
Interdiction in the Early Folding of the p53 DNA-Binding Domain Leads to Its Amyloid-Like Misfolding


Abstract
:1. Introduction
2. Results
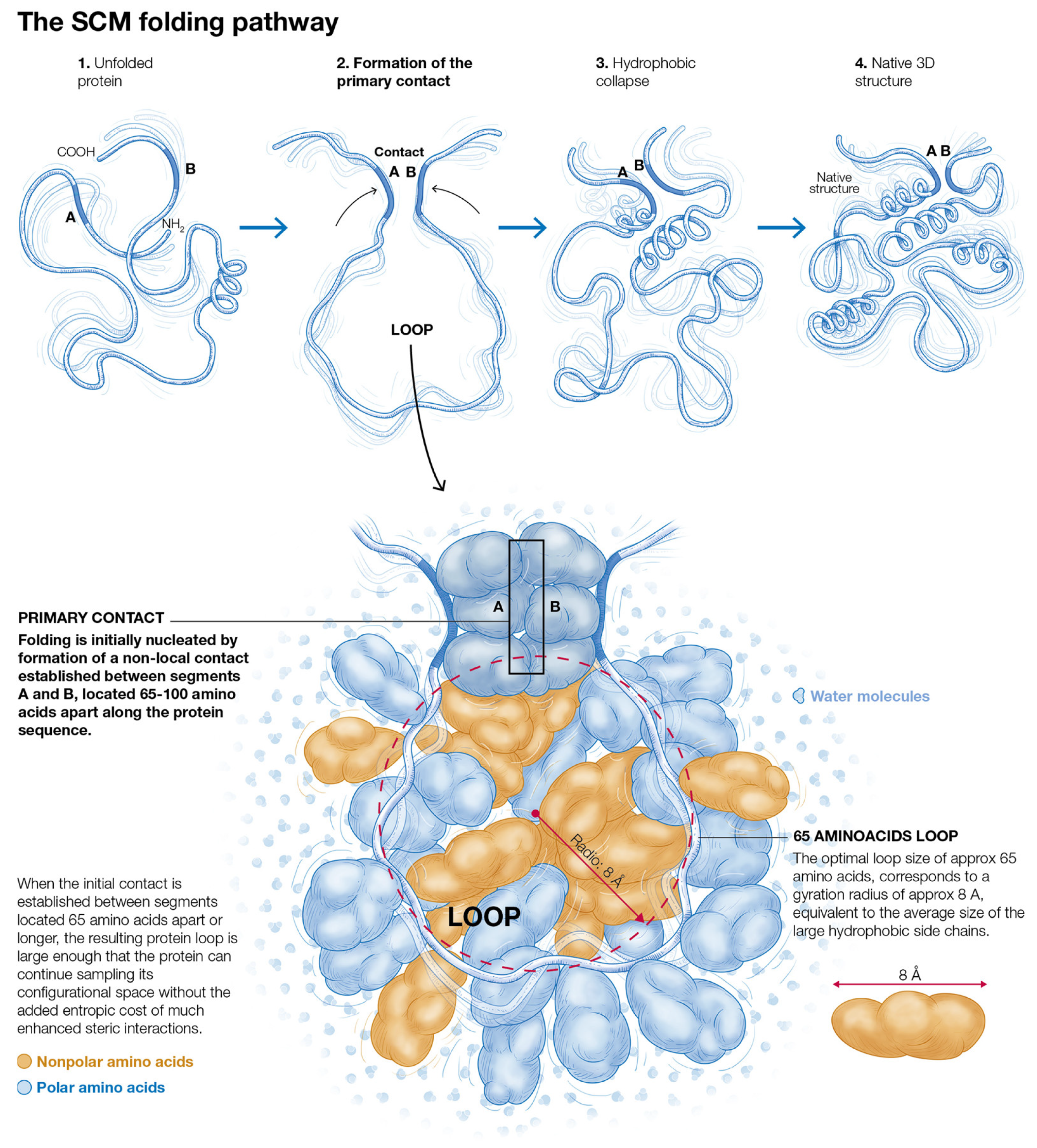
2.1. The Physical Basis of Non-Local Early Contact Formation in the SCM
2.2. Primary Contacts of Core p53
2.3. Comparison of the Predicted Primary Contact Populations with Experimental Data on Core p53 Folding
2.4. Interdiction of the Dominant Folding Pathway through Competition of Peptide P8(250-257) with the Formation of Primary Contact C1
2.5. Possible Coincidence of the p53 and Prion Protein Misfolding Mechanisms
3. Conclusions
4. Methods: Determination of the Primary Contact in the SCM Model
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kress, M.; May, E.; Cassingena, R.; May, P. Simian Virus 40-Transformed Cells Express New Species of Proteins Precipitable by Anti-Simian Virus 40 Tumor Serum. Virol. J. 1979, 31, 472–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.P.; Crawford, L.V. T Antigen is Bound to a Host Protein in SV40-Transformed Cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K Dalton Cellular SV40 Tumor Antigen Present in SV40-Transformed Cells and Uninfected Embryonal Carcinoma Cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- DeLeo, A.B.; Jay, G.; Appella, E.; Dubois, G.C.; Law, L.W.; Old, L.J. Detection of a Transformation-Related Antigen in Chemically Induced Sarcomas and Other Transformed Cells of the Mouse. Proc. Natl. Acad. Sci. USA 1979, 76, 2420–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; van Tuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 Deletions and p53 Mutations in Colorectal Carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Takahashi, T.; Nau, N.M.; Chiba, I.; Birrer, M.J.; Rosenberg, R.K.; Vinocour, M.; Levitt, M.; Pass, H.; Gazdar, A.F.; Minna, J.D. p53: A Frequent Target for Genetic Abnormalities in Lung Cancer. Science 1989, 246, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One Name, Many Proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Wang, X.; Simpson, E.R.; Brown, K.A. p53: Protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res. 2015, 75, 5001–5007. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Lees, A.; Sessler, T.; McDade, S. Dying to survive—The p53 paradox. Cancers 2021, 13, 3257. [Google Scholar] [CrossRef]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G. Restoring p53 in cancer: The promises and the challenges. JMCB 2019, 11, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahalle, A.; Lacroix, M.; De Blasio, C.; Cissé, M.Y.; Linares, L.K.; Le Cam, L. The p53 pathway and metabolism: The tree that hides the forest. Cancers 2021, 13, 133. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Norberg, E.; Vakifahmetoglu-Norberg, H. Mutant p53 as a regulator and target of autophagy. Front. Oncol. 2021, 10, 607149. [Google Scholar] [CrossRef]
- Shi, T.; van Soest, D.M.K.; Polderman, P.E.; Burgering, B.M.T.; Dansen, T.B. DNA damage and oxidant stress activate p53 through differential upstream signaling pathways. Free Radic. Biol. Med. 2021, 172, 298–311. [Google Scholar] [CrossRef]
