
Thermodynamic vs. Kinetic Control in Synthesis of O-Donor 2,5-Substituted Furan and 3,5-Substituted Pyrazole from Heteropropargyl Precursor

 , , ,
, , ,  ,
,
Abstract
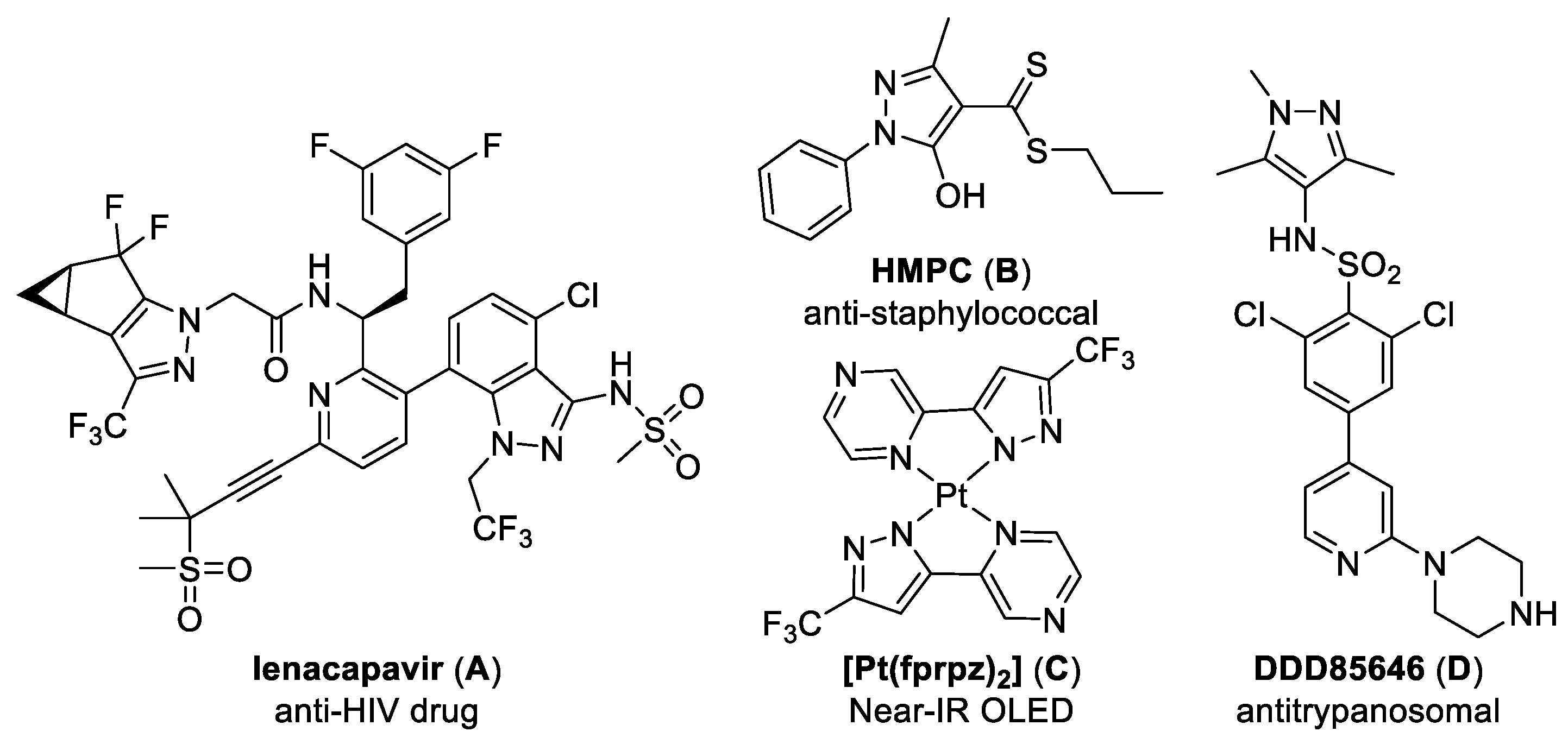
