
New Quinoxaline-Based Derivatives as PARP-1 Inhibitors: Design, Synthesis, Antiproliferative, and Computational Studies

 , and
, and
Abstract
:
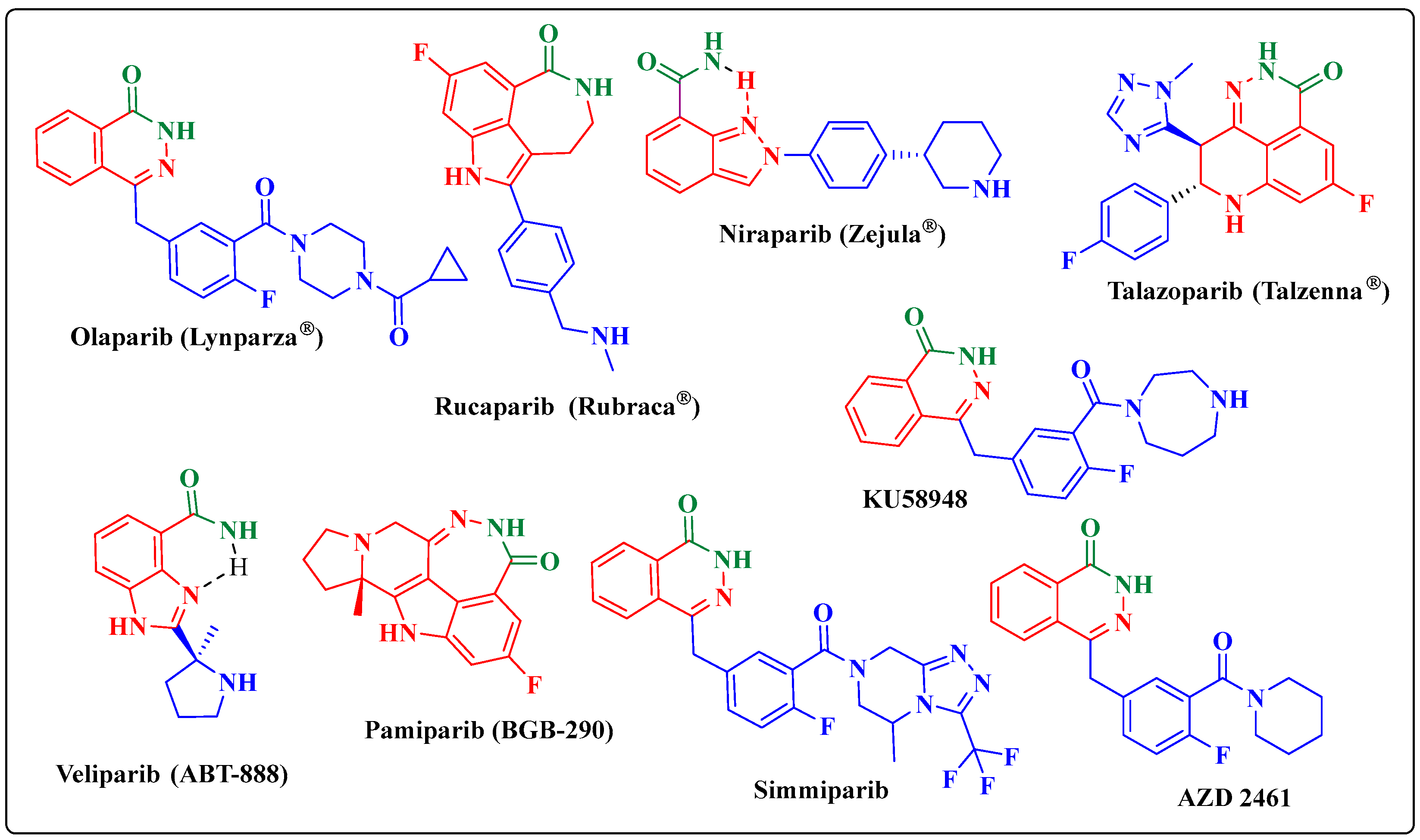
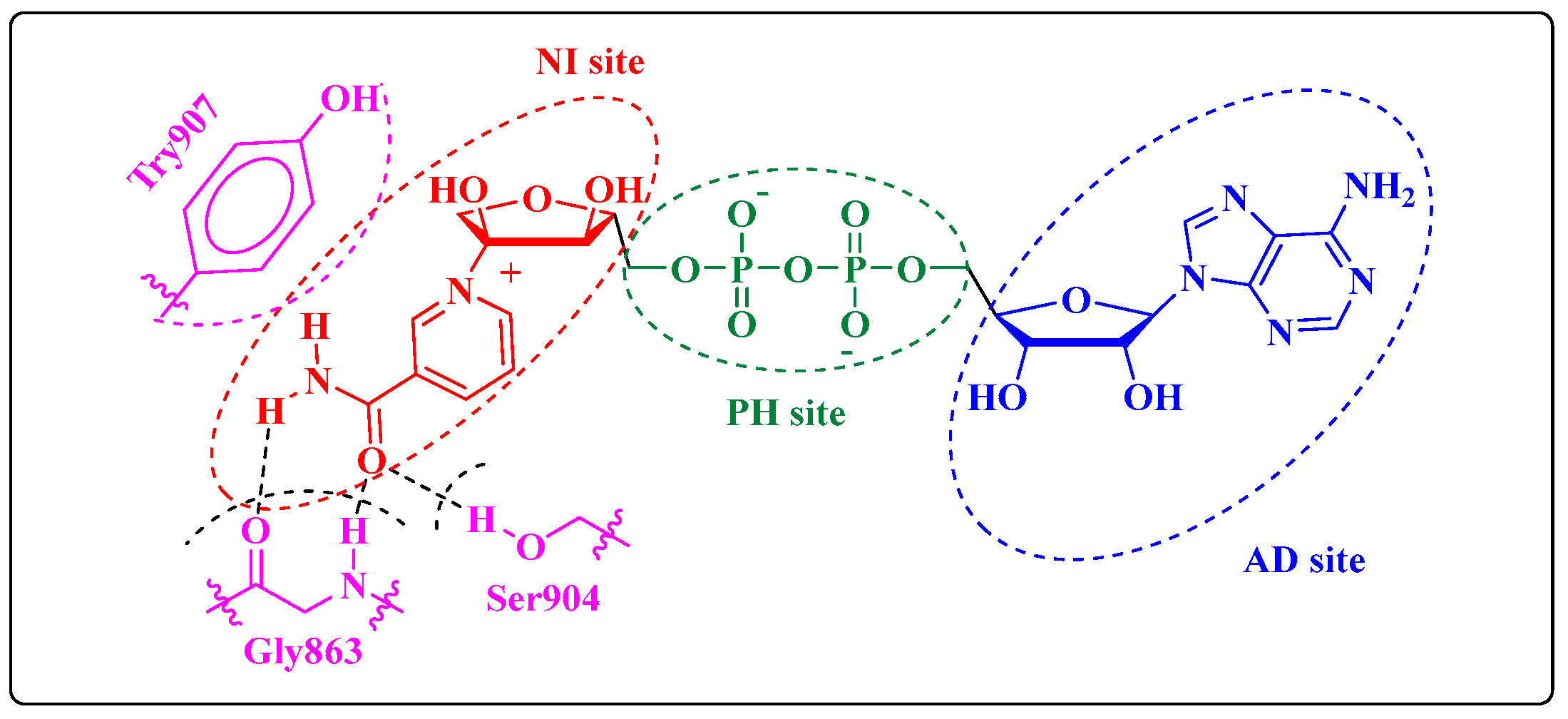
1. Introduction
2. Results
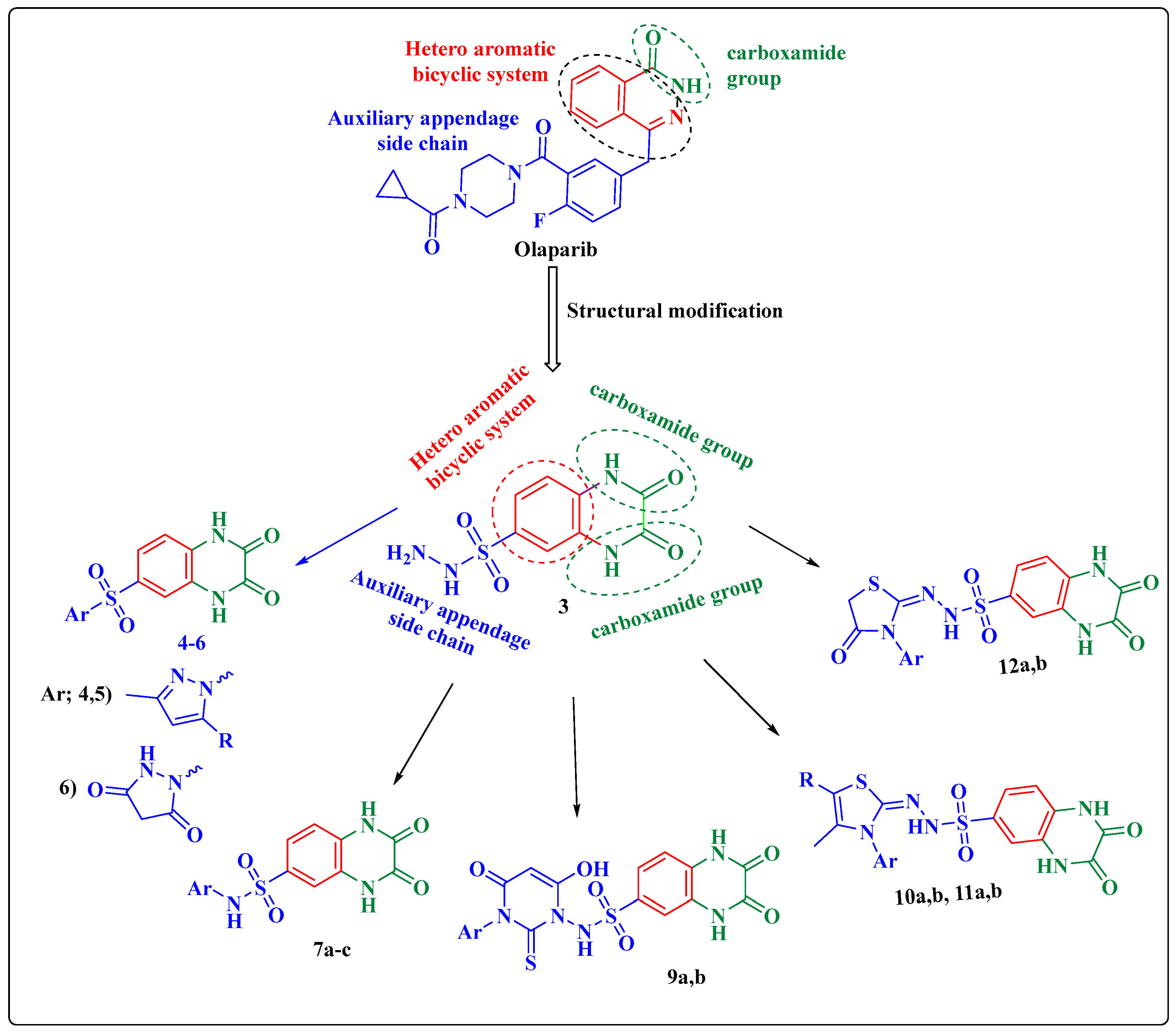
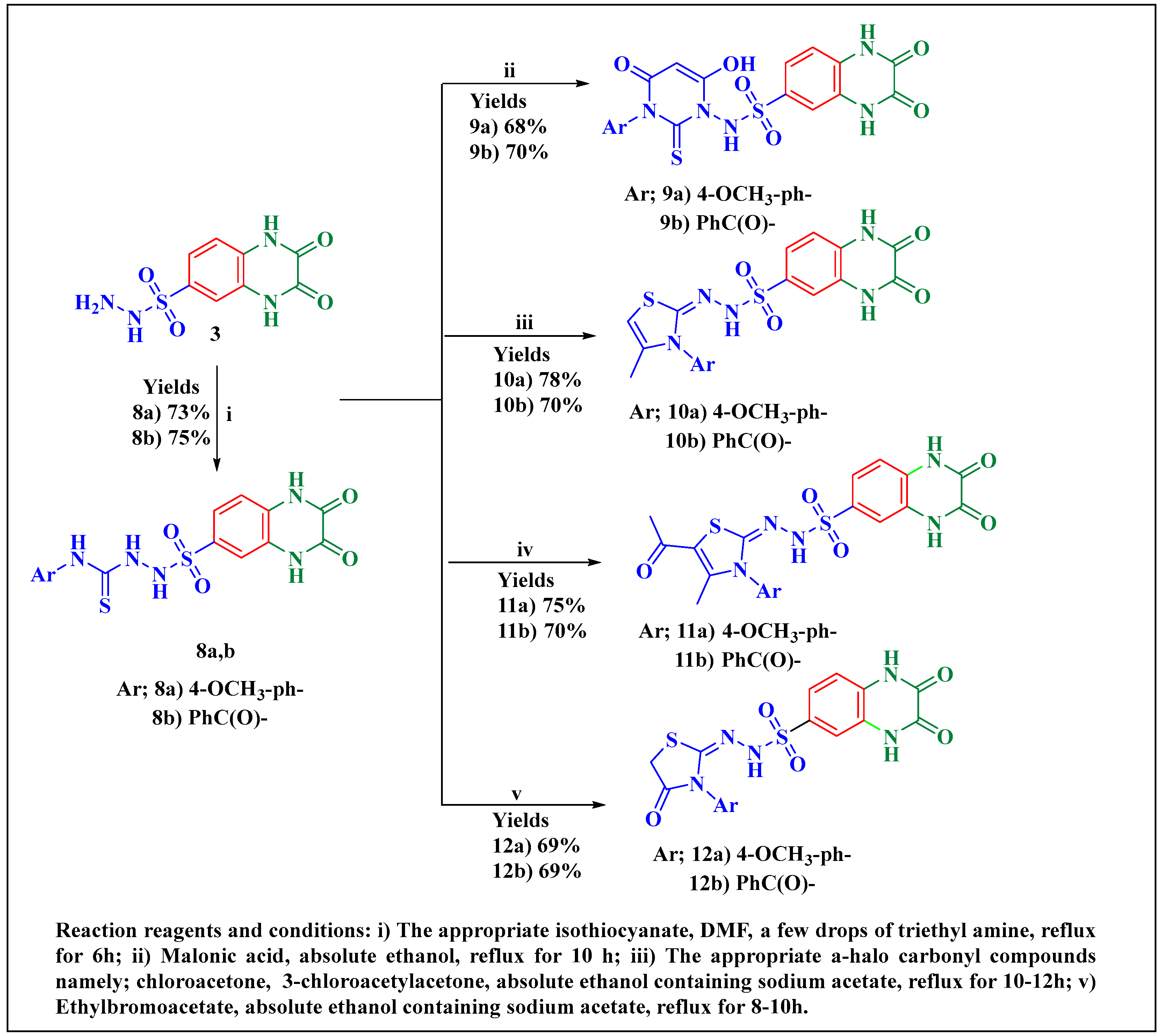
2.1. Chemistry
2.2. Biological Evaluation
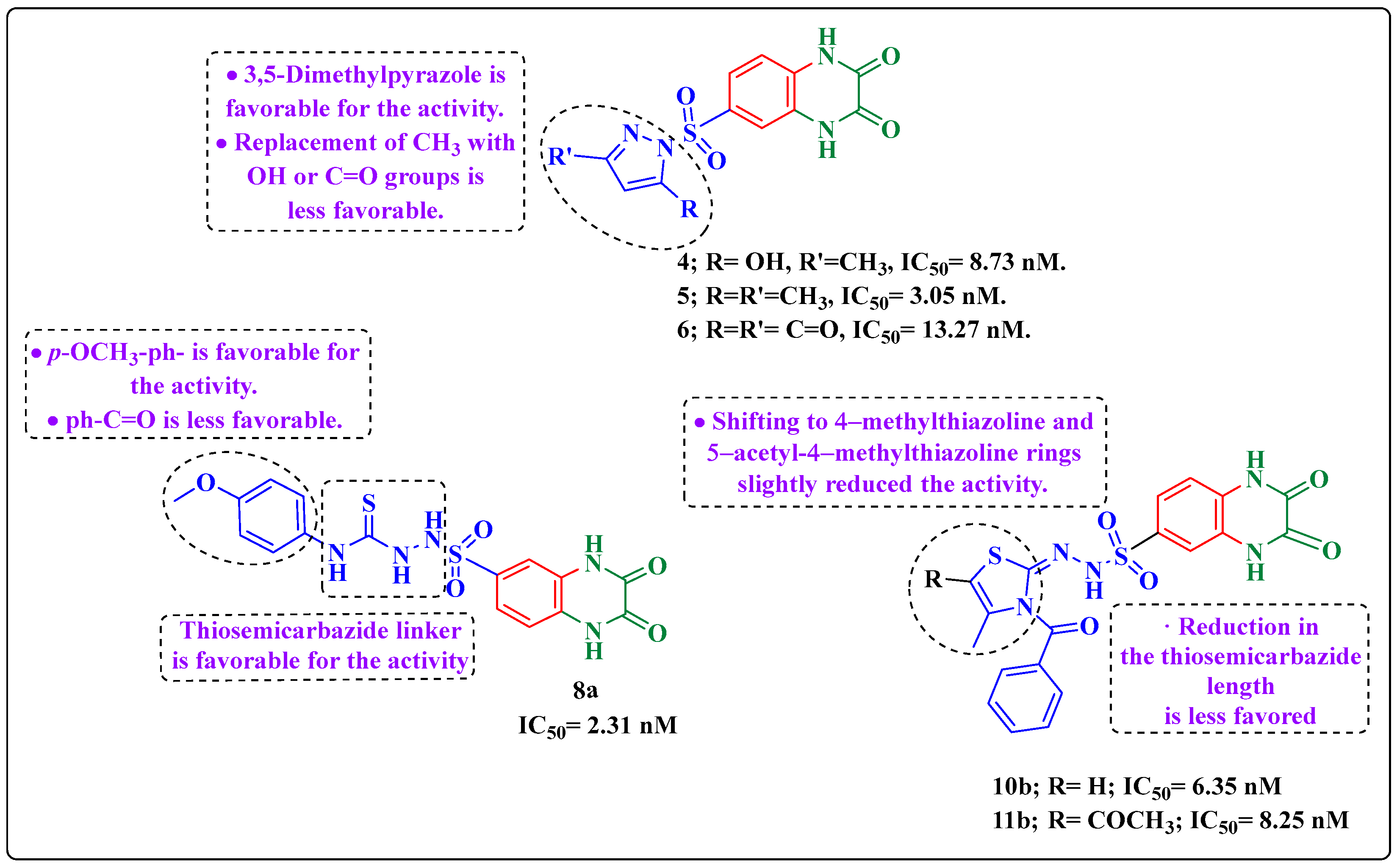
2.2.1. Assessment of PARP-1 Inhibitory Activity of the Target Quinoxaline Compounds
2.2.2. Antiproliferative Activity
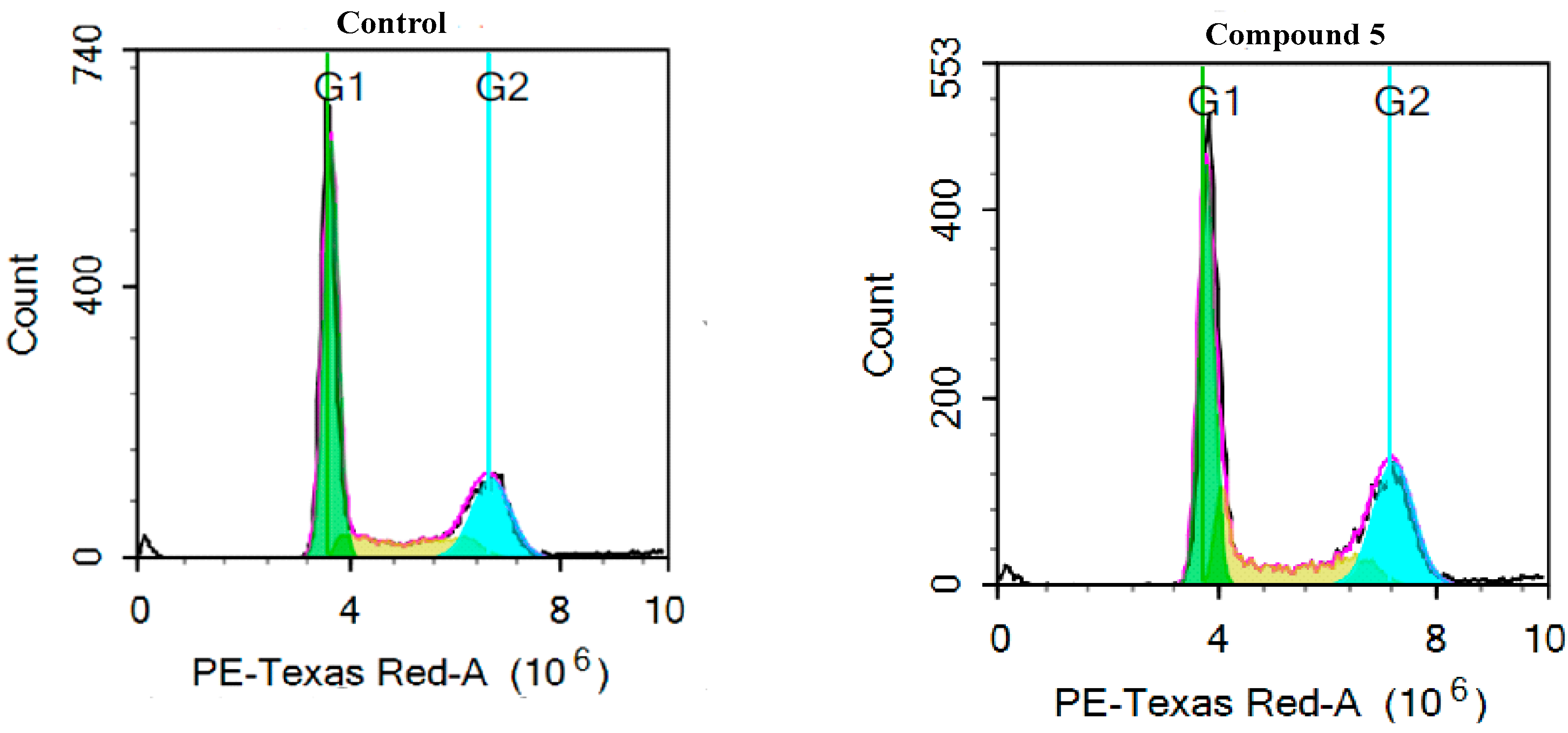
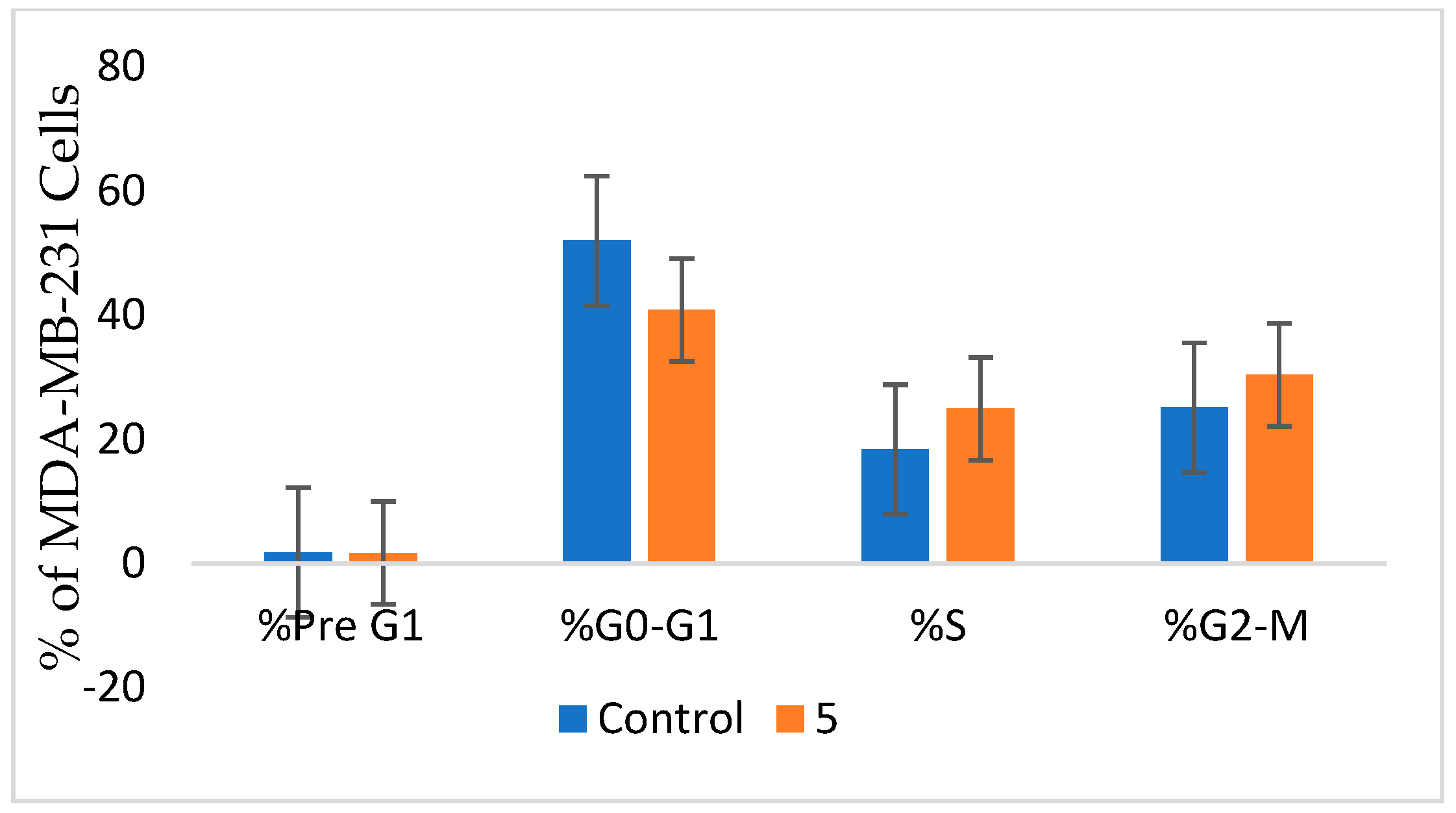
2.2.3. Cell Cycle Analysis in MDA-MB-436
2.2.4. Apoptosis Assay in MDA-MB-436 Cells
2.2.5. Autophagy Assay
2.3. Computational Studies
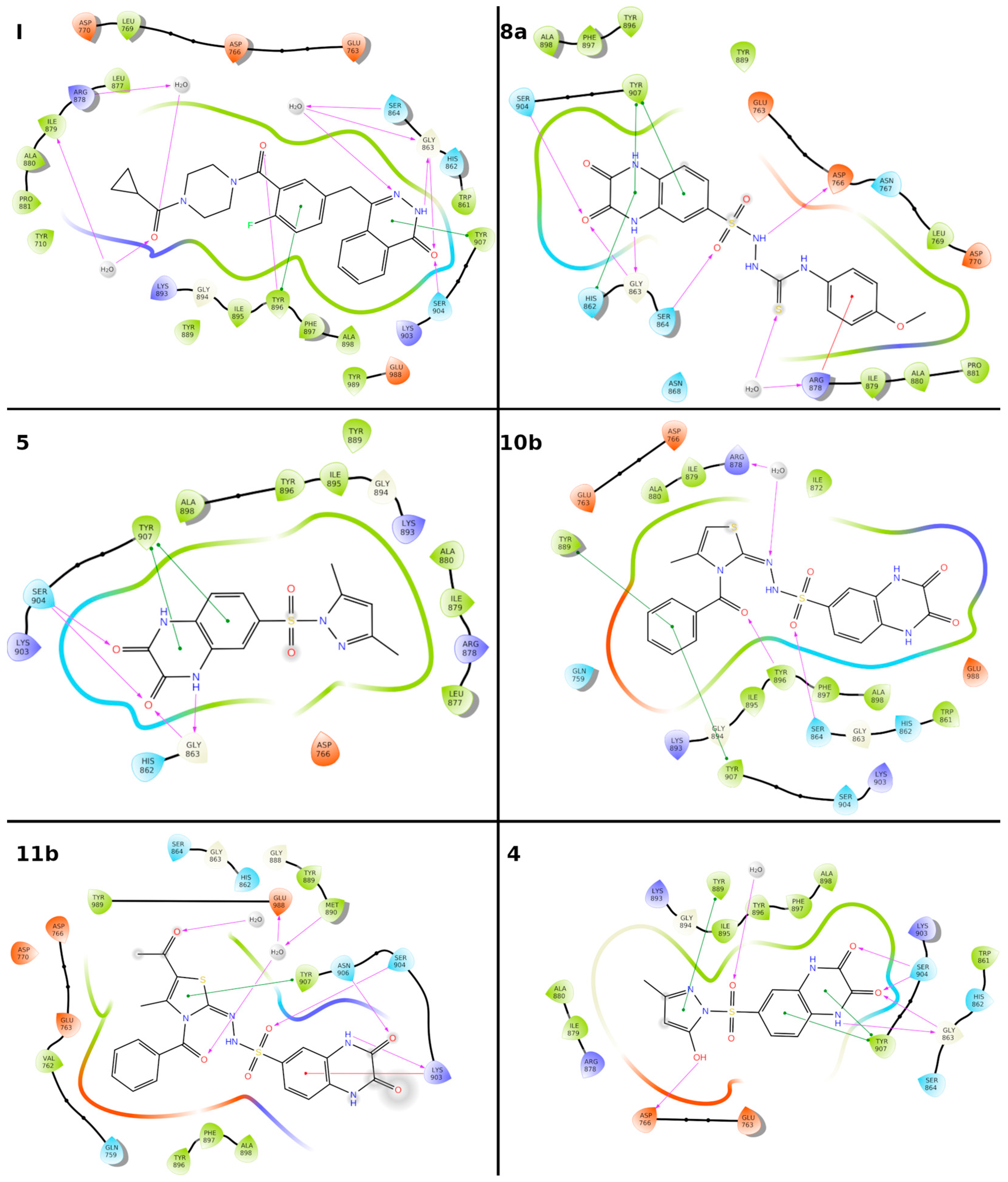
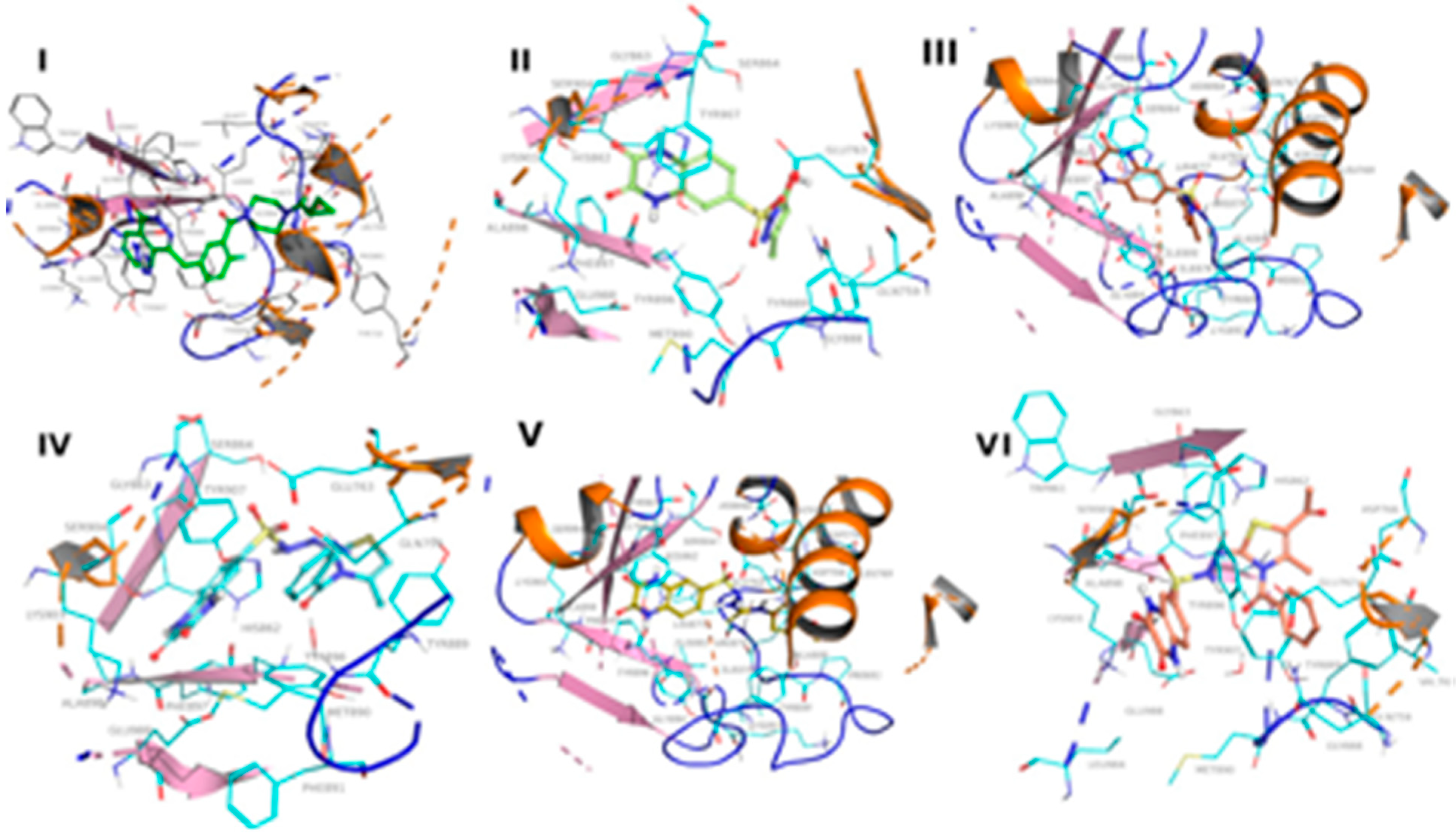
2.3.1. Molecular Docking
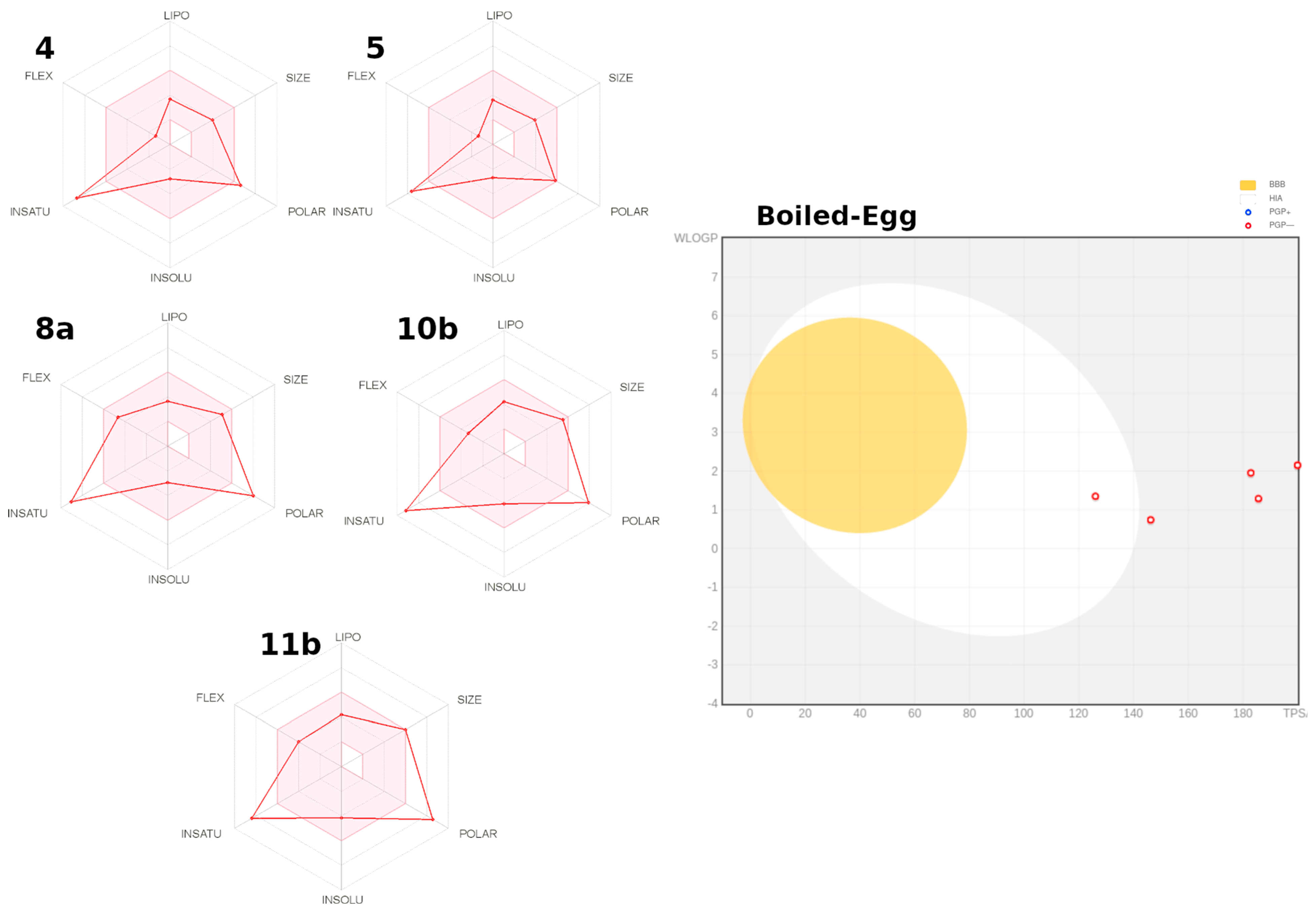
2.3.2. Prediction ADME parameters
3. Materials and Methods
3.1. Chemistry
3.2. PARP-1 Inhibition Assay
3.3. In Vitro Anticancer Screening
3.4. Cell Cycle Analysis
3.5. Apoptosis Analysis
3.6. Autophagy Analysis
3.7. Docking Methodology
3.8. Chemical Synthesis
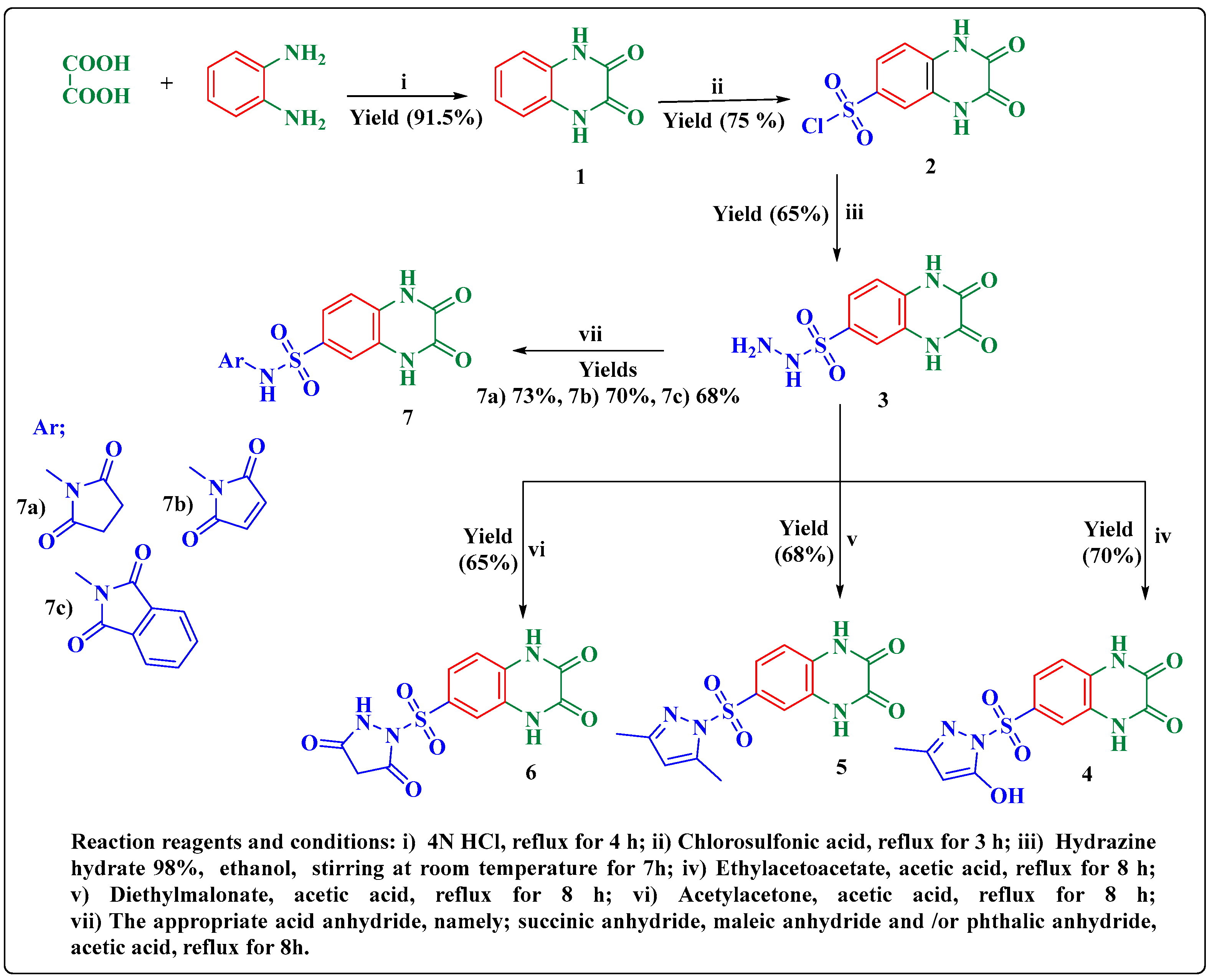
3.8.1. Preparation of Quinoxaline-2,3(1H,4H)-dione (1)
3.8.2. Preparation of 2,3-Dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonyl Chloride (2)
3.8.3. Preparation of 2,3-Dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide (3)
3.8.4. Preparation of 6-(Substituted pyrazolyl)-sulfonyl-1,4-dihydroquinoxaline-2,3-dione Derivatives 4–6
6-((5-Hydroxy-3-methyl-1H-pyrazol-1-yl)sulfonyl)-1,4-dihydroquinoxaline-2,3-dione (4)
6-((3,5-Dimethyl-1H-pyrazol-1-yl)sulfonyl)-1,4-dihydroquinoxaline-2,3-dione (5)
6-((3,5-Dioxopyrazolidin-1-yl)sulfonyl)-1,4-dihydroquinoxaline-2,3-dione (6)
3.8.5. Preparation of N-Substituted-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide Derivatives 7a–7c
N-(2,5-dioxopyrrolidin-1-yl)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (7a)
N-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (7b)
N-(1,3-dioxoisoindolin-2-yl)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (7c)
