
The Distance between Minima of Electron Density and Electrostatic Potential as a Measure of Halogen Bond Strength

 ,
,
Abstract
:
1. Introduction
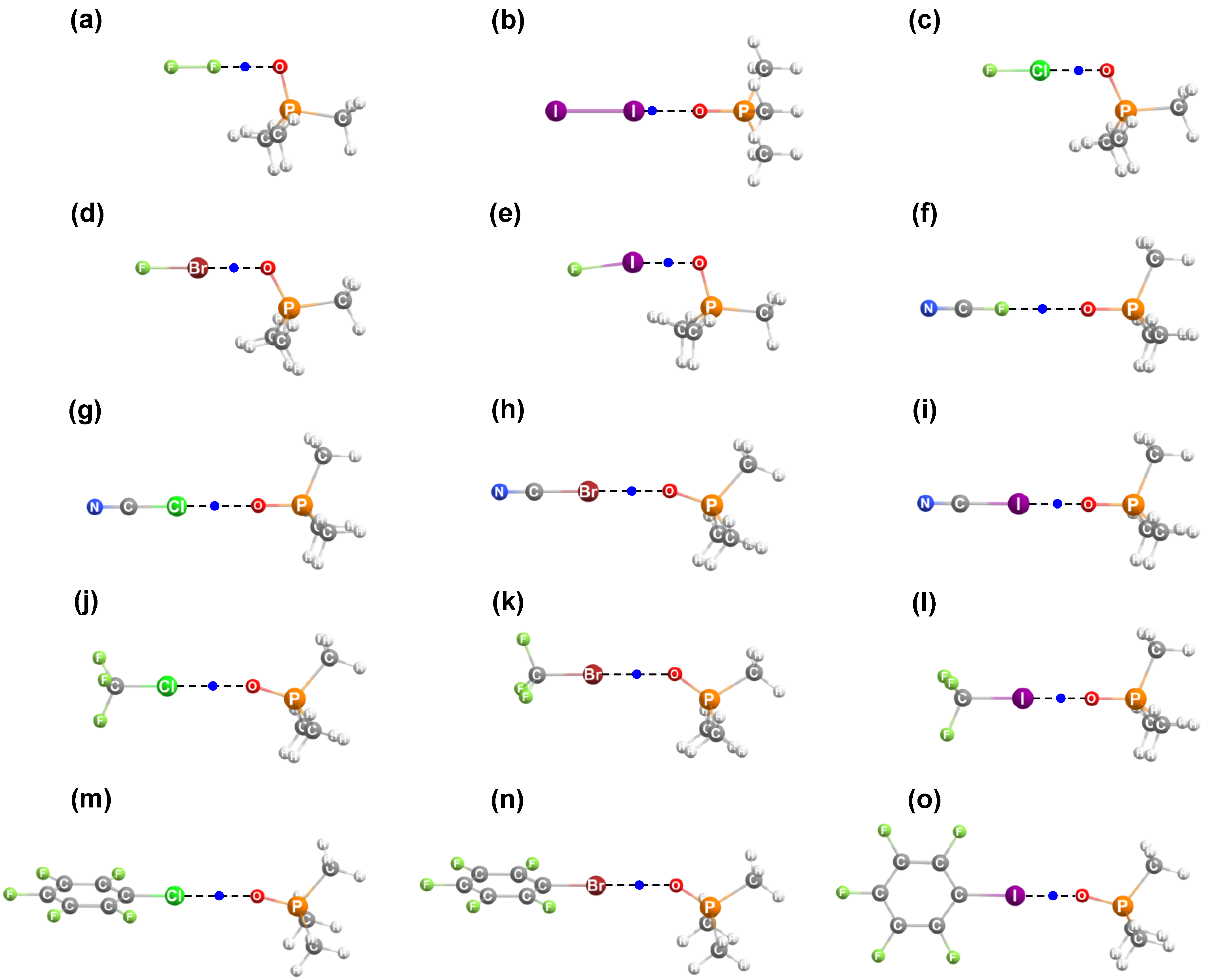
2. Results and Discussion
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An Overview of Halogen Bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Danovich, D.; Mo, Y.; Shaik, S. On the Nature of the Halogen Bond. J. Chem. Theory Comput. 2014, 10, 3726–3737. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wong, M.W. Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective. Molecules 2020, 25, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutar, R.L.; Huber, S.M. Catalysis of Organic Reactions through Halogen Bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Wang, C.G.; Chong, A.M.L.; Pan, H.M.; Sarkar, J.; Tay, X.T.; Goto, A. Recent Development in Halogen-Bonding-Catalyzed Living Radical Polymerization. Polym. Chem. 2020, 11, 5559–5571. [Google Scholar] [CrossRef]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen Bonding Based Recognition Processes: A World Parallel to Hydrogen Bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The Halogen Bond in the Design of Functional Supramolecular Materials: Recent Advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Nemec, V.; Lisac, K.; Bedeković, N.; Fotović, L.; Stilinović, V.; Cinčić, D. Crystal Engineering Strategies towards Halogen-Bonded Metal-Organic Multi-Component Solids: Salts, Cocrystals and Salt Cocrystals. CrystEngComm 2021, 23, 3063–3083. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Bokach, N.A.; Kukushkin, V.Y.; Frontera, A. Metal Centers as Nucleophiles: Oxymoron of Halogen Bond-Involving Crystal Engineering. Chemistry 2022, 28, e202103173. [Google Scholar] [CrossRef]
- Saccone, M.; Catalano, L. Halogen Bonding beyond Crystals in Materials Science. J. Phys. Chem. B 2019, 123, 9281–9290. [Google Scholar] [CrossRef] [Green Version]
- Berger, G.; Frangville, P.; Meyer, F. Halogen Bonding for Molecular Recognition: New Developments in Materials and Biological Sciences. Chem. Commun. 2020, 56, 4970–4981. [Google Scholar] [CrossRef]
- Zheng, J.; Suwardi, A.; Wong, C.J.E.; Loh, X.J.; Li, Z. Halogen Bonding Regulated Functional Nanomaterials. Nanoscale Adv. 2021, 3, 6342–6357. [Google Scholar] [CrossRef]
- Biswas, S.; Das, A. Recent Developments in Polymeric Assemblies and Functional Materials by Halogen Bonding. ChemNanoMat 2021, 7, 748–772. [Google Scholar] [CrossRef]
- Landenberger, K.B.; Bolton, O.; Matzger, A.J. Energetic-Energetic Cocrystals of Diacetone Diperoxide (DADP): Dramatic and Divergent Sensitivity Modifications via Cocrystallization. J. Am. Chem. Soc. 2015, 137, 5074–5079. [Google Scholar] [CrossRef]
- Baldrighi, M.; Cavallo, G.; Chierotti, M.R.; Gobetto, R.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding and Pharmaceutical Cocrystals: The Case of a Widely Used Preservative. Mol. Pharm. 2013, 10, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen Bond: Its Role beyond Drug-Target Binding Affinity for Drug Discovery and Development. J. Chem. Inf. Model. 2014, 54, 69–78. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Wick, C.R.; Clark, T. On Bond-Critical Points in QTAIM and Weak Interactions. J. Mol. Model. 2018, 24, 142. [Google Scholar] [CrossRef] [PubMed]
