
Proteome-Based Serotyping of the Food-Borne Pathogens Salmonella Enterica by Label-Free Mass Spectrometry

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Comprehensive Proteomic Profiling for Different Salmonella Serotypes
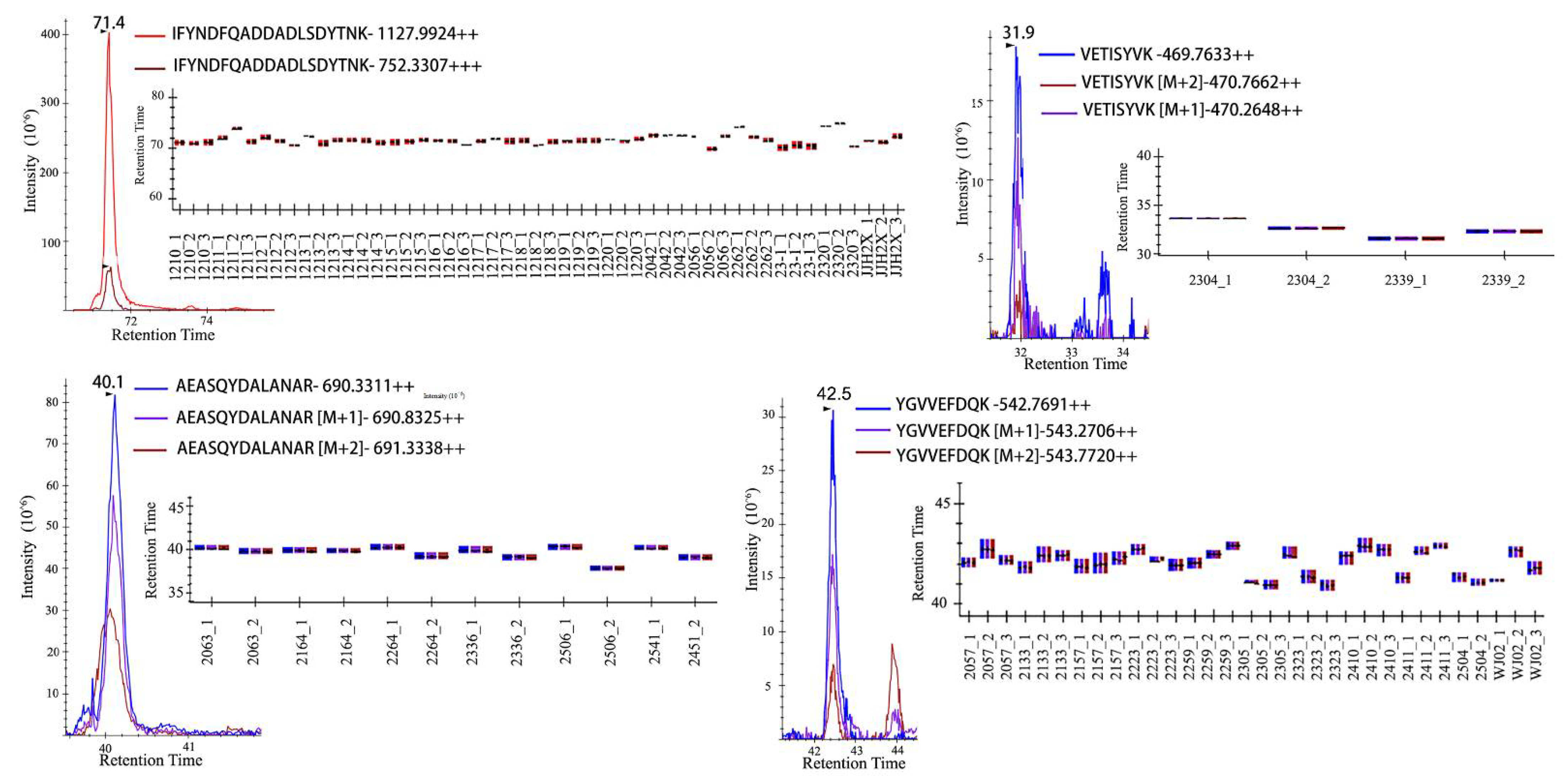
2.2. Peptide Markers for Salmonella enterica Serotyping
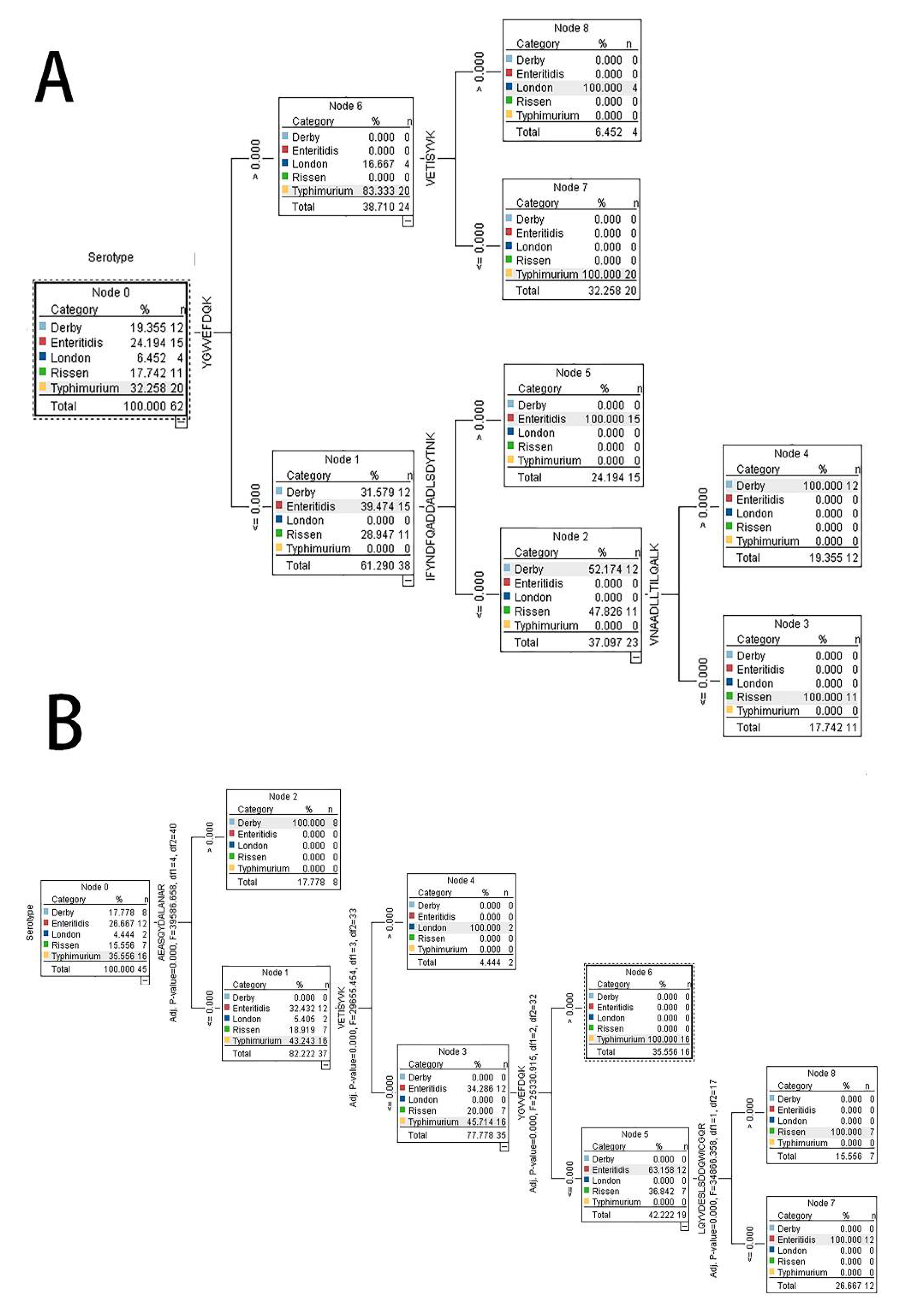
2.3. Accuracy of Models and Important Predictor Variables
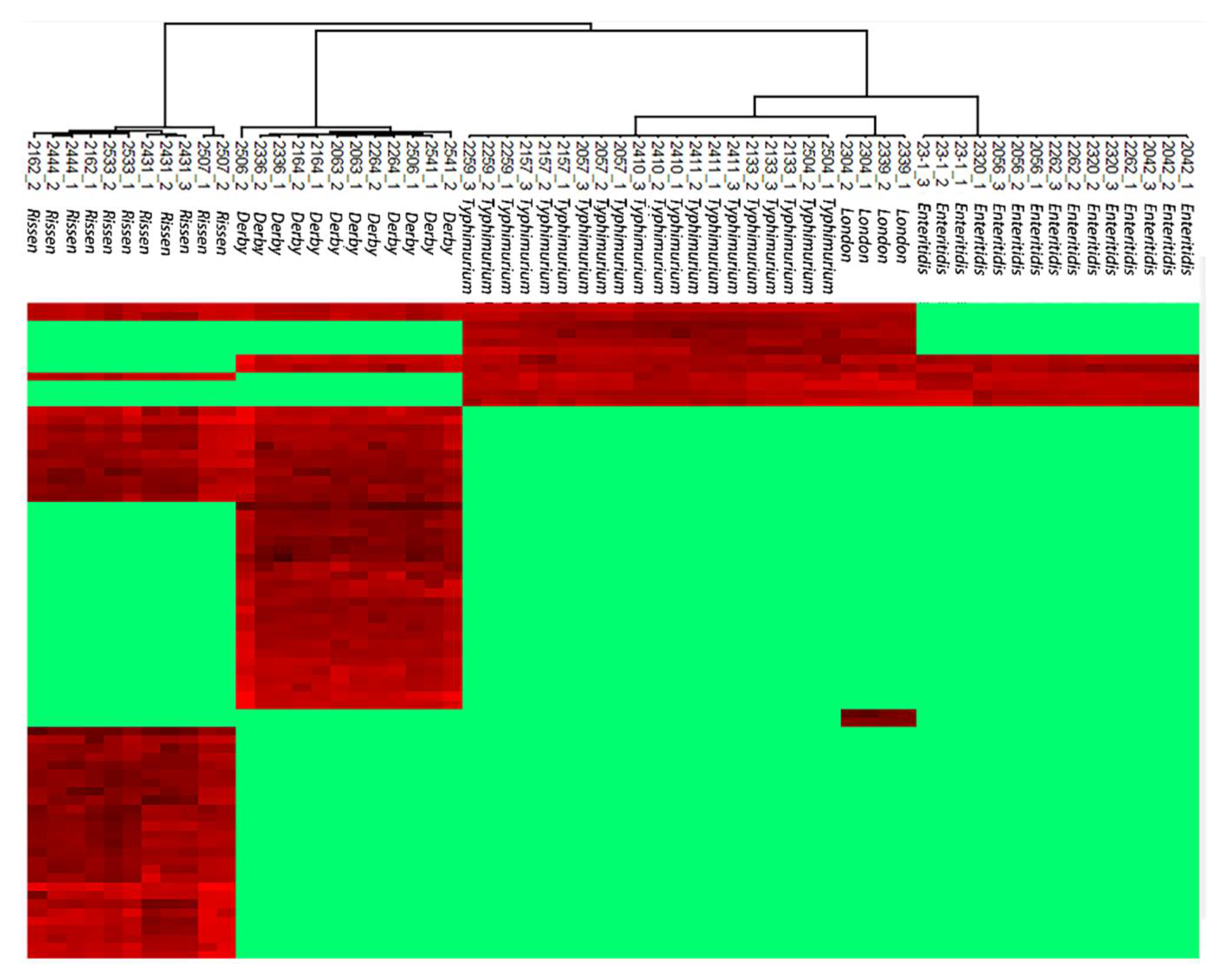
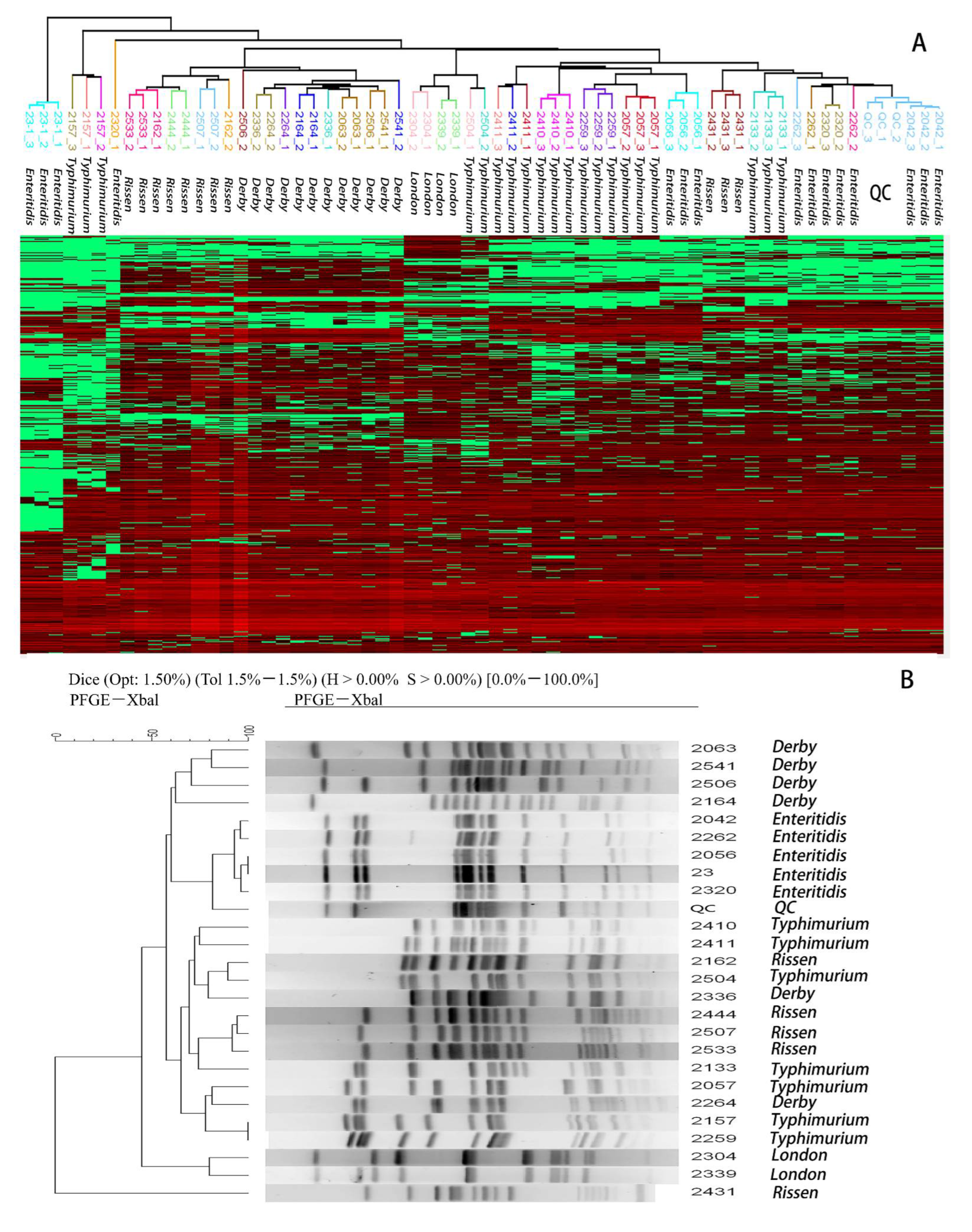
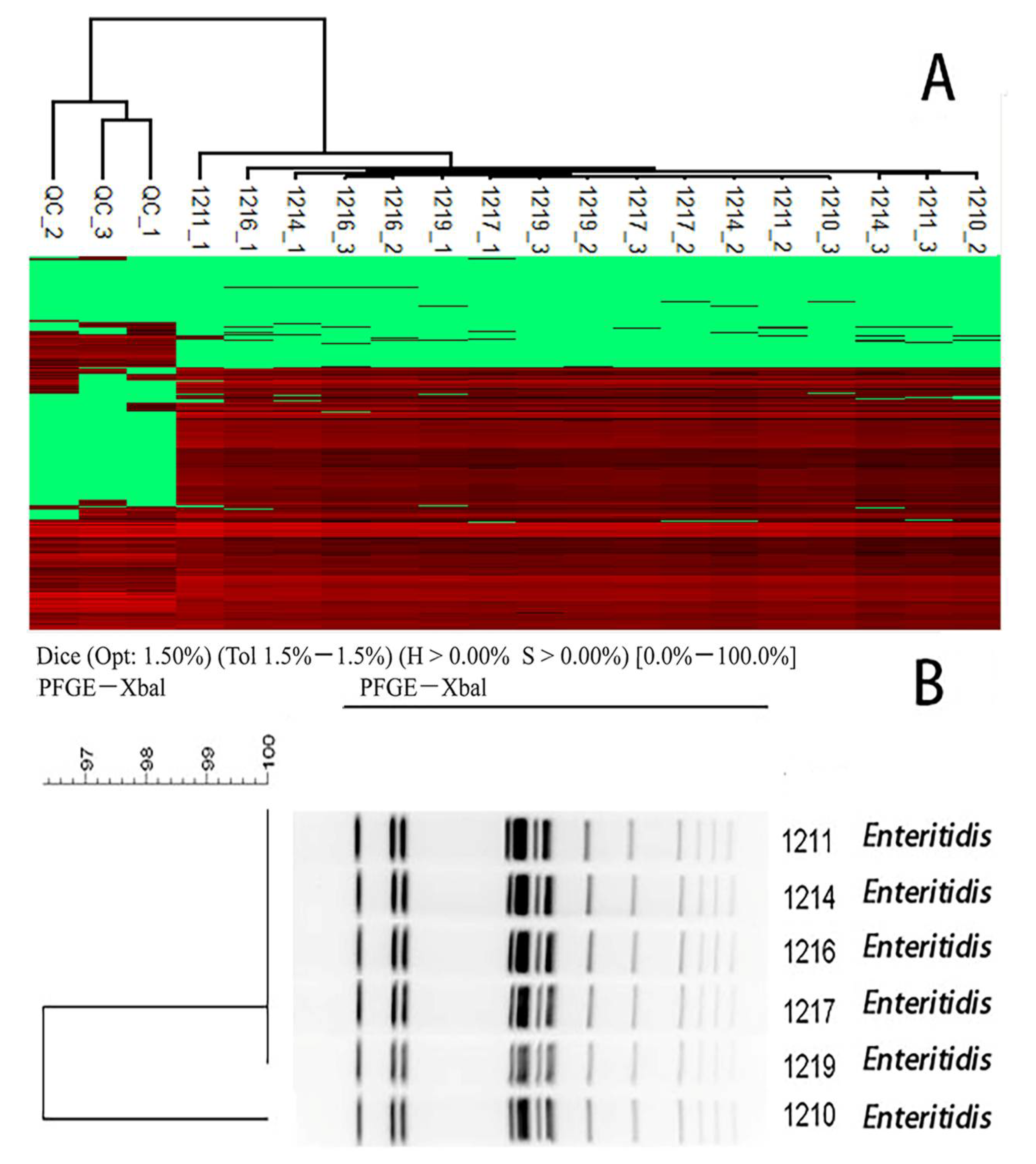
2.4. Hierarchical Clustering to Differentiate Similarity among Salmonella enteric Isolates
2.5. Exploring the Genetic and Biological Explanations for the Distinct Proteomic Profiles among the Salmonella serotypes
2.6. The Serotype-Specific Proteome and Biological Analysis
2.7. The Specificity for the Enteritidis Challenges
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Cell Lysis and Protein Extraction
4.3. Trypsin Digestion and Peptide Enrichment
4.4. Nanoflow High-Performance Liquid Chromatography (HPLC)
4.5. MS Identification
4.6. Pulsed-Field Gel Electrophoresis (PFGE)
4.7. Quantitative Proteomic Analysis and Bioinformatics Methods
4.8. The Predicted Model Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Grimont, P.A.D.; Weill, F.X. Antigenic formulae of the Salmonella serovars. In WHO Collaborating Centre for Reference and Research on Salmonella; Institut Pasteur: Paris, France, 2007. [Google Scholar]
- Chen, B.; Zhang, D.; Wang, X.; Ma, W.; Deng, S.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Proteomics progresses in microbial physiology and clinical antimicrobial therapy. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 403–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, T.; Li, B.; Comi, T.J.; Wu, Y.; Hu, P.; Wu, Y.; Min, Y.; Mitchell, D.A.; Zhao, H.; Sweedler, J.V. Profiling of microbial colonies for high-throughput engineering of multistep enzymatic reactions via optically guided Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. J. Am. Chem. Soc. 2017, 139, 12466–12473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chudejova, K.; Bohac, M.; Skalova, A.; Rotova, V.; Papagiannitsis, C.C.; Hanzlickova, J.; Bergerova, T.; Hrabák, J. Validation of a novel automatic deposition of bacteria and yeasts on MALDI target for MALDI-TOF MS-based identification using MALDI Colonyst robot. PLoS ONE 2017, 12, e0190038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassim, A.; Pflüger, V.; Premji, Z.; Daubenberger, C.; Revathi, G. Comparison of biomarker based Matrix Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry (MALDI-TOF MS) and conventional methods in the identification of clinically relevant bacteria and yeast. BMC Microbiol. 2017, 17, 128. [Google Scholar] [CrossRef]
- Dieckmann, R.; Malorny, B. Rapid screening of epidemiologically important Salmonella enterica subsp. enterica serovars by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl. Environ. Microbiol. 2011, 77, 4136–4146. [Google Scholar] [CrossRef] [Green Version]
- Pauker, V.I.; Thoma, B.R.; Grass, G.; Bleichert, P.; Hanczaruk, M.; Zöller, L.; Zange, S. Improved Discrimination of Bacillus anthracis from Closely Related Species in the Bacillus cereus Sensu Lato Group Based on Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry. J. Clin. Microbiol. 2018, 56, e01900–e01917. [Google Scholar] [CrossRef] [Green Version]
- Fagerquist, C.K.; Zaragoza, W.J. Top-down and middle-down proteomic analysis of Shiga toxin using MALDI-TOF-TOF mass spectrometry. MethodsX 2019, 6, 815–826. [Google Scholar] [CrossRef]