3.8.6. Preparation of 2-((2,3-Dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)-N-substituted Hydrazine-1-carbothioamide 8a,b
2-((2,3-Dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)-N-(4-methoxyphenyl) Hydrazine-1-carbothioamide (8a)
N-(2-((2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)sulfonyl)hydrazine-1-carbonothioyl)benzamide (8b)
3.8.7. Preparation of 4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide Derivatives 9a, 9b
N-(6-hydroxy-3-(4-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (9a)
N-(3-benzoyl-6-hydroxy-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonamide (9b)
3.8.8. Preparation of N′-(3-aryl-4-methylthiazol-2(3H)-ylidene)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide (10a,b) and N′-(5-acetyl-3-aryl-4-methylthiazol-2(3H)-ylidene)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide (11a,b)
N′-(3-(4-methoxyphenyl)-4-methylthiazol-2(3H)-ylidene)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide (10a)
N’-(3-benzoyl-4-methylthiazol-2(3H)-ylidene)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide (10b)
N’-(5-acetyl-3-(4-methoxyphenyl)-4-methylthiazol-2(3H)-ylidene)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide (11a)
N’-(5-acetyl-3-benzoyl-4-methylthiazol-2(3H)-ylidene)-2,3-dioxo-1,2,3,4-tetrahydro Quinoxaline-6-sulfonohydrazide (11b)
3.8.9. Preparation of N’-(3-Aryl-4-oxothiazolidin-2-ylidene)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide derivatives 12a,b
N’-(3-(4-methoxyphenyl)-4-oxothiazolidin-2-ylidene)-2,3-dioxo-1,2,3,4-tetrahydro quinoxaline-6-sulfonohydrazide (12a)
N’-(3-benzoyl-4-oxothiazolidin-2-ylidene)-2,3-dioxo-1,2,3,4-tetrahydroquinoxaline-6-sulfonohydrazide(12b)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Guo, C.; Wang, L.; Li, X.; Wang, S.; Yu, X.; Xu, K.; Zhao, Y.; Luo, J.; Li, X.; Jiang, B.; et al. Discovery of novel bromophenol-thiosemicarbazone hybrids as potent selective inhibitors of poly (ADP-ribose) polymerase-1 (PARP-1) for use in cancer. J. Med. Chem. 2019, 62, 3051–3067. [Google Scholar] [CrossRef]