- Tsai, Y.-Y.; Su, C.-H.; Tarn, W.-Y. p53 activation in genetic disorders: Different routes to the same destination. Int. J. Mol. Sci. 2021, 22, 9307. [Google Scholar] [CrossRef]
- May, P.; May, E. Twenty years of p53 research: Structural and functional aspects of the p53 protein. Oncogene 1999, 18, 7621–7636. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Fersht, A. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Chambers, K.A.; Pabo, C.O. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993, 7, 2556–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Ory, K.; Legros, Y.; Auguin, C.; Soussi, T. Analysis of the most representative tumour-derived p53 mutants reveals that changes in protein conformation are not correlated with loss of transactivation or inhibition of cell proliferation. EMBO J. 1994, 13, 3496–3504. [Google Scholar] [CrossRef]
- Gamble, J.; Milner, J. Evidence that immunological variants of p53 represent alternative protein conformation. Virology 1988, 162, 452–458. [Google Scholar] [CrossRef]
- Bosari, S.; Viale, G.; Roncalli, M.; Graziani, D.; Borsani, G.; Lee, A.K.; Coggi, G. p53 Gene Mutations, p53 Protein Accumulation and Compartmentalization in Colorectal Adenocarcinoma. Am. J. Pathol. 1995, 147, 790–798. [Google Scholar] [PubMed]
- Ishimaru, D.; Andrade, L.R.; Teixeira, L.S.; Quesado, P.A.; Maiolino, L.M.; Lopez, P.M.; Cordeiro, Y.; Costa, L.T.; Heckl, W.M.; Weissmuller, G.; et al. Fibrillar Aggregates of the Tumor Suppresor p53 Core Domain. Biochemistry 2003, 42, 9022–9027. [Google Scholar] [CrossRef] [PubMed]
- Rigacci, S.; Bucciantini, M.; Relini, A.; Pesce, A.; Gliozzi, A.; Berti, A.; Stefani, M. The (1-63) Region of the p53 Transactivation Domain Aggregates in Vitro into Cytotoxic Amyloid Assemblies. Biophys. J. 2008, 94, 3635–3646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bom, A.P.D.; Rangel, L.P.; Costa, D.C.F.; de Oliveira, G.A.P.; Sanchez, D. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Fersht, A.R. Mechanism of initiation of aggregation of p53 revealed by Φ analysis. Proc. Natl. Acad. Sci. USA 2015, 112, 2437–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navalkar, A.; Ghosh, S.; Pandey, S.; Paul, A.; Datta, D.; Maji, S.K. Prion-like amyloids in cancer. Biochemistry 2020, 59, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Galea, C.; DiGiammarino, E.L.; Jun, B.; Murti, G.; Ribeiro, R.C.; Zambetti, G.; Schultz, C.P.; Kriwacki, R.W. Reversible amyloid formation by the p53 tetramerization domain and a cancer-associated mutant. J. Mol. Biol. 2003, 327, 699–709. [Google Scholar] [CrossRef]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Ghosh, D.; Ranganathan, S.; Anoop, A.; Kumar, P.S.; Jha, N.N.; Padinhateeri, R.; Maji, S.K. Investigating the intrinsic aggregation potential of evolutionarily conserved segments in p53. Biochemistry 2014, 53, 5995–6010. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell. Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Forget, K.J.; Tremblay, G.; Roucou, X. p53 aggregates penetrate cells and induce the co-aggregation of intracellular p53. PLoS ONE 2013, 8, e69242. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Prion Biology and Diseases; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2014. [Google Scholar]
