
Crystal Structure and Noncovalent Interactions of Heterocyclic Energetic Molecules

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results
3.1. Crystal and Molecular Structure
3.2. Hirshfeld Surface
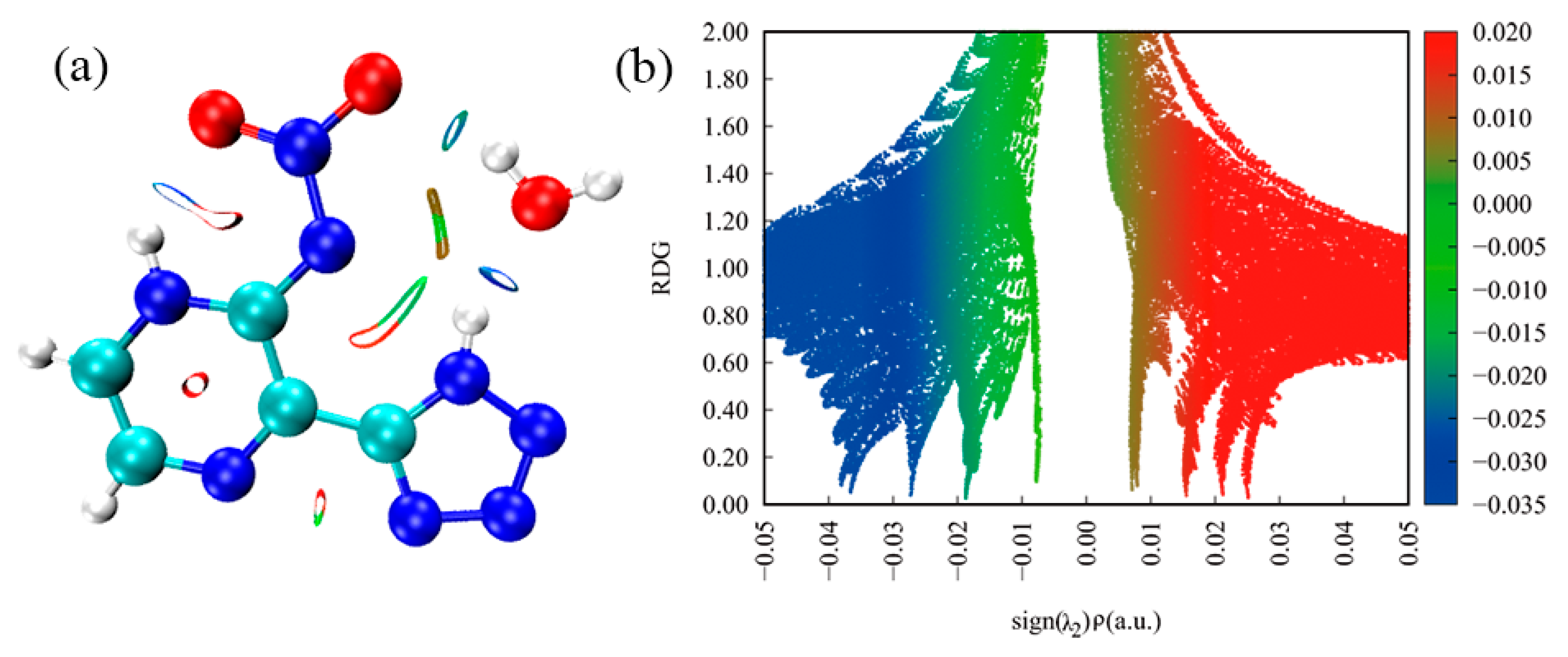
3.3. Reduced Density Gradient Analysis
3.4. Electrostatic Potential Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Millar, D.-I.-A.; Maynard-Casely, H.-E.; Allan, D.-R.; Cumming, A.-S.; Lennie, A.-R.; Mackay, A.-J.; Oswald, I.-D.-H.; Tang, C.-C.; Pulham, C.-R. Crystal engineering of energetic materials: Co-crystal of CL-20. CrystEngComm 2012, 14, 3742–3749. [Google Scholar] [CrossRef] [Green Version]
- Duan, B.-H.; Shu, Y.-J.; Liu, N.; Lu, Y.-Y.; Wang, B.-Z.; Lu, X.-M.; Zhang, J.-Q. Comparative studies on structure, sensitivity and mechanical properties of CL-20/DNDAP cocrystal and composite by molecular dynamics simulation. RSC Adv. 2018, 8, 34690–34698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.-B.; Gong, J.; Hu, X.-Y.; Ju, X. Comparative investigation on the thermostability, sensitivity, and mechanical performance of RDX/HMX energetic cocrystal and its mixture. J. Mol. Model. 2020, 26, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Duan, X.-H.; Li, H.-Z.; Pei, C.-H. A novel nigh-energetic and good-sensitive cocrystal composed of CL-20 and TATB by a rapid solvent/non-solvent method. RSC Adv. 2015, 5, 95764–95770. [Google Scholar] [CrossRef]
- Landenberger, K.-B.; Matzger, A.-J. cocrystal of 1,3,5,7-Tetranitro-1,3,5,7-tetrazacyclooctane (HMX). Cryst. Growth Des. 2012, 12, 3603–3609. [Google Scholar] [CrossRef]
- Sun, T.; Xiao, J.-J.; Liu, Q.; Zhao, F.; Xiao, H.-M. Comparative study on structure, energetic and mechanical properties of a 3-CL-20/HMX cocrystal and its composite with molecular dynamics simulation. J. Mater. Chem. A 2014, 2, 13898–13904. [Google Scholar] [CrossRef]
- Li, S.-J.; Bu, R.-P.; Gou, R.-J.; Zhang, C.-Y. Hirshfeld surface method and its application in energetic crystal. Cryst. Growth Des. 2021, 21, 6619–6634. [Google Scholar] [CrossRef]
- Yang, C.-M.; Chen, J.; Wang, R.-W.; Zhang, M.; Zhang, C.-Y.; Liu, J. Density prediction models for energetic compounds merely using molecular topology. J. Chem. Inf. Model. 2021, 61, 2582–2593. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; He, L.-C.; Shreeve, J.-M. Fused heterocycle-based energetic salts alliance of pyrazole and 1,2,3-triazole. J. Mater. Chem. A. 2016, 4, 1514–1519. [Google Scholar] [CrossRef]
- Klapotke, T.-M.; Schmid, P.-C.; Schnell, S.; Stierstorfer, J. 3,6,7-Triamino-[1,2,4]triazolo[4,3-b][1,2,4]triazole a non-toxic, high-performance energetic building block with excellent stability. Chem. Eur. J. 2015, 21, 9219–9228. [Google Scholar] [CrossRef]
- Luo, Y.-M.; Zheng, W.-W.; Wang, X.-J.; Shen, F. Nitrification progress of nitrogen-rich heterocyclic energetic compounds: A review. Molecules 2022, 27, 1465. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Yao, Y.-S.; Liu, H.-Y.; Ma, Y.-M. Crystalline LiN5 predicted from first-principles as a possible high-energy material. J. Phys. Chem. Lett. 2015, 6, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-J.; Qi, X.-J.; Zhang, W.-Q.; Yin, P.; Cai, Z.-W.; Zhang, Q.-H. Construction of bicyclic 1,2,3-triazine N-oxides from aminocyanides. Org. Lett. 2021, 23, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-X.; Zhang, Q.-H.; Shreeve, J.-M. Fused heterocycle-based energetic materials (2012–2019). J. Mater. Chem. A. 2020, 8, 4193–4216. [Google Scholar] [CrossRef]