- Elmasry, G.F.; Aly, E.E.; Awadallah, F.M.; El-Moghazy, S.M. Design and synthesis of novel PARP-1 inhibitors based on pyridopyridazinone scaffold. Bioorg. Chem. 2019, 87, 655–666. [Google Scholar] [CrossRef]
- Li, S.; Li, X.Y.; Zhang, T.J.; Zhu, J.; Xue, W.H.; Qian, X.H.; Meng, F.H. Design, synthesis and biological evaluation of erythrina derivatives bearing a 1,2,3-triazole moiety as PARP-1 inhibitors. Bioorg. Chem. 2020, 96, 103575. [Google Scholar] [CrossRef]
- Malyuchenko, N.V.; Kotova, E.Y.; Kulaeva, O.I.; Kirpichnikov, M.P.; Studitskiy, V.M. PARP1 Inhibitors: Antitumor drug design. Acta Nat. (Англoязычная Версия) 2015, 7, 27–37. [Google Scholar] [CrossRef]
- Velagapudi, U.K.; Langelier, M.F.; Delgado-Martin, C.; Diolaiti, M.E.; Bakker, S.; Ashworth, A.; Patel, B.A.; Shao, X.; Pascal, J.M.; Talele, T.T. Design and synthesis of poly (ADP-ribose) polymerase inhibitors: Impact of adenosine pocket-binding motif appendage to the 3-oxo-2, 3-dihydrobenzofuran-7-carboxamide on potency and selectivity. J. Med. Chem. 2019, 62, 5330–5357. [Google Scholar] [CrossRef]
- Ramadan, S.K.; Elrazaz, E.Z.; Abouzid, K.A.; El-Naggar, A.M. Design, synthesis and in silico studies of new quinazolinone derivatives as antitumor PARP-1 inhibitors. RSC Adv. 2020, 10, 29475–29492. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Ji, M.; Zhou, J.; Jin, J.; Xue, N.; Chen, J.; Xu, B.; Chen, X. Poly (ADP-ribose) polymerases inhibitor, Zj6413, as a potential therapeutic agent against breast cancer. Biochem. Pharmacol. 2016, 107, 29–40. [Google Scholar] [CrossRef]
- Huang, S.H.; Cao, R.; Lin, Q.W.; Wu, S.Q.; Gao, L.L.; Sun, Q.; Zhu, Q.H.; Zou, Y.; Xu, Y.G.; Wang, S.P. Design, synthesis and mechanism studies of novel dual PARP1/BRD4 inhibitors against pancreatic cancer. Eur. J. Med. Chem. 2022, 230, 114116. [Google Scholar] [CrossRef]
- Curtin, N. PARP inhibitors for anticancer therapy. Biochem. Soc. Trans. 2014, 42, 82–88. [Google Scholar] [CrossRef]
- Liu, J.F.; Konstantinopoulos, P.A.; Matulonis, U.A. PARP inhibitors in ovarian cancer: Current status and future promise. Gynecol. Oncol. 2014, 133, 362–369. [Google Scholar] [CrossRef]
