
Comparison of Vibrational Spectroscopic Techniques for Quantification of Water in Natural Deep Eutectic Solvents

 , , ,
, , ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
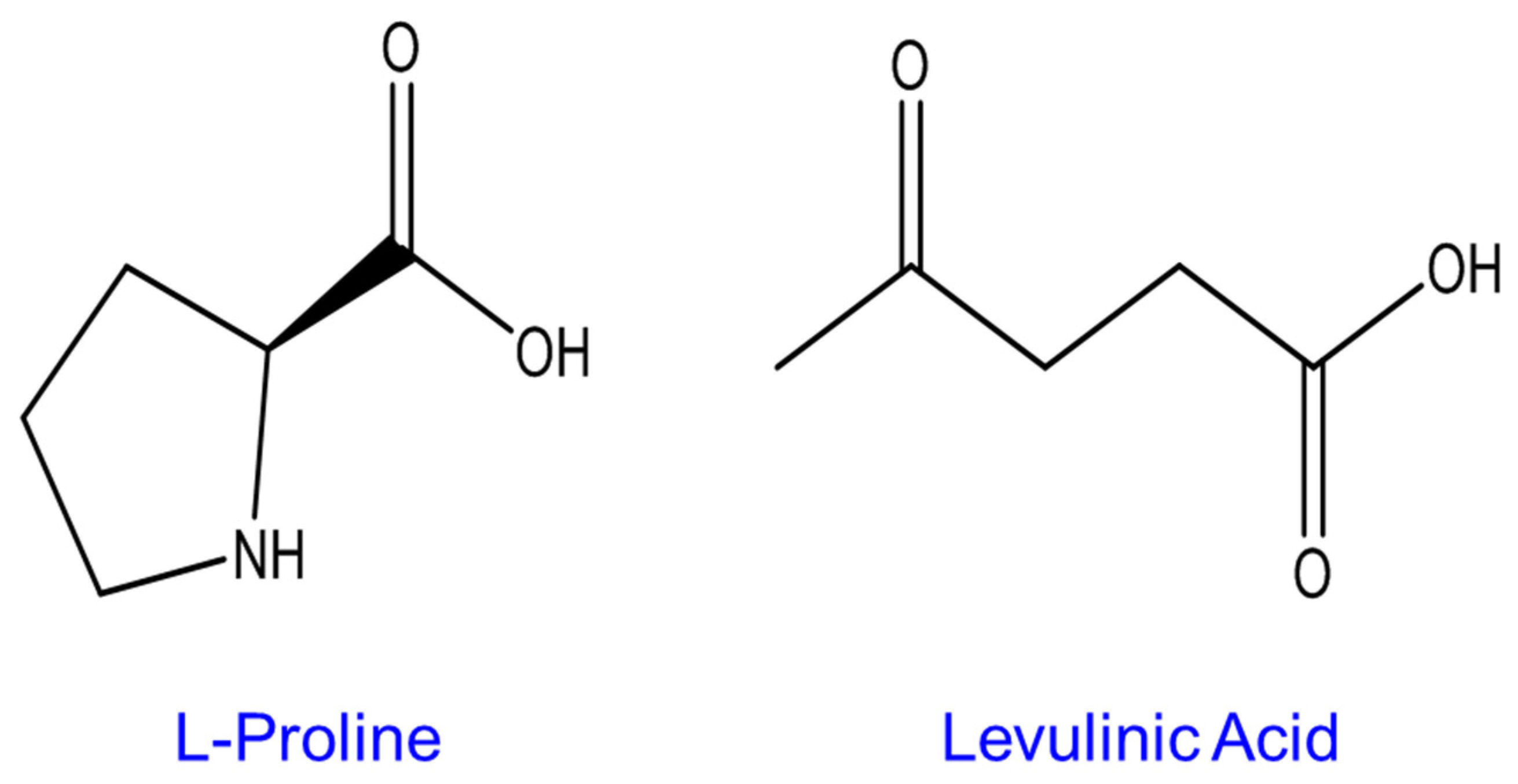
2.2. Preparation of Levulinic Acid/L-Proline (LALP) NADES Samples
2.3. Data Collection
2.3.1. Attenuated Total Reflectance (ATR-IR) Spectroscopy
2.3.2. Benchtop Near Infrared Spectroscopy (NIR-B)
2.3.3. Handheld Near Infrared Spectroscopy (NIR-H)
2.3.4. Benchtop Raman Microscope (Raman-B)
2.4. Data Analysis
3. Results and Discussions
3.1. Construction of Predictive Models and Spectral Characterisation
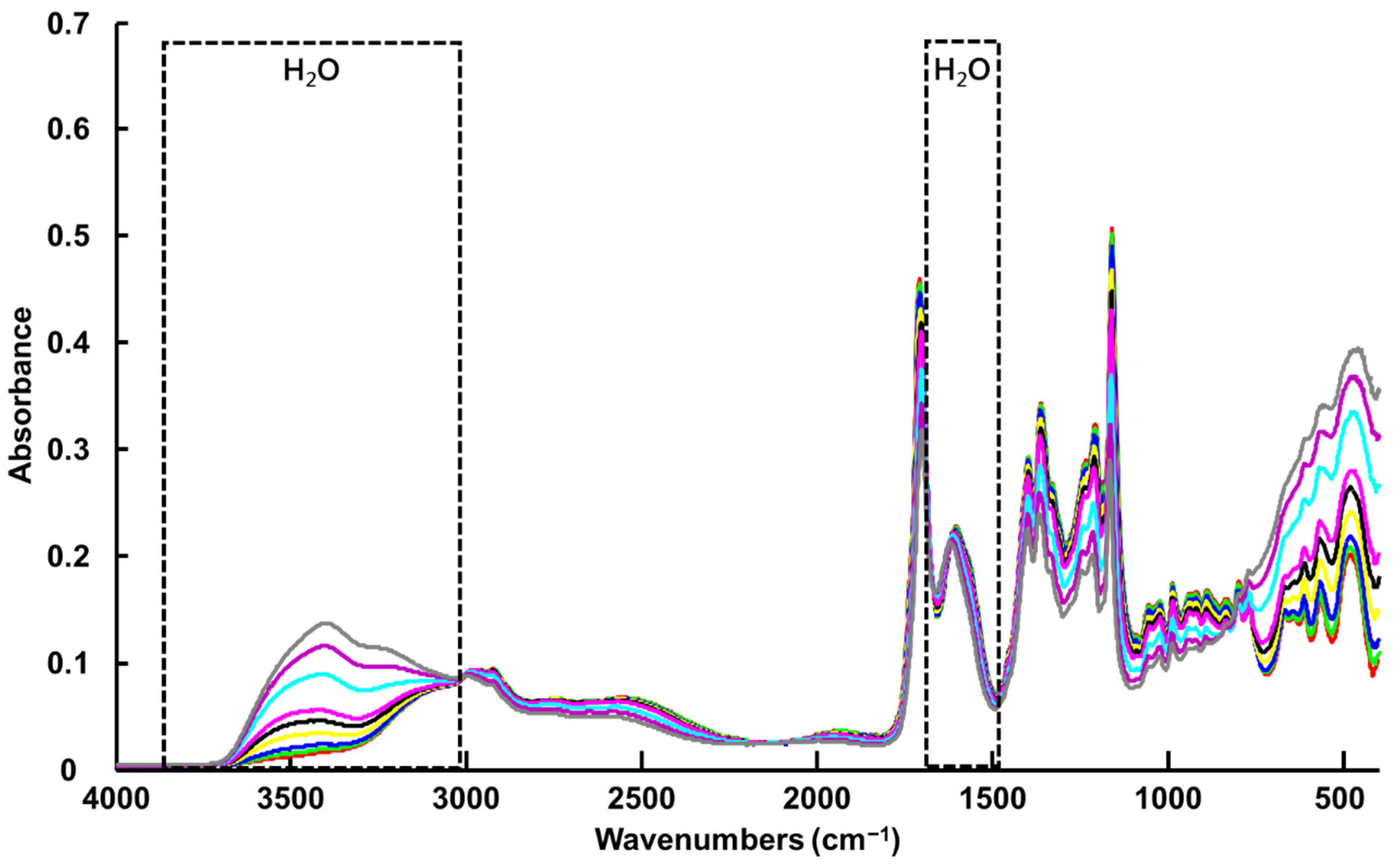
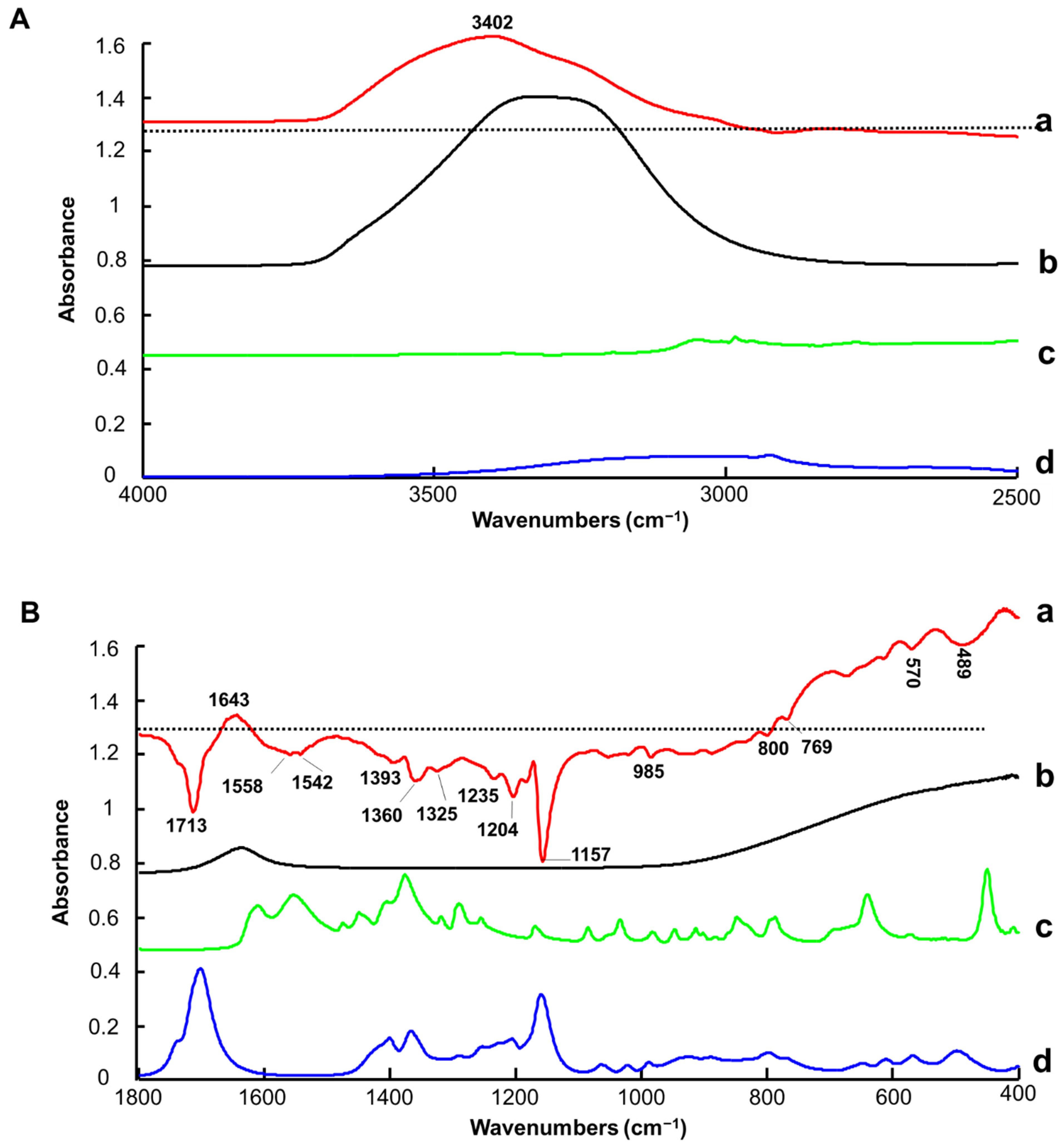
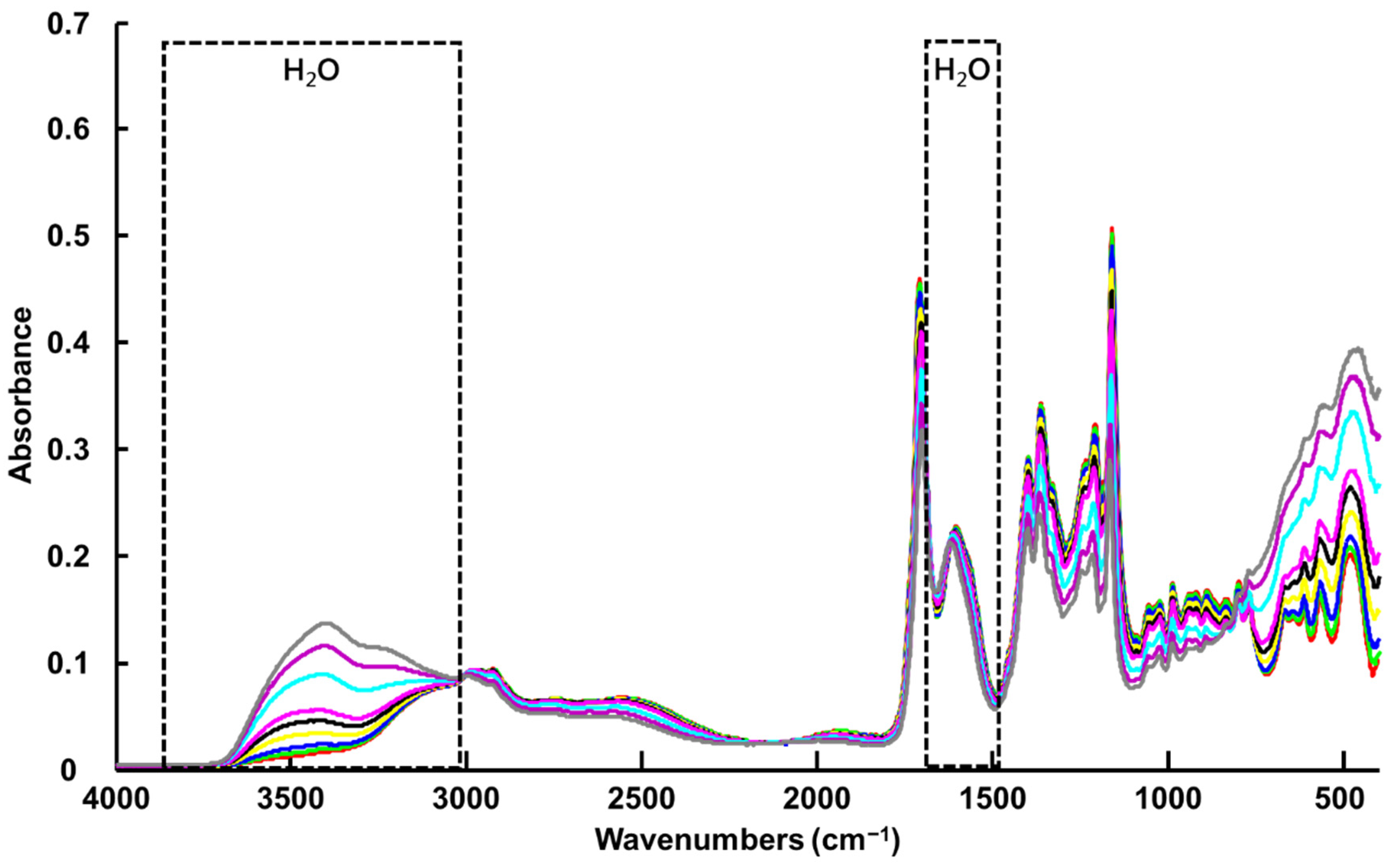
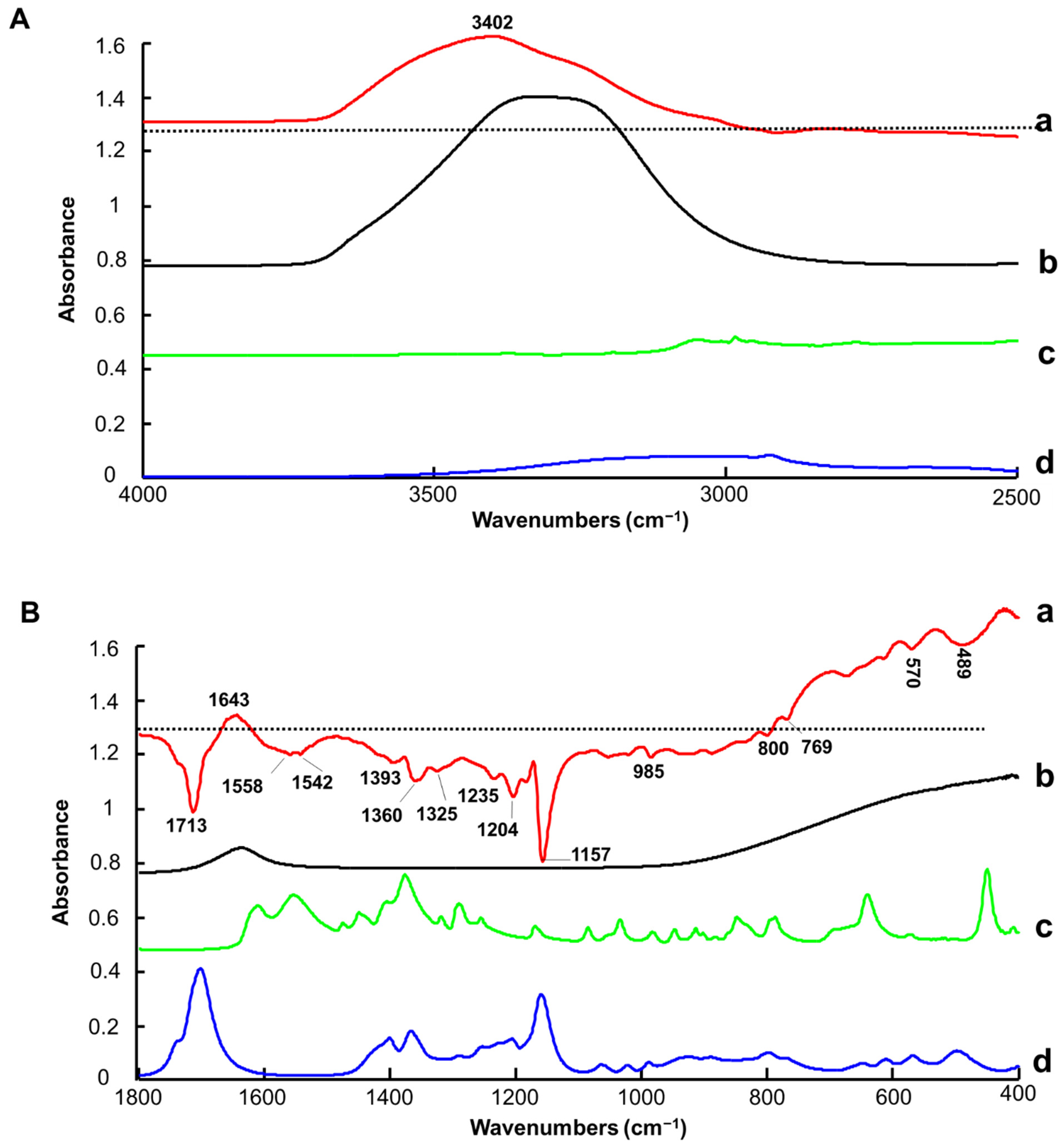
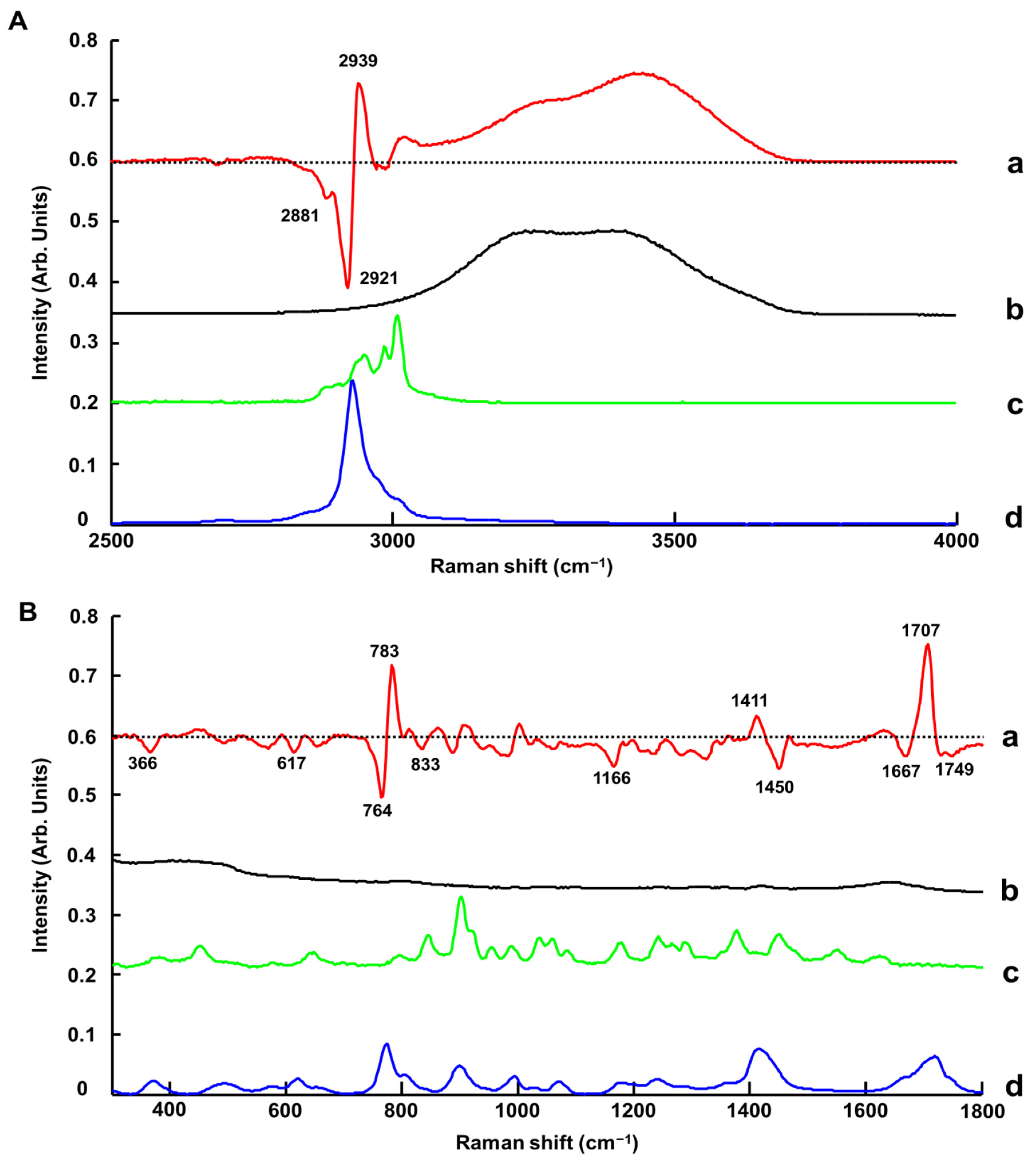
3.1.1. ATR-IR Spectroscopy
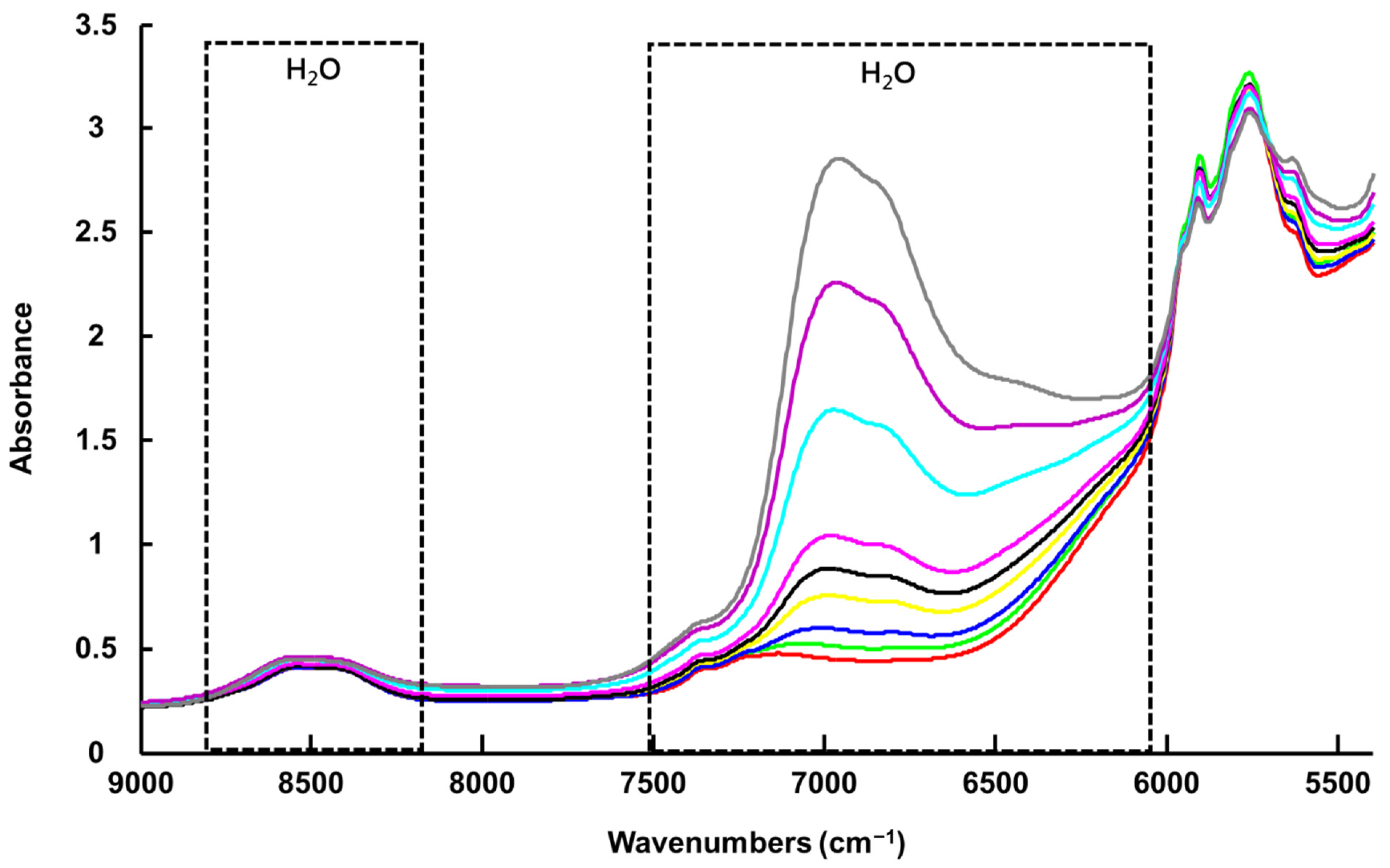
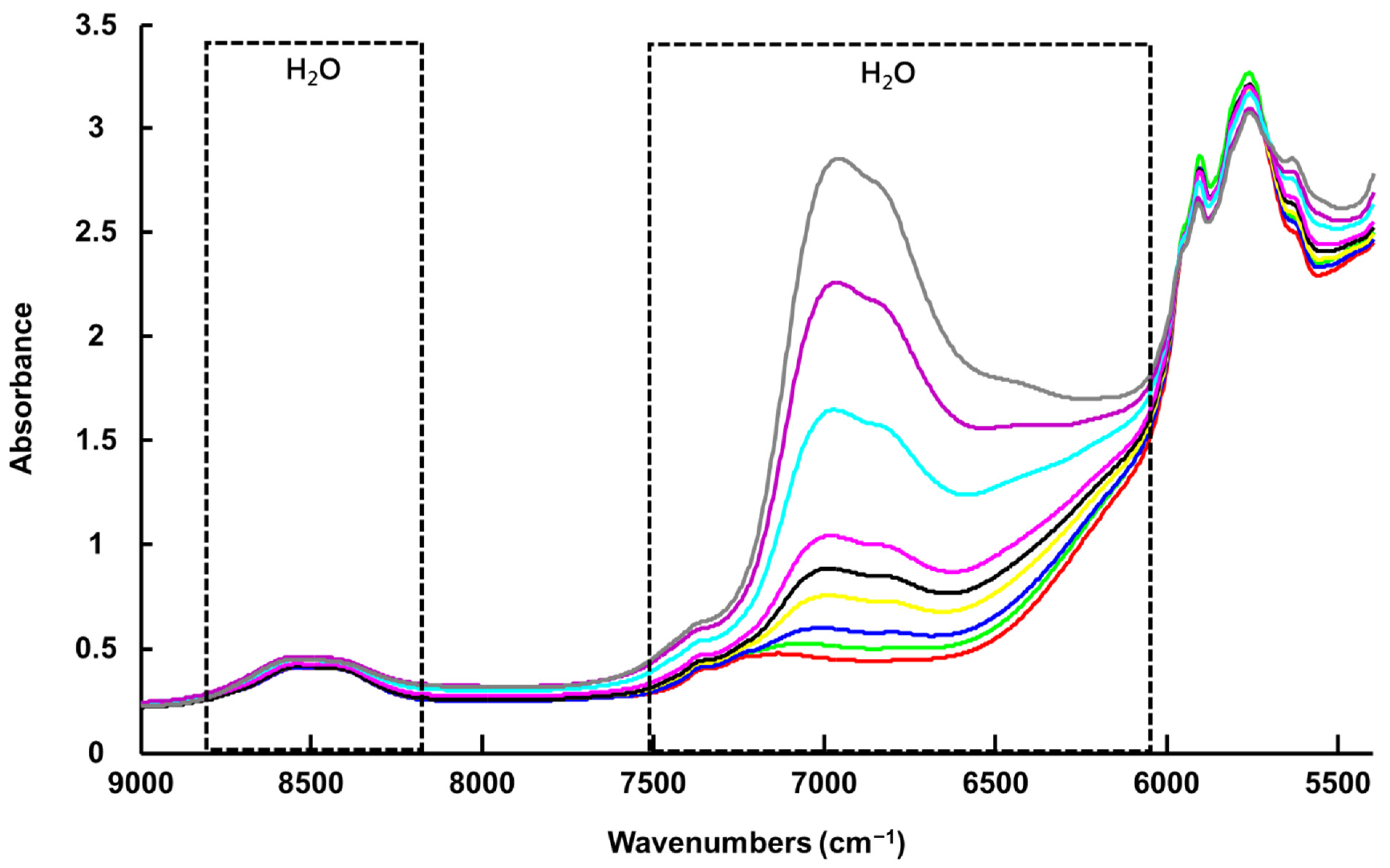
3.1.2. Benchtop NIR Spectroscopy (NIR-B)
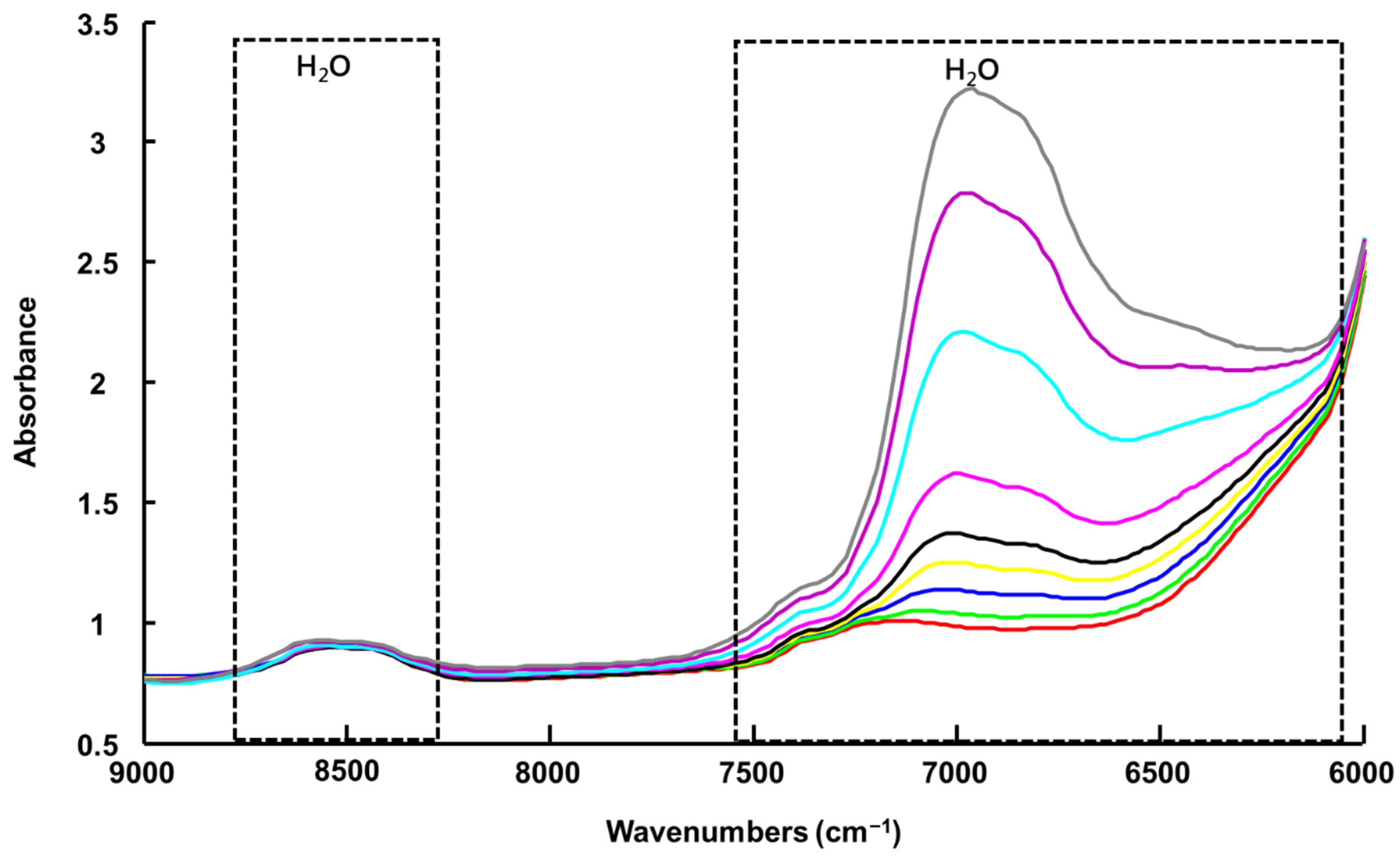
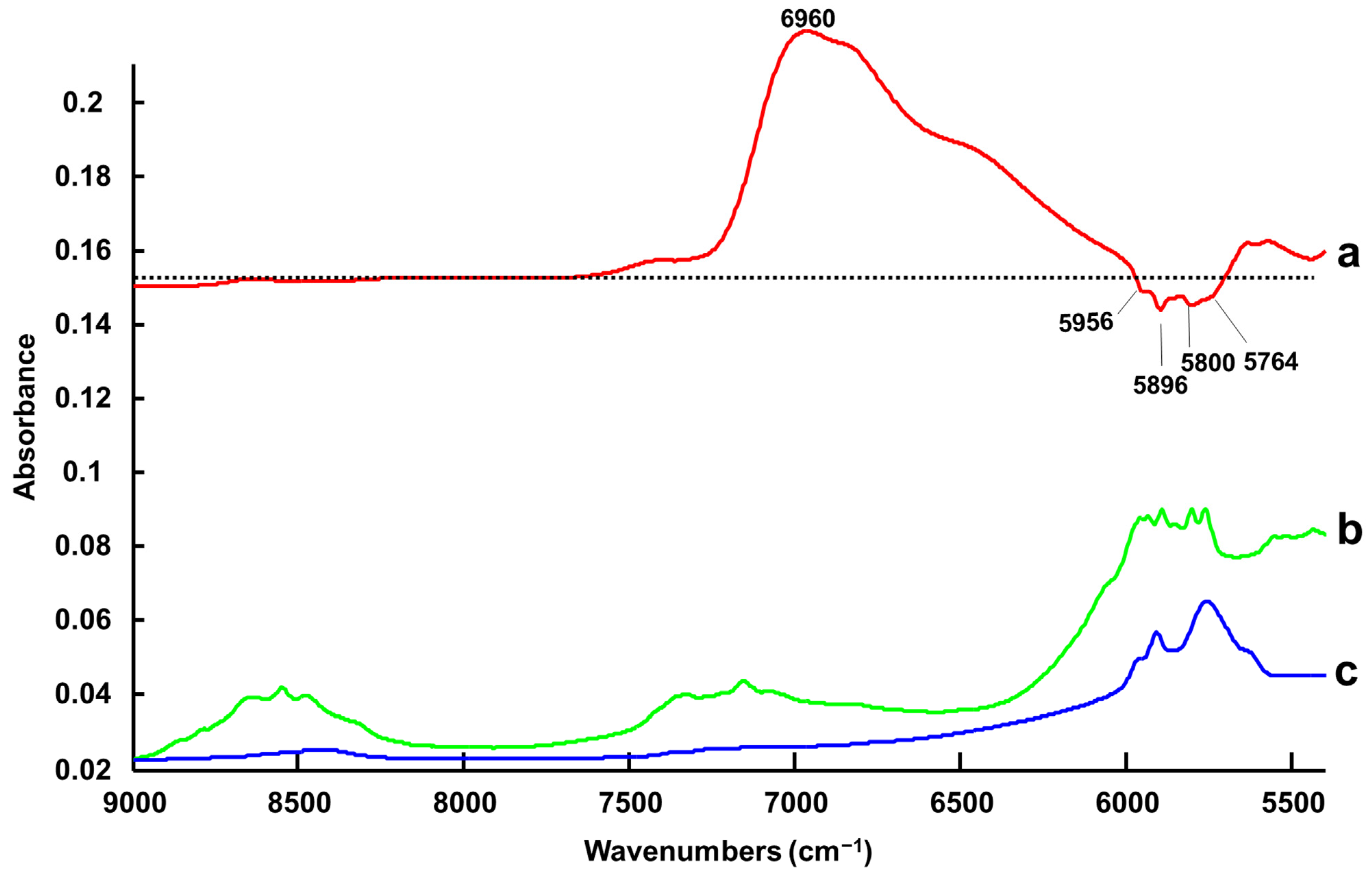
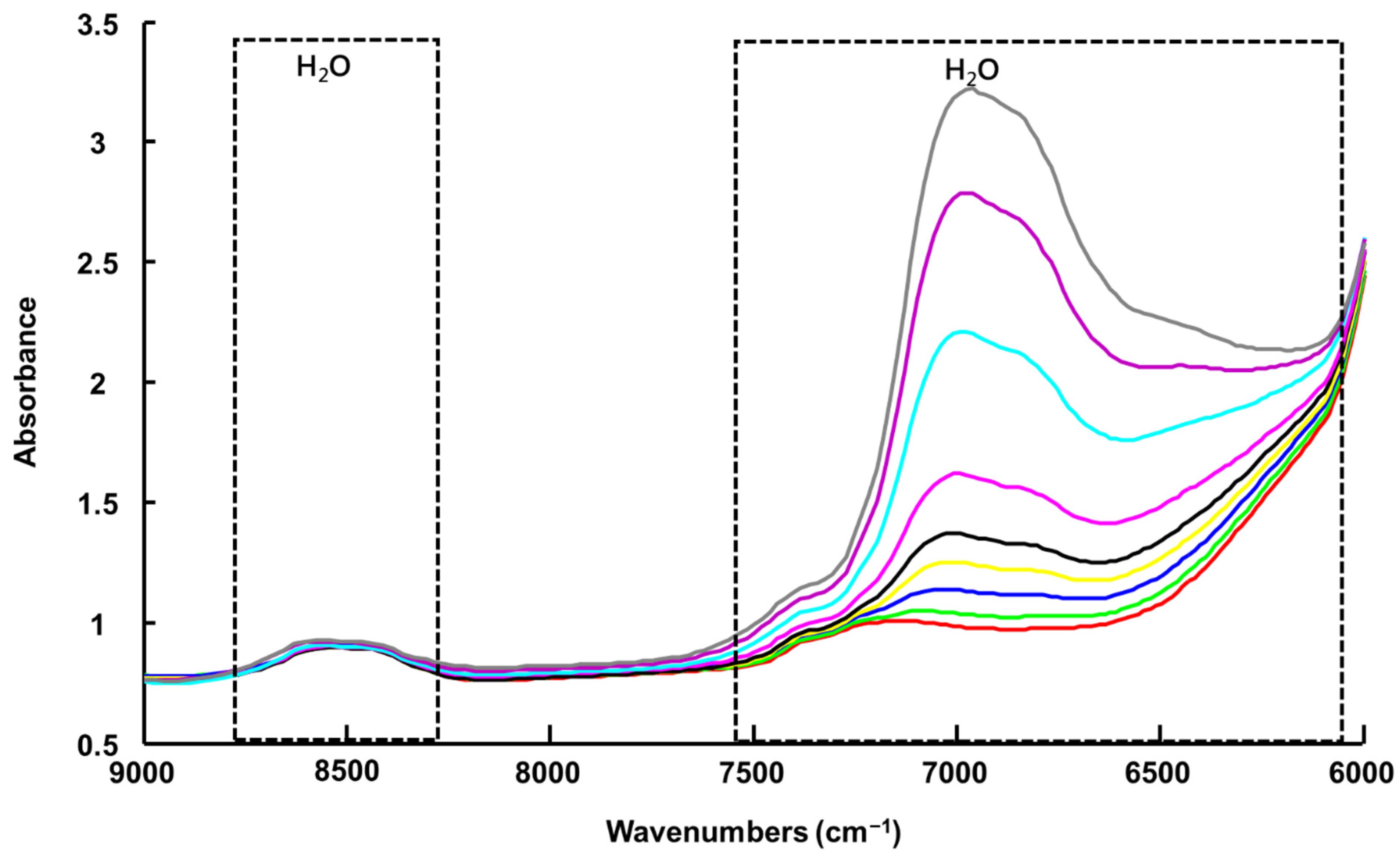
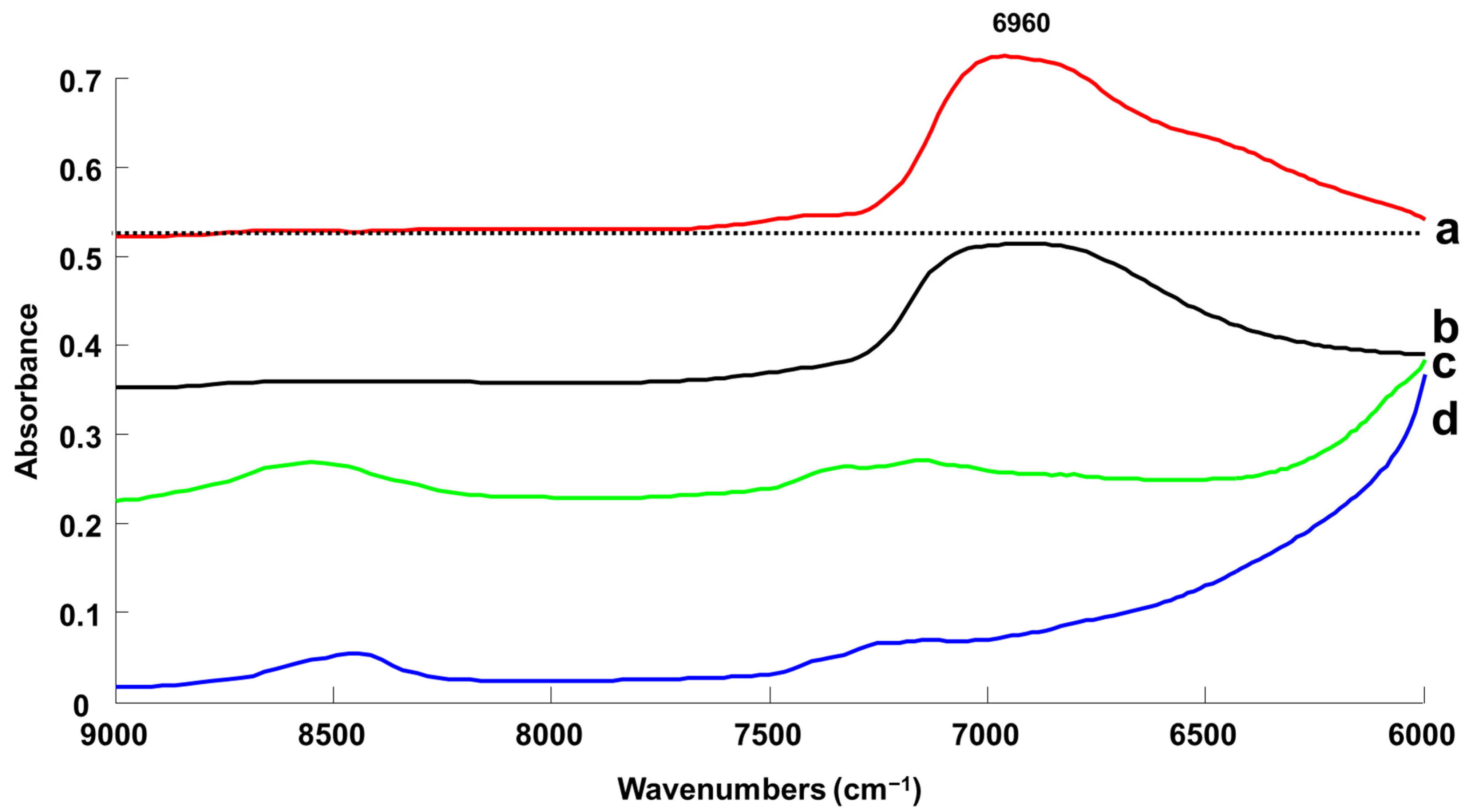
3.1.3. Handheld NIR Spectroscopy (NIR-H)
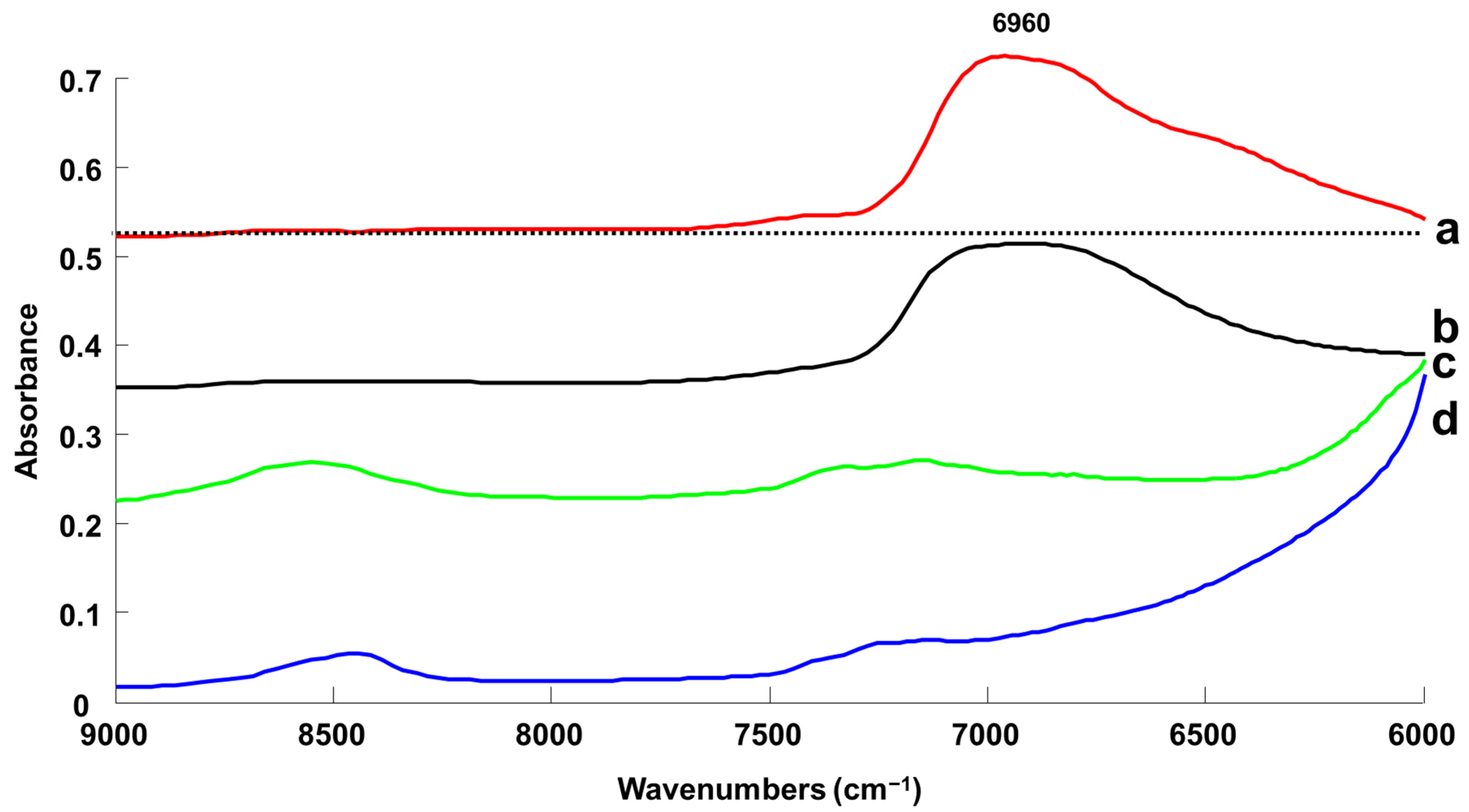
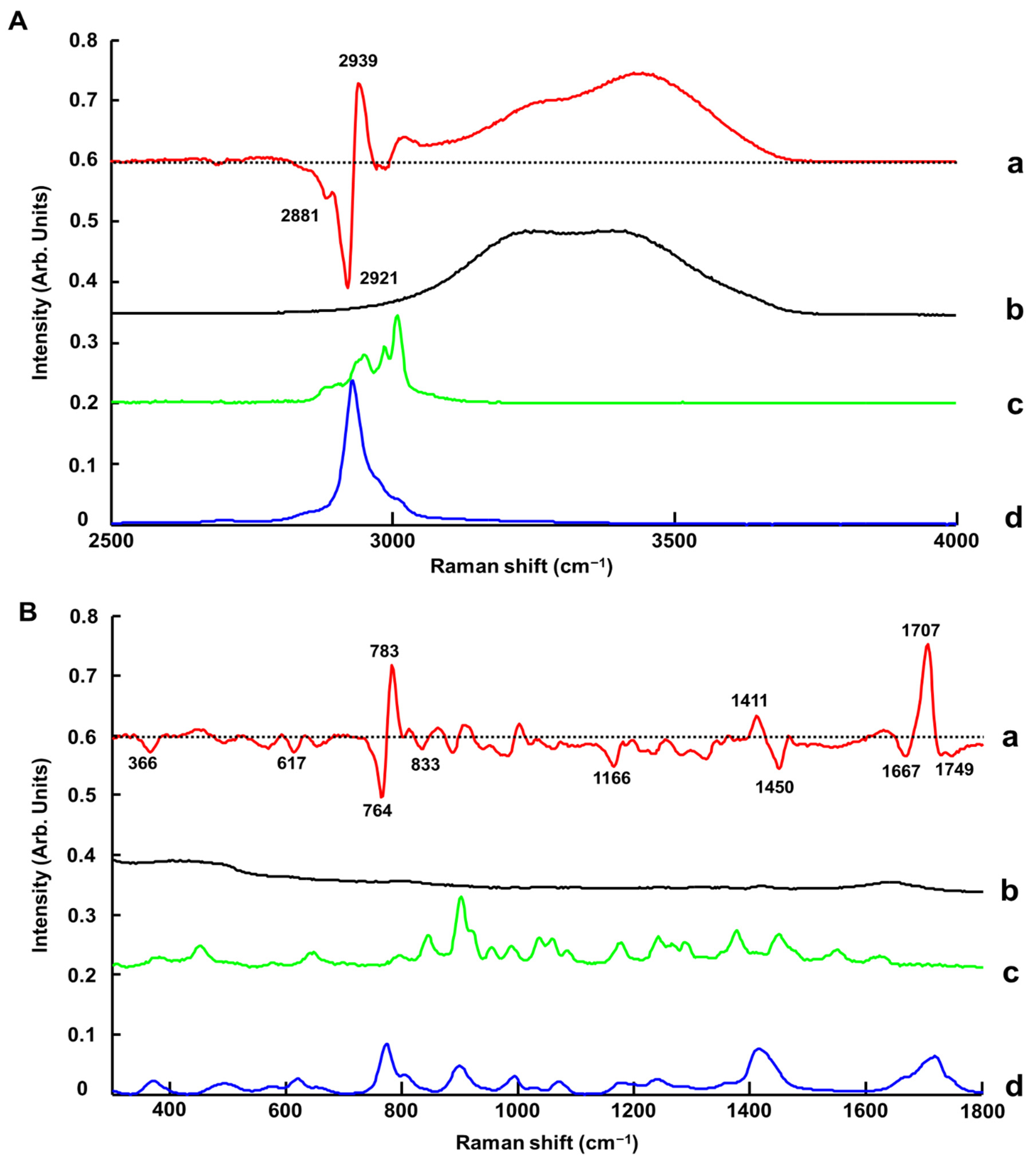
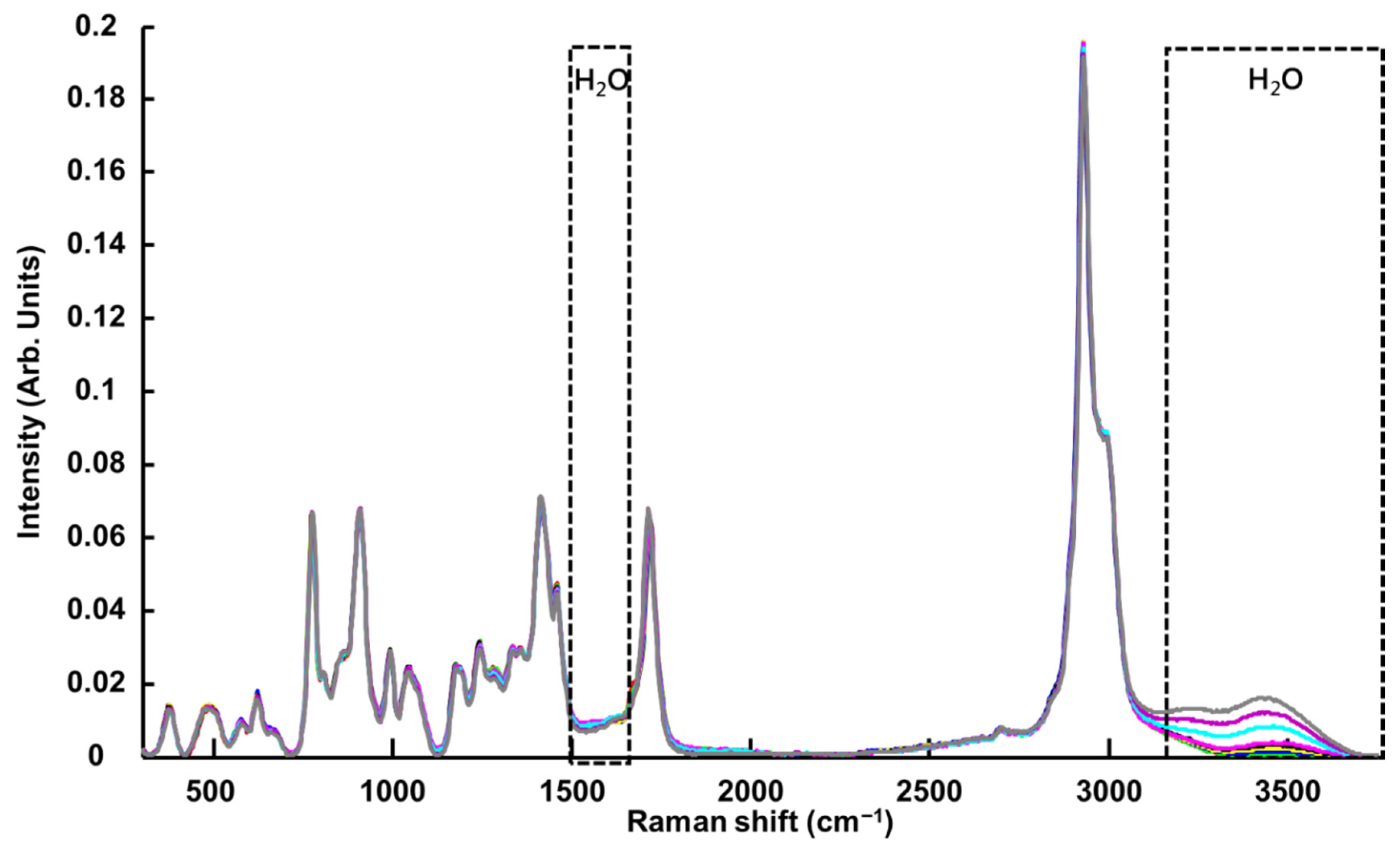
3.1.4. Benchtop Raman Microscope (Raman-B)
3.1.5. Overview of PLSR Cross-Validation Results
3.2. Comparison of Prediction for % w/w Added Water Concentration in Test Sets
3.3. General Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Dai, Y.; van Spronsen, J.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Natural Deep Eutectic Solvents as New Potential Media for Green Technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Socas-Rodríguez, B.; Santana-Mayor, Á.; Herrera-Herrera, A.V.; Rodríguez-Delgado, M.Á. Chapter 5—Deep Eutectic Solvents. In Green Sustainable Process for Chemical and Environmental Engineering and Science; Inamuddin, A.A.M., Kanchi, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 123–177. ISBN 978-0-12-817386-2. [Google Scholar]
- Santana-Mayor, Á.; Rodríguez-Ramos, R.; Herrera-Herrera, A.V.; Socas-Rodríguez, B.; Rodríguez-Delgado, M.Á. Deep Eutectic Solvents. The New Generation of Green Solvents in Analytical Chemistry. TrAC Trends Anal. Chem. 2021, 134, 116108. [Google Scholar] [CrossRef]
- de los Ángeles Fernández, M.; Boiteux, J.; Espino, M.; Gomez, F.J.V.; Silva, M.F. Natural Deep Eutectic Solvents-Mediated Extractions: The Way Forward for Sustainable Analytical Developments. Anal. Chim. Acta 2018, 1038, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Verpoorte, R. Green Solvents for the Extraction of Bioactive Compounds from Natural Products Using Ionic Liquids and Deep Eutectic Solvents. Curr. Opin. Food Sci. 2019, 26, 87–93. [Google Scholar] [CrossRef]
- Liu, Y.; Friesen, J.B.; McAlpine, J.B.; Lankin, D.C.; Chen, S.-N.; Pauli, G.F. Natural Deep Eutectic Solvents: Properties, Applications, and Perspectives. J. Nat. Prod. 2018, 81, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Tailoring Properties of Natural Deep Eutectic Solvents with Water to Facilitate Their Applications. Food Chem. 2015, 187, 14–19. [Google Scholar] [CrossRef]
