
Synthesis of Mixed Arylalkyl Tertiary Phosphines via the Grignard Approach

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
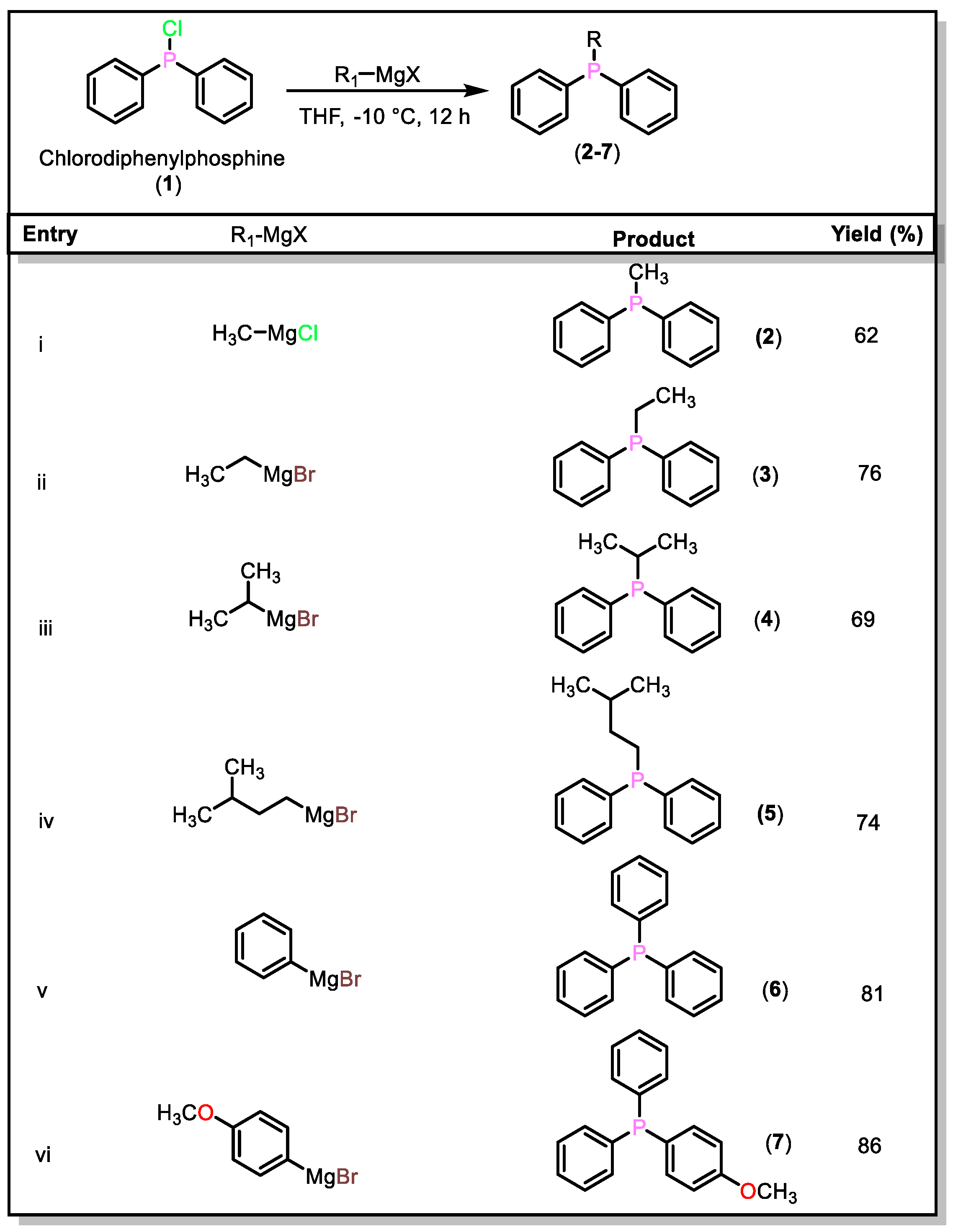
2.1. Synthesis
2.2. Structural Studies
1D- and 2D-NMR Spectroscopy
2.3. Density Functional Theory (DFT) Calculations
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis and Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A
References
- Guo, H.; Fan, Y.C.; Sun, Z.; Wu, Y.; Kwon, O. Phosphine organocatalysis. Chem. Rev. 2018, 118, 10049–10293. [Google Scholar] [CrossRef]
- Haque, A.; Faizi, M.S.H.; Rather, J.A.; Khan, M.S. Next generation NIR fluorophores for tumor imaging and fluorescence-guided surgery: A review. Biorg. Med. Chem. 2017, 25, 2017–2034. [Google Scholar] [CrossRef]
- Haque, A.; Al-Balushi, R.A.; Khan, M.S. σ-Acetylide complexes for biomedical applications: Features, challenges and future directions. J. Organomet. Chem. 2019, 897, 95–106. [Google Scholar] [CrossRef]
- Haque, A.; Al-Balushi, R.A.; Al-Busaidi, I.J.; Khan, M.S.; Raithby, P.R. Rise of Conjugated Poly-ynes and Poly(Metalla-ynes): From Design Through Synthesis to Structure–Property Relationships and Applications. Chem. Rev. 2018, 118, 8474–8597. [Google Scholar] [CrossRef]
- Ho, C.-L.; Yu, Z.-Q.; Wong, W.-Y. Multifunctional polymetallaynes: Properties, functions and applications. Chem. Soc. Rev. 2016, 45, 5264–5295. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.-Y.; Ho, C.-L. Di-, oligo-and polymetallaynes: Syntheses, photophysics, structures and applications. Coord. Chem. Rev. 2006, 250, 2627–2690. [Google Scholar] [CrossRef]
- Pinault, N.; Bruce, D.W. Homogeneous catalysts based on water-soluble phosphines. Coord. Chem. Rev. 2003, 241, 1–25. [Google Scholar] [CrossRef]
- Le Gall, E.; Aïssi, K.B.; Lachaise, I.; Troupel, M. Synthesis of symmetrical and unsymmetrical functionalized arylphosphines from chlorophosphines and organozinc reagents. Synlett 2006, 2006, 954–956. [Google Scholar] [CrossRef]
- Gulyás, H.; Szöllősy, Á.; Hanson, B.E.; Bakos, J. A direct approach to selective sulfonation of triarylphosphines. Tetrahedron Lett. 2002, 43, 2543–2546. [Google Scholar] [CrossRef]
- Maier, L. Preparation and properties of primary, secondary, and tertiary phosphines. Prog. Inorg. Chem. 2009, 5, 27. [Google Scholar]