- Singh, M.; Rajawat, J.; Kuldeep, J.; Shukla, N.; Mishra, D.P.; Siddiqi, M.I. Integrated support vector machine and pharmacophore-based virtual screening driven identification of thiophene carboxamide scaffold containing compound as potential PARP1 inhibitor. J. Biomol. Struct. 2021, 1–14. [Google Scholar] [CrossRef]
- Xin, M.; Sun, J.; Huang, W.; Tang, F.; Liu, Z.; Jin, Q.; Wang, J. Design and synthesis of novel phthalazinone derivatives as potent poly(ADP-ribose)polymerase 1 inhibitors. Future Med. Chem. 2020, 12, 1691–1707. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [Green Version]
- Salech, F.; Ponce, D.P.; SanMartín, C.D.; Rogers, N.K.; Chacón, C.; Henríquez, M.; Behrens, M.I. PARP-1 and p53 regulate the increased susceptibility to oxidative death of lymphocytes from MCI and AD patients. Front. Aging Neurosci. 2017, 9, 310. [Google Scholar] [CrossRef]
- Alluri, S.R.; Riss, P.J. Poly (ADP-ribose) polymerase in neurodegeneration: Radiosynthesis and radioligand binding in ARC-SWE tg mice. ACS Chem. Neurosci. 2018, 9, 1259–1263. [Google Scholar] [CrossRef]
- Tang, L.; Wu, W.; Zhang, C.; Shi, Z.; Chen, D.; Zhai, X.; Jiang, Y. Discovery of the PARP (poly ADP-ribose polymerase) inhibitor 2-(1-(4, 4-difluorocyclohexyl) piperidin-4-yl)-1H-benzo [d] imidazole-4-carboxamide for the treatment of cancer. Bioorg. Chem. 2021, 114, 105026. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Wang, P.Y.; Wang, Y.T.; Yang, G.F.; Zhang, A.; Miao, Z.H. An Update on Poly(ADP-ribose)polymerase-1 (PARP-1) Inhibitors: Opportunities and Challenges in Cancer Therapy. J. Med. Chem. 2016, 59, 9575–9598. [Google Scholar] [CrossRef]
- Jain, P.G.; Patel, B.D. Medicinal chemistry approaches of poly ADP-Ribose polymerase 1 (PARP1) inhibitors as anticancer agents -A recent update. Eur. J. Med. Chem. 2019, 165, 198–215. [Google Scholar] [CrossRef] [PubMed]
- Chen, A. PARP inhibitors: Its role in the treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef]
- Zhu, G.D.; Gong, J.; Gandhi, V.B.; Liu, X.; Shi, Y.; Johnson, E.F.; Donawho, C.K.; Ellis, P.A.; Bouska, J.J.; Osterling, D.J.; et al. Discovery and SAR of orally efficacious tetrahydropyridopyridazinone PARP inhibitors for the treatment of cancer. Bioorg. Med. Chem. 2012, 20, 4635–4645. [Google Scholar] [CrossRef]
