
A Quassinoid Diterpenoid Eurycomanone from Eurycoma longifolia Jack Exerts Anti-Cancer Effect through Autophagy Inhibition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. EN Extraction Method
2.3. Cell Culture
2.4. Cell Viability Assay
2.5. Colony Formation Assay
2.6. Tube Formation Assay
2.7. Transwell Migration Assay
2.8. Flow Cytometry Assay
2.9. Intracellular ROS Level Measurement
2.10. Cellular GSH Level Measurement
2.11. Wound Healing
2.12. Confocal Microscope Assay
2.13. Western Blotting
2.14. In Vivo Study
2.15. H&E Staining Analysis
2.16. Immunohistochemistry
2.17. Statistical Analysis
3. Results
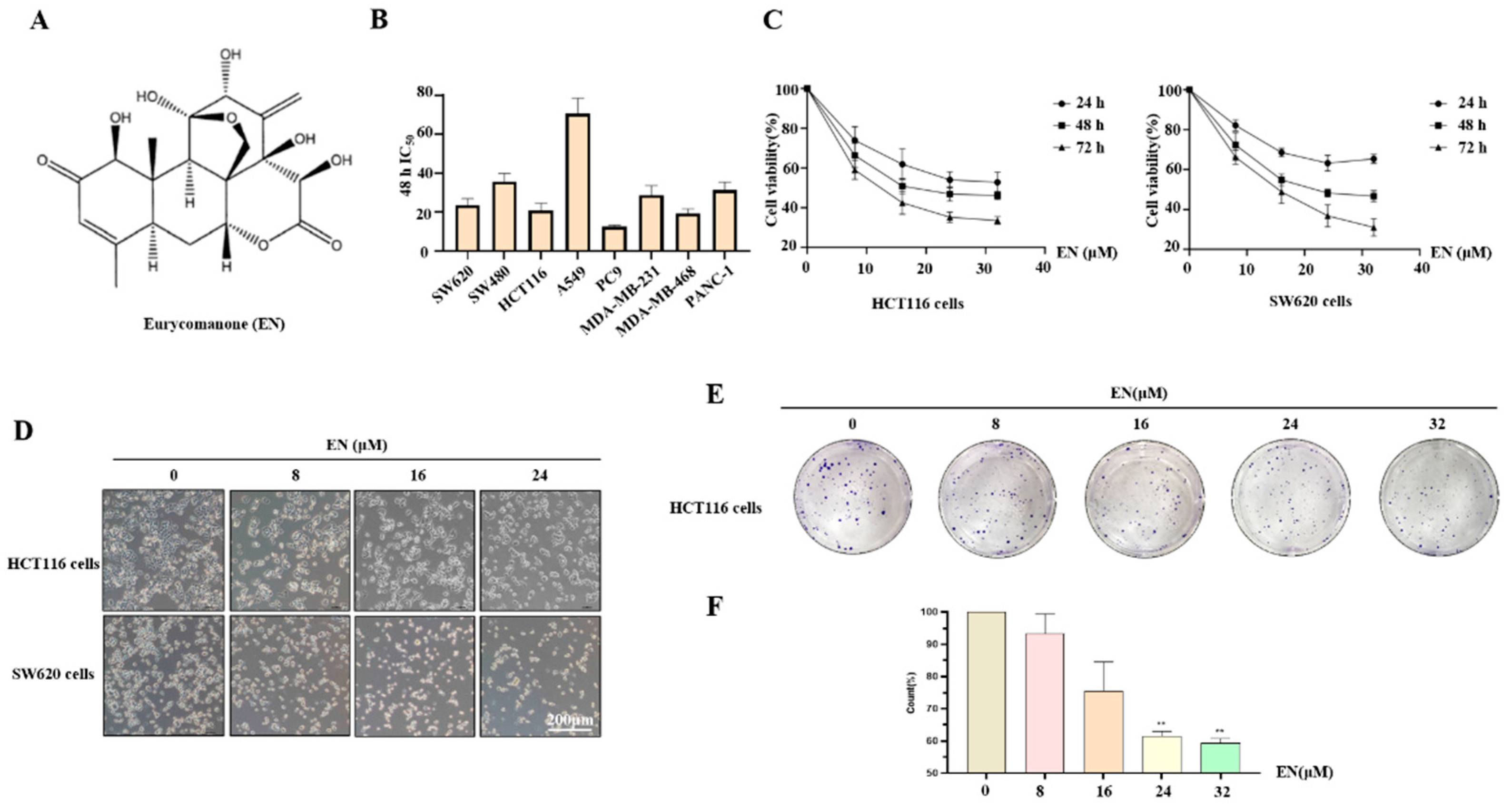
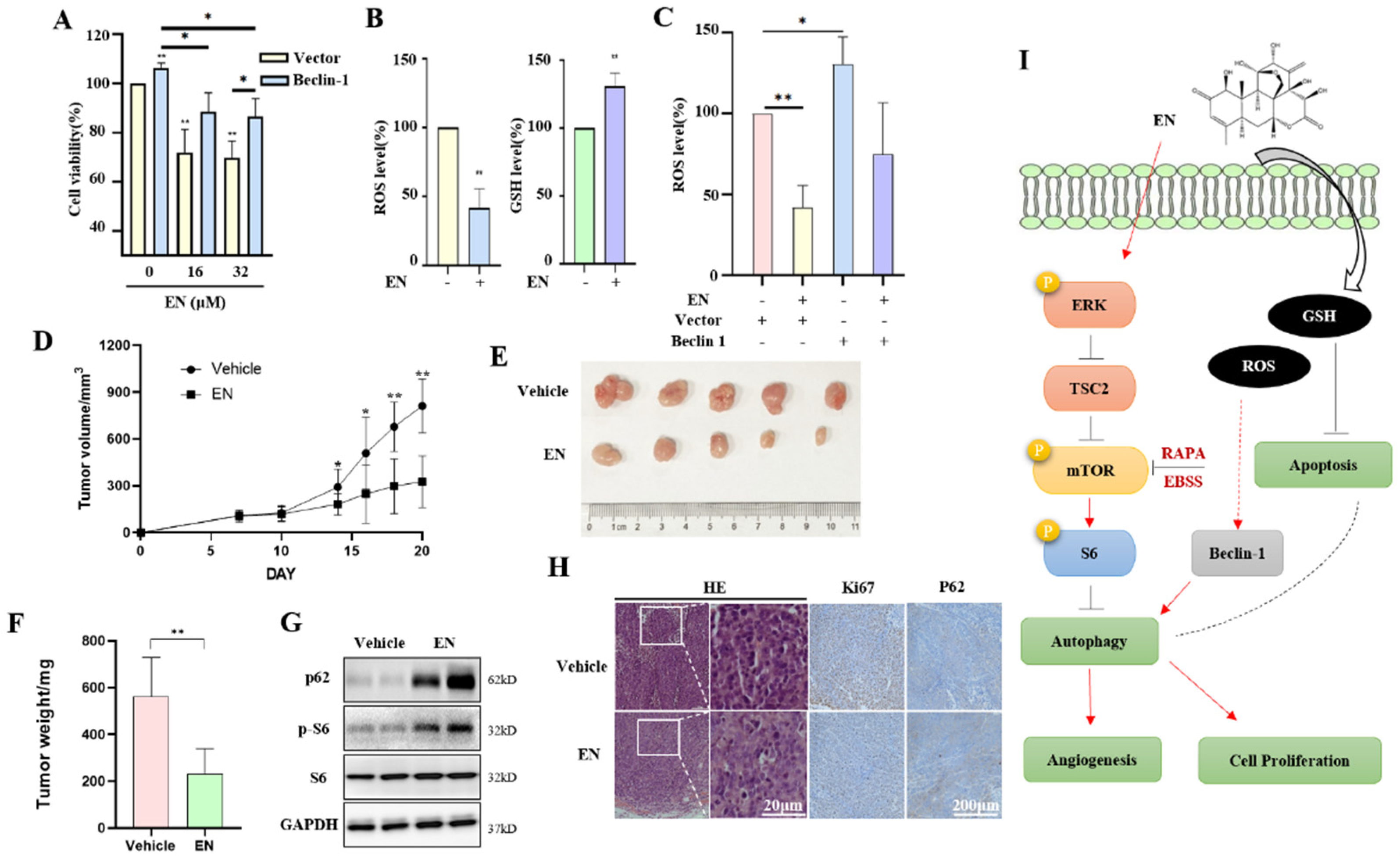
3.1. EN Inhibits Human Colon Cancer Cell Proliferation
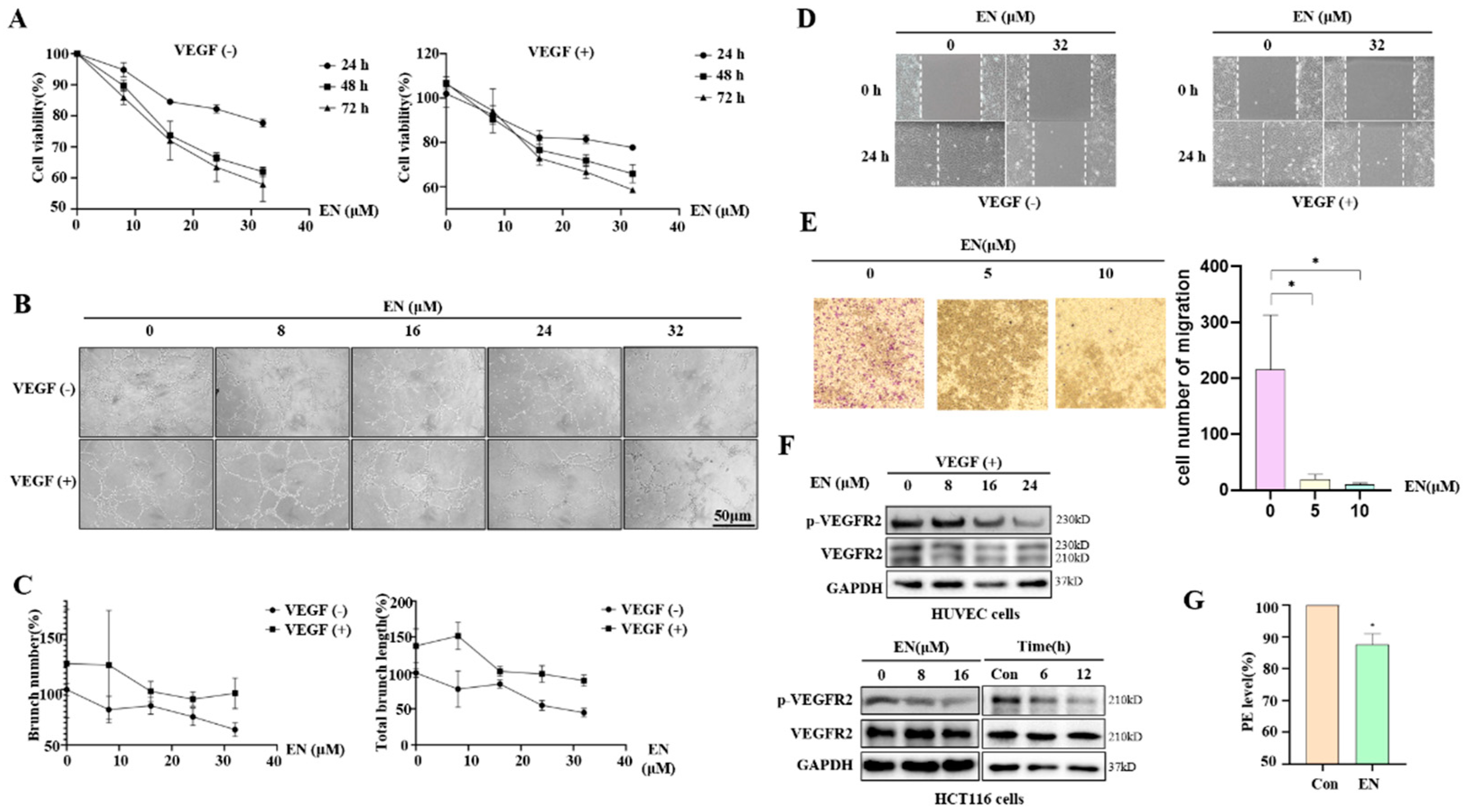
3.2. EN Inhibits Angiogenesis in Human Colon Cancer Cells
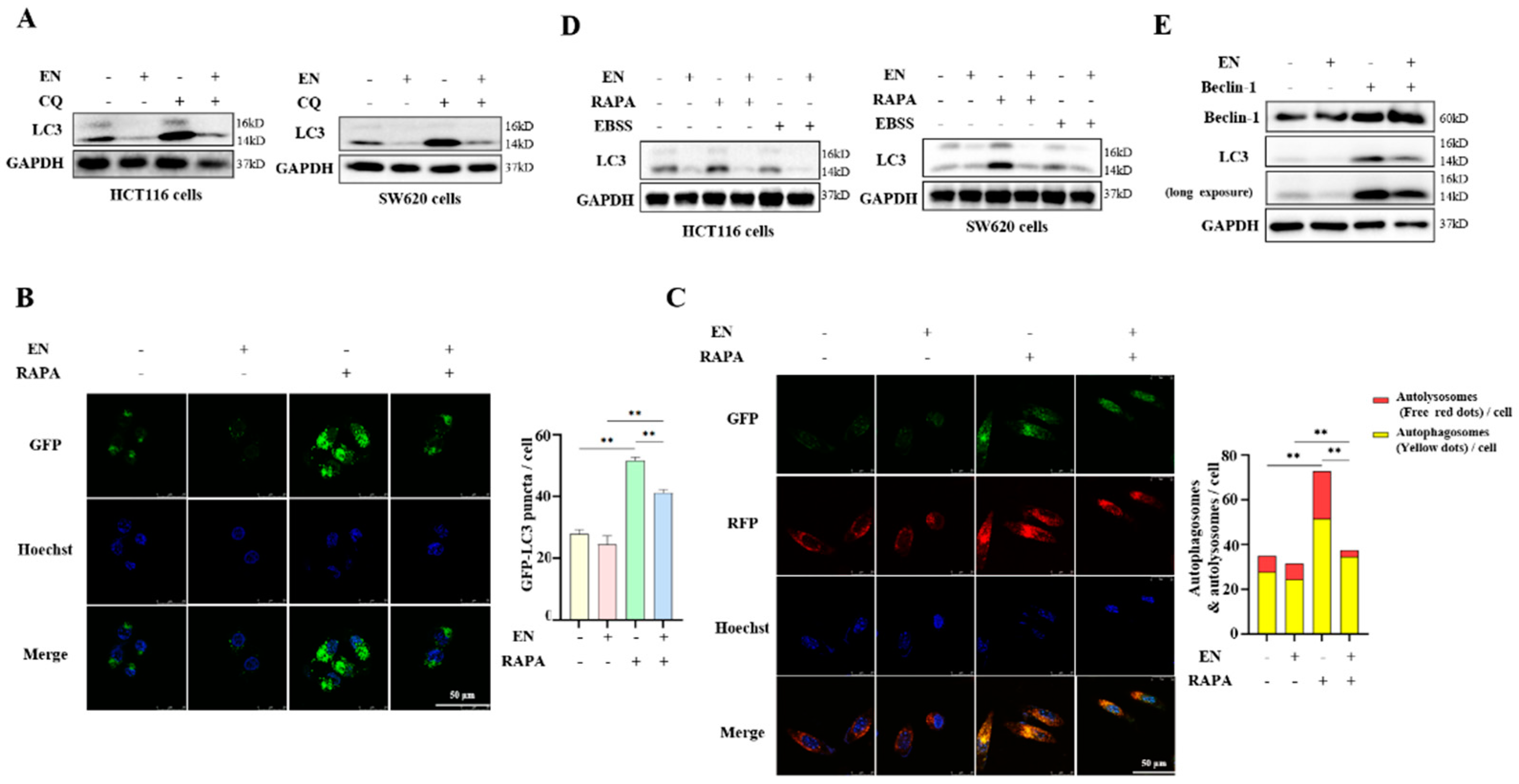
3.3. EN Inhibits Autophagy in Human Colon Cancer Cells
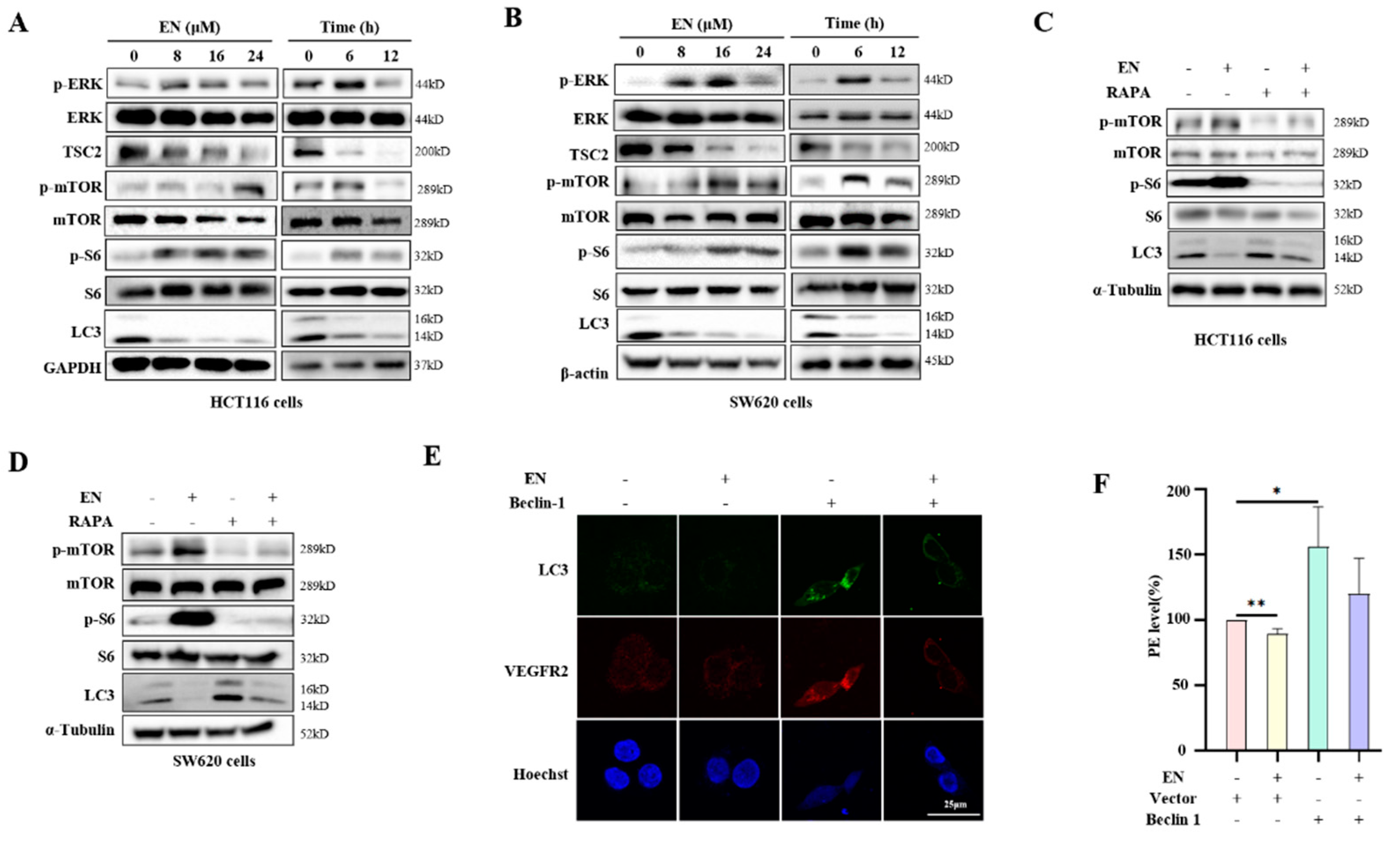
3.4. EN Inhibits Autophagy through Activating the mTOR Pathway
3.5. Autophagy Protects against EN-Caused Cell Death