- Kumar, A.K.; Parikh, B.S.; Liu, L.Z.; Cotta, M.A. Application of Natural Deep Eutectic Solvents in Biomass Pretreatment, Enzymatic Saccharification and Cellulosic Ethanol Production. Mater. Today Proc. 2018, 5, 23057–23063. [Google Scholar] [CrossRef]
- Delaye, P.-O.; Pénichon, M.; Boudesocque-Delaye, L.; Enguehard-Gueiffier, C.; Gueiffier, A. Natural Deep Eutectic Solvents as Sustainable Solvents for Suzuki–Miyaura Cross-Coupling Reactions Applied to Imidazo-Fused Heterocycles. SynOpen 2018, 2, 306–311. [Google Scholar] [CrossRef]
- Mbous, Y.P.; Hayyan, M.; Hayyan, A.; Wong, W.F.; Hashim, M.A.; Looi, C.Y. Applications of Deep Eutectic Solvents in Biotechnology and Bioengineering—Promises and Challenges. Biotechnol. Adv. 2017, 35, 105–134. [Google Scholar] [CrossRef]
- Yang, Z. Natural Deep Eutectic Solvents and Their Applications in Biotechnology. Adv. Biochem. Eng. Biotechnol. 2019, 168, 31–59. [Google Scholar] [CrossRef]
- Ghareh Bagh, F.S.; Shahbaz, K.; Mjalli, F.S.; Hashim, M.A.; AlNashef, I.M. Zinc (II) Chloride-Based Deep Eutectic Solvents for Application as Electrolytes: Preparation and Characterization. J. Mol. Liq. 2015, 204, 76–83. [Google Scholar] [CrossRef]
- Abo-Hamad, A.; Hayyan, M.; AlSaadi, M.A.; Hashim, M.A. Potential Applications of Deep Eutectic Solvents in Nanotechnology. Chem. Eng. J. 2015, 273, 551–567. [Google Scholar] [CrossRef]
- Tohidi, M.; Mahyari, F.A.; Safavi, A. A Seed-Less Method for Synthesis of Ultra-Thin Gold Nanosheets by Using a Deep Eutectic Solvent and Gum Arabic and Their Electrocatalytic Application. RSC Adv. 2015, 5, 32744–32754. [Google Scholar] [CrossRef]
- Liao, H.-G.; Jiang, Y.-X.; Zhou, Z.-Y.; Chen, S.-P.; Sun, S.-G. Shape-Controlled Synthesis of Gold Nanoparticles in Deep Eutectic Solvents for Studies of Structure–Functionality Relationships in Electrocatalysis. Angew. Chem. Int. Ed. 2008, 47, 9100–9103. [Google Scholar] [CrossRef] [Green Version]
- Atilhan, M.; Aparicio, S. Review and Perspectives for Effective Solutions to Grand Challenges of Energy and Fuels Technologies via Novel Deep Eutectic Solvents. Energy Fuels 2021, 35, 6402–6419. [Google Scholar] [CrossRef]
- Chang, S.H. Utilization of Green Organic Solvents in Solvent Extraction and Liquid Membrane for Sustainable Wastewater Treatment and Resource Recovery—A Review. Environ. Sci. Pollut. Res. Int. 2020, 27, 32371–32388. [Google Scholar] [CrossRef]
- Benoit, C.; Virginie, C.; Boris, V. Chapter Twelve—The Use of NADES to Support Innovation in the Cosmetic Industry. In Advances in Botanical Research; Verpoorte, R., Witkamp, G.-J., Choi, Y.H., Eds.; Eutectic Solvents and Stress in Plants; Academic Press: London, UK, 2021; Volume 97, pp. 309–332. [Google Scholar]
- Balakrishnan, I.; Jawahar, N.; Venkatachalam, S.; Datta, D. A Brief Review on Eutectic Mixture and Its Role in Pharmaceutical Field. Int. J. Res. Pharm. Sci. 2020, 11, 3017–3023. [Google Scholar] [CrossRef]
- Mišan, A.; Nađpal, J.; Stupar, A.; Pojić, M.; Mandić, A.; Verpoorte, R.; Choi, Y.H. The Perspectives of Natural Deep Eutectic Solvents in Agri-Food Sector. Crit. Rev. Food Sci. Nutr. 2020, 60, 2564–2592. [Google Scholar] [CrossRef]
