
Dihydroxyquingdainone Induces Apoptosis in Leukaemia and Lymphoma Cells via the Mitochondrial Pathway in a Bcl-2- and Caspase-3-Dependent Manner and Overcomes Resistance to Cytostatic Drugs In Vitro

 ,
,
Abstract
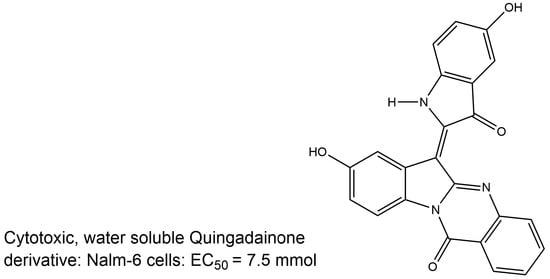
:
1. Introduction
2. Results
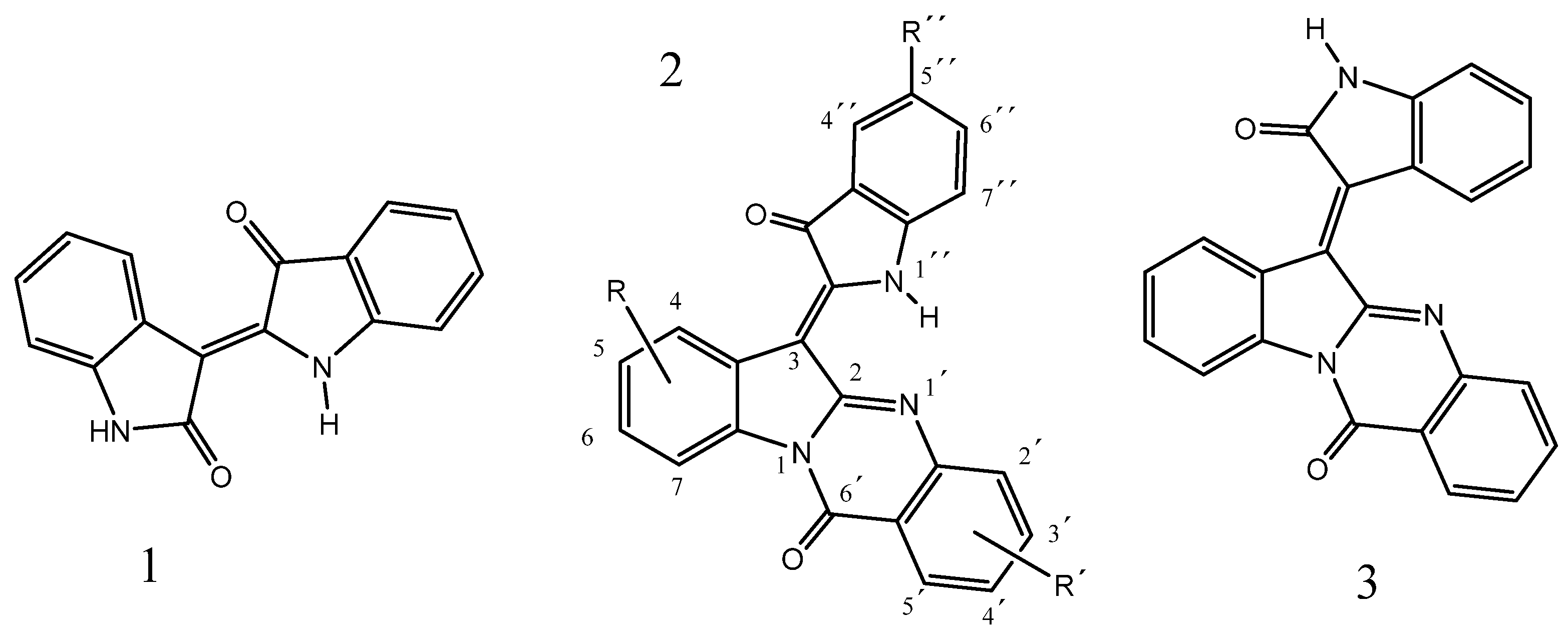
2.1. Chemistry
2.2. Quingdainones Cause Apoptosis in Nalm-6 Cells
2.3. Dihydroxyquingdainone 2g Shows Anti-Proliferative Effects in Nalm-6 Cells
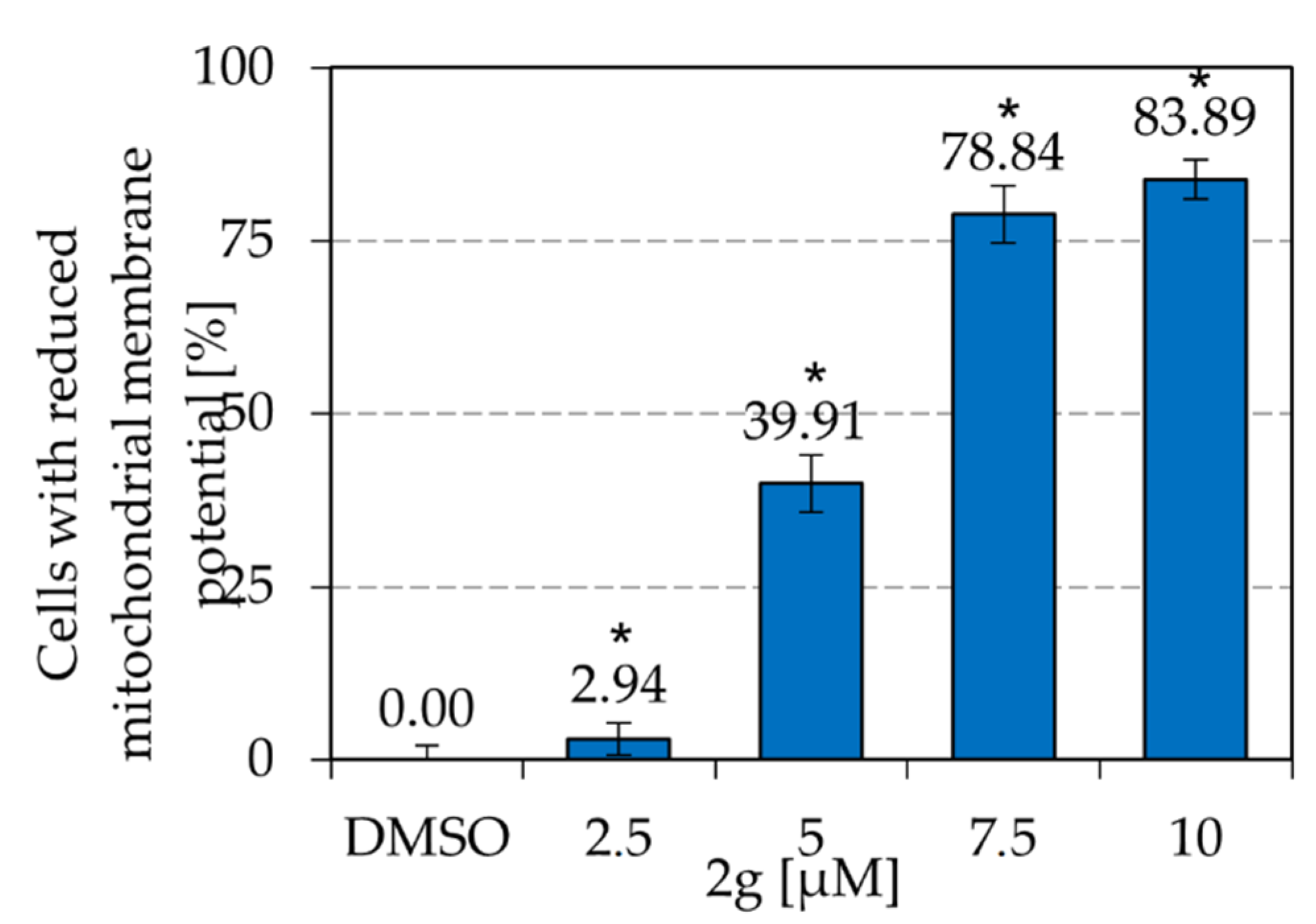
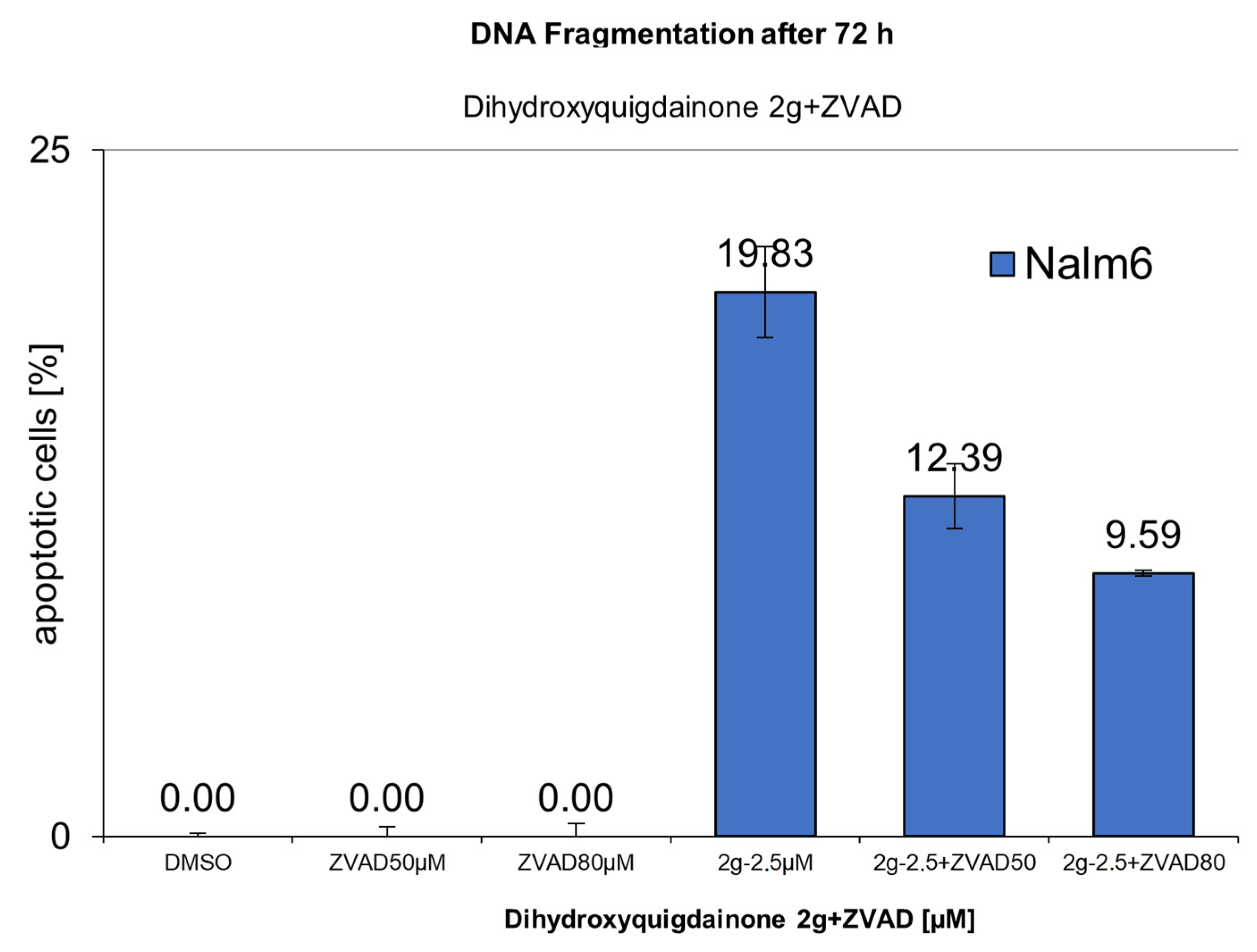
2.4. Dihydroxyquingdainone 2g Involves the Intrinsic Apoptosis Pathway
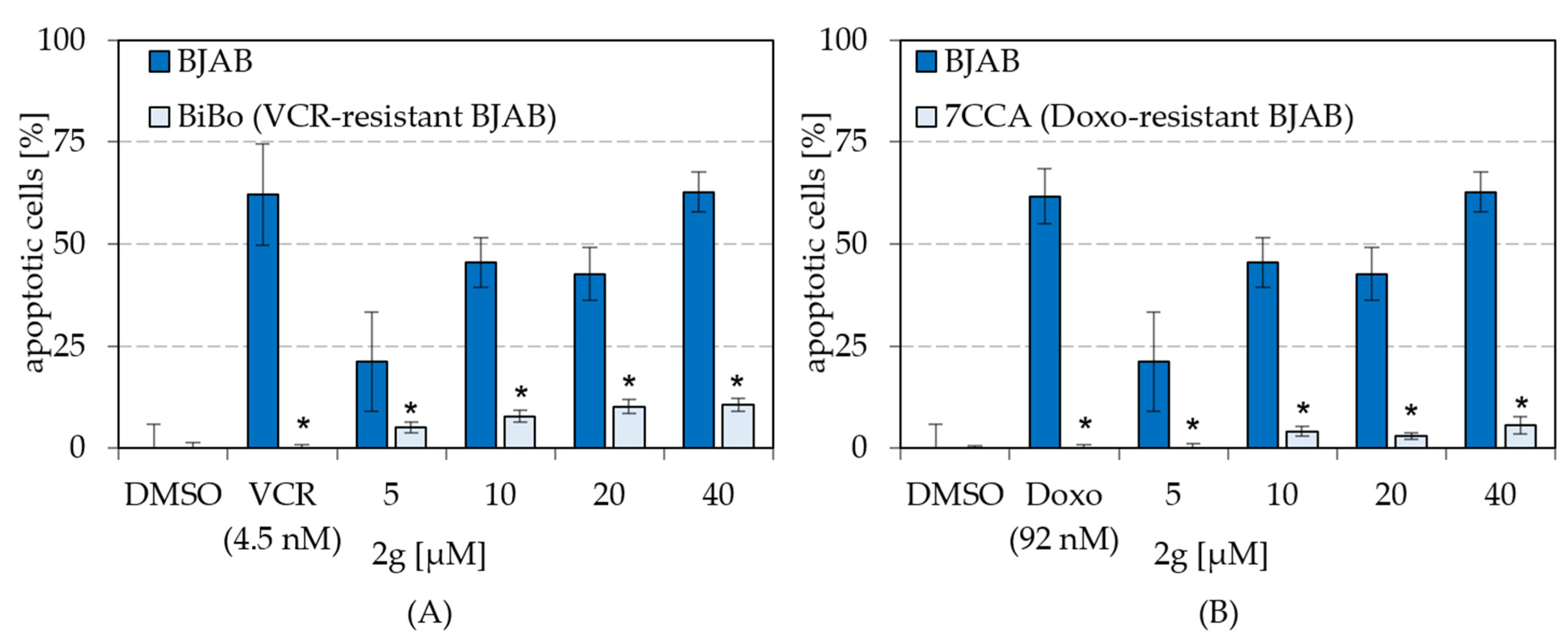
2.5. Dihydroxyquingdainone 2g Overcomes Resistances to Cytostatic Drugs
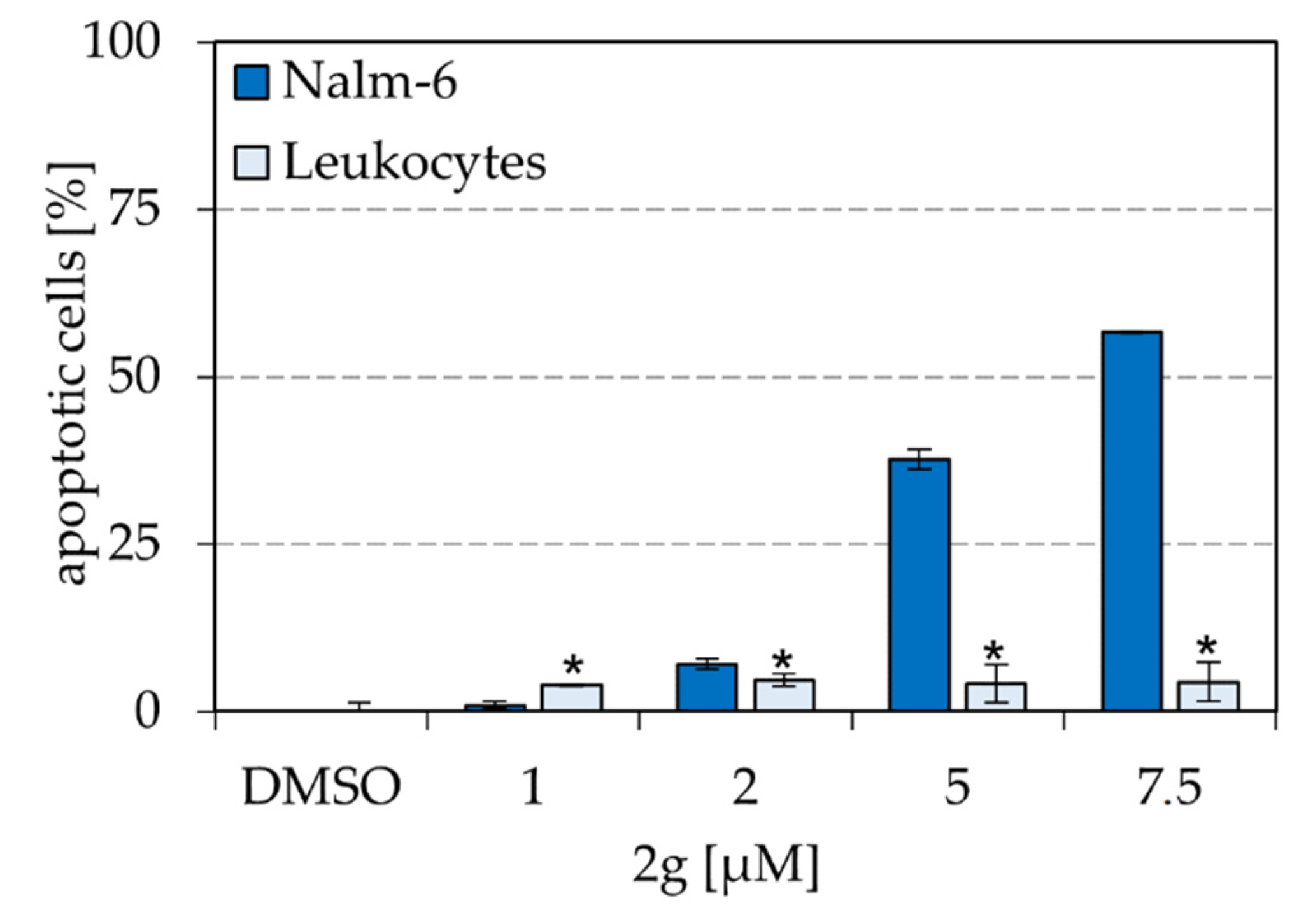
2.6. Dihydroxyquingdainone 2g Causes Significantly Less Apoptosis in Healthy Human Leukocytes Than in Nalm-6
3. Discussion
4. Materials and Methods
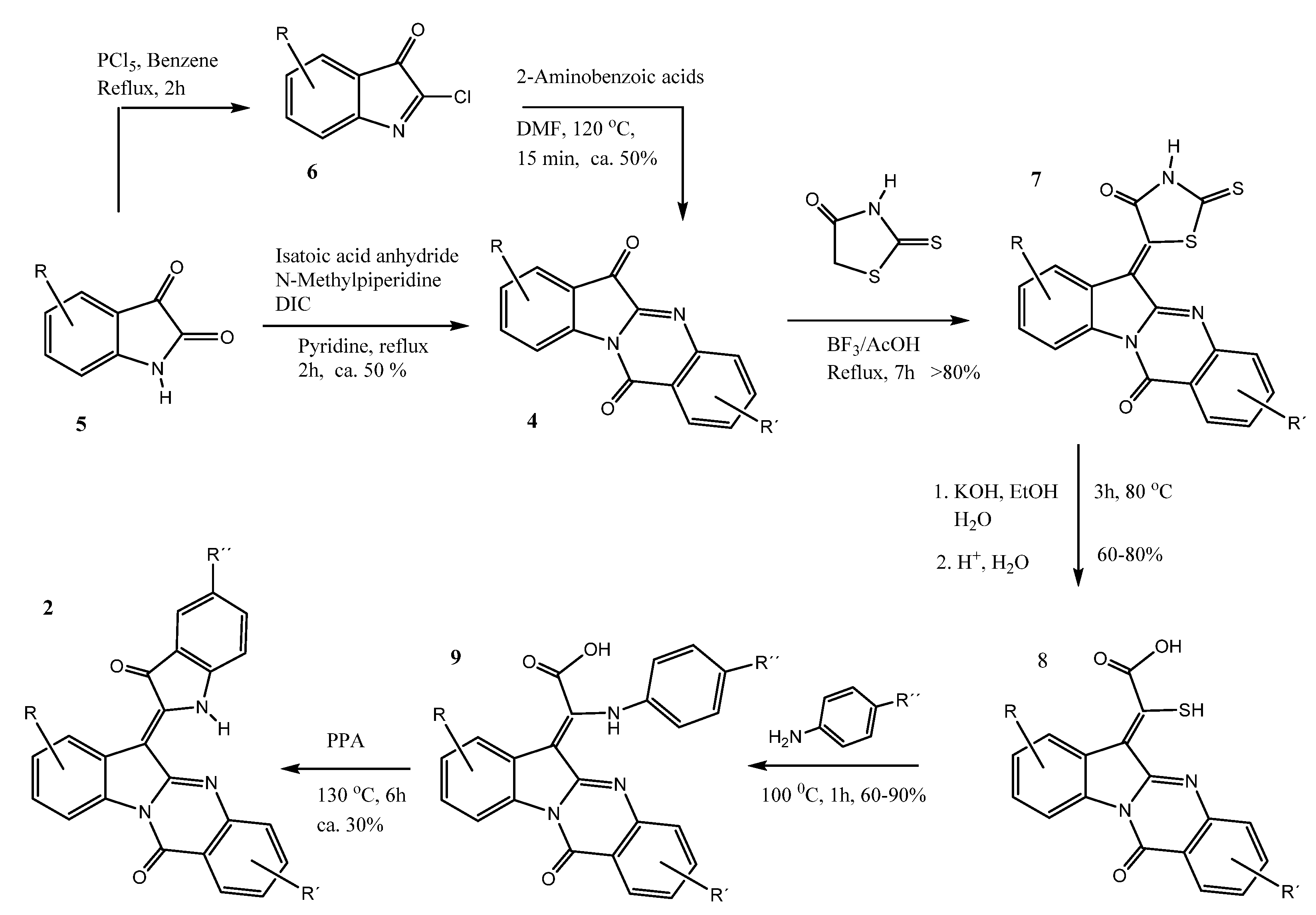
4.1. Synthesis of Quingdainone Derivatives
4.1.1. Synthetic Procedures
General Considerations
General Procedure for the Synthesis of Indolo[2,1-b]quinazoline-6,12-diones (Tryptanthrins)
Indolo[2,1-b]quinazoline-6,12-dione (4a)
8-Chloroindolo[2,1-b]quinazoline-6,12-dione (4b)
8-Bromoindolo[2,1-b]quinazoline-6,12-dione (4c)
8-Methylindolo[2,1-b]quinazoline-6,12-dione (4d)
8-Bromo-6,12-dioxo-6,12-dihydroindolo[2,1-b]quinazoline-3-carboxylic Acid (4e)
8-methoxyindolo[2,1-b]quinazoline-6,12-dione (4f)
General Procedure for the Synthesis of 5-(12-Oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-thioxothiazolidin-4-ones
5-(12-Oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-thioxothiazolidin-4-one (7a)
5-(8-Chloro-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-thioxothiazolidin-4-one (7b)
5-(8-Bromo-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-thioxothiazolidin-4-one (7c)
5-(8-Methyl-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-thioxothiazolidin-4-one (7d)