- Wang, L.; Liu, F.; Jiang, N.; Zhou, W.; Zhou, X.; Zheng, Z. Design, synthesis, and biological evaluation of novel PARP-1 inhibitors based on a 1H-thieno [3,4-d] imidazole-4-carboxamide scaffold. Molecules 2016, 21, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawicki-McKenna, J.M.; Langelier, M.F.; DeNizio, J.E.; Riccio, A.A.; Cao, C.D.; Karch, K.R.; McCauley, M.; Steffen, J.D.; Black, B.E.; Pascal, J.M. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell 2015, 60, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannini, G.; Battistuzzi, G.; Vesci, L.; Milazzo, F.M.; De Paolis, F.; Barbarino, M.; Guglielmi, M.B.; Carollo, V.; Gallo, G.; Artali, R.; et al. Novel PARP-1 inhibitors based on a 2-propanoyl-3H-quinazolin-4-one scaffold. Bioorg. Med. Chem. Lett. 2014, 24, 462–466. [Google Scholar] [CrossRef]
- Ferraris, D.V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors, from concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef] [PubMed]
- Abad, N.; El Bakri, Y.; Lai, C.H.; Karthikeyan, S.; Ramli, Y.; Ferfra, S.; Mague, J.T.; Essassi, E.M. Insights into the crystal structure of two newly synthesized quinoxalines derivatives as a potent inhibitor for c-Jun N-terminal kinases. J. Biomol. Struct. Dyn. 2022, 40, 2797–2814. [Google Scholar] [CrossRef]
- Fatima, S.; Bathini, R.; Sivan, S.K.; Manga, V. Molecular docking and 3D-QSAR studies on inhibitors of DNA damage signaling enzyme human PARP-1. J. Recept. Signal Transduct. Res. 2012, 32, 214–224. [Google Scholar] [CrossRef]
- Iwashita, A.; Hattori, K.; Yamamoto, H.; Ishida, J.; Kido, Y.; Kamijo, K.; Murano, K.; Miyake, H.; Kinoshita, T.; Warizaya, M.; et al. Discovery of quinazolinone and quinoxaline derivatives as potent and selective poly(ADP-ribose) polymerase-1/2 inhibitors. FEBS Lett. 2005, 579, 1389–1393. [Google Scholar] [CrossRef] [Green Version]