- Dai, Y.; Verpoorte, R.; Choi, Y.H. Natural Deep Eutectic Solvents Providing Enhanced Stability of Natural Colorants from Safflower (Carthamus Tinctorius). Food Chem. 2014, 159, 116–121. [Google Scholar] [CrossRef]
- Radošević, K.; Ćurko, N.; Gaurina Srček, V.; Cvjetko Bubalo, M.; Tomašević, M.; Kovačević Ganić, K.; Radojčić Redovniković, I. Natural Deep Eutectic Solvents as Beneficial Extractants for Enhancement of Plant Extracts Bioactivity. Food Sci. Technol. 2016, 73, 45–51. [Google Scholar] [CrossRef]
- Choi, Y.H.; van Spronsen, J.; Dai, Y.; Verberne, M.; Hollmann, F.; Arends, I.W.C.E.; Witkamp, G.-J.; Verpoorte, R. Are Natural Deep Eutectic Solvents the Missing Link in Understanding Cellular Metabolism and Physiology? Plant Physiol. 2011, 156, 1701–1705. [Google Scholar] [CrossRef] [Green Version]
- Vilková, M.; Płotka-Wasylka, J.; Andruch, V. The Role of Water in Deep Eutectic Solvent-Base Extraction. J. Mol. Liq. 2020, 304, 112747. [Google Scholar] [CrossRef]
- Dugoni, G.C.; Mezzetta, A.; Guazzelli, L.; Chiappe, C.; Ferro, M.; Mele, A. Purification of Kraft Cellulose under Mild Conditions Using Choline Acetate Based Deep Eutectic Solvents. Green Chem. 2020, 22, 8680–8691. [Google Scholar] [CrossRef]
- Dantan, N.; Frenzel, W.; Küppers, S. Determination of Water Traces in Various Organic Solvents Using Karl Fischer Method under FIA Conditions. Talanta 2000, 52, 101–109. [Google Scholar] [CrossRef]
- Ronkart, S.N.; Paquot, M.; Fougnies, C.; Deroanne, C.; Van Herck, J.-C.; Blecker, C. Determination of Total Water Content in Inulin Using the Volumetric Karl Fischer Titration. Talanta 2006, 70, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- De Caro, C.A.; Aichert, A.; Walter, C.M. Efficient, Precise and Fast Water Determination by the Karl Fischer Titration. Food Control 2001, 12, 431–436. [Google Scholar] [CrossRef]
- Sanchez, V.; Baeza, R.; Ciappini, C.; Zamora, M.C.; Chirife, J. Comparison between Karl Fischer and Refractometric Method for Determination of Water Content in Honey. Food Control 2010, 21, 339–341. [Google Scholar] [CrossRef]
- Wrolstad, R.E.; Acree, T.E.; Decker, E.A.; Penner, M.H.; Reid, D.S.; Schwartz, S.J.; Shoemaker, C.F.; Smith, D.M.; Sporns, P. Handbook of Food Analytical Chemistry, Volume 1: Water, Proteins, Enzymes, Lipids, and Carbohydrates; John Wiley & Sons: Hoboken, NJ, USA, 2005; ISBN 978-0-471-70909-1. [Google Scholar]
- Scholz, E. Karl Fischer Titration; Chemical Laboratory Practice; Springer: Berlin/Heidelberg, Germany, 1984; ISBN 978-3-642-69991-7. [Google Scholar]
- Isengard, H.-D.; Schultheiß, D.; Radović, B.; Anklam, E. Alternatives to Official Analytical Methods Used for the Water Determination in Honey. Food Control 2001, 12, 459–466. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J. A New Tool for the Evaluation of the Analytical Procedure: Green Analytical Procedure Index. Talanta 2018, 181, 204–209. [Google Scholar] [CrossRef]
- Heinz, A.; Strachan, C.J.; Gordon, K.C.; Rades, T. Analysis of Solid-State Transformations of Pharmaceutical Compounds Using Vibrational Spectroscopy. J. Pharm. Pharmacol. 2009, 61, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Ewing, A.V.; Kazarian, S.G. Recent Advances in the Applications of Vibrational Spectroscopic Imaging and Mapping to Pharmaceutical Formulations. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 197, 10–29. [Google Scholar] [CrossRef]