8-Bromo-12-oxo-6-(4-oxo-2-thioxothiazolidin-5-ylidene)-6,12-dihydroindolo[2,1-b]quinazoline-3-carboxylic Acid (7e)
5-(8-Methoxy-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-thioxothiazolidin-4-one (7f)
General Procedure for the Synthesis of 2-Mercapto-2-(12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)acetic Acids
2-Mercapto-2-(12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)acetic Acid (8a)
2-(Allylthio)-2-(12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)acetic Acid (8a’)
2-(8-Chloro-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-mercaptoacetic Acid (8b)
2-(8-Bromo-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-mercaptoacetic Acid (8c)
2-Mercapto-2-(8-methyl-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)acetic Acid (8d)
2-(Allylthio)-2-(8-methyl-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)acetic Acid (8d’)
8-Bromo-6-(carboxy(mercapto)methylene)-12-oxo-6,12-dihydroindolo[2,1-b]quinazoline-3-carboxylic Acid (8e)
6-((Allylthio)(carboxy)methylene)-8-bromo-12-oxo-6,12-dihydroindolo[2,1-b]quinazoline-3-carboxylic Acid (8e’)
2-Mercapto-2-(8-methoxy-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)acetic Acid (8f)
General Procedure for the Synthesis of 2-(12-Oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-(phenylamino)acetic Acids
2-(8-Chloro-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-(p-tolylamino)acetic Acid (9b)
2-(8-Bromo-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-(p-tolylamino)acetic Acid (9c)
2-(8-Methyl-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-(p-tolylamino)acetic Acid (9d)
2-((4-Chlorophenyl)amino)-2-(8-methyl-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)acetic Acid (9e)
8-Bromo-6-(carboxy(p-tolylamino)methylene)-12-oxo-6,12-dihydroindolo[2,1-b]quinazoline-3-carboxylic Acid (9f)
2-(8-Methoxy-12-oxoindolo[2,1-b]quinazolin-6(12H)-ylidene)-2-((4-methoxyphenyl)amino)acetic Acid (9g)
General Procedure for the Synthesis of (Z)-6-(3-Oxoindolin-2-ylidene)indolo[2,1-b]quinazolin-12(6H)-ones (Quingdainones)