- Fagerquist, C.K.; Sultan, O. A new calibrant for matrix-assisted laser desorption/ionization time-of-flight-time-of-flight post-source decay tandem mass spectrometry of non-digested proteins for top-down proteomic analysis. Rapid Commun. Mass Spectrom. 2012, 26, 1241–1248. [Google Scholar] [CrossRef]
- Fagerquist, C.K.; Garbus, B.R.; Miller, W.G.; Williams, K.E.; Yee, E.; Bates, A.H.; Boyle, S.; Harden, L.A.; Cooley, M.B.; Mandrell, R.E. Rapid identification of protein biomarkers of Escherichia coli O157:H7 by matrix-assisted laser desorption ionization-time-of-flight-time-of-flight mass spectrometry and top-down proteomics. Anal. Chem. 2010, 82, 2717–2725. [Google Scholar] [CrossRef]
- Fagerquist, C.K.; Zaragoza, W.J.; Carter, M.Q. Top-Down proteomic identification of Shiga Toxin 1 and 2 from pathogenic Escherichia coli using MALDI-TOF-TOF tandem mass spectrometry. Microorganisms 2019, 7, 488. [Google Scholar] [CrossRef] [Green Version]
- Blosser, S.J.; Drake, S.K.; Andrasko, J.L.; Henderson, C.M.; Kamboj, K.; Antonara, S.; Mijares, L.; Conville, P.; Frank, K.M.; Harrington, S.M.; et al. Multicenter Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry study for identification of clinically relevant Nocardia spp. J. Clin. Microbiol. 2016, 54, 1251–1258. [Google Scholar] [CrossRef] [Green Version]
- Tammen, H.; Hess, R. Data preprocessing, visualization, and statistical analyses of nontargeted peptidomics data from MALDI-MS. Methods Mol. Biol. 2018, 1719, 187–196. [Google Scholar] [PubMed]
- Sriram, R.; Sahni, A.K.; Dudhat, V.L.; Pujahari, A.K. Matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) for rapid identification of Mycobacterium abscessus. Med. J. Armed Forces India 2018, 74, 22–27. [Google Scholar] [CrossRef]