- Syzgantseva, O.A.; Tognetti, V.; Joubert, L. On the Physical Nature of Halogen Bonds: A QTAIM Study. J. Phys. Chem. A 2013, 117, 8969–8980. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.; Mukhitdinova, S.; Yushina, I.; Tsirelson, V. Electronic Criterion for Categorizing the Chalcogen and Halogen Bonds: Sulfur–Iodine Interactions in Crystals. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 117–126. [Google Scholar] [CrossRef]
- Pavan, M.S.; Durga Prasad, K.; Guru Row, T.N. Halogen Bonding in Fluorine: Experimental Charge Density Study on Intermolecular F⋯F and F⋯S Donor–Acceptor Contacts. Chem. Commun. 2013, 49, 7558. [Google Scholar] [CrossRef]
- Chua, Z.; Zarychta, B.; Gianopoulos, C.G.; Zhurov, V.V.; Pinkerton, A.A. Revisiting the Charge Density Analysis of 2,5-Dichloro-1,4-Benzoquinone at 20 K. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 654–659. [Google Scholar] [CrossRef]
- Eraković, M.; Cinčić, D.; Molčanov, K.; Stilinović, V. A Crystallographic Charge Density Study of the Partial Covalent Nature of Strong N⋯Br Halogen Bonds. Angew. Chem. Int. Ed. 2019, 58, 15702–15706. [Google Scholar] [CrossRef]
- Bianchi, R.; Forni, A.; Pilati, T. Experimental Electron Density Study of the Supramolecular Aggregation between 4,4′-Dipyridyl- N, N′-Dioxide and 1,4-Diiodotetrafluorobenzene at 90 K. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Rosokha, S.V.; Stern, C.L.; Ritzert, J.T. Experimental and Computational Probes of the Nature of Halogen Bonding: Complexes of Bromine-Containing Molecules with Bromide Anions. Chemistry 2013, 19, 8774–8788. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Traversa, A. From Charge Transfer to Electron Transfer in Halogen-Bonded Complexes of Electrophilic Bromocarbons with Halide Anions. Phys. Chem. Chem. Phys. 2015, 17, 4989–4999. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Vinakos, M.K. Hybrid Network Formation via Halogen Bonding of the Neutral Bromo-Substituted Organic Molecules with Anionic Metal–Bromide Complexes. Cryst. Growth Des. 2012, 12, 4149–4156. [Google Scholar] [CrossRef]
- Weinberger, C.; Hines, R.; Zeller, M.; Rosokha, S.V. Continuum of Covalent to Intermolecular Bonding in the Halogen-Bonded Complexes of 1,4-Diazabicyclo[2.2.2]Octane with Bromine-Containing Electrophiles. Chem. Commun. 2018, 54, 8060–8063. [Google Scholar] [CrossRef]
- Coppens, P.; Stevens, E.D. Accurate X-Ray Diffraction and Quantum Chemistry: The Study of Charge Density Distributions. Adv. Quantum Chem. 1977, 10, 1–35. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density ? Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Schwarz, W.H.E.; Ruedenberg, K.; Mensching, L. Chemical Deformation Densities. 1. Principles and Formulation of Quantitative Determination. J. Am. Chem. Soc. 1989, 111, 6926–6933. [Google Scholar] [CrossRef]
- Mensching, L.; Von Niessen, W.; Valtazanos, P.; Ruedenberg, K.; Schwarz, W.H.E. Chemical Deformation Densities. 2. Small Molecules. J. Am. Chem. Soc. 1989, 111, 6933–6941. [Google Scholar] [CrossRef]
