
Dehydrochlorination of PCDDs on SWCN-Supported Ni10 and Ni13 Clusters, a DFT Study




Abstract
:1. Introduction
2. Results and Discussion
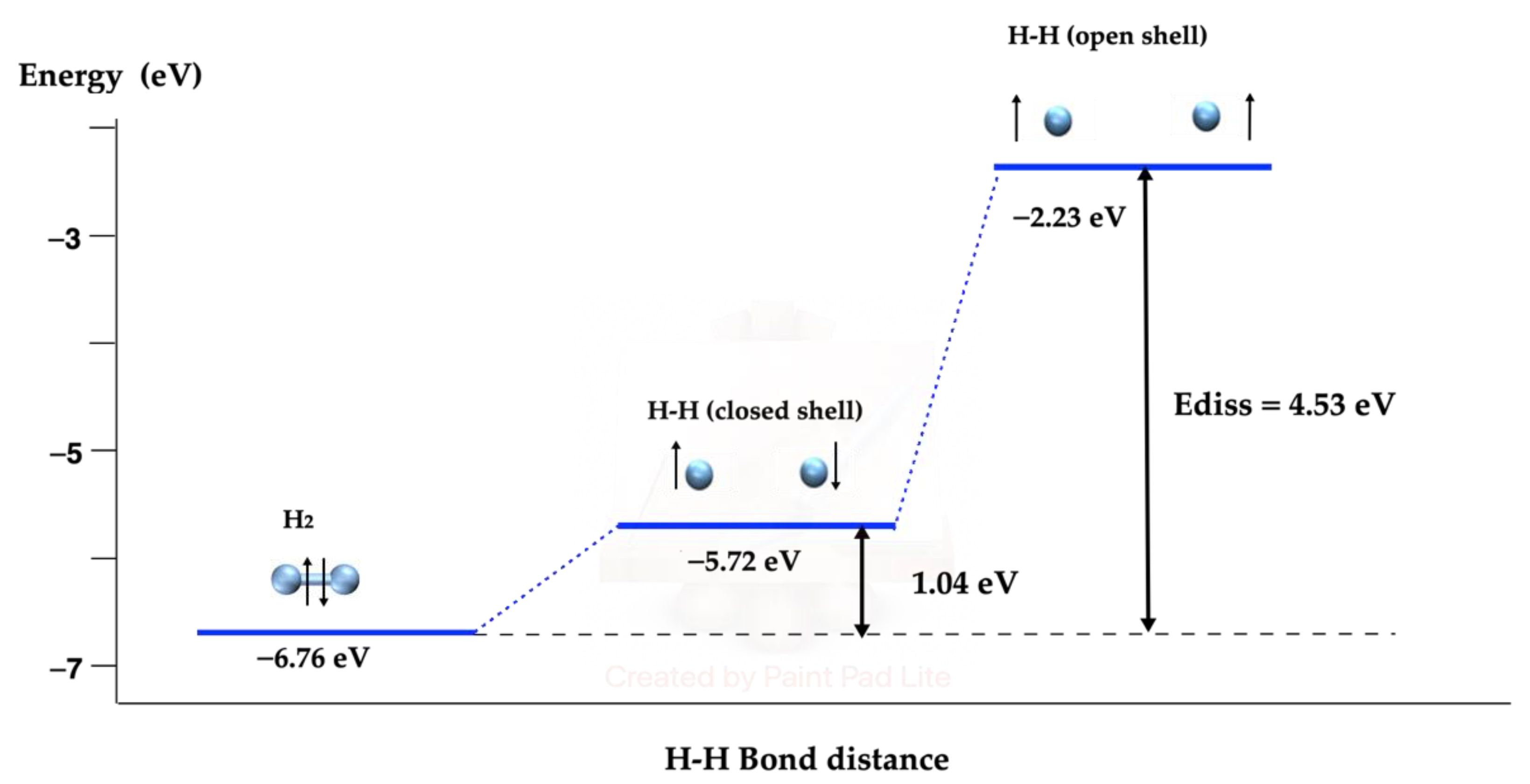
2.1. Free H2 Dissociation
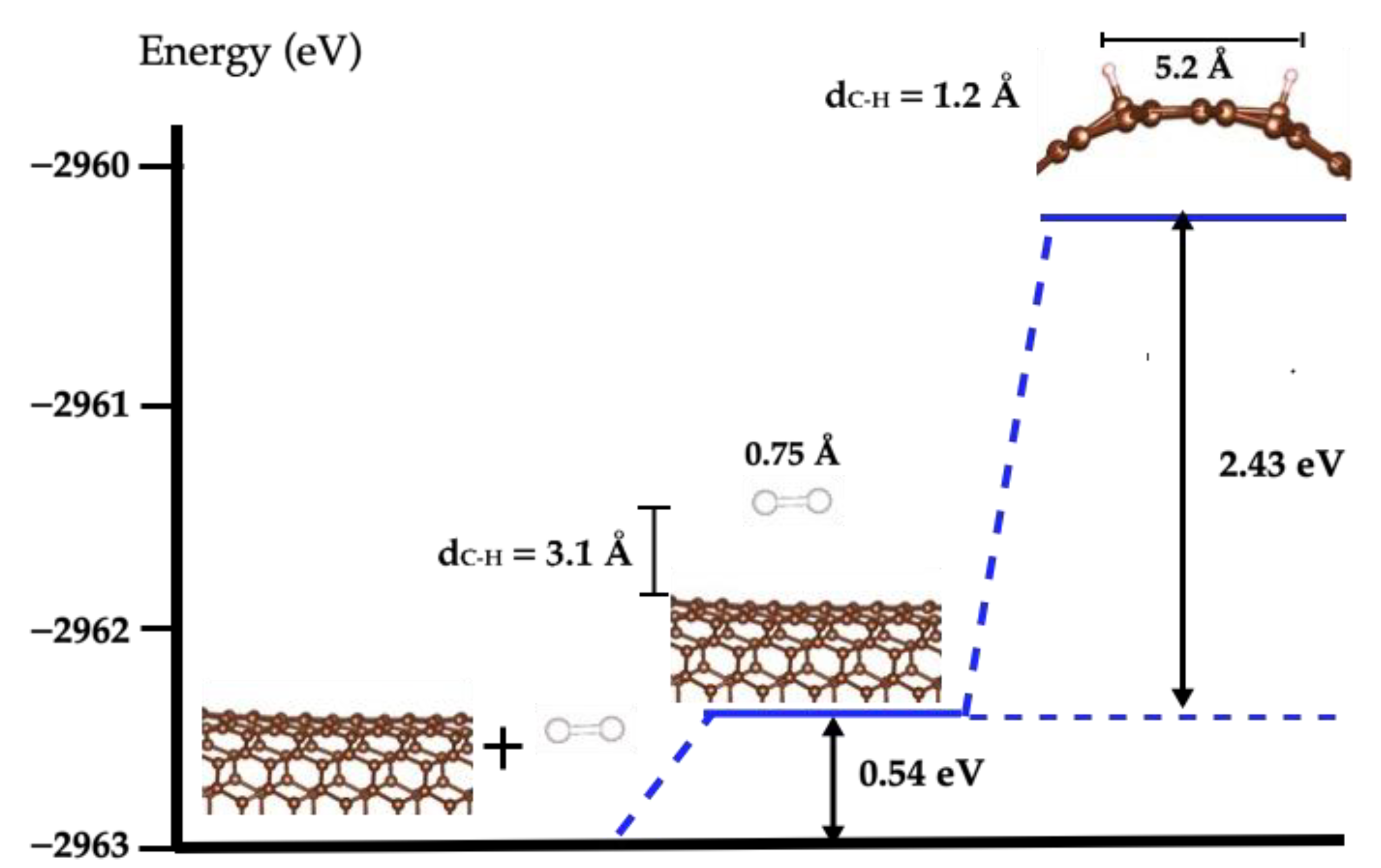
2.2. H2 Dissociation on a SWCN
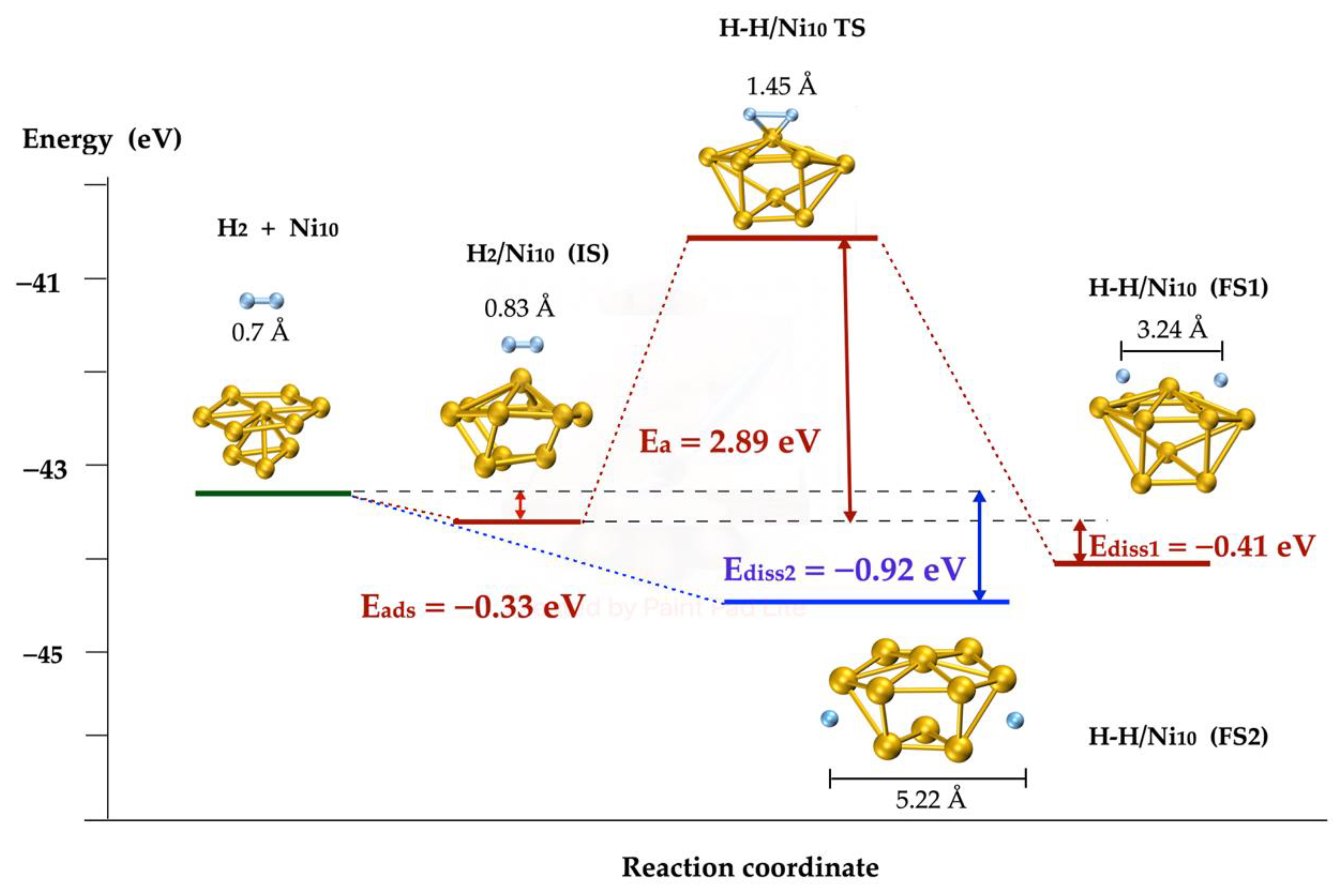
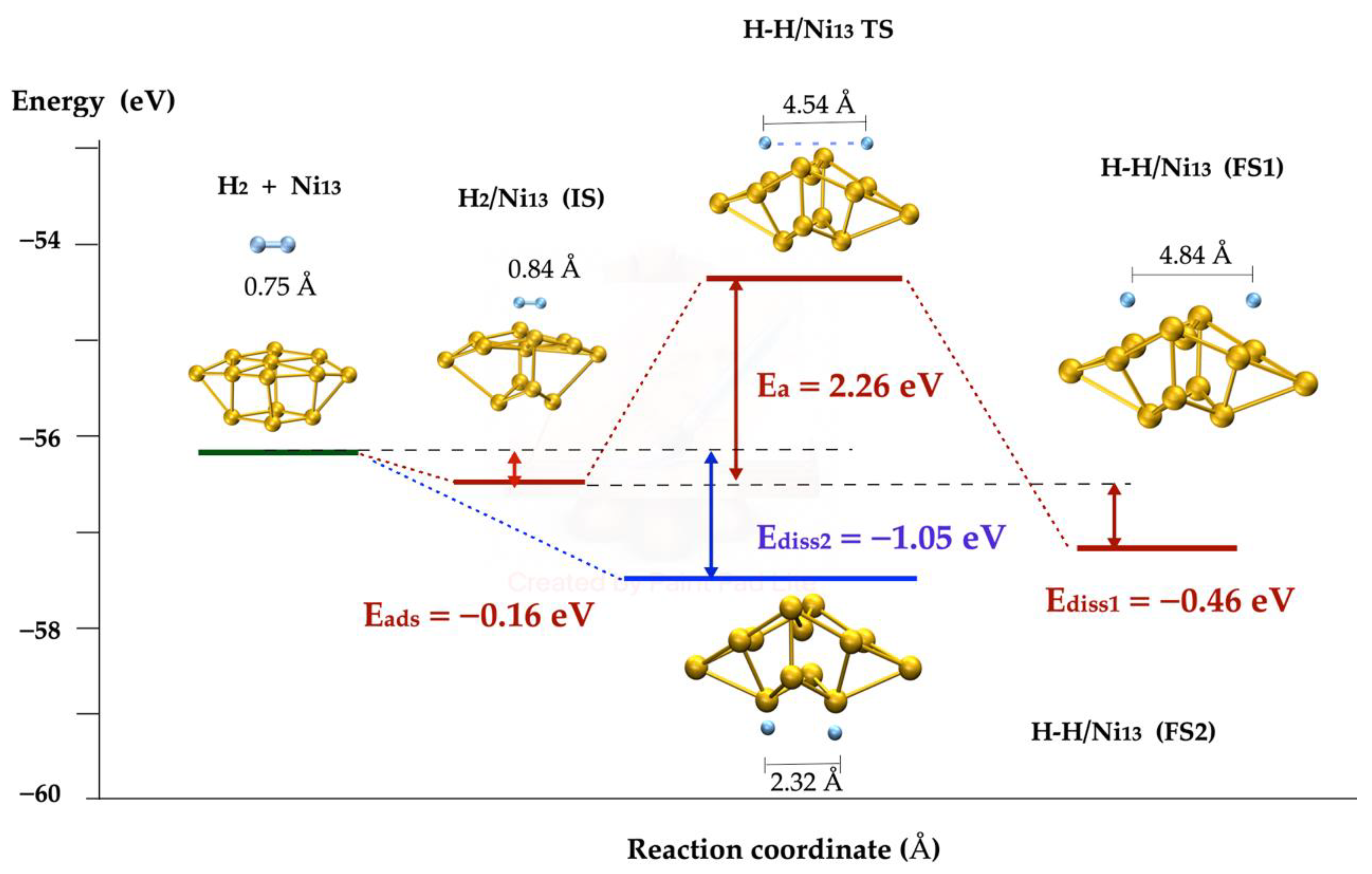
2.3. H2 Dissociation on Ni Metallic Clusters
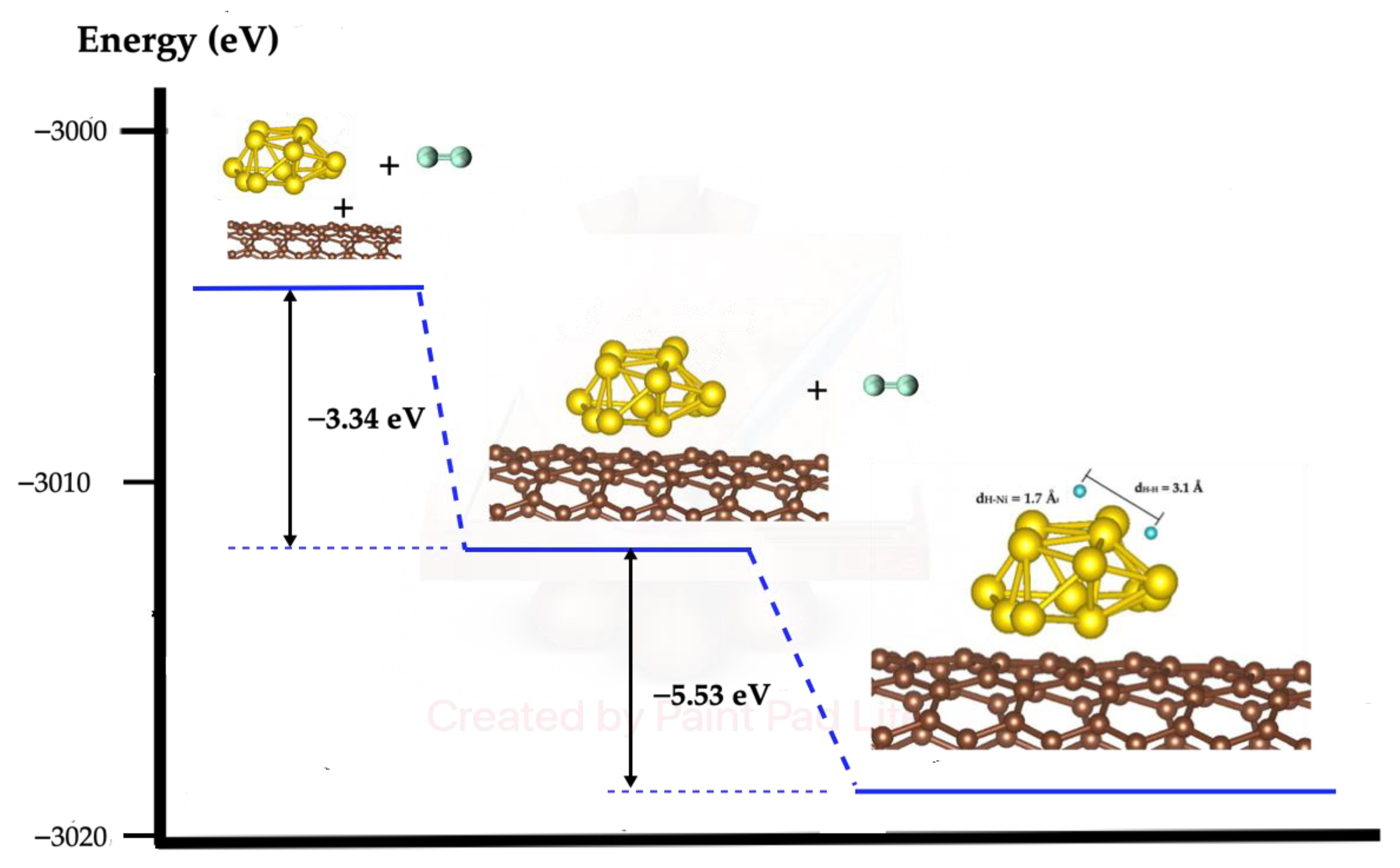
2.4. H2 Dissociation on SWCN Model Functionalized with Ni
2.5. Dehydrochlorination of Polychlorodioxins on a SWCN
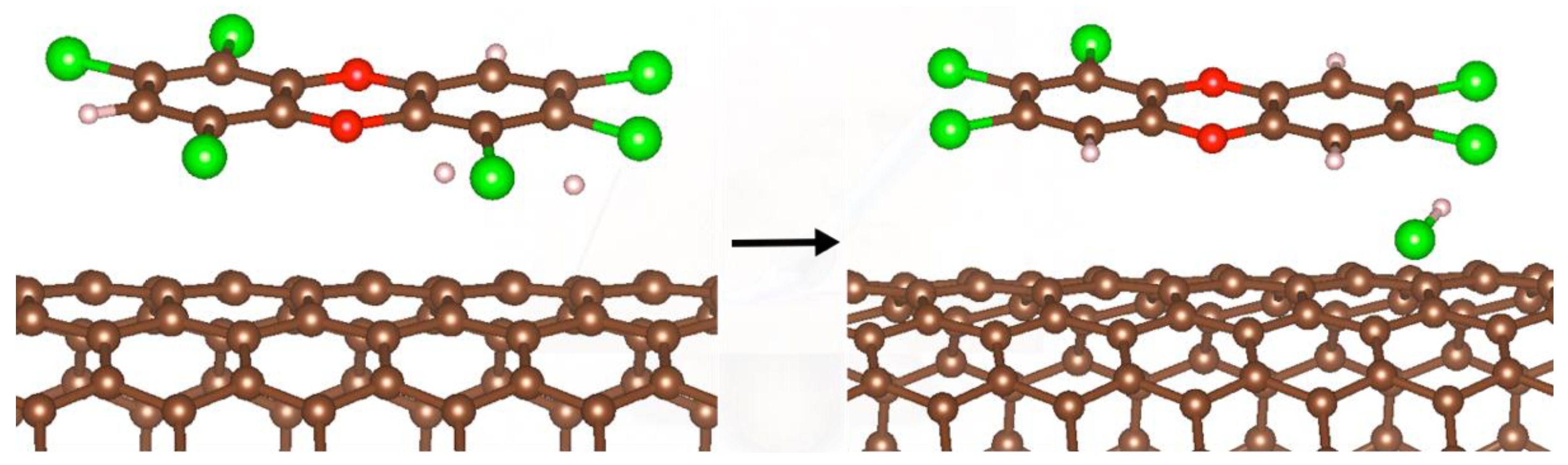
2.6. Dehydrochlorination of Polychlorodioxins on a SWCN with Nix (x = 10, 13) Clusters and H2
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marinković, N.; Pašalić, D.; Ferenčak, G.; Gršković, B.; Rukavina, A.S. Dioxins and Human Toxicity. Arh. Hig. Rada Toksikol. 2010, 61, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimbrough, R.D.; Falk, H.; Stehr, P.; Fries, G. Health Implications of 2,3,7,8-tetrachloro-dibenzodioxin (TCDD) Contamination of Residential Soil. J. Toxicol. Environ. Health 2009, 14, 47–93. [Google Scholar] [CrossRef] [PubMed]
- Lutes, C.C.; Charles, M.J.; Kamens, R.M. The Atmospheric Stability of Polybrominated Dibenzo-p-Dioxins and Dibenzofurans. Chemosphere 1992, 25, 99–102. [Google Scholar] [CrossRef]
- Birnbaum, L.S.; Staskal, D.F.; Diliberto, J.J. Health Effects of Polybrominated Dibenzo-p-Dioxins (PBDDs) and Dibenzofurans (PBDFs). Environ. Int. 2003, 29, 855–860. [Google Scholar] [CrossRef]
- Yoshioka, M.; Regayre, L.A.; Pringle, K.J.; Johnson, J.S.; Mann, G.W.; Partridge, D.G.; Sexton, D.M.H.; Lister, G.M.S.; Schutgens, N.; Stier, P.; et al. Ensembles of Global Climate Model Variants Designed for the Quantification and Constraint of Uncertainty in Aerosols and Their Radiative Forcing. J. Adv. Model. Earth Syst. 2019, 11, 3728–3754. [Google Scholar] [CrossRef] [Green Version]
- Mastalerz, P. The True Story of DDT, PCB, and Dioxins, 1st ed.; Wydawnictwo Chemiczne: Wroclaw, Poland, 2005. [Google Scholar]
- Kulkarni, P.S. Dioxins. Handb. Combust. 2016, 20, 1–28. [Google Scholar] [CrossRef]