:1. Introduction
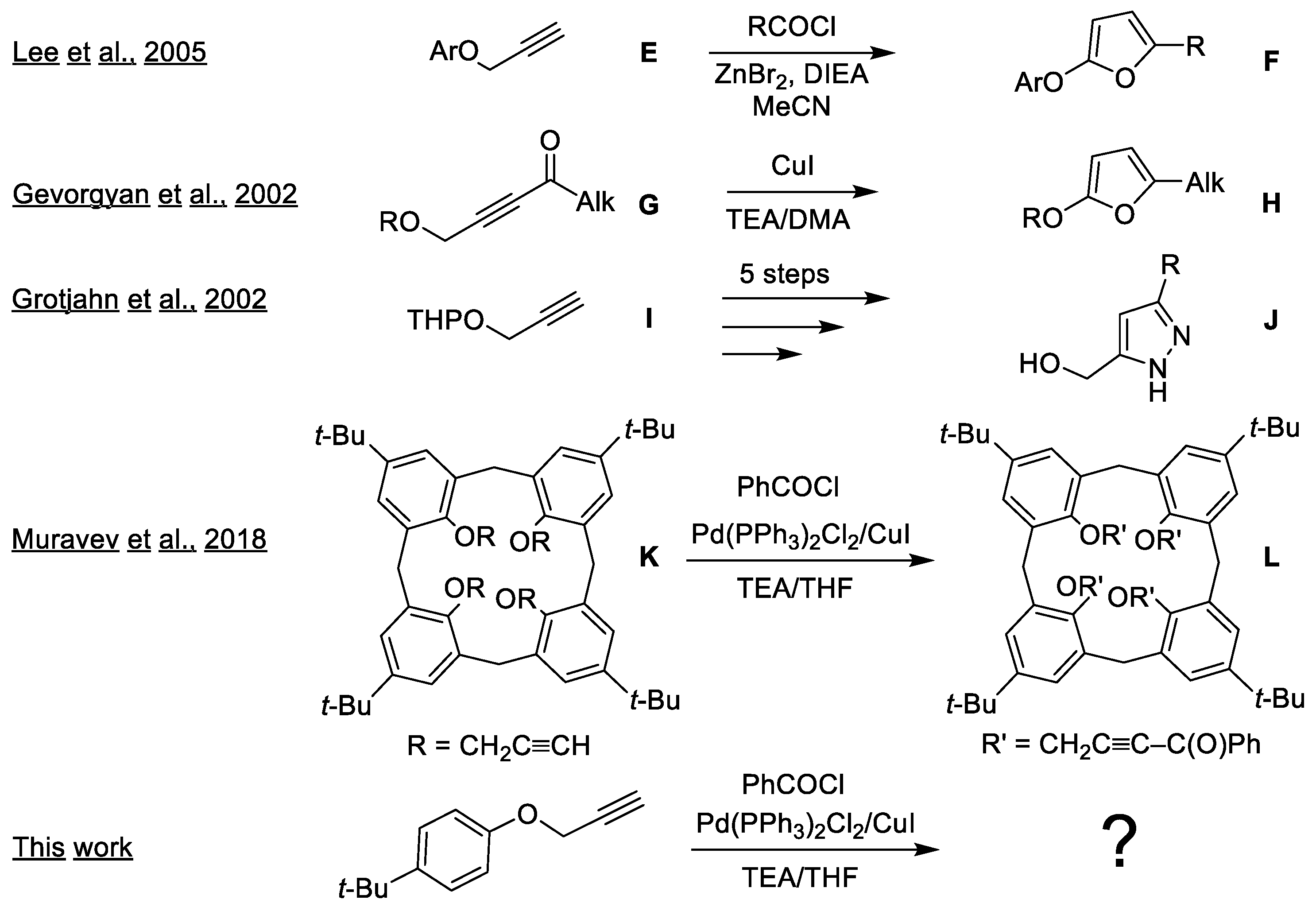
2. Results and Discussion
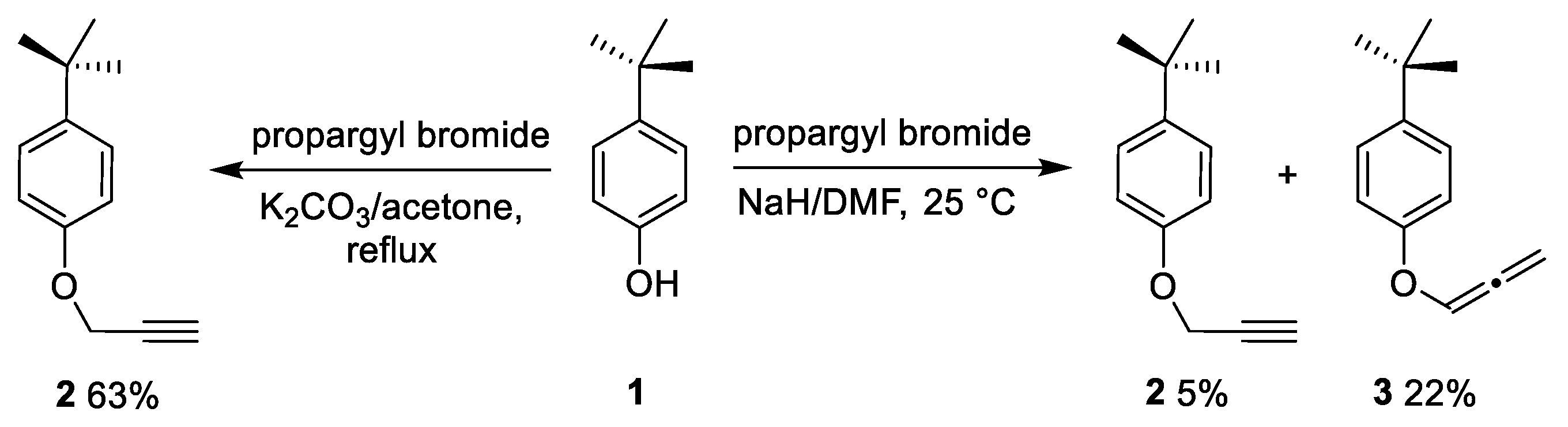