- Nomura, F. Proteome-based bacterial identification using matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS): A revolutionary shift in clinical diagnostic microbiology. Biochim. Biophys. Acta 2015, 1854, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Thouvenot, P.; Vales, G.; Bracq-Dieye, H.; Tessaud-Rita, N.; Maury, M.M.; Moura, A.; Lecuit, M.; Leclercq, A. MALDI-TOF mass spectrometry-based identification of Listeria species in surveillance: A prospective study. J. Microbiol. Methods 2018, 144, 29–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, M.; Tran, V.; Nichol, K.A.; Lagacé-Wiens, P.; Pieroni, P.; Adam, H.J.; Turenne, C.; Walkty, A.J.; Normand, A.C.; Hendrickx, M.; et al. Evaluation of three MALDI-TOF mass spectrometry libraries for the identification of filamentous fungi in three clinical microbiology laboratories in Manitoba, Canada. Mycoses 2018, 61, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.H.; Kim, M.; Bhatt, B.; Choi, J.M.; Roh, J.H. Identification of pathogenic bacteria from public libraries via proteomics analysis. Int. J. Environ. Res. Public Health 2019, 16, 912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauget, M.; Nicolas-Chanoine, M.H.; Cabrolier, N.; Bertrand, X.; Hocquet, D. Matrix-assisted laser desorption ionization-time of flight mass spectrometry assigns Escherichia coli to the phylogroups A, B1, B2 and D. Int. J. Med. Microbiol. 2014, 304, 977–983. [Google Scholar] [CrossRef]
- Kleinteich, J.; Puddick, J.; Wood, S.A.; Hildebrand, F.; Laughinghouse, H.D., IV; Pearce, D.A.; Dietrich, D.R.; Wilmotte, A. Toxic cyanobacteria in svalbard: Chemical diversity of microcystins detected using a liquid chromatography mass spectrometry precursor ion screening method. Toxins 2018, 10, 147. [Google Scholar] [CrossRef] [Green Version]
- Ullberg, M.; Lüthje, P.; Mölling, P.; Strålin, K.; Özenci, V. Broad-range detection of microorganisms directly from bronchoalveolar lavage specimens by PCR/electrospray ionization-mass spectrometry. PLoS ONE 2017, 12, e0170033. [Google Scholar] [CrossRef]