3.6. EN Inhibits the Tumorigenesis of Colon Cancer and Autophagy In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.; Preet, R.; Mohapatra, P.; Satapathy, S.R.; Siddharth, S.; Tamir, T.; Jain, V.; Bharatam, P.V.; Wyatt, M.; Kundu, C.N. 5-Fluorouracil mediated anti-cancer activity in colon cancer cells is through the induction of Adenomatous Polyposis Coli: Implication of the long-patch base excision repair pathway. DNA Repair 2014, 24, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya, I.; Terada, I.; Yoshizumi, T.; Takino, T.; Nagai, N.; Morita, A.; Fushida, S.; Nishimura, G.; Fujimura, T.; Ohta, T.; et al. Anti-metastatic effect of capecitabine on human colon cancer xenografts in nude mouse rectum. Int. J. Cancer 2004, 112, 135–142. [Google Scholar] [CrossRef]
- Fujita, K.-I.; Kubota, Y.; Ishida, H.; Sasaki, Y. Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World J. Gastroenterol. 2015, 21, 12234–12248. [Google Scholar] [CrossRef]
- Comella, P.; Casaretti, R.; Sandomenico, C.; Avallone, A.; Franco, L. Role of oxaliplatin in the treatment of colorectal cancer. Ther. Clin. Risk Manag. 2009, 5, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Bekaii-Saab, T.; Bupathi, M.; Ahn, D.H. Spotlight on bevacizumab in metastatic colorectal cancer: Patient selection and perspectives. Gastrointest. Cancer Targets Ther. 2016, 6, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Verdaguer, H.; Tabernero, J.; Macarulla, T. Ramucirumab