
The Therapeutic Potential of Carnosine as an Antidote against Drug-Induced Cardiotoxicity and Neurotoxicity: Focus on Nrf2 Pathway

 ,
, 
 ,
,  ,
,  and
and {kind=link}
Abstract
:1. Introduction
2. Pharmacological Classes Involved in Cardiotoxicity and Neurotoxicity
2.1. Drug-Induced Cardiotoxicity
2.1.1. Cardiotoxicity Induced by Central Nervous System (CNS) Agents
2.1.2. Cardiotoxicity Induced by Anti-Neoplastic Agents
2.1.3. Cardiotoxicity Induced by Anti-Inflammatory Agents
2.1.4. Cardiotoxicity Induced by Anti-Infective Agents
2.2. Drug-Induced Neurotoxicity
2.2.1. Neurotoxicity Induced by Anti-Neoplastic Agents
2.2.2. Neurotoxicity Induced by Antipsychotic Drugs
2.2.3. Neurotoxicity Induced by Immunosuppressive Drugs
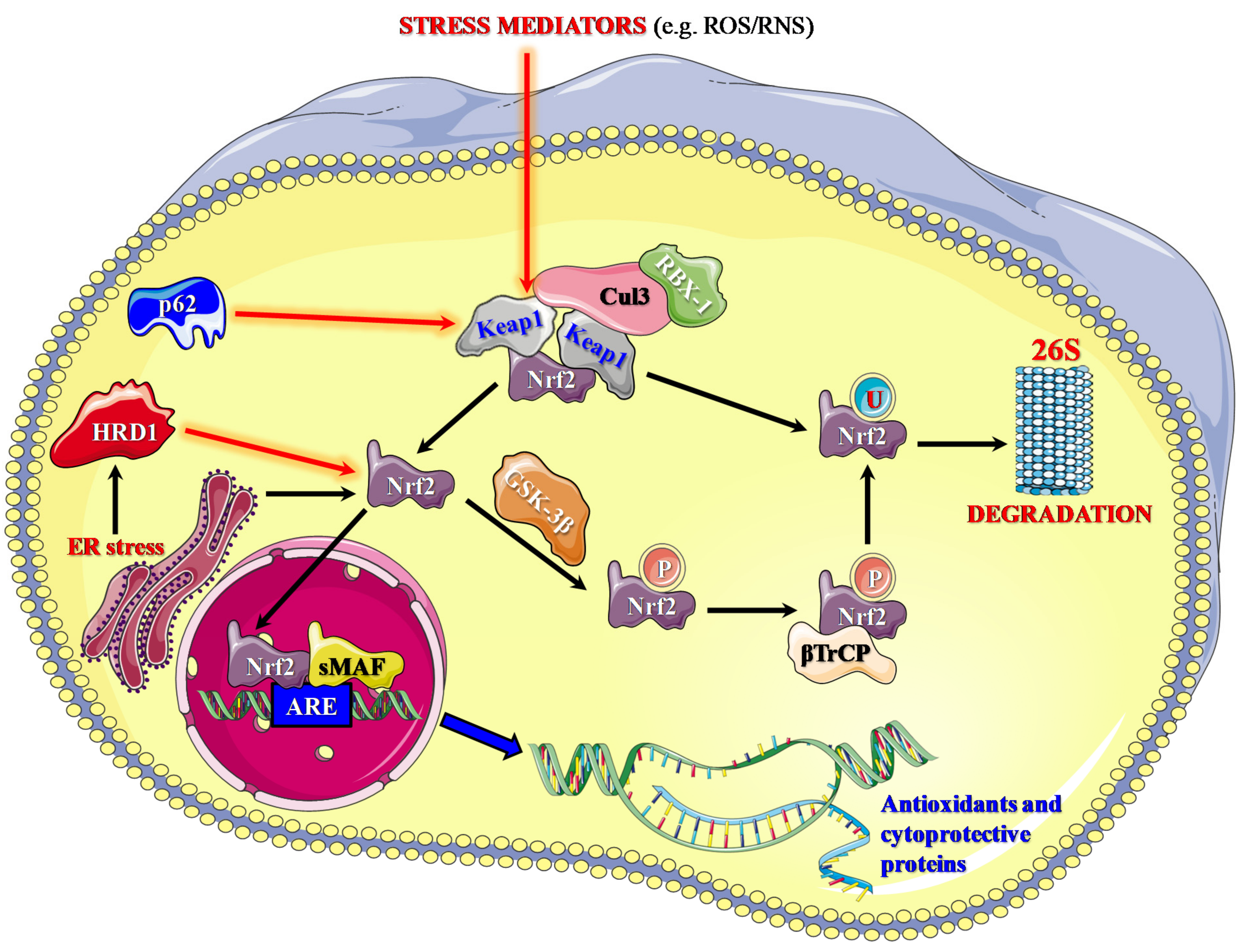
3. The Nrf2 Pathway
4. Nrf2 Pathway Modulation and Its Connection with Cardiotoxicity and Neurotoxicity
5. The Therapeutic Potential of Carnosine as an Antidote in Preventing Drug-Induced Cardiotoxicity and Neurotoxicity through the Modulation of Nrf2
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chacko, B.; Peter, J.V. Antidotes in poisoning. Indian J. Crit. Care Med. 2019, 23, S241–S249. [Google Scholar] [PubMed]
- Mamoshina, P.; Rodriguez, B.; Bueno-Orovio, A. Toward a broader view of mechanisms of drug cardiotoxicity. Cell Rep. Med. 2021, 2, 100216. [Google Scholar] [CrossRef] [PubMed]
- Mohammad Ahmadi Soleimani, S.; Ekhtiari, H.; Cadet, J.L. Drug-induced neurotoxicity in addiction medicine: From prevention to harm reduction. Prog. Brain Res. 2016, 223, 19–41. [Google Scholar] [PubMed]
- Smith, S.W. Drugs and pharmaceuticals: Management of intoxication and antidotes. Mol. Clin. Environ. Toxicol. 2010, 100, 397–460. [Google Scholar]
- Jacobsen, D. The relative efficacy of antidotes. J. Toxicol. Clin. Toxicol. 1995, 33, 705–708. [Google Scholar] [CrossRef]
- Karami, M.; Estachri, M. Principles of toxicotherapy: General and specific therapy. Sch. Acad. J. Pharm. 2015, 4, 153–156. [Google Scholar]
- Zhang, M.Y.; Dugbartey, G.J.; Juriasingani, S.; Sener, A. Hydrogen sulfide metabolite, sodium thiosulfate: Clinical applications and underlying molecular mechanisms. Int. J. Mol. Sci. 2021, 22, 6452. [Google Scholar] [CrossRef]
- Distefano, A.; Caruso, G.; Oliveri, V.; Bellia, F.; Sbardella, D.; Zingale, G.A.; Caraci, F.; Grasso, G. Neuroprotective effect of carnosine is mediated by insulin-degrading enzyme. ACS Chem. Neurosci. 2022, 13, 1588–1593. [Google Scholar] [CrossRef]
- Grasso, M.; Caruso, G.; Godos, J.; Bonaccorso, A.; Carbone, C.; Castellano, S.; Currenti, W.; Grosso, G.; Musumeci, T.; Caraci, F. Improving cognition with nutraceuticals targeting tgf-β1 signaling. Antioxidants 2021, 10, 1075. [Google Scholar] [CrossRef]
- Theofilidis, G.; Bogdanis, G.C.; Koutedakis, Y.; Karatzaferi, C. Monitoring exercise-induced muscle fatigue and adaptations: Making sense of popular or emerging indices and biomarkers. Sports 2018, 6, 153. [Google Scholar] [CrossRef] [Green Version]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and pathophysiology of carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef]
- Derave, W.; Ozdemir, M.S.; Harris, R.C.; Pottier, A.; Reyngoudt, H.; Koppo, K.; Wise, J.A.; Achten, E. Beta-alanine supplementation augments muscle carnosine content and attenuates fatigue during repeated isokinetic contraction bouts in trained sprinters. J. Appl. Physiol. 2007, 103, 1736–1743. [Google Scholar] [CrossRef] [Green Version]
- Trexler, E.T.; Smith-Ryan, A.E.; Stout, J.R.; Hoffman, J.R.; Wilborn, C.D.; Sale, C.; Kreider, R.B.; Jäger, R.; Earnest, C.P.; Bannock, L.; et al. International society of sports nutrition position stand: Beta-alanine. J. Int. Soc. Sports Nutr. 2015, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Spampinato, S.F.; Cardaci, V.; Caraci, F.; Sortino, M.A.; Merlo, S. Β-amyloid and oxidative stress: Perspectives in drug development. Curr. Pharm. Des. 2019, 25, 4771–4781. [Google Scholar] [CrossRef]
- de Campos, R.P.; Siegel, J.M.; Fresta, C.G.; Caruso, G.; da Silva, J.A.; Lunte, S.M. Indirect detection of superoxide in raw 264.7 macrophage cells using microchip electrophoresis coupled to laser-induced fluorescence. Anal. Bioanal. Chem. 2015, 407, 7003–7012. [Google Scholar] [CrossRef]
- Caruso, G.; Godos, J.; Privitera, A.; Lanza, G.; Castellano, S.; Chillemi, A.; Bruni, O.; Ferri, R.; Caraci, F.; Grosso, G. Phenolic acids and prevention of cognitive decline: Polyphenols with a neuroprotective role in cognitive disorders and alzheimer’s disease. Nutrients 2022, 14, 819. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. Nrf2 as a therapeutic target in neurodegenerative diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [Green Version]
- Lipshultz, S.E.; Diamond, M.B.; Franco, V.I.; Aggarwal, S.; Leger, K.; Santos, M.V.; Sallan, S.E.; Chow, E.J. Managing chemotherapy-related cardiotoxicity in survivors of childhood cancers. Pediatric Drugs 2014, 16, 373–389. [Google Scholar] [CrossRef]
- Jain, D.; Ahmad, T.; Cairo, M.; Aronow, W. Cardiotoxicity of cancer chemotherapy: Identification, prevention and treatment. Ann. Transl. Med. 2017, 5, 348. [Google Scholar] [CrossRef] [Green Version]
- Madonna, R. Early diagnosis and prediction of anticancer drug-induced cardiotoxicity: From cardiac imaging to “omics” technologies. Rev. Española Cardiol. 2017, 70, 576–582. [Google Scholar] [CrossRef]
- Sadurska, E. Current views on anthracycline cardiotoxicity in childhood cancer survivors. Pediatric Cardiol. 2015, 36, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Iqubal, A.; Haque, S.E.; Sharma, S.; Ansari, M.A.; Khan, V.; Iqubal, M.K. Clinical updates on drug-induced cardiotoxicity. Int. J. Pharm. Sci. Res. 2018, 9, 16–26. [Google Scholar]
- Fung, M.; Thornton, A.; Mybeck, K.; Wu, J.H.-h.; Hornbuckle, K.; Muniz, E. Evaluation of the characteristics of safety withdrawal of prescription drugs from worldwide pharmaceutical markets-1960 to 1999. Drug Inf. J. 2001, 35, 293–317. [Google Scholar] [CrossRef]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Curto, M.; Girardi, N.; Lionetto, L.; Ciavarella, G.M.; Ferracuti, S.; Baldessarini, R.J. Systematic review of clozapine cardiotoxicity. Curr. Psychiatry Rep. 2016, 18, 68. [Google Scholar] [CrossRef]
- Marano, G.; Traversi, G.; Romagnoli, E.; Catalano, V.; Lotrionte, M.; Abbate, A.; Biondi-Zoccai, G.; Mazza, M. Cardiologic side effects of psychotropic drugs. J. Geriatr. Cardiol. 2011, 8, 243. [Google Scholar]
- Li, K.J.; Gurrera, R.J.; Delisi, L.E. Potentially fatal outcomes associated with clozapine. Schizophr. Res. 2018, 199, 386–389. [Google Scholar] [CrossRef]
- Lee, C.S. Mechanisms of cardiotoxicity and the development of heart failure. Crit. Care Nurs. Clin. N. Am. 2015, 27, 469–481. [Google Scholar] [CrossRef]
- Klimas, J. Drug-Induced Cardiomyopathies; Intech Open Access Publisher: London, UK, 2012. [Google Scholar]