(Z)-6-(5-Methyl-3-oxoindolin-2-ylidene)indolo[2,1-b]quinazolin-12(6H)-one (2a)
(Z)-8-Chloro-6-(5-methyl-3-oxoindolin-2-ylidene)indolo[2,1-b]quinazolin-12(6H)-one (2b)
(Z)-8-Bromo-6-(5-methyl-3-oxoindolin-2-ylidene)indolo[2,1-b]quinazolin-12(6H)-one (2c)
(Z)-8-Methyl-6-(5-methyl-3-oxoindolin-2-ylidene)indolo[2,1-b]quinazolin-12(6H)-one (2d)
(Z)-6-(5-Chloro-3-oxoindolin-2-ylidene)-8-methylindolo[2,1-b]quinazolin-12(6H)-one (2e)
(Z)-8-Bromo-6-(5-methyl-3-oxoindolin-2-ylidene)-12-oxo-6,12-dihydroindolo[2,1-b]quinazoline-3-carboxylic acid (2f)
(Z)-8-Hydroxy-6-(5-hydroxy-3-oxoindolin-2-ylidene)indolo[2,1-b]quinazolin-12(6H)-one (2g)
4.2. Used Cell Lines and Cell Cultivation
4.3. Determination of Cell Concentration
4.4. Measurement of DNA Fragmentation
4.5. Exclusion of Necrosis via LDH Detection
4.6. Measurement of the Mitochondrial Membrane Potential
4.7. Isolation of Healthy Human Leukocytes
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
References
- Marko, D.; Schätzle, S.; Friedel, A.; Genzlinger, A.; Zankl, H.; Meijer, L.; Eisenbrand, G. Inhibition of cyclin-dependent kinase 1 (CDK 1) by indirubin derivatives in human tumor cells. Br. J. Cancer 2001, 84, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.M.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J. The Identity of Candidine and Qingdainone. Phytochemistry 1989, 28, 3547. [Google Scholar] [CrossRef]
- Laatsch, H.; Ludwig-Köhn, H. Isolierung des indigoiden Pigmentes Candidin aus Urin und Hamofiltrat von Uramikern. Liebigs Ann. Chem. 1986, 1847–1853. [Google Scholar] [CrossRef]
- Fiedler, E.; Fiedler, H.P.; Gerhard, A.; Keller-Schierlein, W.; König, W.A.; Zähner, H. Synthese und Biosynthese substituierter Tryptanthrine. Arch. Microbiol. 1976, 107, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Jao, C.W.; Lin, W.C.; Wu, Y.T.; Wu, P.L. Isolation, Structure Elucidation, and Synthesis of Cytotoxic Tryptanthrin Analogues from Phaius mishmensis. J. Nat. Prod. 2008, 71, 1275–1279. [Google Scholar] [CrossRef]