- Vetor, R.; Murray, C.K.; Mende, K.; Melton-Kreft, R.; Akers, K.S.; Wenke, J.; Spirk, T.; Guymon, C.; Zera, W.; Beckius, M.L.; et al. The use of PCR/Electrospray Ionization-Time-of-Flight-Mass Spectrometry (PCR/ESI-TOF-MS) to detect bacterial and fungal colonization in healthy military service members. BMC Infect. Dis. 2016, 22, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, V.; Lassman, M.E.; Chen, Y.; Zhou, H.; Laterza, O.F. Achieving efficient digestion faster with Flash Digest: Potential alternative to multi-step detergent assisted in-solution digestion in quantitative proteomics experiments. Rapid Commun. Mass Spectrom. 2017, 31, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Valletta, M.; Rega, C.; Marasco, R.; Muscariello, L.; Pedone, P.V.; Sacco, M.; Chambery, A. Reliable identification of lactic acid bacteria by targeted and untargeted high-resolution tandem mass spectrometry. Food Chem. 2019, 1, 111–118. [Google Scholar] [CrossRef]
- Yuan, G.; Bai, Y.; Zhang, Y.; Xu, G. Data Mining Mycobacterium tuberculosis Pathogenic Gene Transcription Factors and Their Regulatory Network Nodes. Int. J. Genom. 2018, 2018, 3079730. [Google Scholar]
- Sigurdardottir, A.K.; Jonsdottir, H.; Benediktsson, R. Outcomes of educational interventions in type 2 diabetes: WEKA data-mining analysis. Patient Educ. Couns. 2007, 67, 21–31. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Michael, S.B.; Joshua, B.; Vagisha, S. An Automated pipeline to monitor system performance in Liquid Chromatography–Tandem Mass Spectrometry proteomic experiments. J. Proteome Res. 2016, 15, 4763–4769. [Google Scholar]
- Song, Y.Y.; Lu, Y. Decision tree methods: Applications for classification and prediction. Shanghai Arch. Psychiatry 2015, 27, 130–135. [Google Scholar]