- Ford, G.A.; Wood, S.M.; Daly, A.K. Cyp2d6 and cyp2c19 genotypes of patients with terodiline cardiotoxicity identified through the yellow card system. Br. J. Clin. Pharmacol. 2000, 50, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pavia, P.; Kim, Y.; Restrepo-Cordoba, M.A.; Lunde, I.G.; Wakimoto, H.; Smith, A.M.; Toepfer, C.N.; Getz, K.; Gorham, J.; Patel, P.; et al. Genetic variants associated with cancer therapy-induced cardiomyopathy. Circulation 2019, 140, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Sloan, J.A.; Goldberg, R.M.; Sargent, D.J.; Vargas-Chanes, D.; Nair, S.; Cha, S.S.; Novotny, P.J.; Poon, M.A.; O’Connell, M.J.; Loprinzi, C.L. Women experience greater toxicity with fluorouracil-based chemotherapy for colorectal cancer. J. Clin. Oncol. 2002, 20, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Conrad, S.K.; Catalano, M.C.; Catalano, G. The use of fluoxetine in a patient with takotsubo cardiomyopathy. J. Psychiatr Pract 2016, 22, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Thanacoody, H.K.; Thomas, S.H. Tricyclic antidepressant poisoning: Cardiovascular toxicity. Toxicol. Rev. 2005, 24, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Kecskemeti, V. Cardiovascular side effects of new antidepressants and antipsychotics: New drugs, old concerns? Curr. Pharm. Des. 2004, 10, 2463–2475. [Google Scholar] [CrossRef] [Green Version]
- Christie, L.E.; Picard, J.; Weinberg, G.L. Local anaesthetic systemic toxicity. BJA Educ. 2015, 15, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, B.G.; Rezkalla, S.; Kloner, R.A. Cardiovascular effects of cocaine. Circulation 2010, 122, 2558–2569. [Google Scholar] [CrossRef] [Green Version]
- Graf, B.M. The cardiotoxicity of local anesthetics: The place of ropivacaine. Curr. Top. Med. Chem. 2001, 1, 207–214. [Google Scholar] [CrossRef]
- Horácek, M.; Vymazal, T. Lidocaine not so innocent: Cardiotoxicity after topical anaesthesia for bronchoscopy. Indian J. Anaesth. 2012, 56, 95–96. [Google Scholar] [CrossRef]
- Beach, S.R.; Celano, C.M.; Noseworthy, P.A.; Januzzi, J.L.; Huffman, J.C. Qtc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics 2013, 54, 105–122. [Google Scholar] [CrossRef]
- Waller, E.A.; Kaplan, J. Pergolide-associated valvular heart disease. Compr. Ther. 2006, 32, 94–101. [Google Scholar] [CrossRef]
- Antonini, A.; Poewe, W. Fibrotic heart-valve reactions to dopamine-agonist treatment in parkinson’s disease. Lancet Neurol. 2007, 6, 826–829. [Google Scholar] [CrossRef]
- Janssen, W.; Schymura, Y.; Novoyatleva, T.; Kojonazarov, B.; Boehm, M.; Wietelmann, A.; Luitel, H.; Murmann, K.; Krompiec, D.R.; Tretyn, A.; et al. 5-ht2b receptor antagonists inhibit fibrosis and protect from rv heart failure. Biomed Res. Int. 2015, 2015, 438403. [Google Scholar] [CrossRef]
- Andrejak, M.; Tribouilloy, C. Drug-induced valvular heart disease: An update. Arch. Cardiovasc. Dis. 2013, 106, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Adler, A.; Viskin, S.; Bhuiyan, Z.A.; Eisenberg, E.; Rosso, R. Propoxyphene-induced torsades de pointes. Heart Rhythm. 2011, 8, 1952–1954. [Google Scholar] [CrossRef]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Aminkeng, F.; Ross, C.J.; Rassekh, S.R.; Hwang, S.; Rieder, M.J.; Bhavsar, A.P.; Smith, A.; Sanatani, S.; Gelmon, K.A.; Bernstein, D. Recommendations for genetic testing to reduce the incidence of anthracycline-induced cardiotoxicity. Br. J. Clin. Pharmacol. 2016, 82, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-induced cardiotoxicity: Overview of the roles of oxidative stress. Oxid. Med. Cell. Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef] [Green Version]
- Novo, G.; Cadeddu, C.; Sucato, V.; Pagliaro, P.; Romano, S.; Tocchetti, C.G.; Zito, C.; Longobardo, L.; Nodari, S.; Penco, M. Role of biomarkers in monitoring antiblastic cardiotoxicity. J. Cardiovasc. Med. 2016, 17 (Suppl. S1), S27–S34. [Google Scholar] [CrossRef]
- Varricchi, G.; Ameri, P.; Cadeddu, C.; Ghigo, A.; Madonna, R.; Marone, G.; Mercurio, V.; Monte, I.; Novo, G.; Parrella, P. Antineoplastic drug-induced cardiotoxicity: A redox perspective. Front. Physiol. 2018, 9, 167. [Google Scholar] [CrossRef]
- Popat, R.; Oakervee, H.E.; Hallam, S.; Curry, N.; Odeh, L.; Foot, N.; Esseltine, D.L.; Drake, M.; Morris, C.; Cavenagh, J.D. Bortezomib, doxorubicin and dexamethasone (pad) front-line treatment of multiple myeloma: Updated results after long-term follow-up. Br. J. Haematol. 2008, 141, 512–516. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Gori, T.; Keaney, J.F., Jr.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef] [Green Version]
- Simůnek, T.; Stérba, M.; Popelová, O.; Adamcová, M.; Hrdina, R.; Gersl, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar] [CrossRef]
- Cipriani, P.; Ruscitti, P.; Carubbi, F.; Liakouli, V.; Giacomelli, R. Methotrexate: An old new drug in autoimmune disease. Expert Rev. Clin. Immunol. 2014, 10, 1519–1530. [Google Scholar] [CrossRef]
- Al-Taher, A.Y.; Morsy, M.A.; Rifaai, R.A.; Zenhom, N.M.; Abdel-Gaber, S.A. Paeonol attenuates methotrexate-induced cardiac toxicity in rats by inhibiting oxidative stress and suppressing tlr4-induced nf-κb inflammatory pathway. Mediat. Inflamm. 2020, 2020, 8641026. [Google Scholar] [CrossRef] [Green Version]
- Bălănescu, A.R.; Bojincă, V.C.; Bojincă, M.; Donisan, T.; Bălănescu, S.M. Cardiovascular effects of methotrexate in immune-mediated inflammatory diseases. Exp. Ther. Med. 2019, 17, 1024–1029. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.A.; Antin, J.H.; Guinan, E.C.; Rappeport, J.M. Cyclophosphamide cardiotoxicity: An analysis of dosing as a risk factor. Blood 1986, 68, 1114–1118. [Google Scholar] [CrossRef] [Green Version]
- Madondo, M.T.; Quinn, M.; Plebanski, M. Low dose cyclophosphamide: Mechanisms of t cell modulation. Cancer Treat Rev. 2016, 42, 3–9. [Google Scholar] [CrossRef]
- Feenstra, J.; Grobbee, D.E.; Remme, W.J.; Stricker, B.H. Drug-induced heart failure. J. Am. Coll Cardiol. 1999, 33, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Lewis, W. Cardiomyopathy, nucleoside reverse transcriptase inhibitors and mitochondria are linked through aids and its therapy. Mitochondrion 2004, 4, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Bai, B.; Tian, C.; Li, D.; Yu, H.; Song, B.; Li, B.; Chu, X. The molecular mechanisms of cardiotoxicity induced by her2, vegf, and tyrosine kinase inhibitors: An updated review. Cardiovasc. Drugs Ther. 2021, 36, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.I.; Ai, D.I. Cardiotoxicity associated with targeted cancer therapies. Mol. Clin. Oncol. 2016, 4, 675–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pondé, N.F.; Lambertini, M.; de Azambuja, E. Twenty years of anti-her2 therapy-associated cardiotoxicity. ESMO Open 2016, 1, e000073. [Google Scholar] [CrossRef] [Green Version]
- Mooney, L.; Skinner, M.; Coker, S.J.; Currie, S. Effects of acute and chronic sunitinib treatment on cardiac function and calcium/calmodulin-dependent protein kinase ii. Br. J. Pharmacol. 2015, 172, 4342–4354. [Google Scholar] [CrossRef] [Green Version]