- Huck, C.W. Advances of Vibrational Spectroscopic Methods in Phytomics and Bioanalysis. J. Pharm. Biomed. Anal. 2014, 87, 26–35. [Google Scholar] [CrossRef]
- Depciuch, J.; Kaznowska, E.; Zawlik, I.; Wojnarowska, R.; Cholewa, M.; Heraud, P.; Cebulski, J. Application of Raman Spectroscopy and Infrared Spectroscopy in the Identification of Breast Cancer. Appl. Spectrosc. 2016, 70, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, L.E.; Byrne, H.J. Vibrational Spectroscopy as a Tool for Studying Drug-Cell Interaction: Could High Throughput Vibrational Spectroscopic Screening Improve Drug Development? Vib. Spectrosc. 2017, 91, 16–30. [Google Scholar] [CrossRef] [Green Version]
- Taha Mohamed, H.; Untereiner, V.; Proult, I.; Abdelaziz Ibrahim, S.; Götte, M.; El-Shinawi, M.; Mostafa Mohamed, M.; Sockalingum, G.D.; Brézillon, S. Characterization of Inflammatory Breast Cancer: A Vibrational Microspectroscopy and Imaging Approach at the Cellular and Tissue Level. Analyst 2018, 143, 6103–6112. [Google Scholar] [CrossRef] [Green Version]
- Byrne, H.J.; Bonnier, F.; Casey, A.; Maher, M.; McIntyre, J.; Efeoglu, E.; Farhane, Z. Advancing Raman Microspectroscopy for Cellular and Subcellular Analysis: Towards in Vitro High-Content Spectralomic Analysis. Appl. Opt. 2018, 57, E11–E19. [Google Scholar] [CrossRef] [Green Version]
- Byrne, H.J.; Bonnier, F.; McIntyre, J.; Parachalil, D.R. Quantitative Analysis of Human Blood Serum Using Vibrational Spectroscopy. Clin. Spectrosc. 2020, 2, 100004. [Google Scholar] [CrossRef]
- Makki, A.A.; Bonnier, F.; Respaud, R.; Chtara, F.; Tfayli, A.; Tauber, C.; Bertrand, D.; Byrne, H.J.; Mohammed, E.; Chourpa, I. Qualitative and Quantitative Analysis of Therapeutic Solutions Using Raman and Infrared Spectroscopy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 218, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makki, A.A.; Massot, V.; Byrne, H.J.; Respaud, R.; Bertrand, D.; Mohammed, E.; Chourpa, I.; Bonnier, F. Vibrational Spectroscopy for Discrimination and Quantification of Clinical Chemotherapeutic Preparations. Vib. Spectrosc. 2021, 113, 103200. [Google Scholar] [CrossRef]
- Mazurek, S.; Szostak, R. Quantitative Determination of Prednisone in Tablets by Infrared Attenuated Total Reflection and Raman Spectroscopy. J. AOAC Int. 2012, 95, 744–750. [Google Scholar] [CrossRef]
- Strachan, C.J.; Rades, T.; Gordon, K.C.; Rantanen, J. Raman Spectroscopy for Quantitative Analysis of Pharmaceutical Solids. J. Pharm. Pharmacol. 2007, 59, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Mallah, M.A.; Sherazi, S.T.H.; Bhanger, M.I.; Mahesar, S.A.; Bajeer, M.A. A Rapid Fourier-Transform Infrared (FTIR) Spectroscopic Method for Direct Quantification of Paracetamol Content in Solid Pharmaceutical Formulations. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2015, 141, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Miloudi, L.; Bonnier, F.; Bertrand, D.; Byrne, H.J.; Perse, X.; Chourpa, I.; Munnier, E. Quantitative Analysis of Curcumin-Loaded Alginate Nanocarriers in Hydrogels Using Raman and Attenuated Total Reflection Infrared Spectroscopy. Anal. Bioanal. Chem. 2017, 409, 4593–4605. [Google Scholar] [CrossRef]
