
New Polymethoxyflavones from Hottonia palustris Evoke DNA Biosynthesis-Inhibitory Activity in An Oral Squamous Carcinoma (SCC-25) Cell Line

 ,
,  ,
, 
 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Structural Elucidation of Dibenzoylmethane and 5-Hydroxyflavone Derivatives
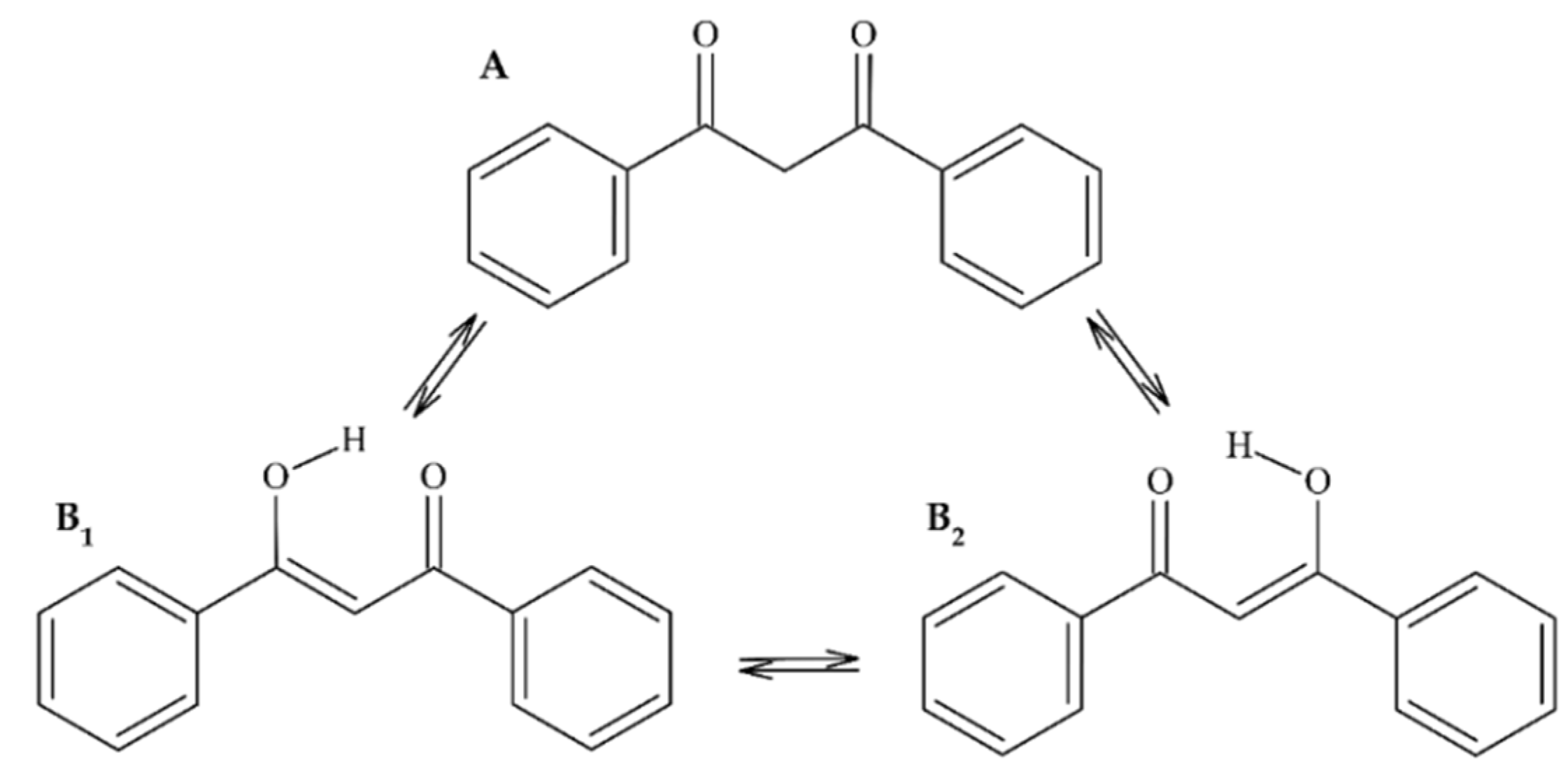
2.1.1. 1,3-Diphenylpropane-1,3-Dione (Dibenzoylmethane) (1)
2.1.2. 5-Hydroxyflavone (Primuletin) (2)
2.1.3. 5-Hydroxy-2′-Methoxyflavone (3)
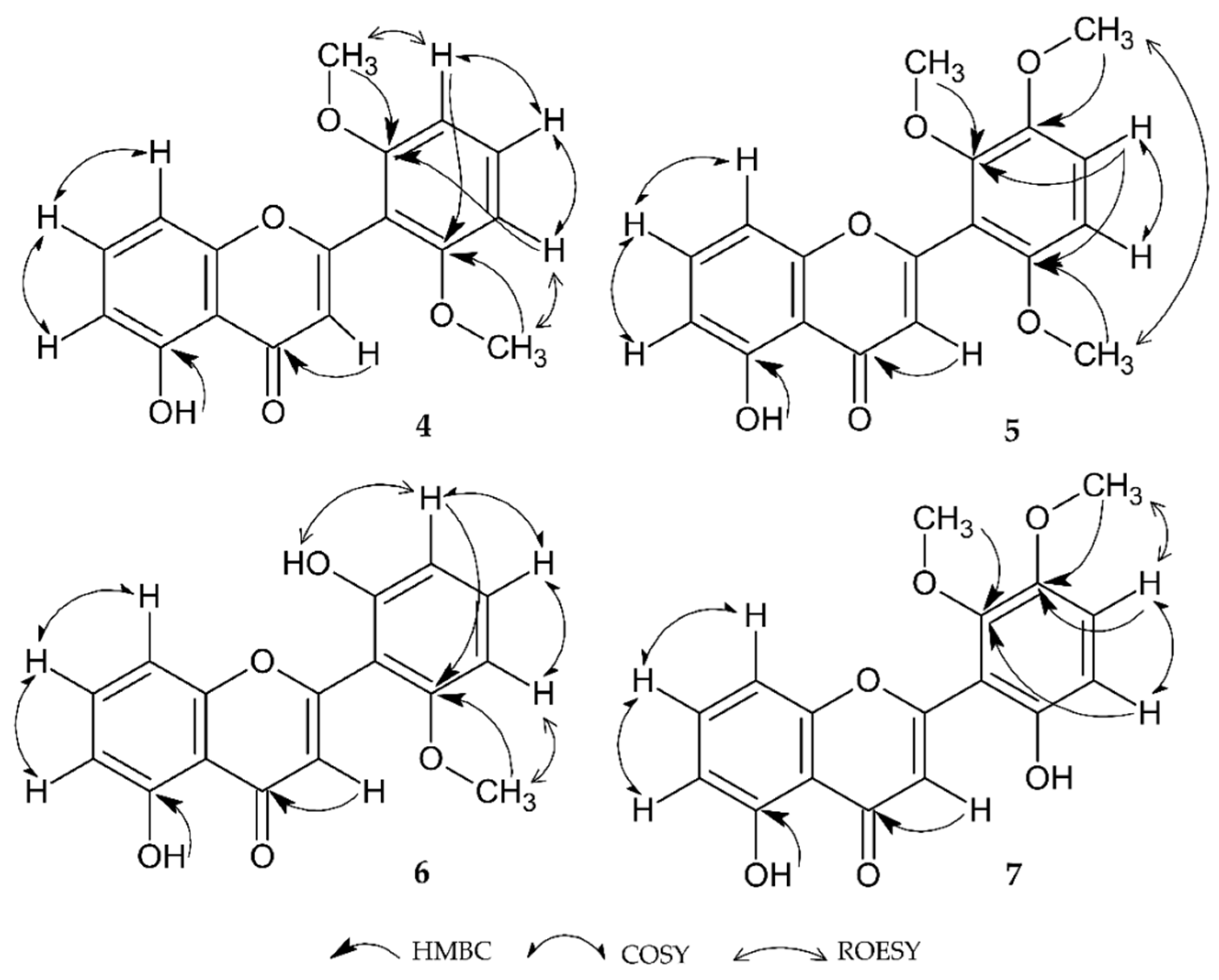
2.1.4. 5-Hydroxy-2′,6′-Dimethoxyflavone (4)
2.1.5. 5-Hydroxy-2′,3′,6′-Trimethoxyflavone (5)
2.1.6. 2′,5-Dihydroxy-6-Methoxyflavone (6)
2.1.7. 5,6′-Dihydroxy-2′,3′-Dimethoxyflavone (7)
2.2. Quantification of Flavones (1–7) in Selected Extracts
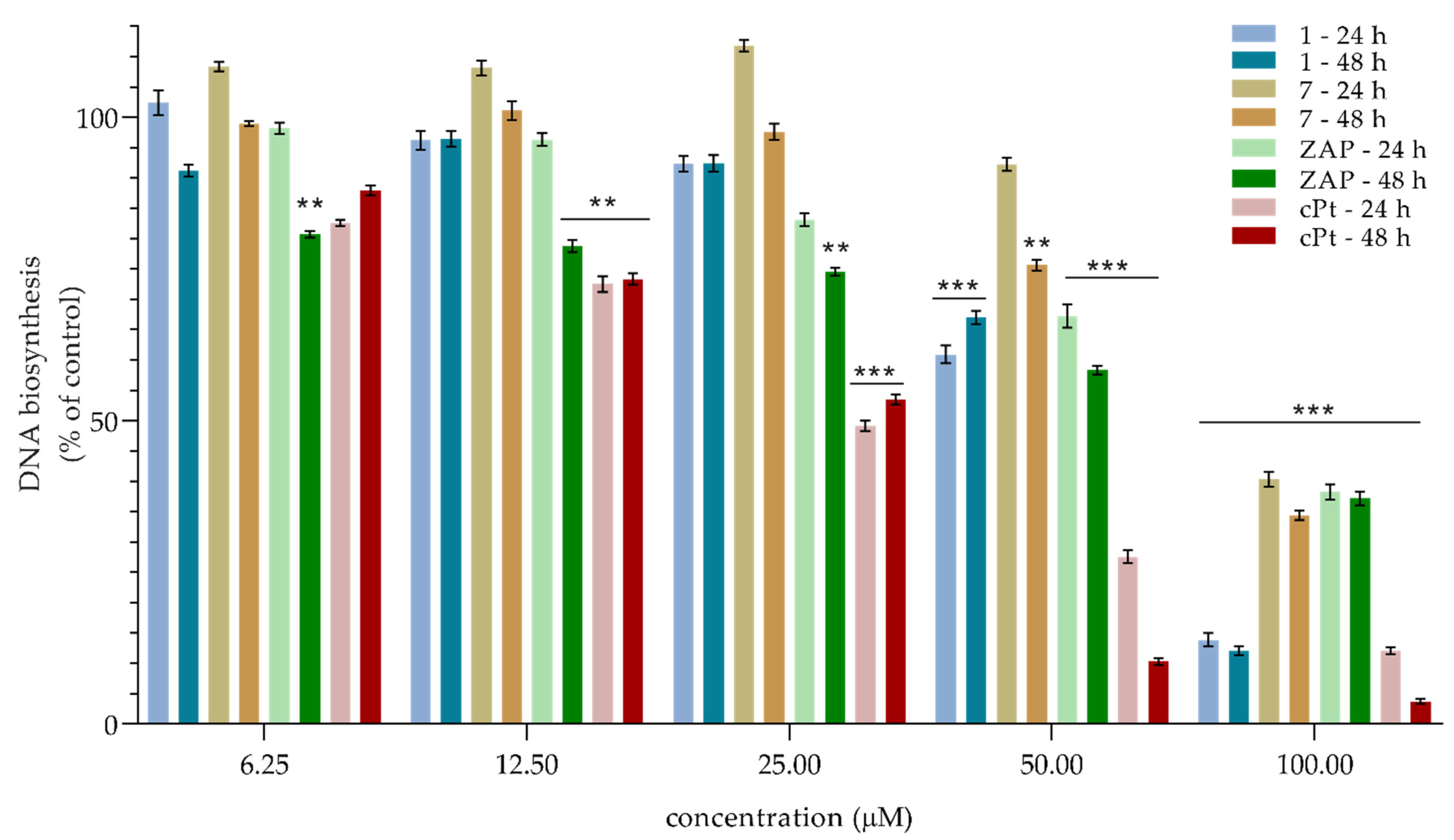
2.3. Cell Viability
3. Materials and Methods
3.1. Chemicals and General Methods
3.2. Plant Material
3.3. Preparation of the Extracts
3.4. Isolation Procedures
3.5. Crystallographic Analysis
3.6. Compound Identification
3.7. Method Development and Flavonoid Quantification in H. palustris by HPLC-PDA
3.7.1. Standard Preparation
3.7.2. Sample Preparation
3.7.3. Analysis Conditions
3.7.4. Specificity, Linearity and Range
3.7.5. Accuracy
3.7.6. Precision
3.7.7. Limit of Detection (LOD) and Limit of Quantification (LOQ)
3.8. Biological Assays
3.8.1. Cell Viability Assay
3.8.2. Proliferation Assay
3.9. Statistical Analysis and Software
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| [α]D | Specific rotation at the sodium D line |
| AlCl3 | Aluminum chloride |
| ANOVA | Analysis of variance |
| c | Concentration |
| C8 | Column contains octyl carbon chain bonded to silica |