- Liu, D.; Yang, C. Application of Quingdainone in the Manufacture of an Antitumor Agent. CN104910160AA, 22 May 2015. [Google Scholar]
- Bergman, J.; Tilstam, U.; Törnroos, K.W. Reduction and Stereochemical Studies through N.M.R. and X-Ray Techniques. J. Chem. Soc. Perkin. Trans. 1987, 1, 519–527. [Google Scholar] [CrossRef]
- Kingi, N.; Bergman, J. Thionation of Tryptanthrin, Rutaecarpine, and Related Molecules with a Reagent Prepared from P4S10 and Pyridine. J. Org. Chem. 2016, 81, 7711–7716. [Google Scholar] [CrossRef] [PubMed]
- Riepl, H.; Urmann, C. Improved Synthesis of Indirubin Derivatives by Sequential Build-Up of the Indoxyl Unit: First Preparation of Fluorescent Indirubins. Helv. Chim. Acta 2012, 95, 1461–1477. [Google Scholar] [CrossRef]
- Gränacher, C.; Mahal, A. Uber die Verwendung des Rhodanins zu organischen Synthesen 111. Derivate des Oxindols. Helv. Chim. Acta 1923, 6, 467–482. [Google Scholar] [CrossRef]
- Boiteau, J.-G.; Arlabosse, J.-M.; Aurelly, M.; Ghillini, A.-L.; Cardineaud, I. Unambigous (Z)-geometry and complete 1H and 13C assignments of candidine. Magn. Res. Chem. 2015, 53, 476. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.-F.; Gagné, M.; Robertson, G.; Collins, P. Reactions of Polyphosphoric Acid and Bitumen Model Compounds with Oxygenated Functional Groups: Where is the Phosphorylation? Energy Fuels 2008, 22, 4151–4157. [Google Scholar] [CrossRef] [Green Version]
- Orlando, C.M.; Wirth, J.G.; Heath, D.R. Methyl Aryl Ether Cleavage in Benzazole. J. Org. Chem. 1970, 35, 3147–3149. [Google Scholar] [CrossRef]
- Shen, Y.; Vignali, P.; Wang, R. Rapid Profiling Cell Cycle by Flow Cytometry Using Concurrent Staining of DNA and Mitotic Markers. Bio-Protocol 2017, 7, 2517. [Google Scholar] [CrossRef] [Green Version]
- Sitnikov, N.; Velder, J.; Abodo, L.; Cuvelier, N.; Neudörfl, J.; Prokop, A.; Krause, G.; Fedorov, A.Y.; Schmalz, H.-G. Total Synthesis of Indole-derived Allocolchicine Analogs Exhibiting Strong Apoptosis-inducing Activity. Chem. Eur. J. 2012, 18, 12096–12102. [Google Scholar] [CrossRef]