- Fernandes, A.R.; Rose, M.; White, S.; Mortimer, D.N.; Gem, M. Dioxins and Polychlorinated Biphenyls (PCBs) in Fish Oil Dietary Supplements: Occurrence and Human Exposure in the UK. Food Addit. Contam. 2011, 23, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, P.S.; Crespo, J.G.; Afonso, C.A.M. Dioxins Sources and Current Remediation Technologies—A Review. Environ. Int. 2008, 34, 139–153. [Google Scholar] [CrossRef]
- Liu, H.Q.; Zeng, T.T.; Wei, G.X.; Zhang, R.; Liu, F.; Wang, H. Comparison of Dioxin Destruction in the Fly Ash and Froths under Microwave Irradiation. Aerosol Air Qual. Res. 2019, 19, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Zhang, P.; Chen, D.; Zhou, B.; Li, J.; Li, X.W. Hydrothermal Treatment of Municipal Solid Waste Incineration Fly Ash for Dioxin Decomposition. J. Hazard. Mater. 2012, 207–208, 79–85. [Google Scholar] [CrossRef]
- Kawasaki, S.I.; Oe, T.; Anjoh, N.; Nakamori, T.; Suzuki, A.; Arai, K. Practical Supercritical Water Reactor for Destruction of High Concentration Polychlorinated Biphenyls (PCB) and Dioxin Waste Streams. Process Saf. Environ. Prot. 2006, 84, 317–324. [Google Scholar] [CrossRef]
- Cagnetta, G.; Hassan, M.M.; Huang, J.; Yu, G.; Weber, R. Dioxins Reformation and Destruction in Secondary Copper Smelting Fly Ash under Ball Milling. Sci. Rep. 2016, 6, 22925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.S. Theoretical Study of Binding of Metal-Doped Graphene Sheet and Carbon Nanotubes with Dioxin. J. Am. Chem. Soc. 2005, 127, 9839–9843. [Google Scholar] [CrossRef] [PubMed]
- Fagan, S.B.; Santos, E.J.G.; Souza Filho, A.G.; Mendes Filho, J.; Fazzio, A. Ab Initio Study of 2,3,7,8-Tetrachlorinated Dibenzo-p-Dioxin Adsorption on Single Wall Carbon Nanotubes. Chem. Phys. Lett. 2007, 437, 79–82. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.; Suman, H.; Shrivastava, S.; Srivastava, A. DFT Analysis of Pristine and Functionalized Zigzag CNT: A Case of H2S Sensing. Chem. Phys. Lett. 2019, 731, 136575–136583. [Google Scholar] [CrossRef]
- Long, R.Q.; Yang, R.T. Carbon Nanotubes as Superior Sorbent for Dioxin Removal. J. Am. Chem. Soc. 2001, 123, 2058–2059. [Google Scholar] [CrossRef]
- Darvish Ganji, M.; Alinezhad, H.; Soleymani, E.; Tajbakhsh, M. Adsorption of TCDD Molecule onto CNTs and BNNTs: Ab Initio van Der Waals Density-Functional Study. Phys. E Low-Dimens. Syst. Nanostruct. 2015, 67, 105–111. [Google Scholar] [CrossRef]