- Saleh, N.M.; El-Gaby, M.S.; El-Adl, K.; Abd El-Sattar, N.E. Design, green synthesis, molecular docking and anticancer evaluations of diazepam bearing sulfonamide moieties as VEGFR-2 inhibitors. Bioorg. Chem. 2020, 104, 104350. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, M.; Alghamdi, K.S.; Ahmed, N.S.; Bakhotmah, D.; Saleh, T.S. Design and green synthesis of novel quinolinone derivatives of potential anti-breast cancer activity against MCF-7 cell line targeting multi-receptor tyrosine kinases. J. Enzyme Inhib. Med. Chem. 2021, 36, 1454–1471. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.M.; Anwar, M.M.; Syam, Y.M.; Khedr, M.; Kamel, M.M.; Kassem, E.M.M. A novel class of substituted spiro [quinazoline-2,1’-cyclohexane] derivatives as effective PARP-1 inhibitors: Molecular modeling, synthesis, cytotoxic and enzyme assay evaluation. Acta. Pol. Pharm. Drug. Res. 2013, 70, 687–708. [Google Scholar]
- Amin, K.M.; Anwar, M.M.; Kamel, M.M.; Kassem, E.M.M.; Syam, Y.M.; El Seginy, S.A. Synthesis, cytotoxic evaluation and molecular docking study of novel quinazoline derivatives as PARP-1 inhibitors. Acta. Pol. Pharm. Drug. Res. 2013, 70, 833–849. [Google Scholar]
- Anwar, M.M.; Abd El-Karim, S.S.; Mahmoud, A.H.; Amr, A.E.G.E.; Al-Omar, M.A. A comparative study of the anticancer activity and PARP-1 inhibiting effect of benzofuran–pyrazole scaffold and its nano-sized particles in human breast cancer cells. Molecules 2019, 24, 2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Wang, P.; Zhang, Z.; Xue, G.; Zha, D.; Wang, J.; Xu, X.; Li, Z. Facile synthesis and anti-proliferative activity evaluation of quinoxaline derivatives. Synth. Commun. 2020, 50, 823–830. [Google Scholar] [CrossRef]
- Obafemi, C.A.; Akinpelu, D.A. Synthesis and antimicrobial activity of some 2(1H)-quinoxalinone-6-sulfonyl derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2005, 180, 1795–1807. [Google Scholar] [CrossRef]
- Acharya, P.T.; Bhavsar, Z.A.; Jethava, D.J.; Patel, D.B.; Patel, H.D. A review on the development of bio-active thiosemicarbazide derivatives: Recent advances. J. Mol. Struct. 2021, 1226, 129268. [Google Scholar] [CrossRef]