- Van De Voort, F.R.; Sedman, J.; Yaylayan, V.; Laurent, C.S.; Mucciardi, C. Quantitative Determination of Moisture in Lubricants by Fourier Transform Infrared Spectroscopy. Appl. Spectrosc. 2004, 58, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Coello, J.; Iturriaga, H.; Maspoch, S.; González, R. Determination of Water in Lubricating Oils by Mid- and near-Infrared Spectroscopy. Mikrochim. Acta 1998, 128, 235–239. [Google Scholar] [CrossRef]
- Hop, E.; Luinge, H.-J.; Hemert, H.V. Quantitative Analysis of Water in Milk by FT-IR Spectrometry. Appl. Spectrosc. 1993, 47, 1180–1182. [Google Scholar] [CrossRef]
- Schiavi, F.; Bolfan-Casanova, N.; Withers, A.C.; Médard, E.; Laumonier, M.; Laporte, D.; Flaherty, T.; Gómez-Ulla, A. Water Quantification in Silicate Glasses by Raman Spectroscopy: Correcting for the Effects of Confocality, Density and Ferric Iron. Chem. Geol. 2018, 483, 312–331. [Google Scholar] [CrossRef]
- Thomas, S.-M.; Thomas, R.; Davidson, P.; Reichart, P.; Koch-Muller, M.; Dollinger, G. Application of Raman Spectroscopy to Quantify Trace Water Concentrations in Glasses and Garnets. Am. Mineral. 2008, 93, 1550–1557. [Google Scholar] [CrossRef]
- Czaja, T.; Kuzawińska, E.; Sobota, A.; Szostak, R. Determining Moisture Content in Pasta by Vibrational Spectroscopy. Talanta 2018, 178, 294–298. [Google Scholar] [CrossRef]
- Czaja, T.; Sobota, A.; Szostak, R. Quantification of Ash and Moisture in Wheat Flour by Raman Spectroscopy. Foods 2020, 9, 280. [Google Scholar] [CrossRef] [Green Version]
- Büning-Pfaue, H. Analysis of Water in Food by near Infrared Spectroscopy. Food Chem. 2003, 82, 107–115. [Google Scholar] [CrossRef]
- Kauppinen, A.; Toiviainen, M.; Korhonen, O.; Aaltonen, J.; Järvinen, K.; Paaso, J.; Juuti, M.; Ketolainen, J. In-Line Multipoint Near-Infrared Spectroscopy for Moisture Content Quantification during Freeze-Drying. Anal. Chem. 2013, 85, 2377–2384. [Google Scholar] [CrossRef]
- Kauppinen, A.; Toiviainen, M.; Lehtonen, M.; Järvinen, K.; Paaso, J.; Juuti, M.; Ketolainen, J. Validation of a Multipoint Near-Infrared Spectroscopy Method for in-Line Moisture Content Analysis during Freeze-Drying. J. Pharm. Biomed. Anal. 2014, 95, 229–237. [Google Scholar] [CrossRef] [PubMed]
- van Kollenburg, G.H.; van Manen, H.-J.; Admiraal, N.; Gerretzen, J.; Jansen, J.J. Low-Cost Handheld NIR Spectroscopy for Identification of Organic Solvents and Low-Level Quantification of Water Contamination. Talanta 2021, 223, 121865. [Google Scholar] [CrossRef]
- Milliken, R.E.; Mustard, J.F. Quantifying Absolute Water Content of Minerals Using Near-Infrared Reflectance Spectroscopy. J. Geophys. Res. Planets 2005, 110, 0148–0227. [Google Scholar] [CrossRef] [Green Version]
- Suh, E.-J.; Woo, Y.-A.; Kim, H.-J. Determination of Water Content in Skin by Using a Ft near Infrared Spectrometer. Arch. Pharm. Res. 2005, 28, 458. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, H.; Egawa, M. Non-Contact Skin Moisture Measurement Based on near-Infrared Spectroscopy. Appl. Spectrosc. 2004, 58, 1439–1446. [Google Scholar] [CrossRef]
- Kilpatrick-Liverman, L.; Kazmi, P.; Wolff, E.; Polefka, T.G. The Use of Near-Infrared Spectroscopy in Skin Care Applications. Skin Res. Technol. 2006, 12, 162–169. [Google Scholar] [CrossRef]