in metastatic colorectal cancer: Evidence to date and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Mazumder, A.; Cerella, C.; Diederich, M. Natural scaffolds in anticancer therapy and precision medicine. Biotechnol. Adv. 2018, 36, 1563–1585. [Google Scholar] [CrossRef] [PubMed]
- Thu, H.E.; Hussain, Z.; Mohamed, I.N.; Shuid, A.N. Eurycoma longifolia, A Potential Phytomedicine for the Treatment of Cancer: Evidence of p53-mediated Apoptosis in Cancerous Cells. Curr. Drug Targets 2018, 19, 1109–1126. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Choe, K.; Yoo, H.H. Review on a Traditional Herbal Medicine, Eurycoma longifolia Jack (Tongkat Ali): Its Traditional Uses, Chemistry, Evidence-Based Pharmacology and Toxicology. Molecules 2016, 21, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Salahi, O.S.A.; Kit-Lam, C.; Majid, A.M.S.A.; Al-Suede, F.S.R.; Saghir, S.A.M.; Abdullah, W.Z.; Ahamed, M.B.K.; Yusoff, N.M. Anti-angiogenic quassinoid-rich fraction from Eurycoma longifolia modulates endothelial cell function. Microvasc. Res. 2013, 90, 30–39. [Google Scholar] [CrossRef]
- Mohamed, A.N.; Vejayan, J.; Yusoff, M.M. Review on Eurycoma longifolia Pharmacological and Phytochemical Properties. J. Appl. Sci. 2015, 15, 831–844. [Google Scholar] [CrossRef] [Green Version]