| cPT | Cisplatin |
| CDCl3 | Deuterated chloroform |
| COSY | Correlation spectroscopy |
| DEPT | Distortionless enhancement by polarization transfer |
| DMSO | Dimethyl sulfoxide |
| DMSO-d6 | Deuterated dimethyl sulfoxide |
| H3BO3 | Boric acid |
| HCL | Hydrochloric acid |
| HMBC | Heteronuclear multiple bond correlation |
| HMQC | Heteronuclear multiple quantum coherence |
| HPLC-PDA | High-pressure liquid chromatograph with a photodiode array detector |
| HRMS | High-resolution mass spectrometry |
| HSQC | Heteronuclear single quantum coherence |
| IC50 | Median inhibitory concentration |
| LC-MS | Liquid chromatography coupled with mass spectrometry |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| LPLC | Low-pressure liquid chromatography |
| mp. | Melting point |
| MTT | Methyl thiazolyl tetrazolinum |
| NaOAc | Sodium acetate |
| NaOH | Sodium hydroxide |
| NaOMe | Sodium methoxide |
| NMR | Nuclear magnetic resonance |
| PBS | Phosphate-buffered saline |
| ROESY | Rotating-frame nuclear overhauser effect spectroscopy |
| SDS | Sodium dodecyl sulfate |
| UV–Vis | Ultraviolet-visible spectroscopy |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, M.; Kapulnik, Y.; Koltai, H. Plant derived substances with anti-cancer activity: From folklore to practice. Front. Plant. Sci. 2015, 6, 799. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.; Pinto, D. Plant secondary metabolites as anticancer agents: Successes in clinical trials and therapeutic application. Int. J. Mol. Sci. 2018, 19, 263. [Google Scholar] [CrossRef] [Green Version]