- Gilheany, D.; Mitchell, C. Preparation of phosphines. In Organophosphorus Compounds (1990) Primary, Secondary and Tertiary Phosphines, Polyphosphines and Heterocyclic Organophosphorus (III) Compounds; Wiley Online Library: Chichester, UK, 1990; Volume 1, pp. 151–190. [Google Scholar]
- Gulyás, H.; Szöllősy, Á.; Szabó, P.; Halmos, P.; Bakos, J. Preparation of new sulfonated triarylphosphanes: Control of the selectivity by structural assistance. Eur. J. Org. Chem. 2003, 2003, 2775–2781. [Google Scholar] [CrossRef]
- Sun, Y.; Hienzsch, A.; Grasser, J.; Herdtweck, E.; Thiel, W.R. Novel phosphine ligands bearing 3 (5)-pyrazolyl and 4-(2-amino) pyrimidinyl groups: Synthesis and coordination chemistry. J. Organomet. Chem. 2006, 691, 291–298. [Google Scholar] [CrossRef]
- Ragaini, F.; Lunardi, L.; Tomasoni, D.; Guglielmi, V. Synthesis of triarylphosphines having para–SH and–SMe groups: Preparation of their complexes and formation of a monolayer on a gold surface. J. Organomet. Chem. 2004, 689, 3621–3630. [Google Scholar]
- Frisch, K.C.; Lyons, H. Silicon-containing Aromatic Phosphorus Derivatives. J. Am. Chem. Soc. 1953, 75, 4078–4079. [Google Scholar] [CrossRef]
- Wei, W.; Yu, B.; Alam, F.; Huang, Y.; Cheng, S.; Jiang, T. Ethylene oligomerization promoted by nickel-based catalysts with silicon-bridged diphosphine amine ligands. Transition Met. Chem. 2019, 44, 125–133. [Google Scholar] [CrossRef]
- Mikolajczyk, M.; Graczyk, P.P. Synthesis and Conformational Behavior of 2-Phosphonio-and 2-Phosphinyl-1, 3-dithianes. Operation of the Generalized Anomeric Effect in the SC-P+ System. J. Org. Chem. 1995, 60, 5190–5208. [Google Scholar] [CrossRef]
- Kuroboshi, M.; Kita, T.; Aono, A.; Katagiri, T.; Kikuchi, S.; Yamane, S.; Kawakubo, H.; Tanaka, H. Reduction of phosphine oxides to the corresponding phosphine derivatives in Mg/Me3SiCl/DMI system. Tetrahedron Lett. 2015, 56, 918–920. [Google Scholar] [CrossRef]
- Jin, S.; Haug, G.C.; Nguyen, V.T.; Flores-Hansen, C.; Arman, H.D.; Larionov, O.V. Decarboxylative phosphine synthesis: Insights into the catalytic, autocatalytic, and inhibitory roles of additives and intermediates. ACS Catal. 2019, 9, 9764–9774. [Google Scholar] [CrossRef]