- Chua, L.S.; Amin, N.A.M.; Neo, J.C.H.; Lee, T.H.; Lee, C.T.; Sarmidi, M.R.; Aziz, R.A. LC–MS/MS-based metabolites of Eurycoma longifolia (Tongkat Ali) in Malaysia (Perak and Pahang). J. Chromatogr. B 2011, 879, 3909–3919. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Huang, F.; Wang, B.-R.; Wang, Y.-G. Role of autophagy in tumorigenesis, metastasis, targeted therapy and drug resistance of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 4643–4651. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, J.; Xie, J.; Liu, Z.; Zhou, Y.; Pan, H.; Han, W. The role of autophagy in colitis-associated colorectal cancer. Signal Transduct. Target. Ther. 2018, 3, 31. [Google Scholar] [CrossRef]
- Xie, C.-M.; Chan, W.Y.; Yu, S.; Zhao, J.; Cheng, C.H. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic. Biol. Med. 2011, 51, 1365–1375. [Google Scholar] [CrossRef]
- Kim, A.D.; Kang, K.A.; Kim, H.S.; Kim, D.H.; Choi, Y.H.; Lee, S.J.; Kim, H.S.; Hyun, J.W. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 2013, 4, e750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakitani, K.; Hirata, Y.; Hikiba, Y.; Hayakawa, Y.; Ihara, S.; Suzuki, H.; Suzuki, N.; Serizawa, T.; Kinoshita, H.; Sakamoto, K.; et al. Inhibition of autophagy exerts