- Forni, C.; Rossi, M.; Borromeo, I.; Feriotto, G.; Platamone, G.; Tabolacci, C.; Mischiati, C.; Beninati, S. Flavonoids: A myth or a reality for cancer therapy? Molecules 2021, 26, 3583. [Google Scholar] [CrossRef]
- Khan, A.U.; Dagur, H.S.; Khan, M.; Malik, N.; Alam, M.; Mushtaque, M. Therapeutic role of flavonoids and flavones in cancer prevention: Current trends and future perspectives. Eur. J. Med. Chem. Reports 2021, 3, 100010. [Google Scholar] [CrossRef]
- Ravishankar, D.; Rajora, A.K.; Greco, F.; Osborn, H.M.I. Flavonoids as prospective compounds for anti-cancer therapy. Int. J. Biochem. Cell Biol. 2013, 45, 2821–2831. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Venegas, G.; Sánchez-Carballido, M.A.; Delmas Suárez, C.; Gómez-Mora, J.A.; Bonneau, N. Effects of flavonoids on tongue squamous cell carcinoma. Cell Biol. Int. 2020, 44, 686–720. [Google Scholar] [CrossRef] [PubMed]
- Stanisław, K.; Grzegorz, K. Flora Polski. Rośliny wodne i bagienne; MULTICO Oficyna Wydawnicza: Warsaw, Poland, 2015. [Google Scholar]
- Włodarczyk, M.; Pasikowski, P.; Osiewała, K.; Frankiewicz, A.; Dryś, A.; Gleńsk, M. In search of high-yielding and single-compound-yielding plants: New sources of pharmaceutically important saponins from the Primulaceae family. Biomolecules 2020, 10, 376. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, S.; Rajan, R.; Santhanam, R. Freshwater Phytopharmaceutical Compounds; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Bach Original Flower Remedies. Available online: https://www.bachremedies.com/en-us/ (accessed on 17 May 2022).