- Mahadevi, A.-S.; Sastry, G.-N. Cooperativity in noncovalent interactions. Chem. Rev. 2016, 116, 2775–2825. [Google Scholar] [CrossRef]
- Prins, L.-J.; Reinhoudt, D.-N.; Timmerman, P. Noncovalent synthesis using hydrogen bonding. Angew. Chem. Int. Ed. 2001, 40, 2382–2426. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Zhao, G.; Yu, Q.; Tang, Y.-X.; Imler, G.-H.; Parrish, D.-A.; Shreeve, J.-M. Intermolecular weak hydrogen bonding (Het-H-N/O): An effective strategy for the synthesis of monosubstituted 1,2,4,5-Tetrazine-based energetic materials with excellent sensitivity. J. Org. Chem. 2019, 84, 16019–16026. [Google Scholar] [CrossRef]
- Bojarska, J.; Remko, M.; Fruzinski, A.; Maniukiewicz, W. The experimental and theoretical landscape of a new antiplatelet drug ticagrelor Insight into supramolecular architecture directed by CH···F, π-π and CH-π interactions. J. Mol. Struct. 2018, 1154, 290–300. [Google Scholar] [CrossRef]
- Desiraju, G.-R.; Ho, P.-S.; Kloo, L.; Legon, A.-C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar]
- Meyer, E.-A.; Castellano, R.-K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in supramolecular chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef]
- Fourmigue, M.; Batail, P. Activation of hydrogen- and halogen-bonding interactions in tetrathiafulvalene-based crystalline molecular conductors. Chem. Rev. 2004, 104, 5379–5418. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Zhang, Q.-H.; Vo, T.-T.; Parrish, D.-A.; Shreeve, J.-M. Energetic salts with π-stacking and hydrogen-bonding interactions lead the way to future energetic materials. J. Am. Chem. Soc. 2015, 137, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.-R.; Turner, M.-J.; McKinnon, J.-J.; Wolff, S.-K.; Grimwood, D.-J.; Jayatilaka, D.; Spackman, M.-A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-G.; Tian, L.-L.; Li, D.-X.; Wang, P.-C.; Lu, M. A series of energetic cyclo-pentazolate salts: Rapid synthesis, characterization, and promising performance. J. Mater. Chem. A. 2019, 7, 12468–12479. [Google Scholar] [CrossRef]
- Liu, Y.; An, C.-W.; Luo, J.; Wang, J.-Y. High-density HNIW/TNT cocrystal synthesized using a green chemical method. Acta Cryst. 2018, B74, 385–393. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.-W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Pant, S.; Zhang, J.; Kim, E.C.; Lam, K.; Chung, H.J.; Tajkhorshid, E. PIP2-dependent coupling of voltage sensor and pore domains in Kv7.2 channel. Commun. Biol. 2021, 4, 1189–1203. [Google Scholar] [CrossRef]
- Wang, K.; Zhu, W.-H. Computational insights into the formation driving force of CL-20 based solvates and their desolvation process. CrystEngComm 2021, 23, 2150–2161. [Google Scholar] [CrossRef]
- Urbelis, J.-H.; Swift, J.-A. Solvent effects on the growth morphology and phase purity of CL-20. Cryst. Growth Des. 2014, 14, 1642–1649. [Google Scholar] [CrossRef]