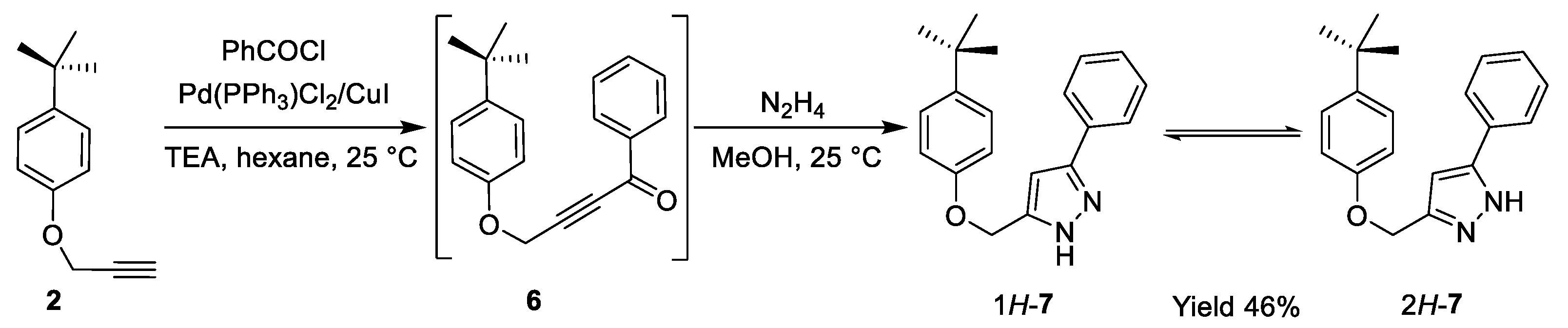
2.1. Optimization of Pyrazole and Furan Synthesis from the