- Xie, Z.; Chen, Y.; Xu, P.; Zhou, Y.; Zhao, Q.; Jiao, H.; Li, Z. Design, synthesis and bio evaluation of 1H-indole4-carboxamide derivatives as potent poly(ADP-ribose) polymerase-1 inhibitors. RSC Adv. 2016, 6, 80784–80796. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Wang, J.; Lenardo, M.J. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J. Cell Sci. 2000, 113, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wouters, B.G. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 1999, 59, 1391–1399. [Google Scholar] [PubMed]
- Bashmail, H.A.; Alamoudi, A.A.; Noorwali, A.; Hegazy, G.A.; Ajabnoor, G.M.; Al-Abd, A.M. Thymoquinone enhances paclitaxel anti-breast cancer activity via inhibiting tumor-associated stem cells despite apparent mathematical antagonism. Molecules 2020, 25, 426. [Google Scholar] [CrossRef] [Green Version]
- Bashmail, H.A.; Alamoudi, A.A.; Noorwali, A.; Hegazy, G.A.; AJabnoor, G.; Choudhry, H.; Al-Abd, A.M. Thymoquinone synergizes gemcitabine anti-breast cancer activity via modulating its apoptotic and autophagic activities. Sci. Rep. 2018, 8, 11674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, K.; Bolaňos, B.; Smith, M.; Palde, P.B.; Cuenca, P.D.; VanArsdale, T.L.; Niessen, S.; Zhang, L.; Behenna, D.; Ornelas, M.A.; et al. Dissecting the molecular determinants of clinical PARP1 inhibitor selectivity for tankyrase1. J. Biol. Chem. 2021, 296, 100251. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-3: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA; Impact, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2018.
- Schrödinger Release 2018-3: Maestro; Schrödinger, LLC: New York, NY, USA, 2018.
- Schrödinger Release 2018-3: LigPrep; Schrödinger, LLC: New York, NY, USA, 2018.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-3: Glide; Schrödinger, LLC: New York, NY, USA, 2018.
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Genet. 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of crystal packing forces in determining protein sidechain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-3: Prime; Schrödinger, LLC: New York, NY, USA, 2018.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | IC50 (mean ± SEM) (nM) | IC50 (mean ± SEM) (µM) | |