- Mantanus, J.; Ziémons, E.; Lebrun, P.; Rozet, E.; Klinkenberg, R.; Streel, B.; Evrard, B.; Hubert, P. Moisture Content Determination of Pharmaceutical Pellets by near Infrared Spectroscopy: Method Development and Validation. Anal. Chim. Acta 2009, 642, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Elderderi, S.; Leman-Loubière, C.; Wils, L.; Henry, S.; Bertrand, D.; Byrne, H.J.; Chourpa, I.; Enguehard-Gueiffier, C.; Munnier, E.; Elbashir, A.A.; et al. ATR-IR Spectroscopy for Rapid Quantification of Water Content in Deep Eutectic Solvents. J. Mol. Liq. 2020, 311, 113361. [Google Scholar] [CrossRef]
- Elderderi, S.; Wils, L.; Leman-Loubière, C.; Henry, S.; Byrne, H.J.; Chourpa, I.; Munnier, E.; Elbashir, A.A.; Boudesocque-Delaye, L.; Bonnier, F. Comparison of Raman and Attenuated Total Reflectance (ATR) Infrared Spectroscopy for Water Quantification in Natural Deep Eutectic Solvent. Anal. Bioanal. Chem. 2021, 413, 4785–4799. [Google Scholar] [CrossRef] [PubMed]
- Elderderi, S.; Wils, L.; Leman-Loubière, C.; Byrne, H.J.; Chourpa, I.; Enguehard-Gueiffier, C.; Munnier, E.; Elbashir, A.A.; Boudesocque-Delaye, L.; Bonnier, F. In Situ Water Quantification in Natural Deep Eutectic Solvents Using Portable Raman Spectroscopy. Molecules 2021, 26, 5488. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Dębosz, M.; Świt, P.; Piech, A.; Kasperek, J.; Kościelniak, P. Reliable Calibration by Nonlinear Standard Addition Method in the Presence of Additive Interference Effects. Mon. Chem.-Chem. Mon. 2018, 149, 1567–1572. [Google Scholar] [CrossRef] [Green Version]
- Byrne, H.J.; Knief, P.; Keating, M.E.; Bonnier, F. Spectral Pre and Post Processing for Infrared and Raman Spectroscopy of Biological Tissues and Cells. Chem. Soc. Rev. 2016, 45, 1865–1878. [Google Scholar] [CrossRef] [Green Version]
- Wartewig, S. IR and Raman Spectroscopy: Fundamental Processing; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 978-3-527-60643-6. [Google Scholar]
- Parachalil, D.R.; Brankin, B.; McIntyre, J.; Byrne, H.J. Raman Spectroscopic Analysis of High Molecular Weight Proteins in Solution—Considerations for Sample Analysis and Data Pre-Processing. Analyst 2018, 143, 5987–5998. [Google Scholar] [CrossRef] [Green Version]
- Heraud, P.; Wood, B.R.; Beardall, J.; McNaughton, D. Effects of Pre-Processing of Raman Spectra on in Vivo Classification of Nutrient Status of Microalgal Cells. J. Chemom. 2006, 20, 193–197. [Google Scholar] [CrossRef]
- Butler, H.J.; Ashton, L.; Bird, B.; Cinque, G.; Curtis, K.; Dorney, J.; Esmonde-White, K.; Fullwood, N.J.; Gardner, B.; Martin-Hirsch, P.L.; et al. Using Raman Spectroscopy to Characterize Biological Materials. Nat. Protoc. 2016, 11, 664–687. [Google Scholar] [CrossRef] [Green Version]
- Gautam, R.; Vanga, S.; Ariese, F.; Umapathy, S. Review of Multidimensional Data Processing Approaches for Raman and Infrared Spectroscopy. EPJ Tech. Instrum. 2015, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Makki, A.A.; Elderderi, S.; Massot, V.; Respaud, R.; Byrne, H.J.; Tauber, C.; Bertrand, D.; Mohammed, E.; Chourpa, I.; Bonnier, F. In Situ Analytical Quality Control of Chemotherapeutic Solutions in Infusion Bags by Raman Spectroscopy. Talanta 2021, 228, 122137. [Google Scholar] [CrossRef]