- Jakimiuk, K.; Sari, S.; Milewski, R.; Supuran, C.T.; Şöhretoğlu, D.; Tomczyk, M. Flavonoids as tyrosinase inhibitors in in silico and in vitro models: Basic framework of SAR using a statistical modelling approach. J. Enzyme Inhib. Med. Chem. 2022, 37, 427–436. [Google Scholar] [CrossRef]
- Strawa, J.; Szoka, Ł.; Tomczyk, M. Preliminary studies of the chemical composition and cytotoxic activity of Hottonia palustris herb. Planta Med. 2019, 85, 1544. [Google Scholar] [CrossRef]
- Strawa, J.W.; Galanty, A.; Jakimiuk, K.; Grabowska, K.; Tomczyk, M. Cytotoxic effect on human melanoma cell lines and tyrosinase inhibition of Hottonia palustris. Planta Med. 2021, 87, 1304. [Google Scholar] [CrossRef]
- Zawadiak, J.; Mrzyczek, M. Correlation of substituted aromatic β-diketones’ characteristic protons chemical shifts with Hammett substituent constants. Magn. Reson. Chem. 2013, 51, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Wojtulewski, S.; Strawa, J.W.; Tomczyk, M.; Gaweł, M.; Brzeziński, K. A new look at two polymorphic crystal structures of dibenzoylmethane: Relationship between the crystal packing and the hydrogen atom position revealed by quantum chemistry and quantum crystallography methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2020, B76, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S. Antitumor-promoting and anti-inflammatory activities of licorice principles and their modified compounds. In Food Phytochemicals for Cancer Prevention II; American Chemical Society: Washington, DC, USA, 1994; Volume 547, pp. 308–321. [Google Scholar]