- Zhou, M.; Chen, Y.; Liu, J.; Huang, G. A predicting model of bone marrow malignant infiltration in 18F-FDG PET/CT images with increased diffuse bone marrow FDG uptake. J. Cancer. 2018, 9, 1737–1744. [Google Scholar] [CrossRef] [Green Version]
- Näsström, E.; Vu Thieu, N.T.; Dongol, S.; Karkey, A.; Voong Vinh, P.; Ha Thanh, T.; Johansson, A.; Arjyal, A.; Thwaites, G.; Dolecek, C.; et al. Salmonella Typhi and Salmonella Paratyphi A elaborate distinct systemic metabolite signatures during enteric fever. eLife 2014, 3, e03100. [Google Scholar] [CrossRef]
- Dong, R.; Wu, Q.P.; Zhang, J.M.; Yu, H.P.; Ma, L.Y.; Guo, W.P. Study on Biomarkers of Foodborne Pathogenic Salmonella Serotypes. Mod. Food Sci. Technol. 2017, 33, 288–292. [Google Scholar]
- Vila Nova, M.; Durimel, K.; La, K.; Felten, A.; Bessières, P.; Mistou, M.Y.; Mariadassou, M.; Radomski, N. Genetic and metabolic signatures of Salmonella enterica subsp. enterica associated with animal sources at the pangenomic scale. BMC Genom. 2019, 20, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 4, D200–D203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Keys, C.E.; Zhao, S.; Meng, J.; Brown, E.W. Enhanced subtyping scheme for Salmonella enteritidis. Emerg. Infect. Dis. 2007, 13, 1932–1935. [Google Scholar] [CrossRef]
- Hunter, S.B.; Vauterin, P.; Lambert-Fair, M.A.; Van Duyne, M.S.; Kubota, K.; Graves, L.; Wrigley, D.; Barrett, T.; Ribot, E. Establishment of a universal size standard strain for use with the PulseNet standardized pulsed-field gel electrophoresis protocols: Converting the national databases to the new size standard. J. Clin. Microbiol. 2005, 43, 1045–1050. [Google Scholar] [CrossRef] [Green Version]
- Dijkshoorn, L.; Towner, K.J.; Struelens, M. New Approaches for the Generation and Analysis of Microbial Typing Data; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- David, H.P.; Fred, C.T.; Randall, T.H.; Margareta, L.; Melissa, B.M.; Frederick, S.N.; Tang, Y.W.; Alex, V.B. Molecular Microbiology: Diagnostic Principles and Practice; ASM Press: Washington, DC, USA, 2011. [Google Scholar]