- Blanco, F.; Alkorta, I.; Rozas, I.; Solimannejad, M.; Elguero, J. A Theoretical Study of the Interactions of NF3 with Neutral Ambidentate Electron Donor and Acceptor Molecules. Phys. Chem. Chem. Phys. 2011, 13, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Solimannejad, M.; Malekani, M.; Alkorta, I. Cooperativity between the Hydrogen Bonding and Halogen Bonding in F3CX⋯NCH(CNH)⋯NCH(CNH) Complexes (X = Cl, Br). Mol. Phys. 2011, 109, 1641–1648. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between Non-Covalent Interactions in Complexes and Crystals with Halogen Bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A Simple Measure of Electron Localization in Atomic and Molecular Systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of Chemical Bonds Based on Topological Analysis of Electron Localization Functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Bulatova, M.; Ivanov, D.M.; Rautiainen, J.M.; Kinzhalov, M.A.; Truong, K.N.; Lahtinen, M.; Haukka, M. Studies of Nature of Uncommon Bifurcated I–I⋯(I–M) Metal-Involving Noncovalent Interaction in Palladium(II) and Platinum(II) Isocyanide Cocrystals. Inorg. Chem. 2021, 60, 13200–13211. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F.; Landis, C.R. Natural Bond Orbitals and Extensions of Localized Bonding Concepts. Chem. Educ. Res. Pract. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Sadlej-Sosnowska, N. Transfer of Electron Density as a Result of Hydrogen Bond Formation. Int. J. Quantum Chem. 2009, 109, 294–300. [Google Scholar] [CrossRef]
- Scrocco, E.; Tomasi, J. Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials. Adv. Quantum Chem. 1978, 11, 115–193. [Google Scholar] [CrossRef]
- Brammer, L.; Bruton, E.A.; Sherwood, P. Understanding the Behavior of Halogens as Hydrogen Bond Acceptors. Cryst. Growth Des. 2001, 1, 277–290. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Revealing the Nature of Intermolecular Interaction and Configurational Preference of the Nonpolar Molecular Dimers (H2)2, (N2)2, and (H2)(N2). J. Mol. Model. 2013, 19, 5387–5395. [Google Scholar] [CrossRef]
- Kolář, M.; Hostaš, J.; Hobza, P. The Strength and Directionality of a Halogen Bond Are Co-Determined by the Magnitude and Size of the σ-Hole. Phys. Chem. Chem. Phys. 2014, 16, 9987–9996. [Google Scholar] [CrossRef]
- Heidrich, J.; Exner, T.E.; Boeckler, F.M. Predicting the Magnitude of σ-Holes Using VmaxPred, a Fast and Efficient Tool Supporting the Application of Halogen Bonds in Drug Discovery. J. Chem. Inf. Model. 2019, 59, 636–643. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface Electrostatic Potentials of Halogenated Methanes as Indicators of Directional Intermolecular Interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. Topological Properties of the Electrostatic Potential in Weak and Moderate N⋯H Hydrogen Bonds. J. Phys. Chem. A 2007, 111, 6425–6433. [Google Scholar] [CrossRef]
- Zelenkov, L.E.; Eliseeva, A.A.; Baykov, S.V.; Suslonov, V.V.; Galmés, B.; Frontera, A.; Kukushkin, V.Y.; Ivanov, D.M.; Bokach, N.A. Electron Belt-to-σ-Hole Switch of Noncovalently Bound Iodine(i) Atoms in Dithiocarbamate Metal Complexes. Inorg. Chem. Front. 2021, 8, 2505–2517. [Google Scholar] [CrossRef]