- Doherty, K.R.; Talbert, D.R.; Trusk, P.B.; Moran, D.M.; Shell, S.A.; Bacus, S. Structural and functional screening in human induced-pluripotent stem cell-derived cardiomyocytes accurately identifies cardiotoxicity of multiple drug types. Toxicol. Appl. Pharmacol. 2015, 285, 51–60. [Google Scholar] [CrossRef]
- Zeglinski, M.; Ludke, A.; Jassal, D.S.; Singal, P.K. Trastuzumab-induced cardiac dysfunction: A ‘dual-hit’. Exp. Clin. Cardiol. 2011, 16, 70–74. [Google Scholar]
- Economopoulou, P.; Kotsakis, A.; Kapiris, I.; Kentepozidis, N. Cancer therapy and cardiovascular risk: Focus on bevacizumab. Cancer Manag. Res. 2015, 7, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Sable, H.; Miller, M.; Nelms, J.; Meyer, A.; Poon, E.; Eubig, P.; Schantz, S. Behavioral pharmacology of cocaine and amphetamine in rats perinatally exposed to polychlorinated biphenyls (pcbs). Neurotoxicol. Teratol. 2014, 43, 88. [Google Scholar] [CrossRef]
- Pallavi, R.; Talebi, S.; Hassen, G.; Visco, F.; Popis-Matejak, B. 1259: Cocaine induced cardiotoxicity. Crit. Care Med. 2014, 42, A1654. [Google Scholar] [CrossRef]
- Liaudet, L.; Calderari, B.; Pacher, P. Pathophysiological mechanisms of catecholamine and cocaine-mediated cardiotoxicity. Heart Fail. Rev. 2014, 19, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Maraj, S.; Figueredo, V.M.; Lynn Morris, D. Cocaine and the heart. Clin. Cardiol. 2010, 33, 264–269. [Google Scholar] [CrossRef]
- Gardner, J.D.; Mouton, A.J. Alcohol effects on cardiac function. Compr. Physiol. 2015, 5, 791–802. [Google Scholar]
- Gardenhire, D.S. Drugs affecting the central nervous system. In Rau’s Respiratory Care Pharmacology; Elsevier: Amsterdam, The Netherlands, 2015; 330p. [Google Scholar]
- Fernandez-Sola, J. Cardiovascular risks and benefits of moderate and heavy alcohol consumption. Nat. Rev. Cardiol. 2015, 12, 576–587. [Google Scholar] [CrossRef]
- Meseeha, M.G.; Kolade, V.O.; Attia, M.N. Partially reversible bortezomib-induced cardiotoxicity: An unusual cause of acute cardiomyopathy. J. Community Hosp. Intern. Med. Perspect. 2015, 5, 28982. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Yin, J.; Wei, J.; Shang, Z. Incidence and risk of cardiotoxicity associated with bortezomib in the treatment of cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e87671. [Google Scholar]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L. Fulminant myocarditis with combination immune checkpoInt. blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef]
- Varricchi, G.; Marone, G.; Mercurio, V.; Galdiero, M.R.; Bonaduce, D.; Tocchetti, C.G. Immune checkpoInt. inhibitors and cardiac toxicity: An emerging issue. Curr. Med. Chem. 2018, 25, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Kourie, H.R.; Klastersky, J. Immune checkpoInt. inhibitors side effects and management. Immunotherapy 2016, 8, 799–807. [Google Scholar] [CrossRef]
- Mason, R.P.; Walter, M.F.; Day, C.A.; Jacob, R.F. A biological rationale for the cardiotoxic effects of rofecoxib: Comparative analysis with other cox-2 selective agents and nsaids. Subcell. Biochem. 2007, 42, 175–190. [Google Scholar]
- Chandra, M.; Kent, P. Cyclo-oxygenase-2 inhibitors and peripheral thrombosis—A case report demonstrating a possible adverse effect. EJVES Extra 2009, 17, 14–16. [Google Scholar] [CrossRef] [Green Version]
- Keserü, G.M. Prediction of herg potassium channel affinity by traditional and hologrAm. qsar methods. Bioorg. Med. Chem. Lett. 2003, 13, 2773–2775. [Google Scholar] [CrossRef]
- Albert, R.K.; Schuller, J.L. Macrolide antibiotics and the risk of cardiac arrhythmias. Am. J. Respir. Crit. Care Med. 2014, 189, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.E.; Mazur, A.; Yang, T.; Roden, D.M. Potassium current antagonist properties and proarrhythmic consequences of quinolone antibiotics. J. Pharmacol. Exp. Ther. 2001, 296, 806–810. [Google Scholar]
- Lewis, W.; Day, B.J.; Copeland, W.C. Mitochondrial toxicity of nrti antiviral drugs: An integrated cellular perspective. Nat. Rev. Drug Discov. 2003, 2, 812–822. [Google Scholar] [CrossRef]
- Nomura, R.; Sato, T.; Sato, Y.; Medin, J.A.; Kushimoto, S.; Yanagisawa, T. Azidothymidine-triphosphate impairs mitochondrial dynamics by disrupting the quality control system. Redox Biol. 2017, 13, 407–417. [Google Scholar] [CrossRef]
- Durante-Mangoni, E.; Parrella, A.; Vitrone, M.; Rago, A.; Pafundi, P.C.; Nigro, G.; Utili, R.; Russo, V. Electrophysiological adverse effects of direct acting antivirals in patients with chronic hepatitis c. J. Clin. Pharmacol. 2017, 57, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Haverkamp, W.; Breithardt, G.; Camm, A.J.; Janse, M.J.; Rosen, M.R.; Antzelevitch, C.; Escande, D.; Franz, M.; Malik, M.; Moss, A.; et al. The potential for qt prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: Clinical and regulatory implications. Report on a policy conference of the european society of cardiology. Cardiovasc. Res. 2000, 47, 219–233. [Google Scholar] [CrossRef]
- Dennis, A.T.; Wang, L.; Wan, H.; Nassal, D.; Deschenes, I.; Ficker, E. Molecular determinants of pentamidine-induced herg trafficking inhibition. Mol. Pharmacol. 2012, 81, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Gibson, J.K.; Yue, Y.; Bronson, J.; Palmer, C.; Numann, R. Human stem cell-derived cardiomyocytes detect drug-mediated changes in action potentials and ion currents. J. Pharmacol. Toxicol. Methods 2014, 70, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Katchman, A.N.; Koerner, J.; Tosaka, T.; Woosley, R.L.; Ebert, S.N. Comparative evaluation of herg currents and qt intervals following challenge with suspected torsadogenic and nontorsadogenic drugs. J. Pharmacol. Exp. Ther. 2006, 316, 1098–1106. [Google Scholar] [CrossRef]
- Bal-Price, A.K.; Hogberg, H.T.; Buzanska, L.; Coecke, S. Relevance of in vitro neurotoxicity testing for regulatory requirements: Challenges to be considered. Neurotoxicol. Teratol. 2010, 32, 36–41. [Google Scholar] [CrossRef]
- Crofton, K.M.; Mundy, W.R.; Shafer, T.J. Developmental neurotoxicity testing: A path forward. Congenit. Anom. 2012, 52, 140–146. [Google Scholar] [CrossRef]
- Schultz, L.; Zurich, M.G.; Culot, M.; da Costa, A.; Landry, C.; Bellwon, P.; Kristl, T.; Hörmann, K.; Ruzek, S.; Aiche, S.; et al. Evaluation of drug-induced neurotoxicity based on metabolomics, proteomics and electrical activity measurements in complementary cns in vitro models. Toxicol. In Vitro 2015, 30, 138–165. [Google Scholar] [CrossRef]
- Coecke, S.; Ahr, H.; Blaauboer, B.J.; Bremer, S.; Casati, S.; Castell, J.; Combes, R.; Corvi, R.; Crespi, C.L.; Cunningham, M.L. Metabolism: A bottleneck in in vitro toxicological test development: The report and recommendations of ecvAm. workshop 54. Altern. Lab. Anim. 2006, 34, 49–84. [Google Scholar] [CrossRef]