- van Belkum, A.; Tassios, P.T.; Dijkshoorn, L.; Haeggman, S.; Cookson, B.; Fry, N.K.; Fussing, V.; Green, J.; Feil, E.; Gerner-Smidt, P.; et al. Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clin. Microbiol. Infect. 2007, 13 (Suppl. S3), 1–46. [Google Scholar] [CrossRef]
- Hyytiä-Trees, E.K.; Cooper, K.; Ribot, E.M.; Gerner-Smidt, P. Recent developments and future prospects in subtyping of foodborne bacterial pathogens. Future Microbiol. 2007, 2, 175–185. [Google Scholar] [CrossRef]
- Fabre, L.; Zhang, J.; Guigon, G.; Le Hello, S.; Guibert, V.; Accou-Demartin, M.; de Romans, S.; Lim, C.; Roux, C.; Passet, V.; et al. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS ONE 2012, 7, e36995. [Google Scholar] [CrossRef]
- Kjeldsen, M.K.; Torpdahl, M.; Pedersen, K.; Nielsen, E.M. Development and comparison of a generic multiple-locus variable-number tandem repeat analysis with pulsed-field gel electrophoresis for typing of Salmonella enterica subsp. enterica. J. Appl. Microbiol. 2015, 119, 1707–1717. [Google Scholar] [CrossRef]
- Everley, R.A.; Mott, T.M.; Toney, D.M.; Croley, T.R. Characterization of Clostridium species utilizing liquid chromatography/mass spectrometry of intact proteins. J. Microbiol. Methods 2009, 77, 152–158. [Google Scholar] [CrossRef]
- Fiori, B.; D’Inzeo, T.; Giaquinto, A.; Menchinelli, G.; Liotti, F.M.; de Maio, F.; De Angelis, G.; Quaranta, G.; Nagel, D.; Tumbarello, M.; et al. Optimized Use of the MALDI BioTyper System and the FilmArray BCID Panel for Direct Identification of Microbial Pathogens from Positive Blood Cultures. J. Clin. Microbiol. 