- Uversky, V.N. α-Synuclein Misfolding and Neurodegenerative Diseases. Curr. Protein Pept. Sci. 2009, 9, 507–540. [Google Scholar] [CrossRef] [Green Version]
- Angot, E.; Steiner, J.A.; Hansen, C.; Li, J.-Y.; Brundin, P. Are synucleinopathies prion-like diseases? Lancet Neurol. 2010, 9, 1128–1138. [Google Scholar] [CrossRef]
- Kondo, J.; Ihara, Y.; Sairoh, T. Molecular Cloning of cDNA Encoding an Unrecognized Component of Amyloid in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11287. [Google Scholar]
- Billant, O.; Friocourt, G.; Roux, P.; Voisset, C. p53, a victim of the prion fashion. Cancers 2021, 13, 269. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Ronneberg, T.A.; Rabitz, H.A. Sequential Collapse Model for Protein Folding Pathways. J. Phys. Chem. B 1999, 103, 9749–9758. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Haas, E.; Rabitz, H.A. Nature’s Shortcut to Protein Folding. J. Phys. Chem. B 2019, 123, 4463–4476. [Google Scholar] [CrossRef] [PubMed]
- Bergasa-Caceres, F.; Rabitz, H.A. Interdiction of Protein Folding for Therapeutic Drug Development in SARS-CoV-2. J. Phys. Chem. B 2020, 124, 8201–8208. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H.A. Protein folding interdiction strategy for therapeutic drug development in viral diseases: Ebola VP40 and Influenza A M1. Int. J. Mol. Sci. 2022, 23, 3906. [Google Scholar] [CrossRef] [PubMed]
- Bergasa-Caceres, F.; Rabitz, H.A. Identification of Two Early Folding Stage Prion Non-Local Contacts Suggested to Serve as Key Steps in Directing the Final Fold to Be Either Native or Pathogenic. Int. J. Mol. Sci. 2021, 22, 8619. [Google Scholar] [CrossRef] [PubMed]
- Berezovsky, I.N.; Trifonov, E.N. Van der Waals Locks: Loop-n-Lock Structure of Globular Proteins. J. Mol. Biol. 2001, 307, 1419–1426. [Google Scholar] [CrossRef]
- Dill, K.A. Dominant Forces in Protein Folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef] [PubMed]
- Kuwajima, K. The molten globule, and two-state vs. non-two-state folding of globular proteins. Biomolecules 2020, 10, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karplus, M.; Weaver, D.L. Diffusion–collision Model for Protein Folding. Biopolymers 1979, 18, 1421–1437. [Google Scholar] [CrossRef]
- Ittah, V.; Haas, E. Nonlocal Interactions Stabilize Long Range Loops in the Initial Folding Intermediates of Reduced Bovine Pancreatic Trypsin Inhibitor. Biochemistry 1995, 34, 4493–4506. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Shakhnovich, E.; Karplus, M. How Does a Protein Fold? Nature 1994, 369, 248–251. [Google Scholar] [PubMed]