- Izakmehri, Z.; Ganji, M.D.; Ardjmand, M. Adsorption of 2, 3, 7, 8-Tetrachlorodibenzo-p-Dioxin (TCDD) on Pristine, Defected and Al-Doped Carbon Nanotube: A Dispersion Corrected DFT Study. Vacuum 2017, 136, 51–59. [Google Scholar] [CrossRef]
- Fan, Y.; Lu, X.; Ni, Y.; Zhang, H.; Zhao, L.; Chen, J.; Sun, C. Destruction of Polychlorinated Aromatic Compounds by Spinel-Type Complex Oxides. Environ. Sci. Technol. 2010, 44, 3079–3084. [Google Scholar] [CrossRef]
- Lokteva, E.S.; Golubina, E.V.; Kachevsky, S.A.; Turakulova, A.O.; Lunin, V.V.; Tundo, P. Heterogeneous Catalysts and Process for Reductive Dechlorination of Polychlorinated Hydrocarbons. Pure Appl. Chem. 2007, 79, 1905–1914. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, X.; Yang, H.; Peng, B. Adsorption and Ability to Carry Catalysts of Carbon Nanotubes for Destructing Dioxins. Recent Pat. Mater. Sci. 2009, 2, 226–231. [Google Scholar] [CrossRef]
- Züttel, A.; Sudan, P.; Mauron, P.; Kiyobayashi, T.; Emmenegger, C.; Schlapbach, L. Hydrogen Storage in Carbon Nanostructures. Int. J. Hydrogen Energy 2002, 27, 203–212. [Google Scholar] [CrossRef]
- Ioannatos, G.E.; Verykios, X.E. H2 Storage on Single- and Multi-Walled Carbon Nanotubes. Int. J. Hydrogen Energy 2010, 35, 622–628. [Google Scholar] [CrossRef]
- Alonso, J.A.; Arellano, J.S.; Molina, L.M.; Rubio, A.; López, M.J. Interaction of Molecular and Atomic Hydrogen with Single-Wall Carbon Nanotubes. IEEE Trans. Nanotechnol. 2004, 3, 304–310. [Google Scholar] [CrossRef]
- An, W.; Wu, X.; Zeng, X.C. Adsorption of O2, H2, CO, NH3, and NO2 on ZnO Nanotube: A Density Functional Theory Study. J. Phys. Chem. C 2008, 112, 5747–5755. [Google Scholar] [CrossRef]
- Rather, S. Ullah. Preparation, Characterization and Hydrogen Storage Studies of Carbon Nanotubes and Their Composites: A Review. Int. J. Hydrogen Energy 2020, 45, 4653–4672. [Google Scholar] [CrossRef]
- Dubot, P.; Cenedese, P. Modeling of Molecular Hydrogen and Lithium Adsorption on Single-Wall Carbon Nanotubes. Phys. Rev. B Condens. Matter Mater. Phys. 2001, 63, 241402. [Google Scholar] [CrossRef]
- Reyhani, A.; Mortazavi, S.Z.; Mirershadi, S.; Moshfegh, A.Z.; Parvin, P.; Golikand, A.N. Hydrogen Storage in Decorated Multiwalled Carbon Nanotubes by Ca, Co, Fe, Ni, and Pd Nanoparticles under Ambient Conditions. J. Phys. Chem. C 2011, 115, 6994–7001. [Google Scholar] [CrossRef]