- Termath, A.O.; Ritter, S.; König, M.; Kranz, D.P.; Neudörfl, J.-P.; Prokop, A.; Schmalz, H.-G. Sythesis of Oxa-B-Ring Analogs of Colchicine through Rh-catalyzed Intramolecular (5 + 2) Cycloaddition. Eur. J. Org. Chem. 2012, 24, 4501–4507. [Google Scholar] [CrossRef]
- Hirschhäuser, C.; Velcicky, J.; Schlawe, D.; Hessler, E.; Majdalani, A.; Neudörfl, J.M.; Prokop, A.; Wieder, T.; Schmalz, H.-G. Nucleoside Analogues with a 1,3-Diene-Fe(CO)3 Substructure: Stereoselective Synthesis, Configurational Assignment, and Apoptosis-Inducing Activity. Chemistry 2013, 39, 13017–13029. [Google Scholar] [CrossRef] [PubMed]
- Sitnikov, N.S.; Sinzov, A.V.; Allegro, D.; Barbier, P.; Onambele, L.A.; Prokop, A.; Schmalz, H.-G.; Fedorov, A.Y. Synthesis of indole-derived allocholchicine congeners exhibiting pronounced anti proliferative and apoptosis inducing properties. Med. Chem. Commun. 2015, 6, 2158. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Karge, B.; Misgeld, R.; Prokop, A.; Brçnstrup, M.; Ott, I. Biscarbene gold(I) complexes: Structure-activity-relationships regarding antibacterial effects, cytotoxicity, TrxR inhibition and cellular bioavailability. Med. Chem. Commun. 2017, 8, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar]
- Reers, M.; Smith, T.W.; Chen, L.B. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 1991, 30, 4480–4486. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Lugmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesmann, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [Google Scholar] [CrossRef]
- Pieters, R.; Klumper, E.; Kaspers, G.J.; Veerman, A.J. Everything you always wanted to know about cellular drug resistance in childhood acute lymphoblastic leukemia. Crit. Rev. Oncol. Hematol. 1997, 25, 11–26. [Google Scholar] [CrossRef]