- Fresta, C.G.; Fidilio, A.; Caruso, G.; Caraci, F.; Giblin, F.J.; Leggio, G.M.; Salomone, S.; Drago, F.; Bucolo, C. A new human blood-retinal barrier model based on endothelial cells, pericytes, and astrocytes. Int. J. Mol. Sci. 2020, 21, 1636. [Google Scholar] [CrossRef] [Green Version]
- Harry, G.J.; Tiffany-Castiglioni, E. Evaluation of neurotoxic potential by use of in vitro systems. Expert Opin. Drug Metab. Toxicol. 2005, 1, 701–713. [Google Scholar] [CrossRef]
- Hallier-Vanuxeem, D.; Prieto, P.; Culot, M.; Diallo, H.; Landry, C.; Tähti, H.; Cecchelli, R. New strategy for alerting central nervous system toxicity: Integration of blood–brain barrier toxicity and permeability in neurotoxicity assessment. Toxicol. In Vitro 2009, 23, 447–453. [Google Scholar] [CrossRef]
- Brenner, G.; Stevens, C.W. Antineoplastic Drugs, Text Book of Pharmacology; Saunders Elsevier: Tulsa, OK, USA, 2010. [Google Scholar]
- Rang, H.; Dale, M.; Ritter, J. Anticancer Drugs, Text Book of Pharmacology; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Hoekman, K.; van der Vijgh, W.J.; Vermorken, J.B. Clinical and preclinical modulation of chemotherapy-induced toxicity in patients with cancer. Drugs 1999, 57, 133–155. [Google Scholar] [CrossRef]
- Remesh, A. Toxicities of anticancer drugs and its management. Int. J. Basic Clin. Pharmacol. 2012, 1, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Youssef, A.A.; Raafat, T.A.; Madney, Y. Child with acute methotrexate related neurotoxicity: Can diffusion weighted mri help? Egypt. J. Radiol. Nucl. Med. 2015, 46, 1149–1153. [Google Scholar] [CrossRef] [Green Version]
- Bhojwani, D.; Sabin, N.D.; Pei, D.; Yang, J.J.; Khan, R.B.; Panetta, J.C.; Krull, K.R.; Inaba, H.; Rubnitz, J.E.; Metzger, M.L. Methotrexate-induced neurotoxicity and leukoencephalopathy in childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2014, 32, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, A.; De Peri, L.; Deste, G.; Barlati, S.; Sacchetti, E. The effect of antipsychotic treatment on cortical gray matter changes in schizophrenia: Does the class matter? A meta-analysis and meta-regression of longitudinal magnetic resonance imaging studies. Biol. Psychiatry 2015, 78, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Górska, A.; Marszałł, M.; Sloderbach, A. The neurotoxicity of pyridinium metabolites of haloperidol. Postepy Hig. Med. Dosw. 2015, 69, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, H.A.; Chen, A.T. Multiple neurotoxic effects of haloperidol resulting in neuronal death. Ann. Clin. Psychiatry 2017, 29, 195–202. [Google Scholar]
- Caruso, G.; Grasso, M.; Fidilio, A.; Tascedda, F.; Drago, F.; Caraci, F. Antioxidant properties of second-generation antipsychotics: Focus on microglia. Pharmaceuticals 2020, 13, 457. [Google Scholar] [CrossRef]
- Batchelor, D.; Lowe, M. Reported neurotoxicity with the lithium/haloperidol combination and other neuroleptics—A literature review. Hum. Psychopharmacol. Clin. Exp. 1990, 5, 275–280. [Google Scholar] [CrossRef]
- Mensink, G.J.; Slooff, C.J. Novel antipsychotics in bipolar and schizoaffective mania. Acta Psychiatr. Scand. 2004, 109, 405–419. [Google Scholar] [CrossRef]
- Hsu, C.W.; Lee, Y.; Lee, C.Y.; Lin, P.Y. Neurotoxicity and nephrotoxicity caused by combined use of lithium and risperidone: A case report and literature review. BMC Pharmacol. Toxicol. 2016, 17, 59. [Google Scholar] [CrossRef] [Green Version]
- Teimouri, A.; Ahmadi, S.R.; Anavri Ardakani, S.; Foroughian, M. Cyclosporine-a-based immunosuppressive therapy-induced neurotoxicity: A case report. Open Access Emerg. Med. 2020, 12, 93–97. [Google Scholar] [CrossRef]
- Pal, P.; Giri, P.P.; Sinha, R. Cyclosporine in resistant systemic arthritis—A cheaper alternative to biologics. Indian J. Pediatr. 2019, 86, 590–594. [Google Scholar] [CrossRef]
- Arslansoyu Camlar, S.; Soylu, A.; Kavukçu, S. Cyclosporine in pediatric nephrology. Iran. J. Kidney Dis. 2018, 12, 319–330. [Google Scholar]
- Shin, H.S.; Grgic, I.; Chandraker, A. Novel targets of immunosuppression in transplantation. Clin. Lab. Med. 2019, 39, 157–169. [Google Scholar] [CrossRef]
- Straathof, K.; Anoop, P.; Allwood, Z.; Silva, J.; Nikolajeva, O.; Chiesa, R.; Veys, P.; Amrolia, P.J.; Rao, K. Long-term outcome following cyclosporine-related neurotoxicity in paediatric allogeneic haematopoietic stem cell transplantation. Bone Marrow Transplant. 2017, 52, 159–162. [Google Scholar] [CrossRef]
- Anghel, D.; Tanasescu, R.; Campeanu, A.; Lupescu, I.; Podda, G.; Bajenaru, O. Neurotoxicity of immunosuppressive therapies in organ transplantation. Maedica 2013, 8, 170–175. [Google Scholar]
- Trullemans, F.; Grignard, F.; Van Camp, B.; Schots, R. Clinical findings and magnetic resonance imaging in severe cyclosporine-related neurotoxicity after allogeneic bone marrow transplantation. Eur. J. Haematol. 2001, 67, 94–99. [Google Scholar] [CrossRef]
- Shrestha, B.M. Two decades of tacrolimus in renal transplant: Basic science and clinical evidences. Exp. Clin. Transplant. 2017, 15, 1–9. [Google Scholar]
- Tolou-Ghamari, Z. Nephro and neurotoxicity of calcineurin inhibitors and mechanisms of rejections: A review on tacrolimus and cyclosporin in organ transplantation. J. Nephropathol. 2012, 1, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Toyama, C.; Deguchi, K.; Masahata, K.; Nomura, M.; Watanabe, M.; Kamiyama, M.; Tazuke, Y.; Bessho, K.; Okuyama, H. Long-term outcome after tacrolimus-related neurotoxicity in pediatric living donor liver transplantation. Transplant. Proc. 2022, 54, 468–471. [Google Scholar] [CrossRef]
- Gao, P.; Du, X.; Liu, L.; Xu, H.; Liu, M.; Guan, X.; Zhang, C. Astragaloside iv alleviates tacrolimus-induced chronic nephrotoxicity via p62-keap1-Nrf2 pathway. Front. Pharmacol. 2020, 11, 610102. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Fresta, C.G.; Grasso, M.; Santangelo, R.; Lazzarino, G.; Lunte, S.M.; Caraci, F. Inflammation as the common biological link between depression and cardiovascular diseases: Can carnosine exert a protective role? Curr. Med. Chem. 2020, 27, 1782–1800. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The keap1-Nrf2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.J.; Cheng, X.D.; Zhang, J.; Zhang, W.D. Dual roles and therapeutic potential of keap1-Nrf2 pathway in pancreatic cancer: A systematic review. Cell Commun. Signal. 2019, 17, 121. [Google Scholar] [CrossRef] [Green Version]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-are signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 69, 393–402. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/cbp augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [Green Version]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind cbp, a creb binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-trcp axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. Rxrα inhibits the Nrf2-are signaling pathway through a direct interaction with the Neh7 domain of Nrf2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Ogura, T.; Tong, K.I.; Mio, K.; Maruyama, Y.; Kurokawa, H.; Sato, C.; Yamamoto, M. Keap1 is a forked-stem dimer structure with two large spheres. enclosing the intervening, double glycine repeat, and c-terminal domains. Proc. Natl. Acad. Sci. USA 2010, 107, 2842–2847. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; McMahon, M. Nrf2 and keap1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the keap1–Nrf2 pathway by p62/sqstm1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Um, H.C.; Jang, J.H.; Kim, D.H.; Lee, C.; Surh, Y.J. Nitric oxide activates Nrf2 through s-nitrosylation of keap1 in pc12 cells. Nitric Oxide 2011, 25, 161–168. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via s-sulfhydration of keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Zipper, L.M.; Mulcahy, R.T. The keap1 btb/poz dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor keap1 functions as an adaptor for cul3-based e3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Dayalan Naidu, S.; Muramatsu, A.; Saito, R.; Asami, S.; Honda, T.; Hosoya, T.; Itoh, K.; Yamamoto, M.; Suzuki, T.; Dinkova-Kostova, A.T. C151 in keap1 is the main cysteine sensor for the cyanoenone class of Nrf2 activators, irrespective of molecular size or shape. Sci. Rep. 2018, 8, 8037. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. Scf/β-trcp promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, G.; Grasso, M.; Fidilio, A.; Torrisi, S.A.; Musso, N.; Geraci, F.; Tropea, M.R.; Privitera, A.; Tascedda, F.; Puzzo, D.; et al. Antioxidant activity of fluoxetine and vortioxetine in a non-transgenic animal model of alzheimer’s disease. Front. Pharmacol. 2021, 12, 809541. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Torrisi, S.A.; Mogavero, M.P.; Currenti, W.; Castellano, S.; Godos, J.; Ferri, R.; Galvano, F.; Leggio, G.M.; Grosso, G.; et al. Polyphenols and neuroprotection: Therapeutic implications for cognitive decline. Pharmacol. Ther. 2022, 232, 108013. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and nf-κb response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The ikk nf-kappa b system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevskaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.Y.; Bach, F.H. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar] [CrossRef] [Green Version]
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and nf-κb pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.G.; Zhang, Y.Q.; Wu, Z.Z.; Hsieh, C.W.; Chu, C.S.; Wung, B.S. Peanut arachidin-1 enhances Nrf2-mediated protective mechanisms against tnf-α-induced icam-1 expression and nf-κb activation in endothelial cells. Int. J. Mol. Med. 2018, 41, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Arinze, I.J.; Kawai, Y. Transcriptional activation of the human galphai2 gene promoter through nuclear factor-kappab and antioxidant response elements. J. Biol. Chem. 2005, 280, 9786–9795. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by nf-κb and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [Green Version]
- Senoner, T.; Dichtl, W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, Y.M.; Menze, E.T.; Azab, S.S.; Awad, A.S. Involvement of Nrf2/ho-1 antioxidant signaling and nf-κb inflammatory response in the potential protective effects of vincamine against methotrexate-induced nephrotoxicity in rats: Cross talk between nephrotoxicity and neurotoxicity. Arch. Toxicol. 2019, 93, 1417–1431. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.U.; Cho, H.K.; Min, K.J.; Woo, S.M.; Kim, S.; Park, J.W.; Kim, S.H.; Choi, Y.H.; Keum, Y.S.; Hyun, J.W.; et al. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c-flip and mcl-1 expression. Cell Death Dis. 2017, 8, e2599. [Google Scholar] [CrossRef]
- Zeng, D.; Wang, Y.; Chen, Y.; Li, D.; Li, G.; Xiao, H.; Hou, J.; Wang, Z.; Hu, L.; Wang, L.; et al. Angelica polysaccharide antagonizes 5-fu-induced oxidative stress injury to reduce apoptosis in the liver through Nrf2 pathway. Front. Oncol. 2021, 11, 720620. [Google Scholar] [CrossRef]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic drugs and mitochondrial dysfunction: Focus on doxorubicin, trastuzumab, and sunitinib. Oxid. Med. Cell. Longev. 2018, 2018, 7582730. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, X.; Pi, W.; Zhang, Y.; Yu, L.; Xu, C.; Sun, Z.; Jiang, J. Fisetin attenuates doxorubicin-induced cardiomyopathy in vivo and in vitro by inhibiting ferroptosis through sirt1/Nrf2 signaling pathway activation. Front. Pharmacol. 2021, 12, 808480. [Google Scholar] [CrossRef]
- Zhang, W.B.; Lai, X.; Guo, X.F. Activation of Nrf2 by mir-152 inhibits doxorubicin-induced cardiotoxicity via attenuation of oxidative stress, inflammation, and apoptosis. Oxid. Med. Cell. Longev. 2021, 2021, 8860883. [Google Scholar] [CrossRef]
- Fang, Z.Y.; Zhang, M.; Liu, J.N.; Zhao, X.; Zhang, Y.Q.; Fang, L. Tanshinone iia: A review of its anticancer effects. Front. Pharmacol. 2020, 11, 611087. [Google Scholar] [CrossRef]
- Guo, Z.; Yan, M.; Chen, L.; Fang, P.; Li, Z.; Wan, Z.; Cao, S.; Hou, Z.; Wei, S.; Li, W.; et al. Nrf2-dependent antioxidant response mediated the protective effect of tanshinone iia on doxorubicin-induced cardiotoxicity. Exp. Ther. Med. 2018, 16, 3333–3344. [Google Scholar] [CrossRef] [Green Version]
- Yarmohammadi, F.; Rezaee, R.; Karimi, G. Natural compounds against doxorubicin-induced cardiotoxicity: A review on the involvement of Nrf2/are signaling pathway. Phytother. Res. 2021, 35, 1163–1175. [Google Scholar] [CrossRef]
- Sharma, A.; Parikh, M.; Shah, H.; Gandhi, T. Modulation of Nrf2 by quercetin in doxorubicin-treated rats. Heliyon 2020, 6, e03803. [Google Scholar] [CrossRef]
- Hassanein, E.H.M.; Abd El-Ghafar, O.A.M.; Ahmed, M.A.; Sayed, A.M.; Gad-Elrab, W.M.; Ajarem, J.S.; Allam, A.A.; Mahmoud, A.M. Edaravone and acetovanillone upregulate Nrf2 and pi3k/akt/mtor signaling and prevent cyclophosphamide cardiotoxicity in rats. Drug Des. Dev. Ther. 2020, 14, 5275–5288. [Google Scholar] [CrossRef]
- O’Grady, K. Progress in applications of magnetic nanoparticles in biomedicine. J. Phys. D Appl. Phys. 2009, 42, 220301. [Google Scholar] [CrossRef] [Green Version]
- Emara, A.M.; El Kelany, R.S.; Moustafa, K.A. Comparative study of the protective effect between deferoxamine and deferiprone on chronic iron overload induced cardiotoxicity in rats. Hum. Exp. Toxicol. 2006, 25, 375–385. [Google Scholar] [CrossRef]
- Elgharabawy, R.M.; Elgharbawy, D.M.; Emara, A.M. Activation of the molecular and functional effects of Nrf2 against chronic iron oxide nanorod overload-induced cardiotoxicity. Hum. Exp. Toxicol. 2018, 37, 870–885. [Google Scholar] [CrossRef]
- Devarajan, N.; Manjunathan, R.; Ganesan, S.K. Tumor hypoxia: The major culprit behind cisplatin resistance in cancer patients. Crit. Rev. Oncol. Hematol. 2021, 162, 103327. [Google Scholar] [CrossRef]