- Peng, Y.; Bishop, K.S.; Quek, S.Y. Compositional analysis and aroma evaluation of Feijoa essential oils from New Zealand grown cultivars. Molecules 2019, 24, 2053. [Google Scholar] [CrossRef] [Green Version]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The systematic identification of flavonoids; Springer: Berlin/Heidelberg, Germany, 1970. [Google Scholar]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant flavonoids: Chemical characteristics and biological activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef]
- Nguyen, T.K.P.; Nguyen, K.P.P.; Kamounah, F.S.; Zhang, W.; Hansen, P.E. NMR of a series of novel hydroxyflavothiones. Magn. Reson. Chem. 2009, 47, 1043–1054. [Google Scholar] [CrossRef]
- Ambrozin, A.R.P.; Vieira, P.C.; Fernandes, J.B.; Silva, M.F.d.G.F.d.; Albuquerque, S.d. New pyranoflavones and trypanocidal activity of compounds isolated from Conchocarpus heterophyllus. Quim. Nova 2008, 31, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Tokalov, S.V.; Kind, B.; Wollenweber, E.; Gutzeit, H.O. Biological effects of epicuticular flavonoids from Primula denticulata on human leukemia cells. J. Agric. Food Chem. 2004, 52, 239–245. [Google Scholar] [CrossRef]
- Zakharov, A.M.; Glyzin, V.I.; Ban’kovskii, A.I. Flavonoids from Primula turkestanica. Chem. Nat. Compd. 1971, 7, 814. [Google Scholar] [CrossRef]
- Boltenkov, E.V.; Rybin, V.G.; Zarembo, E.V. Flavones from callus tissue of Iris ensata. Chem. Nat. Compd. 2005, 41, 539–541. [Google Scholar] [CrossRef]
- Wollenweber, E.; Iinuma, M.; Tanaka, T.; Mizuno, M. 5-Hydroxy-6,2′-dimethoxyflavone from Primula denticulata. Phytochemistry 1990, 29, 633–637. [Google Scholar] [CrossRef]
- Somayaji, A.; Shastry, C.S. Interference of antioxidant flavonoids with MTT tetrazolium assay in a cell free system. J. Pharm. Res. Int. 2021, 76–83. [Google Scholar] [CrossRef]