- Valls, E.; Suades, J.; Donadieu, B.; Mathieu, R. Study of the complexing properties toward Ru II of new polydentate amphiphilic phosphines containing polyether chains. Unprecedented η 3 mode of bonding of the new ligand PPh [(CH 2) 3 CHMe 2][(CH 2 CH 2 O) 3 Me] and study of its hemilabile character. Chem. Commun. 1996, 771–772. [Google Scholar] [CrossRef]
- Russell, M.G.; Warren, S. Wittig reactions in water. Synthesis of new water-soluble phosphonium salts and their reactions with substituted benzaldehydes. Tetrahedron Lett. 1998, 39, 7995–7998. [Google Scholar] [CrossRef]
- Stevens, M.A.; Hashim, F.H.; Gwee, E.S.; Izgorodina, E.I.; Mulvey, R.E.; Blair, V.L. Contrasting synergistic heterobimetallic (Na–Mg) and homometallic (Na or Mg) bases in metallation reactions of dialkylphenylphosphines and dialkylanilines: Lateral versus ring selectivities. Chem. A Eur. J. 2018, 24, 15669–15677. [Google Scholar] [CrossRef] [Green Version]
- Kovács, T.; Urbanics, A.; Csatlós, F.; Keglevich, G. A study on the deoxygenation of trialkyl-, dialkyl-phenyl-and alkyl-diphenyl phosphine oxides by hydrosilanes. Heteroat. Chem 2017, 28, e21376. [Google Scholar] [CrossRef]
- Nowrouzi, N.; Keshtgar, S.; Jahromi, E.B. Ligand-free palladium catalyzed phosphorylation of aryl iodides. Tetrahedron Lett. 2016, 57, 348–350. [Google Scholar] [CrossRef]
- Li, C.-J.; Lü, J.; Zhang, Z.-X.; Zhou, K.; Li, Y.; Qi, G.-H. Copper-catalyzed C–P cross-coupling of secondary phosphines with (hetero) aromatic bromide. Res. Chem. Intermed. 2018, 44, 4547–4562. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, C.; Shen, Z.; Dai, B.; Chen, J. Efficient potassium hydroxide promoted P-arylation of aryl halides with diphenylphosphine. J. Organomet. Chem. 2021, 949, 121932. [Google Scholar] [CrossRef]
- Huang, W.; Tian, X.; Jiao, H.; Jackstell, R.; Beller, M. Iridium-Catalyzed Domino Hydroformylation/Hydrogenation of Olefins to Alcohols: Synergy of Two Ligands. Chem. A Eur. J. 2021, 28, e202104012. [Google Scholar] [CrossRef]
- Kaljurand, I. UV-VIS Spectra of Neutral Bases and Their Protonated Conjugate Cationic Acids in Acetonitrile. Available online: http://tera.chem.ut.ee/~manna/spe/base.htm (accessed on 1 May 2022).