|---|---|---|---|
| PARP-1 | MDA-MB-436 | WI-38 | |
| 3 | 12.86 ± 4.73 | ||
| 4 | 8.73 ± 0.44 | 30.30 ± 1.78 | |
| 5 | 3.05 ± 0.16 | 2.57 ± 0.15 SI = 31.77 | 81.67 ± 1.70 |
| 6 | 13.27 ± 0.68 | ||
| 7a | 57.14 ± 2.91 | ||
| 7b | 35.82 ± 1.80 | ||
| 7c | 43.40 ± 0.10 | ||
| 8a | 2.31 ± 0.30 | 10.70 ± 0.63 SI = 6.58 | 70.46 ± 0.43 |
| 8b | 11.06 ± 0.56 | ||
| 9a | 57.35 ± 2.90 | ||
| 9b | 35.71 ± 1.82 | ||
| 10a | 21.63 ± 1.10 | ||
| 10b | 6.35 ± 0.32 | 9.62 ± 0.56 SI = 7.86 | 75.66 ± 0.51 |
| 11a | 19.45 ± 0.99 | ||
| 11b | 8.25 ± 0.42 | 11.50 ± 0.67 SI = 6.96 | 80.12 ± 0.82 |
| 12a | 36.11 ± 1.84 | ||
| 12b | 40.54 ± 2.06 | ||
| Olaparib | 4.40 ± 0.30 | 8.63 ± 1.25 | |
| Compd No. | Binding Free Energy (kcal/mol) | Hydrogen-Bond | Water-Bridged Hydrogen Bond | pi-pi/Cation-pi |
|---|---|---|---|---|
| 4 | −54.3 | Ser904, Gly863, Asp766 | Tyr907, Tyr889. | |
| 5 | −60.9 | Gly863, Ser904 | Tyr907 | |
| 8a | −79.3 | Ser904, His862, Asp766, Ser864, | - | Tyr907, His862, Arg848 |
| 10b | −63.9 | Tyr896, Ser894, | Arg873, | Tyr907, Tyr889 |
| 11b | −54.4 | Ser904, Asn906, Lys903, | Met890, Glu988 | Tyr907, Lys903. |
| Olaparib | −93 | Ser904, Gly863, Ser864, Tyr896 | Arg878, Ile879 | Tyr896 and Tyr907 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syam, Y.M.; Anwar, M.M.; Abd El-Karim, S.S.; Elokely, K.M.; Abdelwahed, S.H. New Quinoxaline-Based Derivatives as PARP-1 Inhibitors: Design, Synthesis, Antiproliferative, and Computational Studies. Molecules 2022, 27, 4924. https://doi.org/10.3390/molecules27154924
Syam YM, Anwar MM, Abd El-Karim SS, Elokely KM, Abdelwahed SH. New Quinoxaline-Based Derivatives as PARP-1 Inhibitors: Design, Synthesis, Antiproliferative, and Computational Studies. Molecules. 2022; 27(15):4924. https://doi.org/10.3390/molecules27154924
Chicago/Turabian StyleSyam, Yasmin M., Manal M. Anwar, Somaia S. Abd El-Karim, Khaled M. Elokely, and Sameh H. Abdelwahed. 2022. "New Quinoxaline-Based Derivatives as PARP-1 Inhibitors: Design, Synthesis, Antiproliferative, and Computational Studies" Molecules 27, no. 15: 4924. https://doi.org/10.3390/molecules27154924