- Habtemariam, S. Catechols and quercetin reduce MTT through iron ions: A possible artefact in cell viability assays. Phyther. Res. 1995, 9, 603–605. [Google Scholar] [CrossRef]
- Strawa, J.W.; Jakimiuk, K.; Tomczyk, M. Zapotin, a polymethoxyflavone, with potential therapeutic attributes. Int. J. Mol. Sci. 2021, 22, 13227. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.A.; Abd El-Wahab, M.F.; Shaheen, U.; Mohammed, A.E.-S.I.; Abdalla, A.N.; Ragab, E.A. Isolation, characterization, complete structural assignment and anticancer activities of the methoxylated flavonoids from Rhamnus disperma roots. Molecules 2021, 26, 5827. [Google Scholar] [CrossRef]
- Jackson, K.M.; DeLeon, M.; Verret, C.R.; Harris, W.B. Dibenzoylmethane induces cell cycle deregulation in human prostate cancer cells. Cancer Lett. 2002, 178, 161–165. [Google Scholar] [CrossRef]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as anticancer agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef] [Green Version]
- Tutin, T.G.; Heywood, V.H.; Burges, N.A.; Moore, D.M.; Valentine, D.H.; Walters, S.M.; Webb, D.A. Flora Europaea; Cambridge University Press: Cambridge, UK, 1972. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Centre Cambridge Crystallographic Data The Cambridge Crystallographic Data Centre (CCDC). Available online: www.ccdc.cam.ac.uk/data_request/cif (accessed on 7 May 2022).
- ICH Harmonised Tripartte Guideline Q2(R1) Validation of Analytical Procedures: Text and Methodology. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29Guideline.pdf (accessed on 7 May 2022).