anti-colon cancer effects via apoptosis induced by p53 activation and ER stress. BMC Cancer 2015, 15, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, H.; Song, B.; Xu, X.; Zhu, W.; Ju, J. Inhibition of autophagy and tumor growth in colon cancer by miR-502. Oncogene 2012, 32, 1570–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M. Angiogenesis. Circulation 2005, 111, 1556–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, T.M.; Sood, A.K.; Coleman, R.L. Contemporary use of bevacizumab in ovarian cancer. Expert Opin. Biol. Ther. 2012, 13, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Sun, Q.; Brown, M.F.; Dudgeon, C.; Chandler, J.; Xu, X.; Shu, Y.; Zhang, L.; Yu, J. The Multi-Targeted Kinase Inhibitor Sunitinib Induces Apoptosis in Colon Cancer Cells via PUMA. PLoS ONE 2012, 7, e43158. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.C.; Tournigand, C.; Swieboda-Sadlej, A.; Barone, C.A.; Wainberg, Z.A.; Kim, J.G.; Pericay, C.; Pastorelli, D.; Tarazi, J.; Rosbrook, B.; et al. Axitinib or Bevacizumab Plus FOLFIRI or Modified FOLFOX-6 After Failure of First-Line Therapy for Metastatic Colorectal Cancer: A Randomized Phase II Study. Clin. Color. Cancer 2013, 12, 239–247. [Google Scholar] [CrossRef]