- Devi, N.R.; Gayathri, V. Effect of Structural Defects on the Hydrogen Adsorption in Promising Nanostructures. Comput. Mater. Sci. 2015, 96, 284–289. [Google Scholar] [CrossRef]
- Nikitin, A.; Li, X.; Zhang, Z.; Ogasawara, H.; Dai, H.; Nilsson, A. Hydrogen Storage in Carbon Nanotubes through the Formation of Stable C-H Bonds. Nano Lett. 2008, 8, 162–167. [Google Scholar] [CrossRef]
- Beheshtian, J.; Ahmadi Peyghan, A.; Bagheri, Z. Hydrogen Dissociation on Diene-Functionalized Carbon Nanotubes. J. Mol. Model. 2013, 19, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Au, C.-T.; Ng, C.-F.; Liao, M.-S. Methane Dissociation and Syngas Formation on Ru, Os, Rh, Ir, Pd, Pt, Cu, Ag, and Au: A Theoretical Study. J. Catal. 1999, 22, 12–22. [Google Scholar] [CrossRef]
- Bertolini, J.-C. Model Catalysis by Metals and Alloys: From Single-Crystal Surfaces to Well-Defined Nano-Particles. Catal. Today 2008, 138, 84–96. [Google Scholar] [CrossRef]
- Whitten, J.L.; Yang, H. Theory of Chemisorption and Reactions on Metal Surfaces. Surf. Sci. Rep. 1996, 24, 55–124. [Google Scholar] [CrossRef]
- Li, J.; He, H.; Hu, C.; Zhao, J. The Abatement of Major Pollutants in Air and Water by Environmental Catalysis. Front. Environ. Sci. Eng. 2013, 7, 302–325. [Google Scholar] [CrossRef]
- Liu, B.; Lusk, M.T.; Ely, J.F. Influence of Nickel Catalyst Geometry on the Dissociation Barriers of H2 and CH4: Ni13 versus Ni(111). J. Phys. Chem. C 2009, 113, 13715–13722. [Google Scholar] [CrossRef]
- Seenivasan, H.; Tiwari, A.K. Enhancing Methane Dissociation with Nickel Nanoclusters. Comput. Theor. Chem. 2015, 1064, 7–14. [Google Scholar] [CrossRef]
- Andriani, K.F.; Mucelini, J.; Da Silva, J.L.F. Methane Dehydrogenation on 3d 13-Atom Transition-Metal Clusters: A Density Functional Theory Investigation Combined with Spearman Rank Correlation Analysis. Fuel 2020, 275, 117790–117803. [Google Scholar] [CrossRef]
- Gödde, J.; Merko, M.; Xia, W.; Muhler, M. Nickel Nanoparticles Supported on Nitrogen–Doped Carbon Nanotubes Are a Highly Active, Selective and Stable CO2 Methanation Catalyst. J. Energy Chem. 2021, 54, 323–331. [Google Scholar] [CrossRef]
- Lyalin, A.; Shimizu, K.I.; Taketsugu, T. Interface Effects in Hydrogen Elimination Reaction from Isopropanol by Ni13 Cluster on θ-Al2O3(010) Surface. J. Phys. Chem. C 2017, 121, 3488–3495. [Google Scholar] [CrossRef]
