)(2) complexes. Inorg. Chim. Acta 2017, 464, 23–30. [Google Scholar] [CrossRef]
- Szijjarto, C.; Pershagen, E.; Borbas, K.E. Functionalisation of lanthanide complexes via microwave-enhanced Cu(I)-catalysed azide-alkyne cycloaddition. Dalton Trans. 2012, 41, 7660–7669. [Google Scholar] [CrossRef]
- Mohanan, K.; Kumari, B.S.; Rijulal, G. Microwave assisted synthesis, spectroscopic, thermal, and antifungal studies of some lanthanide(III) complexes with a heterocyclic bishydrazone. J. Rare Earths 2008, 26, 16–21. [Google Scholar] [CrossRef]
- Sonnauer, A.; Stock, N. High-throughput and microwave investigation of rare earth phosphonatoethanesulfonates-Ln(O3P-C2H4-SO3) (Ln = Ho, Er, Tm, Yb, Lu, Y). J. Solid State Chem. 2008, 181, 3065–3070. [Google Scholar] [CrossRef]
- Yates’ Algorithm. In The Concise Encyclopedia of Statistics; Springer: New York, NY, USA, 2008; pp. 579–581. [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabano, E.; Ravera, M. Microwave-Assisted Synthesis: Can Transition Metal Complexes Take Advantage of This “Green” Method? Molecules 2022, 27, 4249. https://doi.org/10.3390/molecules27134249
Gabano E, Ravera M. Microwave-Assisted Synthesis: Can Transition Metal Complexes Take Advantage of This “Green” Method? Molecules. 2022; 27(13):4249. https://doi.org/10.3390/molecules27134249
Chicago/Turabian StyleGabano, Elisabetta, and Mauro Ravera. 2022. "Microwave-Assisted Synthesis: Can Transition Metal Complexes Take Advantage of This “Green” Method?" Molecules 27, no. 13: 4249. https://doi.org/10.3390/molecules27134249
APA StyleGabano, E., & Ravera, M. (2022). Microwave-Assisted Synthesis: Can Transition Metal Complexes Take Advantage of This “Green” Method? Molecules, 27(13), 4249. https://doi.org/10.3390/molecules27134249