- Selvakumaran, M.; Amaravadi, R.K.; Vasilevskaya, I.A.; O’Dwyer, P.J. Autophagy Inhibition Sensitizes Colon Cancer Cells to Antiangiogenic and Cytotoxic Therapy. Clin. Cancer Res. 2013, 19, 2995–3007. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wang, H.; Yang, H.; Zhou, Y.; Tang, L. Autophagy induction by rapamycin ameliorates experimental colitis and improves intestinal epithelial barrier function in IL-10 knockout mice. Int. Immunopharmacol. 2019, 81, 105977. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar] [PubMed]
- Yusuf, H.; Satria, D.; Zulkarnain. The activity of Eurycomanone derivatives on cancer cell lines. Int. J. Pharm. Sci. Res. 2019, 40, 2947–2950. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.-X.; Russell, R.C.; Guan, K.-L. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2013, 9, 1983–1995. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Azad, M.; Chen, Y.; Gibson, S.B. Regulation of Autophagy by Reactive Oxygen Species (ROS): Implications for Cancer Progression and Treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Gibson, S.B. A matter of balance between life and death: Targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy. Autophagy 2010, 6, 835–837. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Panayiotidis, M.I.; Cidlowski, J.A. Glutathione Depletion Is Necessary for Apoptosis in Lymphoid Cells Independent of Reactive Oxygen Species Formation. J. Biol. Chem. 2007, 282, 30452–30465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, P.; Jiang, B.; Li, Y.; Liu, Z.; Zhang, P.; Zhang, M.; Huang, X.; Xiao, X. Autophagy promotes angiogenesis via AMPK/Akt/mTOR signaling during the recovery of heat-denatured endothelial cells. Cell Death Dis. 2018, 9, 1152. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Song, X.; Yang, D.; Bai, D.; Yao, Y.; Lu, N. Anlotinib inhibits angiogenesis via suppressing the activation of VEGFR2, PDGFRβ and FGFR1. Gene 2018, 654, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Wan, X.; Quan, H.; Zheng, M.; Fu, L.; Li, Y.; Lou, L. Preclinical characterization of anlotinib, a highly potent and selective vascular endothelial growth factor receptor-2 inhibitor. Cancer Sci. 2018, 109, 1207–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Li, P.; Luo, Z.; Chen, X.; Zhang, J.; Wang, C.; Chen, P.; Dong, Z. Chloroquine inhibits hepatocellular carcinoma cell growth in vitro and in vivo. Oncol. Rep. 2015, 35, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Ezeldien, S.; Khalil, W.F.; Fayez, M.; Abdel-Daim, M.M. Chloroquine and Gemifloxacin Potentiate the Anticancer Effect of Doxorubicin: In-Vitro and In-Vivo Models. Biomed. Pharmacol. J. 2019, 12, 1613–1620. [Google Scholar] [CrossRef]