- Stein, A.; Thomopoulou, P.H.N.; Frias, C.; Hopff, S.M.; Varela, P.; Wilke, N.; Mariappan, A.; Neudörfl, J.-M.; Fedorov, A.Y.; Gopalakrishnan, J.; et al. B-nor-methylene Colchicinoid PT-100 Selectively Induces Apoptosis in Multidrug-Resistant Human Cancer Cells via an Intrinsic Pathway in a Caspase-Independent Manner. ACS Omega 2022, 7, 2591–2603. [Google Scholar] [CrossRef] [PubMed]
- Spychala, J. Selective cytostatic and cytotoxic anticancer effects of bisfunctional agents: A strategy for the design of DNA binding agents. Cancer Lett. 2009, 281, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Greenwell, M.; Rahman, P.K. Medicinal Plants: Their Use in Anticancer Treatment. Int. J. Pharm. Sci. Res. 2015, 6, 4103–4112. [Google Scholar]
- Bocca, C. Taxol: A short history of a promising anticancer drug. Minerva Biotecnol 1998, 10, 81. [Google Scholar]
- Kumar, A. Vincristine and Vinblastine: A Review. Int. J. Med. Pharm. 2016, 6, 23–30. [Google Scholar]
- Gu, S.; Xue, Y.; Gao, Y.; Shen, S.; Zhang, Y.; Chen, K.; Xue, S.; Pan, J.; Tang, Y.; Zhu, H.; et al. Mechanisms of indigo naturalis on treating ulcerative colitis explored by GEO gene chips combined with network pharmacology and molecular docking. Sci. Rep. 2020, 10, 15204. [Google Scholar] [CrossRef] [PubMed]
- Vougogiannopoulou, K.; Skaltsounis, A.-L. From Tyrian purple to kinase modulators: Naturally halogenated indirubins and synthetic analogues. Planta Med. 2012, 78, 1515–1528. [Google Scholar] [CrossRef] [Green Version]
- Bergman, J.; Lindström, J.O.; Tilstam, U. The structure and properties of some indolic constituents in Couroupita guianensis Aubl. Tetrahedron 1985, 41, 2879–2881. [Google Scholar] [CrossRef]
- Grimshaw, J.; Begley, W.J. Synthesis of 2-anilino-3h-indol-3-one derivatives. Synthesis 1974, 7, 496–498. [Google Scholar] [CrossRef]
- Onambele, L.A.; Riepl, H.; Fischer, R.; Pradel, G.; Prokop, A.; Aminake, M.N. Synthesis and evaluation of the antiplasmodial activity of tryptanthrin derivatives. Int. J. Parasitol. 2015, 5, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Prokop, A.; Riepl, H.; Wieder, T. 8-Nitro-tryptanthrin and Other Tryptanthrine Derivatives Used for the Treatment of Diseases Caused by Highly Proliferating Cells. WO2004087164A1, 14 October 2004. [Google Scholar]