- Vendruscolo, M.; Paci, E.; Dobson, C.M.; Karplus, M. Three Key Residues Form a Critical Contact Network in a Protein Folding Transition State. Nature 2001, 409, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Alm, E.; Baker, D. Prediction of Protein-Folding Mechanisms from Free-energy Landscapes Derived from Native Structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11305–11310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, V.; Eaton, W.A. A Simple Model for Calculating the Kinetics of Protein Folding from Three-dimensional Structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11311–11316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clementi, C.; Jennings, P.A.; Onuchic, J.N. How Native-state Topology Affects the Folding of Dihydrofolate Reductase and Interleukin-1ß. Proc. Natl. Acad. Sci. USA 2000, 97, 5871–5876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarov, D.E.; Plaxco, K.W. The Topomer Search Model: A Simple, Quantitative Theory of Two-State Protein Folding Kinetics. Protein Sci. 2003, 12, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Rosengarth, A.; Luecke, H. Structure of the human p53 core domain in the absence of DNA. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 276–281. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S.; et al. Mol* viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H.A. Role of topology in the cooperative collapse of the protein core in the sequential collapse model. Folding pathway of α-lactalbumin and hen lysozyme. J. Phys. Chem. B 2001, 105, 2874–2880. [Google Scholar] [CrossRef]
- Butler, J.S.; Loh, S.N. Kinetic partitioning during folding of the p53 DNA binding domain. J. Mol. Biol. 2005, 350, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.S.; Loh, S.N. Folding and misfolding mechanism of the p53 DNA binding domain at physiological temperature. Prot. Sci. 2006, 15, 2457–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergasa-Caceres, F.; Rabitz, H.A. Relating contact order to the rate of cooperative collapse in the sequential collapse model for protein folding pathways. Chem. Phys. Lett. 2003, 376, 612–617. [Google Scholar] [CrossRef]
- Bullock, A.N.; Henckel, J.; De Decker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, H.C.; Joerger, A.C.; Mayer, S.; Fersht, A.R. Effects of common cancer mutations on stability and DNA binding of full length p53 compared with isolated core domains. J. Biol. Chem. 2006, 281, 21934–21941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dea, A.J.; Michaels, T.C.T.; Meisl, G.; Klenerman, D.; Wu, S.; Perrett, S.; Linse, S.; Dobson, C.M.; Knowles, T.P.J. Kinetic diversity of amyloid oligomers. Proc. Natl. Acad. Sci. USA 2020, 22, 12087–12094. [Google Scholar]
- Singh, J.; Udgaonkar, J.B. Molecular Mechanism of the Misfolding and Oligomerization of the Prion Protein: Current Understanding and Its Implication. J. Phys. Chem. B 2015, 54, 4431–4442. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Udgaonkar, J.B. The Pathogenic Mutation T182A Converts the Prion Protein into a Molten Globule-like Conformation Whose Misfolding to Oligomers but Not to Fibrils is Drastically Accelerated. J. Phys. Chem. B 2015, 55, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.A.; Coffey, H.A.; Moprin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.L.; Cino, E.A.; Soares, I.N.; Ferreira, V.F.; de Oliveira, G.A.P. Targeting the prion-like aggregation of mutant p53 to combat cancer. Acc. Chem. Res. 2018, 51, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Poulson, B.G.; Szczepski, K.; Lachowicz, J.I.; Jaremko, L.; Emwas, A.