- Célino, A.; Gonçalves, O.; Jacquemin, F.; Fréour, S. Qualitative and Quantitative Assessment of Water Sorption in Natural Fibres Using ATR-FTIR Spectroscopy. Carbohydr. Polym. 2014, 101, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Libnau, F.O.; Kvalheim, O.M.; Christy, A.A.; Toft, J. Spectra of Water in the Near- and Mid-Infrared Region. Vib. Spectrosc. 1994, 7, 243–254. [Google Scholar] [CrossRef]
- Zhao, W.; Li, Y.; Song, C.; Liu, S.; Li, X.; Long, J. Intensified Levulinic Acid/Ester Production from Cassava by One-Pot Cascade Prehydrolysis and Delignification. Appl. Energy 2017, 204, 1094–1100. [Google Scholar] [CrossRef]
- Dwivedi, A.; Pandey, A.; Bajpai, A. Comparative Study of Structural, Vibrational, Electronic Properties of Pentanoic Acid (Valeric Acid) and Its Derivative 4-Oxopentanoic Acid (Levulinic Acid) by Density Functional Theory. J. Sci. Res. Adv. 2014, 1, 18–24. [Google Scholar]
- Mary, Y.S.; Ushakumari, L.; Harikumar, B.; Varghese, H.T.; Panicker, C.Y. FT-IR, FT-Raman and SERS Spectra of L-Proline. J. Iran. Chem. Soc. 2009, 6, 138–144. [Google Scholar] [CrossRef]
- Schwanninger, M.; Rodrigues, J.C.; Fackler, K. A Review of Band Assignments in near Infrared Spectra of Wood and Wood Components. J. Infrared Spectrosc. 2011, 19, 287–308. [Google Scholar] [CrossRef]
- Westad, F.; Schmidt, A.; Kermit, M. Incorporating Chemical Band-Assignment in near Infrared Spectroscopy Regression Models. J. Infrared Spectrosc. 2008, 16, 265–273. [Google Scholar] [CrossRef]
- Numata, Y.; Iida, Y.; Tanaka, H. Quantitative Analysis of Alcohol–Water Binary Solutions Using Raman Spectroscopy. J. Quant. Spectrosc. Radiat. Transf. 2011, 112, 1043–1049. [Google Scholar] [CrossRef]
- Carey, D.M.; Korenowski, G.M. Measurement of the Raman Spectrum of Liquid Water. J. Chem. Phys. 1998, 108, 2669–2675. [Google Scholar] [CrossRef]
- Kim, T.; Assary, R.S.; Curtiss, L.A.; Marshall, C.L.; Stair, P.C. Vibrational Properties of Levulinic Acid and Furan Derivatives: Raman Spectroscopy and Theoretical Calculations. J. Raman Spectrosc. 2011, 42, 2069–2076. [Google Scholar] [CrossRef]
- Panda, S.; Kundu, K.; Kiefer, J.; Umapathy, S.; Gardas, R.L. Molecular-Level Insights into the Microstructure of a Hydrated and Nanoconfined Deep Eutectic Solvent. J. Phys. Chem. B 2019, 123, 3359–3371. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, R.; Hemmateenejad, B.; Safavi, A.; Shojaeifard, Z.; Shahsavar, A.; Mohajeri, A.; Dokoohaki, M.H.; Zolghadr, A.R. Deep Eutectic–Water Binary Solvent Associations Investigated by Vibrational Spectroscopy and Chemometrics. Phys. Chem. Chem. Phys. 2018, 20, 18463–18473. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.E.; Tortora, M.; Bottari, C.; Colombo Dugoni, G.; Pivato, R.V.; Rossi, B.; Paolantoni, M.; Mele, A. In Competition for Water: Hydrated Choline Chloride:Urea vs Choline Acetate:Urea Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2021, 9, 12262–12273. [Google Scholar] [CrossRef]
- He, Y.; Tang, L.; Wu, X.; Hou, X.; Lee, Y. Spectroscopy: The Best Way Toward Green Analytical Chemistry? Appl. Spectrosc. Rev. 2007, 42, 119–138. [Google Scholar] [CrossRef]