- Lee, S.-Y.; Hille, A.; Kitanovic, I.; Jesse, P.; Henze, G.; Wölfl, S.; Gust, S.; Prokop, A. [Fe(III)(salophene)Cl], a potent iron salophene complex overcomes multiple drug resistance in lymphoma and leukemia cells. Leuk. Res. 2011, 35, 387–393. [Google Scholar] [CrossRef]
- Mulcahy, S.P.; Gründler, K.; Frias, C.; Wagner, L.; Prokop, A.; Meggers, E. Discovery of a strongly apoptotic ruthenium complex through combinatorial coordination chemistry. Dalton Trans. 2010, 39, 8177–8182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Majumder, P.; Sanyal, S.; Singh, J.; Jana, K.; Das, C.; Dasgupta, D. The DNA intercalators ethidium bromide and propidium iodide also bind to core histones. FEBS 2014, 4, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef]
- Chan, F.K.; Koriwaki, K.; de Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar]
- Maino, G.; la Gattuta, M.; Thompson, T.E. Cellular death and necrosis: Chemical, physical and morphologic changes in rat liver. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 1960, 333, 421–465. [Google Scholar]
- Lambert, I.H.; Hoffmann, E.K.; Jorgensen, F. Membrane potential, anion and cation conductances in Ehrlich ascites tumor cell. J. Membr. Biol. 1989, 111, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 30, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.A.; Merino, S.P. Application of Density Gradient Ultracentrifugation Using Zonal Rotors in the Large-Scale Purification of Biomolecules. In Downstream Processing of Proteins; Humana Press: Totowa, NJ, USA, 2000; pp. 59–72. [Google Scholar]
- Teetson, W.; Cartwright, C.; Dreiling, B.J.; Steinberg, M.H. The Leukocyte Composition of Peripheral Blood Buffy Coat. Am. J. Clin. Pathol. 1983, 79, 500–501. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | R | R’ | R’’ |