- Bu, R.-P.; Xiong, Y.; Wei, X.-F.; Li, H.-Z.; Zhang, C.-Y. Hydrogen bonding in CHON-containing energetic crystals a review. Cryst. Growth Des. 2019, 19, 5981–5997. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Wang, G.-X. Configuration and stability of 1,1-diamino-2,2-dinitroethylene (FOX-7) embedded in graphene. Bull. Korean Chem. Sos. 2016, 37, 1571–1576. [Google Scholar] [CrossRef]
- Liu, G.-R.; Wei, S.-H.; Zhang, C.-Y. Review of the intermolecular interactions in energetic molecular cocrystals. Cryst. Growth Des. 2020, 20, 7056–7079. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Wang, X.-C.; Huang, H. π-stacked interactions in explosive crystals: Buffers against external mechanical stimuli. J. Am. Chem. Sos. 2008, 130, 8359–8365. [Google Scholar] [CrossRef]
- Wu, J.-T.; Xu, J.; Li, W.; Li, H.-B. Coplanar fused heterocycle-based energetic materials. Propellants Explos. Pyrotech. 2020, 45, 1–11. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, A.-B.; Zhang, C.-H.; Jiang, D.-J.; Zhu, Y.-Q.; Zhang, C.-H. Crystal packing of low-sensitivity and high-energy explosives. Cryst. Growth Des. 2014, 14, 4703–4713. [Google Scholar] [CrossRef]
- Yin, P.; Mitchell, L.-A.; Parrish, D.-A.; Shreeve, J.-M. Energetic N-nitramino/N-oxyl-functionalized pyrazoles with versatile π-π stacking: Structure-property relationships of high-performance energetic materials. Angew. Chem. 2016, 128, 1–4. [Google Scholar] [CrossRef]
- Boddu, V.-M.; Viswanath, D.-S.; Ghosh, T.-K.; Damavarapu, R. 2,4,6-Triamino-1,3,5-trinitrobenzene (TATB) and TATB-based formulations-a review. J. Hazard. Mater. 2010, 181, 1–8. [Google Scholar] [CrossRef]
- Duan, B.-H.; Shu, Y.-J.; Liu, N.; Wang, B.-Z.; Lu, X.-M.; Lu, Y.-Y. Direct insight into the formation driving force, sensitivity and detonation performance of the observed CL-20-based energetic cocrystals. CrystEngComm 2018, 20, 5790–5800. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.-S. The Fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Rice, B.-M.; Hare, J.-J. Improved prediction of heats of formation of energetic materials using quantum mechanical calculations. J. Phys. Chem. A. 2002, 106, 1770–1783. [Google Scholar] [CrossRef]
- Kuklja, M.-M.; Rashkeev, S.-N. Shear-strain-induced chemical reactivity of layered molecular crystals. Appl. Phys. Lett. 2007, 90, 151913–151916. [Google Scholar] [CrossRef]
- Zeman, S.; Friedl, Z. A new approach to the application of molecular surface electrostatic potential in the study of detonation. Propellants. Explos. Pyrotech. 2012, 37, 609–613. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Fan, J.; Xue, Z.; Lu, Y.; Zhao, J.; Hui, W. Crystal Structure and Noncovalent Interactions of Heterocyclic Energetic Molecules. Molecules 2022, 27, 4969. https://doi.org/10.3390/molecules27154969
Liu Y, Fan J, Xue Z, Lu Y, Zhao J, Hui W. Crystal Structure and Noncovalent Interactions of Heterocyclic Energetic Molecules. Molecules. 2022; 27(15):4969. https://doi.org/10.3390/molecules27154969
Chicago/Turabian StyleLiu, Yan, Jiake Fan, Zhongqing Xue, Yajing Lu, Jinan Zhao, and Wenyan Hui. 2022. "Crystal Structure and Noncovalent Interactions of Heterocyclic Energetic Molecules" Molecules 27, no. 15: 4969. https://doi.org/10.3390/molecules27154969