- Alexandre, J.; Moslehi, J.J.; Bersell, K.R.; Funck-Brentano, C.; Roden, D.M.; Salem, J.E. Anticancer drug-induced cardiac rhythm disorders: Current knowledge and basic underlying mechanisms. Pharmacol. Ther. 2018, 189, 89–103. [Google Scholar] [CrossRef]
- Jia, Y.; Guo, H.; Cheng, X.; Zhang, Y.; Si, M.; Shi, J.; Ma, D. Hesperidin protects against cisplatin-induced cardiotoxicity in mice by regulating the p62-keap1-Nrf2 pathway. Food Funct. 2022, 13, 4205–4215. [Google Scholar] [CrossRef]
- Yardim, A.; Kandemir, F.M.; Ozdemir, S.; Kucukler, S.; Comakli, S.; Gur, C.; Celik, H. Quercetin provides protection against the peripheral nerve damage caused by vincristine in rats by suppressing caspase 3, nf-κb, atf-6 pathways and activating Nrf2, akt pathways. Neurotoxicology 2020, 81, 137–146. [Google Scholar] [CrossRef]
- Miao, H.; Xu, J.; Xu, D.; Ma, X.; Zhao, X.; Liu, L. Nociceptive behavior induced by chemotherapeutic paclitaxel and beneficial role of antioxidative pathways. Physiol. Res. 2019, 68, 491–500. [Google Scholar] [CrossRef]
- Shin, D.H.; Park, H.M.; Jung, K.A.; Choi, H.G.; Kim, J.A.; Kim, D.D.; Kim, S.G.; Kang, K.W.; Ku, S.K.; Kensler, T.W.; et al. The Nrf2-heme oxygenase-1 system modulates cyclosporin a-induced epithelial-mesenchymal transition and renal fibrosis. Free Radic. Biol. Med. 2010, 48, 1051–1063. [Google Scholar] [CrossRef] [Green Version]
- Han, H.Y.; Choi, M.S.; Yoon, S.; Ko, J.W.; Kim, S.K.; Kim, T.W. Investigation of ifosfamide toxicity induces common upstreAm. regulator in liver and kidney. Int. J. Mol. Sci. 2021, 22, 12201. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, B.; Liu, J.; Qiao, C.; Liu, Y.; Li, S.; Lv, H. Isoorientin exerts a protective effect against 6-ohda-induced neurotoxicity by activating the ampk/akt/Nrf2 signalling pathway. Food Funct. 2020, 11, 10774–10785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, G.; He, J.; Yang, Q.; Li, D.; Li, J.; Zhang, F. Icariin attenuates neuroinflammation and exerts dopamine neuroprotection via an Nrf2-dependent manner. J. Neuroinflamm. 2019, 16, 92. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Han, C.; Ma, K.; Xia, Y.; Wan, F.; Yin, S.; Kou, L.; Sun, Y.; Wu, J.; Hu, J.; et al. Hydralazine protects nigrostriatal dopaminergic neurons from mpp(+) and mptp induced neurotoxicity: Roles of Nrf2-are signaling pathway. Front. Neurol. 2019, 10, 271. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.D.; Chen, G.S.; Liu, J.R.; Hsieh, C.E.; Chern, J.W. Acrylamide functional group incorporation improves drug-like properties: An example with egfr inhibitors. ACS Med. Chem. Lett. 2019, 10, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Davuljigari, C.B.; Ekuban, F.A.; Zong, C.; Fergany, A.A.M.; Morikawa, K.; Ichihara, G. Nrf2 activation attenuates acrylamide-induced neuropathy in mice. Int. J. Mol. Sci. 2021, 22, 5995. [Google Scholar] [CrossRef]
- Ekuban, F.A.; Zong, C.; Takikawa, M.; Morikawa, K.; Sakurai, T.; Ichihara, S.; Itoh, K.; Yamamoto, M.; Ohsako, S.; Ichihara, G. Genetic ablation of Nrf2 exacerbates neurotoxic effects of acrylamide in mice. Toxicology 2021, 456, 152785. [Google Scholar] [CrossRef]
- Collins, S.E.; Kirouac, M. Alcohol consumption. In Encyclopedia of Behavioral Medicine; Gellman, M.D., Turner, J.R., Eds.; Springer: New York, NY, USA, 2013; pp. 61–65. [Google Scholar]
- Sun, J.; Hong, Z.; Shao, S.; Li, L.; Yang, B.; Hou, Y.; Wang, H.; Xu, Y.; Zhang, Q.; Pi, J.; et al. Liver-specific Nrf2 deficiency accelerates ethanol-induced lethality and hepatic injury in vivo. Toxicol. Appl. Pharmacol. 2021, 426, 115617. [Google Scholar] [CrossRef]
- Ali, T.; Rehman, S.U.; Shah, F.A.; Kim, M.O. Acute dose of melatonin via Nrf2 dependently prevents acute ethanol-induced neurotoxicity in the developing rodent brain. J. Neuroinflamm. 2018, 15, 119. [Google Scholar] [CrossRef]
- Gulewitsch, W.; Amiradžibi, S. Ueber das carnosin, eine neue organische base des fleischextractes. Ber. Dtsch. Chem. Ges. 1900, 33, 1902–1903. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Godos, J.; Castellano, S.; Micek, A.; Murabito, P.; Galvano, F.; Ferri, R.; Grosso, G.; Caraci, F. The therapeutic potential of carnosine/anserine supplementation against cognitive decline: A systematic review with meta-analysis. Biomedicines 2021, 9, 253. [Google Scholar] [CrossRef]
- Kalyankar, G.D.; Meister, A. Enzymatic synthesis of carnosine and related β-alanyl and γ-aminobutyryl peptides. J. Biol. Chem. 1959, 234, 3210–3218. [Google Scholar] [CrossRef]
- Winnick, R.; Winnick, T. Carnosine-anserine synthetase of muscle i. Preparation and properties of a soluble enyzme from chick muscle. Biochim. Biophys. Acta 1959, 31, 47–55. [Google Scholar] [CrossRef]
- Caruso, G.; Musso, N.; Grasso, M.; Costantino, A.; Lazzarino, G.; Tascedda, F.; Gulisano, M.; Lunte, S.M.; Caraci, F. Microfluidics as a novel tool for biological and toxicological assays in drug discovery processes: Focus on microchip electrophoresis. Micromachines 2020, 11, 593. [Google Scholar] [CrossRef]
- Gariballa, S.E.; Sinclair, A.J. Carnosine: Physiological properties and therapeutic potential. Age Ageing 2000, 29, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Hipkiss, A.R.; Preston, J.E.; Himsworth, D.T.; Worthington, V.C.; Keown, M.; Michaelis, J.; Lawrence, J.; Mateen, A.; Allende, L.; Eagles, P.A.; et al. Pluripotent protective effects of carnosine, a naturally occurring dipeptide. Ann. N. Y. Acad. Sci. 1998, 854, 37–53. [Google Scholar] [CrossRef]
- Lenney, J.F.; George, R.P.; Weiss, A.M.; Kucera, C.M.; Chan, P.W.; Rinzler, G.S. Human serum carnosinase: Characterization, distinction from cellular carnosinase, and activation by cadmium. Clin. Chim. Acta 1982, 123, 221–231. [Google Scholar] [CrossRef]
- Lenney, J.F.; Peppers, S.C.; Kucera-Orallo, C.M.; George, R.P. Characterization of human tissue carnosinase. Biochem. J. 1985, 228, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Stvolinskiĭ, S.L.; Dobrota, D.; Mezeshova, V.; Liptaĭ, T.; Pronaĭova, N.; Zalibera, L.; Boldyrev, A.A. Carnosine and anserine in working muscles--study using proton nmr spectroscopy. Biokhimiia 1992, 57, 1317–1323. [Google Scholar]
- Sale, C.; Artioli, G.G.; Gualano, B.; Saunders, B.; Hobson, R.M.; Harris, R.C. Carnosine: From exercise performance to health. Amino Acids 2013, 44, 1477–1491. [Google Scholar] [CrossRef]
- Severin, S.E.; Kirzon, M.V.; Kaftanova, T.M. Effect of carnosine and anserine on action of isolated frog muscles. Dokl. Akad. Nauk. SSSR 1953, 91, 691–694. [Google Scholar]
- Boldyrev, A.A.; Petukhov, V.B. Localization of carnosine effect on the fatigued muscle preparation. Gen. Pharmacol. 1978, 9, 17–20. [Google Scholar] [CrossRef]
- Brisola, G.M.P.; de Souza Malta, E.; Santiago, P.R.P.; Vieira, L.H.P.; Zagatto, A.M. Β-alanine supplementation’s improvement of high-intensity game activities in water polo. Int. J. Sports Physiol. Perform. 2018, 13, 1208–1214. [Google Scholar] [CrossRef]