- Schott, C.; Ludwig, L.; Mutsaers, A.J.; Foster, R.A.; Wood, G.A. The autophagy inhibitor spautin-1, either alone or combined with doxorubicin, decreases cell survival and colony formation in canine appendicular osteosarcoma cells. PLoS ONE 2018, 13, e0206427. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, G.; Xu, M.; Shu, Y.; Sun, X.; Mai, Y.; Hong, Y.; Zhang, J.; Tian, J. A Quassinoid Diterpenoid Eurycomanone from Eurycoma longifolia Jack Exerts Anti-Cancer Effect through Autophagy Inhibition. Molecules 2022, 27, 4398. https://doi.org/10.3390/molecules27144398
Ye G, Xu M, Shu Y, Sun X, Mai Y, Hong Y, Zhang J, Tian J. A Quassinoid Diterpenoid Eurycomanone from Eurycoma longifolia Jack Exerts Anti-Cancer Effect through Autophagy Inhibition. Molecules. 2022; 27(14):4398. https://doi.org/10.3390/molecules27144398
Chicago/Turabian StyleYe, Guiqin, Mengting Xu, Yuhan Shu, Xin Sun, Yuanyuan Mai, Yupeng Hong, Jianbin Zhang, and Jingkui Tian. 2022. "A Quassinoid Diterpenoid Eurycomanone from Eurycoma longifolia Jack Exerts Anti-Cancer Effect through Autophagy Inhibition" Molecules 27, no. 14: 4398. https://doi.org/10.3390/molecules27144398