2016, 54, 576–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhns, M.; Zautner, A.E.; Rabsch, W.; Zimmermann, O.; Weig, M.; Bader, O.; Groß, U. Rapid discrimination of Salmonella enterica serovar Typhi from other serovars by MALDI-TOF mass spectrometry. PLoS ONE 2012, 7, e40004. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Bielow, C.; Mastrobuoni, G.; Kempa, S. Proteomics quality control: Quality control software for MaxQuant results. J. Proteome Res. 2016, 15, 777–787. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Serovar | No. of Strains | Source(s) |
|---|---|---|---|
| Enteritidis (9,12:g,m:-) | 5 | Human, food | |
| Typhimurium (4,5,12:i:1,2) | 7 | Human, food | |
| Training | Derby (4,5,12:f,g:-) | 6 | Human, food |
| Rissen(6,7:f,g:-) | 5 | Human, food | |
| London (3,10:l,v:1,6) | 2 | Human, food | |
| Enteritidis (9,12:g,m:-) | 6 | Human, food | |
| Typhimurium (4,5,12:i:1,2) | 3 | Human, food | |
| Testing | Derby (4,5,12:f,g:-) | 2 | Human, food |
| Rissen (6,7:f,g:-) | 2 | Human, food | |
| London (3,10:l,v:1,6) | 1 | Human, food | |
| Sagona. (4,5,12:f,g,s:-) | 1 | Human, food |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Chen, C.; Yang, Y.; Wang, L.; Li, M.; Zhang, P.; Deng, S.; Liang, S. Proteome-Based Serotyping of the Food-Borne Pathogens Salmonella Enterica by Label-Free Mass Spectrometry. Molecules 2022, 27, 4334. https://doi.org/10.3390/molecules27144334
Wang X, Chen C, Yang Y, Wang L, Li M, Zhang P, Deng S, Liang S. Proteome-Based Serotyping of the Food-Borne Pathogens Salmonella Enterica by Label-Free Mass Spectrometry. Molecules. 2022; 27(14):4334. https://doi.org/10.3390/molecules27144334
Chicago/Turabian StyleWang, Xixi, Chen Chen, Yang Yang, Lian Wang, Ming Li, Peng Zhang, Shi Deng, and Shufang Liang. 2022. "Proteome-Based Serotyping of the Food-Borne Pathogens Salmonella Enterica by Label-Free Mass Spectrometry" Molecules 27, no. 14: 4334. https://doi.org/10.3390/molecules27144334