- Tsirelson, V.G.; Avilov, A.S.; Lepeshov, G.G.; Kulygin, A.K.; Stahn, J.; Pietsch, U.; Spence, J.C.H. Quantitative Analysis of the Electrostatic Potential in Rock-Salt Crystals Using Accurate Electron Diffraction Data. J. Phys. Chem. B 2002, 105, 5068–5074. [Google Scholar] [CrossRef]
- Tsirelson, V.G.; Shishkina, A.V.; Stash, A.I.; Parsons, S. The Experimental and Theoretical QTAIMC Study of the Atomic and Molecular Interactions in Dinitrogen Tetroxide. Acta Crystallogr. Sect. B Struct. Sci. 2009, 65, 647–658. [Google Scholar] [CrossRef]
- Dabranskaya, U.; Ivanov, D.M.; Novikov, A.S.; Matveychuk, Y.V.; Bokach, N.A.; Kukushkin, V.Y. Metal-Involving Bifurcated Halogen Bonding C-Br⋯η2(Cl-Pt). Cryst. Growth Des. 2019, 19, 1364–1376. [Google Scholar] [CrossRef]
- Lamberts, K.; Handels, P.; Englert, U.; Aubert, E.; Espinosa, E. Stabilization of Polyiodide Chains via Anion⋯anion Interactions: Experiment and Theory. CrystEngComm 2016, 18, 3832–3841. [Google Scholar] [CrossRef]
- Kashina, M.V.; Ivanov, D.M.; Kinzhalov, M.A. The Isocyanide Complexes Cis-[MCl2(CNC6H4-4-X)2] (M = Pd, Pt; X = Cl, Br) as Tectons in Crystal Engineering Involving Halogen Bonds. Crystals 2021, 11, 799. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Matveychuk, Y.V.; Mukhitdinova, S.E.; Sobalev, S.A.; Khrenova, M.G.; Tsirelson, V.G. The Common Trends for the Halogen, Chalcogen, and Pnictogen Bonds via Sorting Principles and Local Bonding Properties. Theor. Chem. Acc. 2020, 139, 26. [Google Scholar] [CrossRef]
- Bartashevich, E.; Matveychuk, Y.; Tsirelson, V. Identification of the Tetrel Bonds between Halide Anions and Carbon Atom of Methyl Groups Using Electronic Criterion. Molecules 2019, 24, 1083. [Google Scholar] [CrossRef] [Green Version]
- Abramov, Y.A. On the Possibility of Kinetic Energy Density Evaluation from the Experimental Electron-Density Distribution. Acta Crystallogr. Sect. A Found. Crystallogr. 1997, 53, 264–272. [Google Scholar] [CrossRef]
- Mayer, U.; Gutmann, V.; Gerger, W. The Acceptor Number—A Quantitative Empirical Parameter for the Electrophilic Properties of Solvents. Mon. Chem. 1975, 106, 1235–1257. [Google Scholar] [CrossRef]
- Beckett, M.A.; Brassington, D.S.; Coles, S.J.; Hursthouse, M.B. Lewis Acidity of Tris(Pentafluorophenyl) Borane: Crystal and Molecular Structure of B(C6F5)3·OPEt3. Inorg. Chem. Commun. 2000, 3, 530–533. [Google Scholar] [CrossRef]
- McCune, J.A.; He, P.; Petkovic, M.; Coleman, F.; Estager, J.; Holbrey, J.D.; Seddon, K.R.; Swadźba-Kwaśny, M. Brønsted Acids in Ionic Liquids: How Acidity Depends on the Liquid Structure. Phys. Chem. Chem. Phys. 2014, 16, 23233–23243. [Google Scholar] [CrossRef] [Green Version]
- Osegovic, J.P.; Drago, R.S. Measurement of the Global Acidity of Solid Acids by 31P MAS NMR of Chemisorbed Triethylphosphine Oxide. J. Phys. Chem. B 2000, 104, 147–154. [Google Scholar] [CrossRef]
- Zheng, A.; Liu, S.B.; Deng, F. 31P NMR Chemical Shifts of Phosphorus Probes as Reliable and Practical Acidity Scales for Solid and Liquid Catalysts. Chem. Rev. 2017, 117, 12475–12531. [Google Scholar] [CrossRef]