- Felício-Sousa, P.; Andriani, K.F.; Da Silva, J.L.F. Ab Initio Investigation of the Role of the d-States Occupation on the Adsorption Properties of H2, CO, CH4 and CH3OH on the Fe13, Co13, Ni13 and Cu13 Clusters. Phys. Chem. Chem. Phys. 2021, 23, 8739–8751. [Google Scholar] [CrossRef] [PubMed]
- Verdinelli, V.; Germán, E.; Luna, C.R.; Marchetti, J.M.; Volpe, M.A.; Juan, A. Theoretical Study of Hydrogen Adsorption on Ru-Decorated (8,0) Single-Walled Carbon Nanotube. J. Phys. Chem. C 2014, 118, 27672–27680. [Google Scholar] [CrossRef]
- Singh, N.B.; Bhattacharya, B.; Mondal, R.; Sarkar, U. Nickel Cluster Functionalised Carbon Nanotube for CO Molecule Detection: A Theoretical Study. Mol. Phys. 2015, 114, 671–680. [Google Scholar] [CrossRef]
- Safina, L.R.; Krylova, K.A.; Murzaev, R.T.; Baimova, J.A.; Mulyukov, R.R. Crumpled Graphene-Storage Media for Hydrogen and Metal Nanoclusters. Materials 2021, 14, 2098. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Bhethanabotla, V.R.; Joseph, B. DFT Calculations of Hydrogen Interactions with Pd and Pd/Ni Chain Functionalized Single-Walled Carbon Nanotubes for Sensor Applications. In Proceedings of the AIChE Annual Meeting, Conference Proceedings, San Francisco, CA, USA, 12–17 November 2006. [Google Scholar]
- Zhang, Z.; Cho, K. Ab Initio Study of Hydrogen Interaction with Pure and Nitrogen-Doped Carbon Nanotubes. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 075420–075425. [Google Scholar] [CrossRef]
- Seenithurai, S.; Kodi Pandyan, R.; Vinodh Kumar, S.; Mahendran, M. H2 Adsorption in Ni and Passivated Ni Doped 4 Å Single Walled Carbon Nanotube. Int. J. Hydrogen Energy 2013, 38, 7376–7381. [Google Scholar] [CrossRef]
- Wang, B.; Jin, C.; Shao, S.; Yue, Y.; Zhang, Y.; Wang, S.; Chang, R.; Zhang, H.; Zhao, J.; Li, X. Electron-deficient Cu site catalyzed acetylene hydrochlorination. Green Energy Environ. 2022. In press. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, B.; Zhang, Y.; Li, M.; Sun, Y.; Zhao, J.; Li, X.; Zhang, H. Regulation of the liquid–solid interface of Cs catalysts for the synthesis of 1,1-Dichloroethylene from 1,1,2-Trichloroethane. Appl. Surf. Sci. 2022, 599, 154033. [Google Scholar] [CrossRef]