- Salehpour, S.; Dubé, M.A. Reaction Monitoring of Glycerol Step-Growth Polymerization Using ATR-FTIR Spectroscopy. Macromol. React. Eng. 2012, 6, 85–92. [Google Scholar] [CrossRef]
- Alimagham, F.; Winterburn, J.; Dolman, B.; Domingues, P.M.; Everest, F.; Platkov, M.; Basov, S.; Izakson, G.; Katzir, A.; Elliott, S.R.; et al. Real-Time Bioprocess Monitoring Using a Mid-Infrared Fibre-Optic Sensor. Biochem. Eng. J. 2021, 167, 107889. [Google Scholar] [CrossRef]
- Bloomfield, M.; Andrews, D.; Loeffen, P.; Tombling, C.; York, T.; Matousek, P. Non-Invasive Identification of Incoming Raw Pharmaceutical Materials Using Spatially Offset Raman Spectroscopy. J. Pharm. Biomed. Anal. 2013, 76, 65–69. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Pre-Processing | Cross-Validation | ||
|---|---|---|---|---|
| LV | R2 +/− SD | RMSECV+/− SD (% w/w Added) | ||
| ATR_IR | Raw data | 4 | 0.9990 +/− 0.0016 | 0.27 +/− 0.17 |
| NIR-B | Raw data | 3 | 0.9982 +/− 0.0029 | 0.35 +/− 0.08 |
| NIR-H | Raw data | 6 | 0.9988 +/− 0.0010 | 0.36 +/− 0.10 |
| Raman -B | RBVN | 5 | 0.9977 +/− 0.0033 | 0.43 +/− 0.11 |
| Technique | Pre-Processing | Test Set | |
|---|---|---|---|
| R2 +/− SD | RMSEP +/− SD (% w/w Added) | ||
| ATR-IR | Raw data | 0.9993 +/− 0.0004 | 0.27 +/− 0.08 |
| NIR-B | Raw data | 0.9969 +/− 0.0004 | 0.56 +/− 0.03 |
| NIR-H | Raw data | 0.9984 +/− 0.0002 | 0.68 +/− 0.08 |
| Raman-B | RBVN | 0.9955 +/− 0.0015 | 0.67 +/− 0.11 |
| Technique | % Relative Error | Mean % RE | % RE min–max Values | ||||||
|---|---|---|---|---|---|---|---|---|---|
| <1 | ˂2.5 | <5 | <7.5 | <10 | >10 | ||||
| Number of Samples | ATR-IR | 7 | 8 | 7 | - | 1 | 1 | 2.59 | 0.02–14.84 |
| NIR-B | 1 | 7 | 8 | 4 | 1 | 3 | 5.13 | 0.50–20.85 | |
| NIR-H | 2 | 4 | 7 | 5 | 1 | 5 | 6.23 | 0.88–17.21 | |
| Raman-B | 4 | 5 | 2 | 4 | 3 | 6 | 6.75 | 0.07–20.87 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elderderi, S.; Sacré, P.-Y.; Wils, L.; Chourpa, I.; Elbashir, A.A.; Hubert, P.; Byrne, H.J.; Boudesocque-Delaye, L.; Ziemons, E.; Bonnier, F. Comparison of Vibrational Spectroscopic Techniques for Quantification of Water in Natural Deep Eutectic Solvents. Molecules 2022, 27, 4819. https://doi.org/10.3390/molecules27154819
Elderderi S, Sacré P-Y, Wils L, Chourpa I, Elbashir AA, Hubert P, Byrne HJ, Boudesocque-Delaye L, Ziemons E, Bonnier F. Comparison of Vibrational Spectroscopic Techniques for Quantification of Water in Natural Deep Eutectic Solvents. Molecules. 2022; 27(15):4819. https://doi.org/10.3390/molecules27154819
Chicago/Turabian StyleElderderi, Suha, Pierre-Yves Sacré, Laura Wils, Igor Chourpa, Abdalla A. Elbashir, Philippe Hubert, Hugh J. Byrne, Leslie Boudesocque-Delaye, Eric Ziemons, and Franck Bonnier. 2022. "Comparison of Vibrational Spectroscopic Techniques for Quantification of Water in Natural Deep Eutectic Solvents" Molecules 27, no. 15: 4819. https://doi.org/10.3390/molecules27154819
APA StyleElderderi, S., Sacré, P.-Y., Wils, L., Chourpa, I., Elbashir, A. A., Hubert, P., Byrne, H. J., Boudesocque-Delaye, L., Ziemons, E., & Bonnier, F. (2022). Comparison of Vibrational Spectroscopic Techniques for Quantification of Water in Natural Deep Eutectic Solvents. Molecules, 27(15), 4819. https://doi.org/10.3390/molecules27154819