- de Andrade Kratz, C.; de Salles Painelli, V.; de Andrade Nemezio, K.M.; da Silva, R.P.; Franchini, E.; Zagatto, A.M.; Gualano, B.; Artioli, G.G. Beta-alanine supplementation enhances judo-related performance in highly-trained athletes. J. Sci. Med. Sport 2017, 20, 403–408. [Google Scholar] [CrossRef] [Green Version]
- Furst, T.; Massaro, A.; Miller, C.; Williams, B.T.; LaMacchia, Z.M.; Horvath, P.J. Β-alanine supplementation increased physical performance and improved executive function following endurance exercise in middle aged individuals. J. Int. Soc. Sports Nutr. 2018, 15, 32. [Google Scholar] [CrossRef] [Green Version]
- Glenn, J.M.; Smith, K.; Moyen, N.E.; Binns, A.; Gray, M. Effects of acute beta-alanine supplementation on anaerobic performance in trained female cyclists. J. Nutr. Sci. Vitaminol. 2015, 61, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Tiedje, K.; Stevens, K.; Barnes, S.; Weaver, D. Β-alanine as a small molecule neurotransmitter. Neurochem. Int. 2010, 57, 177–188. [Google Scholar] [CrossRef]
- Fresta, C.G.; Fidilio, A.; Lazzarino, G.; Musso, N.; Grasso, M.; Merlo, S.; Amorini, A.M.; Bucolo, C.; Tavazzi, B.; Lazzarino, G.; et al. Modulation of pro-oxidant and pro-inflammatory activities of m1 macrophages by the natural dipeptide carnosine. Int. J. Mol. Sci. 2020, 21, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, G.; Fresta, C.G.; Fidilio, A.; O’Donnell, F.; Musso, N.; Lazzarino, G.; Grasso, M.; Amorini, A.M.; Tascedda, F.; Bucolo, C.; et al. Carnosine decreases pma-induced oxidative stress and inflammation in murine macrophages. Antioxidants 2019, 8, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mal’tseva, V.V.; Sergienko, V.V.; Stvolinskii, S.L. The effect of carnosine on hematopoietic stem cell activity in irradiated animals. Biokhimiia 1992, 57, 1378–1382. [Google Scholar] [PubMed]
- Caruso, G.; Fresta, C.G.; Siegel, J.M.; Wijesinghe, M.B.; Lunte, S.M. Microchip electrophoresis with laser-induced fluorescence detection for the determination of the ratio of nitric oxide to superoxide production in macrophages during inflammation. Anal. Bioanal. Chem. 2017, 409, 4529–4538. [Google Scholar] [CrossRef]
- Fresta, C.G.; Hogard, M.L.; Caruso, G.; Melo Costa, E.E.; Lazzarino, G.; Lunte, S.M. Monitoring carnosine uptake by RAW 264.7 macrophage cells using microchip electrophoresis with fluorescence detection. Anal. Methods 2017, 9, 402–408. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Fresta, C.G.; Martinez-Becerra, F.; Antonio, L.; Johnson, R.T.; de Campos, R.P.S.; Siegel, J.M.; Wijesinghe, M.B.; Lazzarino, G.; Lunte, S.M. Carnosine modulates nitric oxide in stimulated murine raw 264.7 macrophages. Mol. Cell. Biochem. 2017, 431, 197–210. [Google Scholar] [CrossRef]
- Caruso, G.; Benatti, C.; Musso, N.; Fresta, C.G.; Fidilio, A.; Spampinato, G.; Brunello, N.; Bucolo, C.; Drago, F.; Lunte, S.M.; et al. Carnosine protects macrophages against the toxicity of aβ1-42 oligomers by decreasing oxidative stress. Biomedicines 2021, 9, 477. [Google Scholar] [CrossRef]
- Pepper, E.D.; Farrell, M.J.; Nord, G.; Finkel, S.E. Antiglycation effects of carnosine and other compounds on the long-term survival of escherichia coli. Appl. Environ. Microbiol. 2010, 76, 7925–7930. [Google Scholar] [CrossRef] [Green Version]
- Boldyrev, A.A.; Gallant, S.C.; Sukhich, G.T. Carnosine, the protective, anti-aging peptide. Biosci. Rep. 1999, 19, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Hasanein, P.; Felegari, Z. Chelating effects of carnosine in ameliorating nickel-induced nephrotoxicity in rats. Can. J. Physiol. Pharmacol. 2017, 95, 1426–1432. [Google Scholar] [CrossRef]
- Brown, C.E.; Antholine, W.E. Chelation chemistry of carnosine. Evidence that mixed complexes may occur in vivo. J. Phys. Chem. 1979, 83, 3314–3319. [Google Scholar] [CrossRef]
- Ouyang, L.; Tian, Y.; Bao, Y.; Xu, H.; Cheng, J.; Wang, B.; Shen, Y.; Chen, Z.; Lyu, J. Carnosine decreased neuronal cell death through targeting glutamate system and astrocyte mitochondrial bioenergetics in cultured neuron/astrocyte exposed to ogd/recovery. Brain Res. Bull. 2016, 124, 76–84. [Google Scholar] [CrossRef]
- Wang-Eckhardt, L.; Bastian, A.; Bruegmann, T.; Sasse, P.; Eckhardt, M. Carnosine synthase deficiency is compatible with normal skeletal muscle and olfactory function but causes reduced olfactory sensitivity in aging mice. J. Biol. Chem. 2020, 295, 17100–17113. [Google Scholar] [CrossRef]
- Gonçalves, L.S.; Sales, L.P.; Saito, T.R.; Campos, J.C.; Fernandes, A.L.; Natali, J.; Jensen, L.; Arnold, A.; Ramalho, L.; Bechara, L.R.G.; et al. Histidine dipeptides are key regulators of excitation-contraction coupling in cardiac muscle: Evidence from a novel carns1 knockout rat model. Redox Biol. 2021, 44, 102016. [Google Scholar] [CrossRef]
- Wang-Eckhardt, L.; Becker, I.; Wang, Y.; Yuan, J.; Eckhardt, M. Absence of endogenous carnosine synthesis does not increase protein carbonylation and advanced lipoxidation end products in brain, kidney or muscle. Amino Acids 2022, 54, 1013–1023. [Google Scholar] [CrossRef]
- Caruso, G. Unveiling the hidden therapeutic potential of carnosine, a molecule with a multimodal mechanism of action: A position paper. Molecules 2022, 27, 3303. [Google Scholar] [CrossRef]
- Albrecht, T.; Schilperoort, M.; Zhang, S.; Braun, J.D.; Qiu, J.; Rodriguez, A.; Pastene, D.O.; Krämer, B.K.; Köppel, H.; Baelde, H.; et al. Carnosine attenuates the development of both type 2 diabetes and diabetic nephropathy in btbr ob/ob mice. Sci. Rep. 2017, 7, 44492. [Google Scholar] [CrossRef]
- Everaert, I.; Mooyaart, A.; Baguet, A.; Zutinic, A.; Baelde, H.; Achten, E.; Taes, Y.; De Heer, E.; Derave, W. Vegetarianism, female gender and increasing age, but not cndp1 genotype, are associated with reduced muscle carnosine levels in humans. Amino Acids 2011, 40, 1221–1229. [Google Scholar] [CrossRef]
- Masuoka, N.; Yoshimine, C.; Hori, M.; Tanaka, M.; Asada, T.; Abe, K.; Hisatsune, T. Effects of anserine/carnosine supplementation on mild cognitive impairment with APOE4. Nutrients 2019, 11, 1626. [Google Scholar] [CrossRef] [Green Version]
- Araminia, B.; Shalbafan, M.; Mortezaei, A.; Shirazi, E.; Ghaffari, S.; Sahebolzamani, E.; Mortazavi, S.H.; Shariati, B.; Ardebili, M.E.; Aqamolaei, A.; et al. L-carnosine combination therapy for major depressive disorder: A randomized, double-blind, placebo-controlled trial. J. Affect. Disord. 2020, 267, 131–136. [Google Scholar] [CrossRef]
- Houjeghani, S.; Kheirouri, S.; Faraji, E.; Jafarabadi, M.A. L-carnosine supplementation attenuated fasting glucose, triglycerides, advanced glycation end products, and tumor necrosis factor-alpha levels in patients with type 2 diabetes: A double-blind placebo-controlled randomized clinical trial. Nutr. Res. 2018, 49, 96–106. [Google Scholar] [CrossRef]