- Ukisu, Y.; Miyadera, T. Hydrogen-Transfer Hydrodechlorination of Polychlorinated Dibenzo-p-Dioxins and Dibenzofurans Catalyzed by Supported Palladium Catalysts. Appl. Catal. B Environ. 2003, 40, 141–149. [Google Scholar] [CrossRef]
- Dag, S.; Ozturk, Y.; Ciraci, S.; Yildirim, T. Adsorption and Dissociation of Hydrogen Molecules on Bare and Functionalized Carbon Nanotubes. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 72, 155404–155411. [Google Scholar] [CrossRef] [Green Version]
- Vuckovic, S.; Wagner, L.O.; Mirtschink, A.; Gori-Giorgi, P. Hydrogen Molecule Dissociation Curve with Functionals Based on the Strictly Correlated Regime. J. Chem. Theory Comput. 2015, 11, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.O.; Gori-Giorgi, P. Electron Avoidance: A Nonlocal Radius for Strong Correlation. Phys. Rev. A 2014, 90, 052512. [Google Scholar] [CrossRef] [Green Version]
- Peach, M.J.G.; Teale, A.M.; Tozer, D.J. Modeling the Adiabatic Connection in H2. J. Chem. Phys. 2007, 126, 244104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teale, A.M.; Coriani, S.; Helgaker, T. Accurate Calculation and Modeling of the Adiabatic Connection in Density Functional Theory. J. Chem. Phys. 2010, 132, 164115. [Google Scholar] [CrossRef]
- Grüning, M.; Gritsenko, O.V.; Baerends, E.J. Exchange-Correlation Energy and Potential as Approximate Functionals of Occupied and Virtual Kohn–Sham Orbitals: Application to Dissociating H2. J. Chem. Phys. 2003, 118, 7183–7192. [Google Scholar] [CrossRef] [Green Version]
- Gritsenko, O.V.; Braïda, B.; Baerends, E.J. Physical Interpretation and Evaluation of the Kohn–Sham and Dyson Components of the ε–I Relations between the Kohn–Sham Orbital Energies and the Ionization Potentials. J. Chem. Phys. 2003, 119, 1937–1950. [Google Scholar] [CrossRef] [Green Version]
- Baerends, E.J. Exact Exchange-Correlation Treatment of Dissociated H2 in Density Functional Theory. Phys. Rev. Lett. 2001, 87, 133004. [Google Scholar] [CrossRef] [Green Version]
- Rohr, D.R.; Toulouse, J.; Pernal, K. Combining Density-Functional Theory and Density-Matrix-Functional Theory. Phys. Rev. A At. Mol. Opt. Phys. 2010, 82, 052502. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, M.; Niquet, Y.-M.; Gonze, X.; Burke, K. Describing Static Correlation in Bond Dissociation by Kohn–Sham Density Functional Theory. J. Chem. Phys. 2005, 122, 094116. [Google Scholar] [CrossRef] [Green Version]