- Szoka, L.; Nazaruk, J.; Stocki, M.; Isidorov, V. Santin and cirsimaritin from Betula pubescens and Betula pendula buds induce apoptosis in human digestive system cancer cells. J. Cell. Mol. Med. 2021, 25, 11085–11096. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C No. | 4 A | 5 A | 6 B | 7 B | ||||
|---|---|---|---|---|---|---|---|---|
| δ C | δ H | δ C | δ H | δ C | δ H | δ C | δ H | |
| 2 | 162.15 | - | 161.71 | - | 163.08 | - | 162.61 | - |
| 3 | 113.47 | 6.36, s | 113.13 | 6.38, s | 112.68 | 6.4, s | 112.34 | 6.46, s |
| 4 | 183.74 | - | 183.55 | - | 183.01 | - | 182.91 | - |
| 5 | 160.83 | - | 160.86 | - | 159.96 | - | 159.97 | - |
| 6 | 110.76 | 6.8 (dd, J = 8.03, 0.75) | 110.95 | 6.81 (dd, J = 8.28, 0.75) | 110.76 | 6.83 (dd, J = 8.5, 0.85) | 110.86 | 6.84 (d, J = 8.28) |
| 7 | 134.9 | 7.51 (t, J = 8.28) | 135.07 | 7.52 (t, J = 8.28) | 135.91 | 7.65 (t, J = 8.3) | 136.0 | 7.67 (t, J = 8.5) |
| 8 | 107.26 | 6.91 (dd, J = 8.50, 1.0) | 107.15 | 6.91 (dd, J = 8.53, 1.0) | 107.46 | 7.05 (dd, J = 8.5, 0.8) | 107.44 | 7.08 (d, J = 8.28) |
| 9 | 157.53 | - | 157.29 | - | 156.82 | - | 156.64 | - |
| 10 | 110.76 | - | 111.01 | - | 110.05 | - | 110.04 | - |
| 5-OH | - | 12.69, s | - | 12.66, s | - | 12.66, s | - | 12.65, s |
| 1′ | 111.04 | - | 117.18 | - | 109.01 | - | 115.02 | - |
| 2′ | 158.5 | - | 146.99 | - | 156.53 | - | 147.45 | - |
| 3′ | 103.93 | 6.65 (d, J = 8.53) | 148.40 | - | 108.74 | 6.62 (dd, J = 8.3, 1.8) | 145.15 | - |
| 4′ | 132.41 | 7.42 (t, J = 8.5) | 115.49 | 7.03 (d, J = 9.03) | 132.49 | 7.32 (t, J = 8.3) | 117.22 | 7.12, (d, J = 9.03) |
| 5′ | 103.93 | 6.65 (d, J = 8.53) | 106.16 | 6.69 (d, J = 9.03) | 102.23 | 6.62 (dd, J = 8.3, 1.8) | 110.83 | 6.7, (d, J = 9.03) |
| 6′ | 158.5 | - | 151.56 | - | 158.21 | - | 149.47 | - |
| 2′-OH | - | - | - | - | - | 10.13, s | - | - |
| 6′-OH | - | - | - | - | - | - | - | 9.74, s |
| 2′-OCH3 | 55.98 | 3.81, s | 61.48 | 3.88, s | - | - | 60.94 | 3.77, s |
| 3′-OCH3 | - | - | 56.53 | 3.87, s | - | - | 56.55 | 3.78, s |
| 6′-OCH3 | 55.98 | 3.81, s | 56.17 | 3.77, s | 55.95 | 3,75, s | - | - |
| Content of Compounds Expressed as µg/mg of Dry Extract | ||||
|---|---|---|---|---|
| Compounds | HP1A | HP6A | HP7A | HP8A |
| 1 | 0.81 ± 0.01 | 2.7 ± 0.01 | 4.98 ± 0.01 | 0.26 ± 0.01 |
| 2 | 0.29 ± 0.01 | 10.04 ± 0.02 | 1.76 ± 0.01 | 0.22 ± 0.01 |
| 3 | 1.31 ± 0.01 | 58.82 ± 0.44 | 6.02 ± 0.03 | 0.86 ± 0.01 |