- Rezende, N.S.; Swinton, P.; de Oliveira, L.F.; da Silva, R.P.; da Eira Silva, V.; Nemezio, K.; Yamaguchi, G.; Artioli, G.G.; Gualano, B.; Saunders, B.; et al. The muscle carnosine response to beta-alanine supplementation: A systematic review with bayesian individual and aggregate data e-max model and meta-analysis. Front. Physiol. 2020, 11, 913. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Mancuso, C.; Pennisi, G.; Calafato, S.; Bellia, F.; Bates, T.E.; Giuffrida Stella, A.M.; Schapira, T.; Dinkova Kostova, A.T.; et al. Cellular stress response: A novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neurochem. Res. 2008, 33, 2444–2471. [Google Scholar] [CrossRef]
- Calabrese, V.; Scuto, M.; Salinaro, A.T.; Dionisio, G.; Modafferi, S.; Ontario, M.L.; Greco, V.; Sciuto, S.; Schmitt, C.P.; Calabrese, E.J.; et al. Hydrogen sulfide and carnosine: Modulation of oxidative stress and inflammation in kidney and brain axis. Antioxidants 2020, 9, 1303. [Google Scholar] [CrossRef]
- Smith, R.E.; Tran, K.; Smith, C.C.; McDonald, M.; Shejwalkar, P.; Hara, K. The role of the Nrf2/are antioxidant system in preventing cardiovascular diseases. Diseases 2016, 4, 34. [Google Scholar] [CrossRef]
- Aldini, G.; de Courten, B.; Regazzoni, L.; Gilardoni, E.; Ferrario, G.; Baron, G.; Altomare, A.; D’Amato, A.; Vistoli, G.; Carini, M. Understanding the antioxidant and carbonyl sequestering activity of carnosine: Direct and indirect mechanisms. Free Radic. Res. 2021, 55, 321–330. [Google Scholar] [CrossRef]
- Boldyrev, A.A. Carnosine and Oxidative Stress in Cells and Tissues; Nova Publishers: Hauppauge, NY, USA, 2007. [Google Scholar]
- Gardner, M.L.; Illingworth, K.M.; Kelleher, J.; Wood, D. Intestinal absorption of the intact peptide carnosine in man, and comparison with intestinal permeability to lactulose. J. Physiol. 1991, 439, 411–422. [Google Scholar] [CrossRef]
- Goto, K.; Maemura, H.; Takamatsu, K.; Ishii, N. Hormonal responses to resistance exercise after ingestion of carnosine and anserine. J. Strength Cond. Res. 2011, 25, 398–405. [Google Scholar] [CrossRef]
- Dolu, N.; Acer, H.; Kara, A.Y. Investigation of dose-related effects of carnosine on anxiety with sympathetic skin response and t-maze. Acta Med. 2014, 57, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Décombaz, J.; Beaumont, M.; Vuichoud, J.; Bouisset, F.; Stellingwerff, T. Effect of slow-release β-alanine tablets on absorption kinetics and paresthesia. Amino Acids 2012, 43, 67–76. [Google Scholar] [CrossRef]
- Salatto, R.W.; McGinnis, G.R.; Davis, D.W.; Carrier, B.; Manning, J.W.; DeBeliso, M.; Navalta, J.W. Effects of acute beta-alanine ingestion and immersion-plus-exercise on connectedness to nature and perceived pain. Int. J. Environ. Res. Public Health 2021, 18, 8134. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.G.; Knatko, E.V.; Casey, A.M.; Hukelmann, J.L.; Dayalan Naidu, S.; Brenes, A.J.; Ekkunagul, T.; Baker, C.; Higgins, M.; Tronci, L.; et al. Nrf2 activation reprograms macrophage intermediary metabolism and suppresses the type i interferon response. iScience 2022, 25, 103827. [Google Scholar] [CrossRef] [PubMed]
- Alsheblak, M.M.; Elsherbiny, N.M.; El-Karef, A.; El-Shishtawy, M.M. Protective effects of l-carnosine on ccl4 -induced hepatic injury in rats. Eur. Cytokine Netw. 2016, 27, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Leon, B.M.; Maddox, T.M. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 2015, 6, 1246–1258. [Google Scholar] [CrossRef]
- Ahshin-Majd, S.; Zamani, S.; Kiamari, T.; Kiasalari, Z.; Baluchnejadmojarad, T.; Roghani, M. Carnosine ameliorates cognitive deficits in streptozotocin-induced diabetic rats: Possible involved mechanisms. Peptides 2016, 86, 102–111. [Google Scholar] [CrossRef]
- Zieba, R.; Wagrowska-Danilewicz, M. Influence of carnosine on the cardiotoxicity of doxorubicin in rabbits. Pol. J. Pharmacol. 2003, 55, 1079–1087. [Google Scholar]
- Zhao, K.; Li, Y.; Wang, Z.; Han, N.; Wang, Y. Carnosine protects mouse podocytes from high glucose induced apoptosis through pi3k/akt and Nrf2 pathways. BioMed Res. Int. 2019, 2019, 4348973. [Google Scholar] [CrossRef]
- Scuto, M.; Trovato Salinaro, A.; Modafferi, S.; Polimeni, A.; Pfeffer, T.; Weigand, T.; Calabrese, V.; Schmitt, C.P.; Peters, V. Carnosine activates cellular stress response in podocytes and reduces glycative and lipoperoxidative stress. Biomedicines 2020, 8, 177. [Google Scholar] [CrossRef]
- Afifi, N.A.; Ramadan, A.; Erian, E.Y.; Sedik, A.A.; Amin, M.M.; Hassan, A.; Saleh, D.O. Synergistic effect of aminoguanidine and l-carnosine against thioacetamide-induced hepatic encephalopathy in rats: Behavioral, biochemical, and ultrastructural evidence. Can. J. Physiol. Pharmacol. 2021, 99, 332–347. [Google Scholar] [CrossRef]
- Caruso, G.; Caraci, F.; Jolivet, R.B. Pivotal role of carnosine in the modulation of brain cells activity: Multimodal mechanism of action and therapeutic potential in neurodegenerative disorders. Prog. Neurobiol. 2019, 175, 35–53. [Google Scholar] [CrossRef]
- Ibrahim, N.; El Said, H.; Choukair, A. Zinc carnosine-based modified bismuth quadruple therapy vs. standard triple therapy for helicobacter pylori eradication: A randomized controlled study. World J. Clin. Cases 2022, 10, 227–235. [Google Scholar] [CrossRef]
- Gao, X.; Al-Baadani, M.A.; Wu, M.; Tong, N.; Shen, X.; Ding, X.; Liu, J. Study on the local anti-osteoporosis effect of polaprezinc-loaded antioxidant electrospun membrane. Int. J. Nanomed. 2022, 17, 17–29. [Google Scholar] [CrossRef]
- Yehia, R.; Saleh, S.; El Abhar, H.; Saad, A.S.; Schaalan, M. L-carnosine protects against oxaliplatin-induced peripheral neuropathy in colorectal cancer patients: A perspective on targeting nrf-2 and nf-κb pathways. Toxicol. Appl. Pharmacol. 2019, 365, 41–50. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caruso, G.; Privitera, A.; Antunes, B.M.; Lazzarino, G.; Lunte, S.M.; Aldini, G.; Caraci, F. The Therapeutic Potential of Carnosine as an Antidote against Drug-Induced Cardiotoxicity and Neurotoxicity: Focus on Nrf2 Pathway. Molecules 2022, 27, 4452. https://doi.org/10.3390/molecules27144452
Caruso G, Privitera A, Antunes BM, Lazzarino G, Lunte SM, Aldini G, Caraci F. The Therapeutic Potential of Carnosine as an Antidote against Drug-Induced Cardiotoxicity and Neurotoxicity: Focus on Nrf2 Pathway. Molecules. 2022; 27(14):4452. https://doi.org/10.3390/molecules27144452
Chicago/Turabian StyleCaruso, Giuseppe, Anna Privitera, Barbara Moura Antunes, Giuseppe Lazzarino, Susan Marie Lunte, Giancarlo Aldini, and Filippo Caraci. 2022. "The Therapeutic Potential of Carnosine as an Antidote against Drug-Induced Cardiotoxicity and Neurotoxicity: Focus on Nrf2 Pathway" Molecules 27, no. 14: 4452. https://doi.org/10.3390/molecules27144452
APA StyleCaruso, G., Privitera, A., Antunes, B. M., Lazzarino, G., Lunte, S. M., Aldini, G., & Caraci, F. (2022). The Therapeutic Potential of Carnosine as an Antidote against Drug-Induced Cardiotoxicity and Neurotoxicity: Focus on Nrf2 Pathway. Molecules, 27(14), 4452. https://doi.org/10.3390/molecules27144452