-H.; Jaremko, M. Aggregation of biologically important peptides and proteins: Inhibition or acceleration depending on protein and metal ion concentrations. RSC Adv. 2020, 10, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Emwas, A.H.; Alghrably, M.; Dhahri, M.; Sharfalddin, A.; Alsiary, R.; Jaremko, M.; Faa, G.; Campagna, M.; Congiu, T.; Piras, M.; et al. Living with the enemy: From protein-misfolding pathologies we know, to those we want to know. Ageing Res. Rev. 2021, 70, 101391. [Google Scholar] [CrossRef]
- Jaremko, M.; Jaremko, L.; Kim, H.-Y.; Cho, M.-K.; Schwieters, C.D.; Giller, K.; Becker, S.; Zweckstetter, M. Cold denaturation of a protein dimer monitored at atomic resolution. Nat. Chem. Biol. 2013, 9, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Bergasa-Caceres, F.; Rabitz, H.A. Low Entropic Barrier to the Hydrophobic Collapse of the Prion Protein: Effects of Intermediate States and Conformational Flexibility. J. Phys. Chem. A 2010, 114, 6978–6982. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Fu, H.; Fryar, K.L.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of Hydrogen Bonds to Protein Stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Fauchere, J.L.; Pliska, V. Hydrophobic Parameters II of Amino-Acid Side Cains from the Partitioning of N-Acetyl-Amino-Acid Amides. Eur. J. Med. Chem. 1983, 18, 369–375. [Google Scholar]
- Ozkan, S.B.; Wu, G.A.; Chodera, J.D.; Dill, K.A. Protein Folding by Zipping and Assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 11987–11992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fersht, A.R. Nucleation mechanisms in protein folding. Curr. Op. Struct. Biol. 1997, 7, 3–9. [Google Scholar] [CrossRef]
- Nishikawa, N.; Sakae, Y.; Gouda, T.; Tsujimura, Y.; Okamoto, Y. Structural analysis of a trimer of ß2-microglobulin fragment by molecular dynamics simulations. Biophys. J. 2019, 116, 781–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contact | Stability | 3D Structure | |
|---|---|---|---|
| C1 | 159AMAIY163 on 251ILTII255 | 8.9 ± 0.3 | Native |
| C2 | 143VQLWV147 on 216VVVPY220 | 8.2 ± 0.4 | Non-Native |
| C3 | 143VQLWV147 on 234YNYMC238 | 7.8 ± 0.4 | Non-Native |
| C4 | 133MFCQL137 on 216VVVPY220 | 7.8 ± 0.4 | Non-native |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergasa-Caceres, F.; Rabitz, H.A. Interdiction in the Early Folding of the p53 DNA-Binding Domain Leads to Its Amyloid-Like Misfolding. Molecules 2022, 27, 4810. https://doi.org/10.3390/molecules27154810
Bergasa-Caceres F, Rabitz HA. Interdiction in the Early Folding of the p53 DNA-Binding Domain Leads to Its Amyloid-Like Misfolding. Molecules. 2022; 27(15):4810. https://doi.org/10.3390/molecules27154810
Chicago/Turabian StyleBergasa-Caceres, Fernando, and Herschel A. Rabitz. 2022. "Interdiction in the Early Folding of the p53 DNA-Binding Domain Leads to Its Amyloid-Like Misfolding" Molecules 27, no. 15: 4810. https://doi.org/10.3390/molecules27154810
APA StyleBergasa-Caceres, F., & Rabitz, H. A. (2022). Interdiction in the Early Folding of the p53 DNA-Binding Domain Leads to Its Amyloid-Like Misfolding. Molecules, 27(15), 4810. https://doi.org/10.3390/molecules27154810