- Darling, G.R.; Holloway, S. H2 Dissociation Dynamics on Metals: Where Do We Stand? In The Chemical Physics of Solid Surfaces; Elsevier: Amsterdam, The Netherlands, 2003; Volume 11, pp. 27–49. [Google Scholar] [CrossRef]
- Lee, C.-Y.; DePristo, A.E. Dissociative Chemisorption of H2 on Ni Surfaces: Dependence on Incident Angles and Rovibrational States. J. Chem. Phys. 1987, 87, 1401–1404. [Google Scholar] [CrossRef]
- Yang, H.; Whitten, J.L. Chemisorption of Hydrogen on the Nickel (111) Surface. J. Chem. Phys. 1988, 89, 5329–5334. [Google Scholar] [CrossRef]
- Bourcet, A.; Tantardini, G.F. Theoretical Study of the Adsorption Dynamics of Hydrogen on Ni(111) Surface. J. Elec. Spectros. Relat. Phenom. 1994, 69, 55–64. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Depristo, A.E. Dissociative Chemisorption Dynamics of H2 on Ni and Cu Surfaces: Morphology and Surface Temperature Effects. J. Chem. Phys. 1986, 85, 4161–4171. [Google Scholar] [CrossRef]
- Kresse, G. Dissociation and Sticking of H2 on the Ni(111), (100), and (110) Substrate. Phys. Rev. B Condens. Matter Mater. Phys. 2000, 62, 8295–8305. [Google Scholar] [CrossRef]
- Nobuhara, K.; Kasai, H.; Diño, W.A.; Nakanishi, H. H2 Dissociative Adsorption on Mg, Ti, Ni, Pd and La Surfaces. Surf. Sci. 2004, 566–568, 703–707. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Cheng, C.H.; Kim, J.T.; Stanojevic, J.; Eyler, E.E. Dissociation Energies of Molecular Hydrogen and the Hydrogen Molecular Ion. Phys. Rev. Lett. 2004, 92, 203003–203006. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Dickenson, G.D.; Niu, M.L.; Salumbides, E.J.; Komasa, J.; Eikema, K.S.E.; Pachucki, K.; Ubachs, W. Fundamental Vibration of Molecular Hydrogen. Phys. Rev. Lett. 2013, 110, 193601. [Google Scholar] [CrossRef] [Green Version]
- Luniakov, Y.V. First Principle Simulations of the Surface Diffusion of Si and Me Adatoms on the Si(111) Al, Ga, In, Pb. Surf. Sci. 2011, 605, 1866–1871. [Google Scholar] [CrossRef]
- Henkelman, G.; Jonsson, H. A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B. Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Accelrys Software Inc. Materials Studio, Biovia. 2011. Available online: https://www.3ds.com/products-services/biovia (accessed on 31 July 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | PCDD Abbreviation | IUPAC Nomenclature |