- Bracker, M.; Helmecke, L.; Kleinschmidt, M.; Czekelius, C.; Marian, C.M. Visible light-induced homolytic cleavage of perfluoroalkyl iodides mediated by phosphines. Molecules 2020, 25, 1606. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.; Al-Balushi, R.; Al-Busaidi, I.J.; Al-Rasbi, N.K.; Al-Bahri, S.; Al-Suti, M.K.; Abou-Zied, O.K.; Khan, M.S.; Skelton, J.M.; Raithby, P.R. Two is Better Than One? Investigating the Effect of Incorporating Re(CO)3Cl Side-Chains into Pt(II) Di-ynes and Poly-ynes. Inorg. Chem. 2021, 60, 745–759. [Google Scholar] [CrossRef]
- Alshammari, M.M.; Soury, R.; Alenezi, K.M.; Mushtque, M.; Rizvi, M.M.A.; Haque, A. Synthesis, characterization, anticancer and in silico studies of a pyrazole-tethered thiazolidine-2,4-dione derivative. J. Biomol. Struct. Dyn. 2021, 1–8. [Google Scholar] [CrossRef]
- Al-Busaidi, I.J.; Haque, A.; Husband, J.; Al Rasbi, N.K.; Abou-Zied, O.K.; Al Balushi, R.; Khan, M.S.; Raithby, P.R. Electronic and steric effects of platinum (ii) di-yne and poly-yne substituents on the photo-switching behaviour of stilbene: Experimental and theoretical insights. Dalton Trans. 2021, 50, 2555–2569. [Google Scholar] [CrossRef]
- Mushtaque, M.; Avecilla, F.; Haque, A.; Yab, Z.; Rizvi, M.M.A.; Khan, M.S. Synthesis, structural and biological activity of N-substituted 2-methyl-4-/5-nitroimidazole derivatives. J. Mol. Struct. 2019, 1185, 440–449. [Google Scholar] [CrossRef]
- Al-Balushi, R.A.; Haque, A.; Jayapal, M.; Al-Suti, M.K.; Husband, J.; Khan, M.S.; Skelton, J.M.; Molloy, K.C.; Raithby, P.R. Impact of the alkyne substitution pattern and metalation on the photoisomerization of azobenzene-based platinum(II) diynes and polyynes. Inorg. Chem. 2016, 55, 10955–10967. [Google Scholar] [CrossRef] [Green Version]
- Al-Balushi, R.A.; Haque, A.; Jayapal, M.; Al-Suti, M.K.; Husband, J.; Khan, M.S.; Koentjoro, O.F.; Molloy, K.C.; Skelton, J.M.; Raithby, P.R. Experimental and Theoretical Investigation for the Level of Conjugation in Carbazole-Based Precursors and Their Mono-, Di-, and Polynuclear Pt(II) Complexes. Inorg. Chem. 2016, 55, 6465–6480. [Google Scholar] [CrossRef] [Green Version]
- Mushtaque, M.; Ahamad, S.; Jahan, M.; Hussain, K.; Khan, M.S. Azole-based compounds as antiamoebic agents: A perspective using theoretical calculations. RSC Adv. 2016, 6, 815–824. [Google Scholar] [CrossRef]
- Stewart, B.; Harriman, A.; Higham, L.J. Predicting the air stability of phosphines. Organometallics 2011, 30, 5338–5343. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Montgomery, J., Jr.; Vreven, T.; Kudin, K.; Burant, J. Gaussian 03, Revision C. 02; Gaussian, Inc.: Wallingford, CT, USA, 2004.
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.; Radom, L.; Schleyer, P.v.R.; Pople, J.A. Ab initio Molecular Orbital Theory; John Wiley and Sons: Hoboken, NJ, USA, 1986. [Google Scholar]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Absorption Maxima (nm) | Band Gap (Eg) 1 | |
|---|---|---|---|
| Calc. 1 | Exp. 2 | ||
| PBu3 | 199 | 216 | 5.8 |
| (10) | 238 | 220 | 3.5 |
| (5) | 238, 330 | 225, 263 | 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, A.; Alenezi, K.M.; Moll, H.E.; Khan, M.S.; Wong, W.-Y. Synthesis of Mixed Arylalkyl Tertiary Phosphines via the Grignard Approach. Molecules 2022, 27, 4253. https://doi.org/10.3390/molecules27134253
Haque A, Alenezi KM, Moll HE, Khan MS, Wong W-Y. Synthesis of Mixed Arylalkyl Tertiary Phosphines via the Grignard Approach. Molecules. 2022; 27(13):4253. https://doi.org/10.3390/molecules27134253
Chicago/Turabian StyleHaque, Ashanul, Khalaf M. Alenezi, Hani El Moll, Muhammad S. Khan, and Wai-Yeung Wong. 2022. "Synthesis of Mixed Arylalkyl Tertiary Phosphines via the Grignard Approach" Molecules 27, no. 13: 4253. https://doi.org/10.3390/molecules27134253
APA StyleHaque, A., Alenezi, K. M., Moll, H. E., Khan, M. S., & Wong, W.-Y. (2022). Synthesis of Mixed Arylalkyl Tertiary Phosphines via the Grignard Approach. Molecules, 27(13), 4253. https://doi.org/10.3390/molecules27134253