|---|---|---|---|
| 2a | H | H | 5’’ -CH3 |
| 2b | 5-Cl | H | 5’’ -CH3 |
| 2c | 5-CH3 | H | 5’’ -CH3 |
| 2d | 5-Br | H | 5’’ -CH3 |
| 2e | 5-CH3 | H | 5’’ -Cl |
| 2f | 5-Br | 3’-COOH | 5’’ -CH3 |
| 2g | 5-OH | H | 5’’ -OH |
| Substance | AC50 |
|---|---|
| 2a | >50 µM |
| 2b | >50 µM |
| 2c | / |
| 2d | / |
| 2e | / |
| 2f | / |
| 2g Quingdainone (R=R’=R’’= H) | <7.5 µM / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baas, J.; Bieringer, S.; Frias, C.; Frias, J.; Soehnchen, C.; Urmann, C.; Ritter, S.; Riepl, H.; Prokop, A. Dihydroxyquingdainone Induces Apoptosis in Leukaemia and Lymphoma Cells via the Mitochondrial Pathway in a Bcl-2- and Caspase-3-Dependent Manner and Overcomes Resistance to Cytostatic Drugs In Vitro. Molecules 2022, 27, 5038. https://doi.org/10.3390/molecules27155038
Baas J, Bieringer S, Frias C, Frias J, Soehnchen C, Urmann C, Ritter S, Riepl H, Prokop A. Dihydroxyquingdainone Induces Apoptosis in Leukaemia and Lymphoma Cells via the Mitochondrial Pathway in a Bcl-2- and Caspase-3-Dependent Manner and Overcomes Resistance to Cytostatic Drugs In Vitro. Molecules. 2022; 27(15):5038. https://doi.org/10.3390/molecules27155038
Chicago/Turabian StyleBaas, Jennifer, Sebastian Bieringer, Corazon Frias, Jerico Frias, Carolina Soehnchen, Corinna Urmann, Steffi Ritter, Herbert Riepl, and Aram Prokop. 2022. "Dihydroxyquingdainone Induces Apoptosis in Leukaemia and Lymphoma Cells via the Mitochondrial Pathway in a Bcl-2- and Caspase-3-Dependent Manner and Overcomes Resistance to Cytostatic Drugs In Vitro" Molecules 27, no. 15: 5038. https://doi.org/10.3390/molecules27155038
APA StyleBaas, J., Bieringer, S., Frias, C., Frias, J., Soehnchen, C., Urmann, C., Ritter, S., Riepl, H., & Prokop, A. (2022). Dihydroxyquingdainone Induces Apoptosis in Leukaemia and Lymphoma Cells via the Mitochondrial Pathway in a Bcl-2- and Caspase-3-Dependent Manner and Overcomes Resistance to Cytostatic Drugs In Vitro. Molecules, 27(15), 5038. https://doi.org/10.3390/molecules27155038