- Ostras, A.S.; Ivanov, D.M.; Novikov, A.S.; Tolstoy, P.M. Phosphine Oxides as Spectroscopic Halogen Bond Descriptors: IR and NMR Correlations with Interatomic Distances and Complexation Energy. Molecules 2020, 25, 1406. [Google Scholar] [CrossRef] [Green Version]
- Kostin, M.A.; Pylaeva, S.A.; Tolstoy, P.M. Phosphine Oxides as NMR and IR Spectroscopic Probes for the Estimation of the Geometry and Energy of PO⋯H–A Hydrogen Bonds. Phys. Chem. Chem. Phys. 2022, 24, 7121–7133. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X-H⋯F-Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Alkorta, I.; Legon, A.C. Nucleophilicities of Lewis Bases b and Electrophilicities of Lewis Acids a Determined from the Dissociation Energies of Complexes B⋯A Involving Hydrogen Bonds, Tetrel Bonds, Pnictogen Bonds, Chalcogen Bonds and Halogen Bonds. Molecules 2017, 22, 1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikbaeva, Z.M.; Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Bokach, N.A.; Kukushkin, V.Y. Electrophilic-Nucleophilic Dualism of Nickel(II) toward Ni⋯I Noncovalent Interactions: Semicoordination of Iodine Centers via Electron Belt and Halogen Bonding via σ-Hole. Inorg. Chem. 2017, 56, 13562–13578. [Google Scholar] [CrossRef]
- Van Der Bondi, A. Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Becke, A.D. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Barbieri, P.L.; Fantin, P.A.; Jorge, F.E. Gaussian Basis Sets of Triple and Quadruple Zeta Valence Quality for Correlated Wave Functions. Mol. Phys. 2006, 104, 2945–2954. [Google Scholar] [CrossRef]
- MacHado, S.F.; Camiletti, G.G.; Neto, A.C.; Jorge, F.E.; Jorge, R.S. Gaussian Basis Set of Triple Zeta Valence Quality for the Atoms from K to Kr: Application in DFT and CCSD(T) Calculations of Molecular Properties. Mol. Phys. 2009, 107, 1713–1727. [Google Scholar] [CrossRef]
- Campos, C.T.; Jorge, F.E. Triple Zeta Quality Basis Sets for Atoms Rb through Xe: Application in CCSD(T) Atomic and Molecular Property Calculations. Mol. Phys. 2013, 111, 167–173. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Version 1.8. Available online: www.chemcraftprog.com (accessed on 24 July 2022).
- Origin, Version 2018; OriginLab Corporation: Northampton, MA, USA, 2018.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakalov, E.R.; Tupikina, E.Y.; Ivanov, D.M.; Bartashevich, E.V.; Tolstoy, P.M. The Distance between Minima of Electron Density and Electrostatic Potential as a Measure of Halogen Bond Strength. Molecules 2022, 27, 4848. https://doi.org/10.3390/molecules27154848
Chakalov ER, Tupikina EY, Ivanov DM, Bartashevich EV, Tolstoy PM. The Distance between Minima of Electron Density and Electrostatic Potential as a Measure of Halogen Bond Strength. Molecules. 2022; 27(15):4848. https://doi.org/10.3390/molecules27154848
Chicago/Turabian StyleChakalov, Edem R., Elena Yu. Tupikina, Daniil M. Ivanov, Ekaterina V. Bartashevich, and Peter M. Tolstoy. 2022. "The Distance between Minima of Electron Density and Electrostatic Potential as a Measure of Halogen Bond Strength" Molecules 27, no. 15: 4848. https://doi.org/10.3390/molecules27154848