| 4 | 1.05 ± 0.01 | 39.19 ± 0.09 | 8.44 ± 0.05 | 0.75 ± 0.01 |
| 5 | 0.67 ± 0.01 | 29.57 ± 0.02 | 5.34 ± 0.02 | 0.51 ± 0.01 |
| 6 | 0.9 ± 0.01 | 24.84 ± 0.15 | 10.21 ± 0.03 | 0.9 ± 0.01 |
| 7 | 1.26 ± 0.01 | 18.76 ± 0.36 | 7.83 ± 0.07 | 0.66 ± 0.01 |
| ZAP | 8.13 ± 0.06 | 154.77 ± 0.19 | 31.73 ± 0.13 | 3.84 ± 0.04 |
| Sum | 14.42 ± 0.01 | 338.68 ± 0.16 | 76.31 ± 0.04 | 7.99 ± 0.01 |
| 24 h | 48 h | |||
|---|---|---|---|---|
| Sample | SCC-25 | Fibroblasts | SCC-25 | Fibroblasts |
| HP1 A | 37.3 ± 1.86 | >200 | 30.3 ± 1.15 | >200 |
| HP6 A | 50.88 ± 2.54 | >200 | 14.9 ± 0.74 | >200 |
| HP7 A | 48.4 ± 2.40 | >200 | 16.7 ± 0.83 | 58.90 ± 2.94 |
| HP8 A | >200 | - | >200 | - |
| 1 B | 39.98 ± 1.89 | >200 | 29.1 ± 1.45 | >200 |
| 2 B | >200 | - | >200 | - |
| 3 B | >200 | - | >200 | - |
| 4 B | >200 | - | >200 | - |
| 5 B | >200 | - | >200 | - |
| 6 B | >200 | - | >200 | - |
| 7 B | 78.2 ± 3.25 | >200 | 40.6 ± 1.65 | >200 |
| ZAP B | 64.94 ± 3.24 | >200 | 20.33 ± 1.01 | >200 |
| cPT B,C | 26.1 ± 1.03 | >200 | 7.58 ± 0.37 | 40.50 ± 2.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strawa, J.W.; Jakimiuk, K.; Szoka, Ł.; Brzezinski, K.; Drozdzal, P.; Pałka, J.A.; Tomczyk, M. New Polymethoxyflavones from Hottonia palustris Evoke DNA Biosynthesis-Inhibitory Activity in An Oral Squamous Carcinoma (SCC-25) Cell Line. Molecules 2022, 27, 4415. https://doi.org/10.3390/molecules27144415
Strawa JW, Jakimiuk K, Szoka Ł, Brzezinski K, Drozdzal P, Pałka JA, Tomczyk M. New Polymethoxyflavones from Hottonia palustris Evoke DNA Biosynthesis-Inhibitory Activity in An Oral Squamous Carcinoma (SCC-25) Cell Line. Molecules. 2022; 27(14):4415. https://doi.org/10.3390/molecules27144415
Chicago/Turabian StyleStrawa, Jakub W., Katarzyna Jakimiuk, Łukasz Szoka, Krzysztof Brzezinski, Paweł Drozdzal, Jerzy A. Pałka, and Michał Tomczyk. 2022. "New Polymethoxyflavones from Hottonia palustris Evoke DNA Biosynthesis-Inhibitory Activity in An Oral Squamous Carcinoma (SCC-25) Cell Line" Molecules 27, no. 14: 4415. https://doi.org/10.3390/molecules27144415
APA StyleStrawa, J. W., Jakimiuk, K., Szoka, Ł., Brzezinski, K., Drozdzal, P., Pałka, J. A., & Tomczyk, M. (2022). New Polymethoxyflavones from Hottonia palustris Evoke DNA Biosynthesis-Inhibitory Activity in An Oral Squamous Carcinoma (SCC-25) Cell Line. Molecules, 27(14), 4415. https://doi.org/10.3390/molecules27144415