|---|---|---|
| OCDD | OCDD | Octachlorodibenzodioxin |
| HpCDD | 1,2,3,4,6,7,8-HpCDD | 1,2,3,4,6,7,8-Heptachlorodibenzodioxin |
| Hx4CDD | 1,2,3,4,7,8-HxCDD | 1,2,3,4,7,8-Hexachlorodibenzodioxin |
| Hx6CDD | 1,2,3,6,7,8-HxCDD | 1,2,3,6,7,8-Hexachlorodibenzodioxin |
| Hx7CDD | 1,2,3,7,8,9-HxCDD | 1,2,3,7,8,9-Hexachlorodibenzodioxin |
| PeCDD | 1,2,3,7,8-PeCDD | 1,2,3,7,8-Pentachlorodibenzodioxin |
| Pe4CDD | 1,2,4,7,8-PeCDD | 1,2,4,7,8-Pentachlorodibenzodioxin |
| TCDD | 2,3,7,8-TCDD | 2,3,7,8-Tetrachlorodibenzodioxin |
| T1CDD | 1,3,7,8-TCDD | 1,3,7,8-Tetrachlorodibenzodioxin |
| TrCDD | 2,3,7-TrCDD | 2,3,7-Trichlorodibenzodioxin |
| Reactants | Products | dPCDD–HCl (Å) | Eads (eV) | Erxn (eV) |
|---|---|---|---|---|
| OCDD/SWCN + H-H | HpCDD/SWCN + HCl | 3.27 | −0.60 | −10.83 |
| HpCDD/SWCN + H-H | Hx4CDD/SWCN + HCl | 3.99 | −0.41 | −9.99 |
| HpCDD/SWCN + H-H | Hx6CDD/SWCN + HCl | 4.08 | −9.99 | |
| HpCDD/SWCN + H-H | Hx7CDD/SWCN + HCl | 4.07 | −9.99 | |
| Hx4CDD/SWCN + H-H | Pe4CDD/SWCN + H-Cl | 3.97 | −1.10 | −10.23 |
| Hx6CDD/SWCN + H-H | PeCDD/SWCN + H-Cl | 4.03 | −1.10 | −10.20 |
| Hx7CDD/SWCN + H-H | PeCDD/SWCN + H-Cl | 4.01 | −1.10 | −10.20 |
| PeCDD/SWCN + H-H | TCDD/SWCN + H-Cl | 3.98 | −1.05 | −10.21 |
| TCDD/SWCN + H-H | TrCDD/SWCN + H-Cl | 4.22 | −0.96 | −10.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, S.; Porras, M.; Jimbo, A.; Zambrano, C.H. Dehydrochlorination of PCDDs on SWCN-Supported Ni10 and Ni13 Clusters, a DFT Study. Molecules 2022, 27, 5074. https://doi.org/10.3390/molecules27165074
González S, Porras M, Jimbo A, Zambrano CH. Dehydrochlorination of PCDDs on SWCN-Supported Ni10 and Ni13 Clusters, a DFT Study. Molecules. 2022; 27(16):5074. https://doi.org/10.3390/molecules27165074
Chicago/Turabian StyleGonzález, Silvia, Martha Porras, Arianna Jimbo, and Cesar H. Zambrano. 2022. "Dehydrochlorination of PCDDs on SWCN-Supported Ni10 and Ni13 Clusters, a DFT Study" Molecules 27, no. 16: 5074. https://doi.org/10.3390/molecules27165074
APA StyleGonzález, S., Porras, M., Jimbo, A., & Zambrano, C. H. (2022). Dehydrochlorination of PCDDs on SWCN-Supported Ni10 and Ni13 Clusters, a DFT Study. Molecules, 27(16), 5074. https://doi.org/10.3390/molecules27165074