Heteropropargyl Precursor
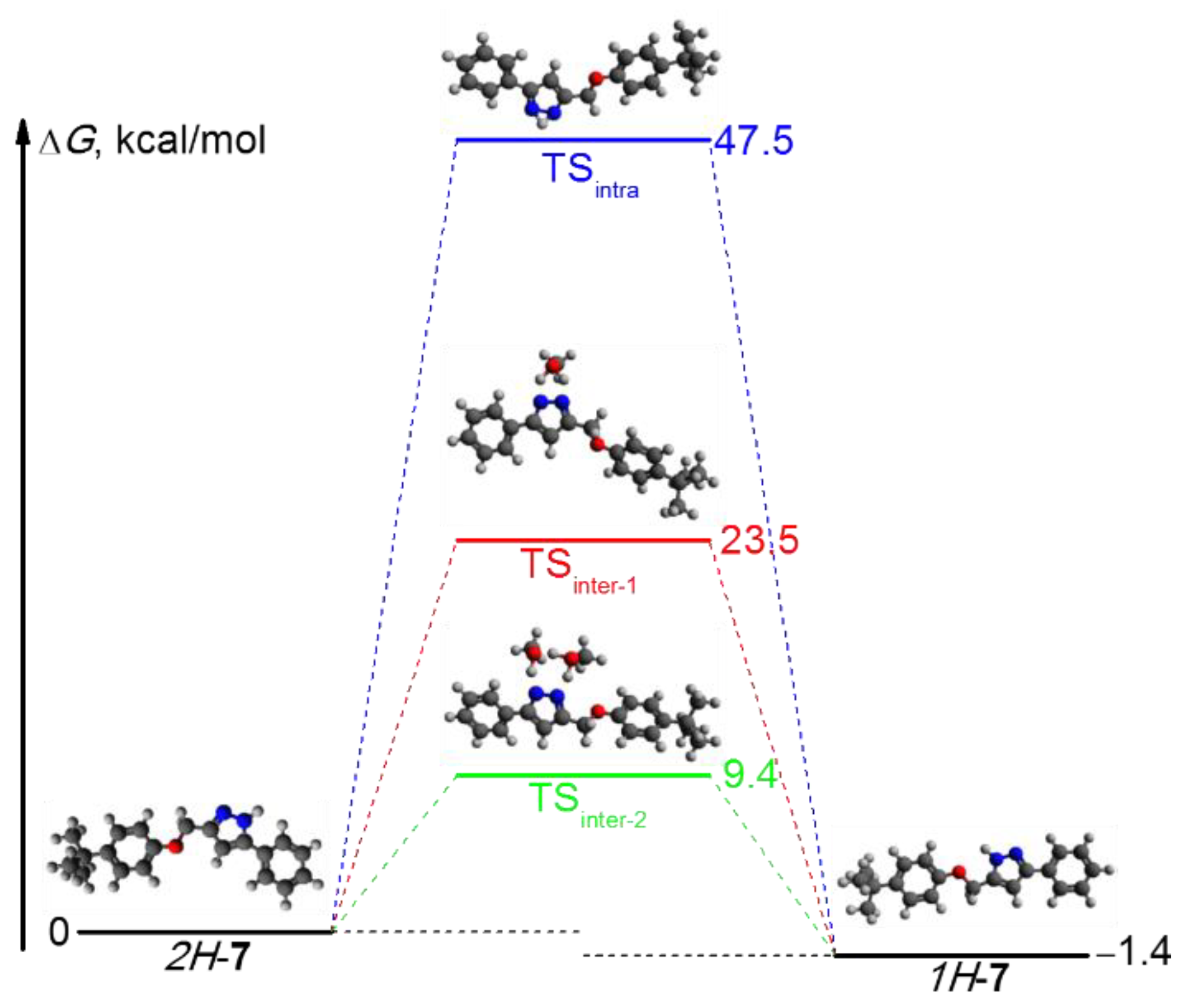
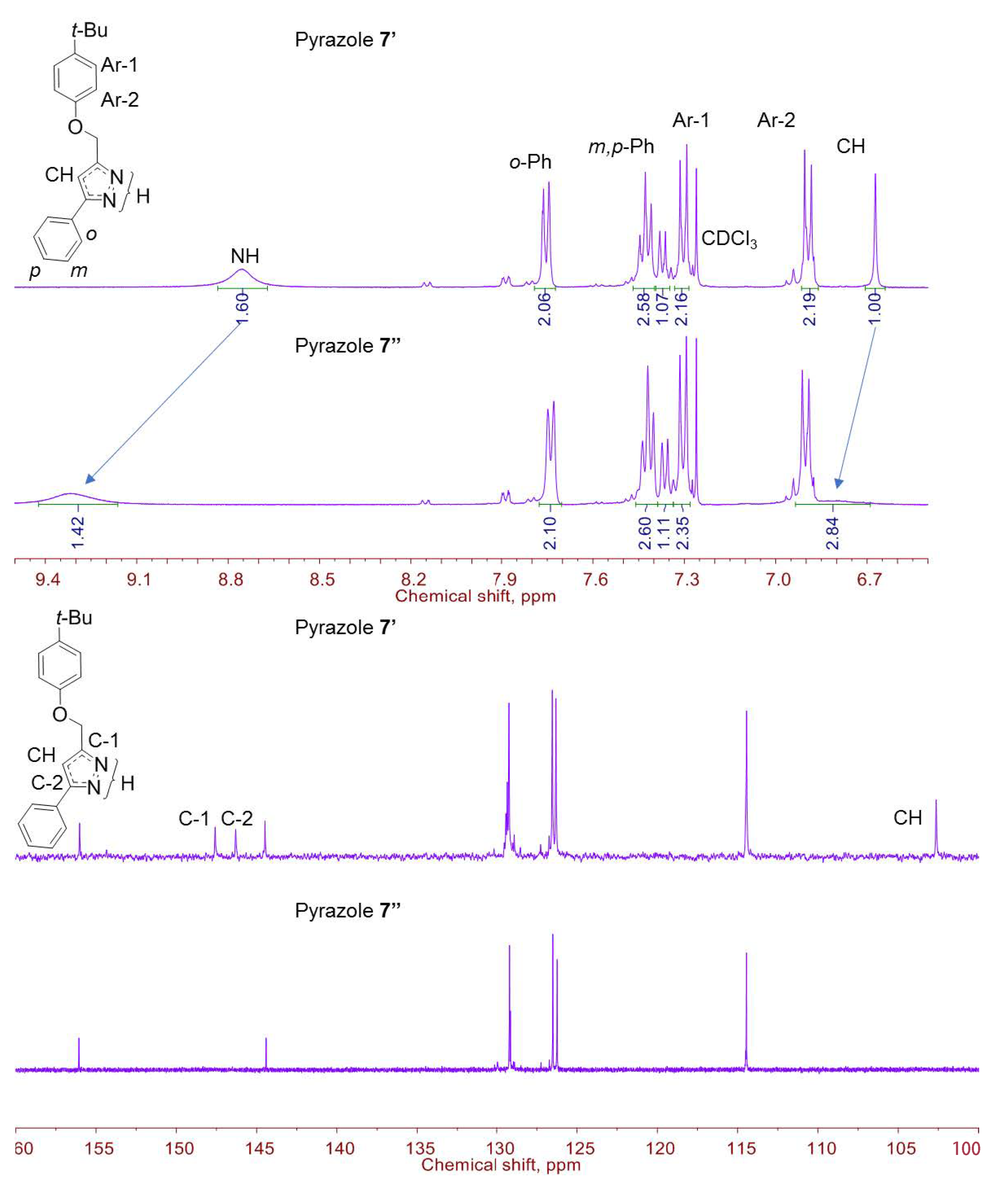
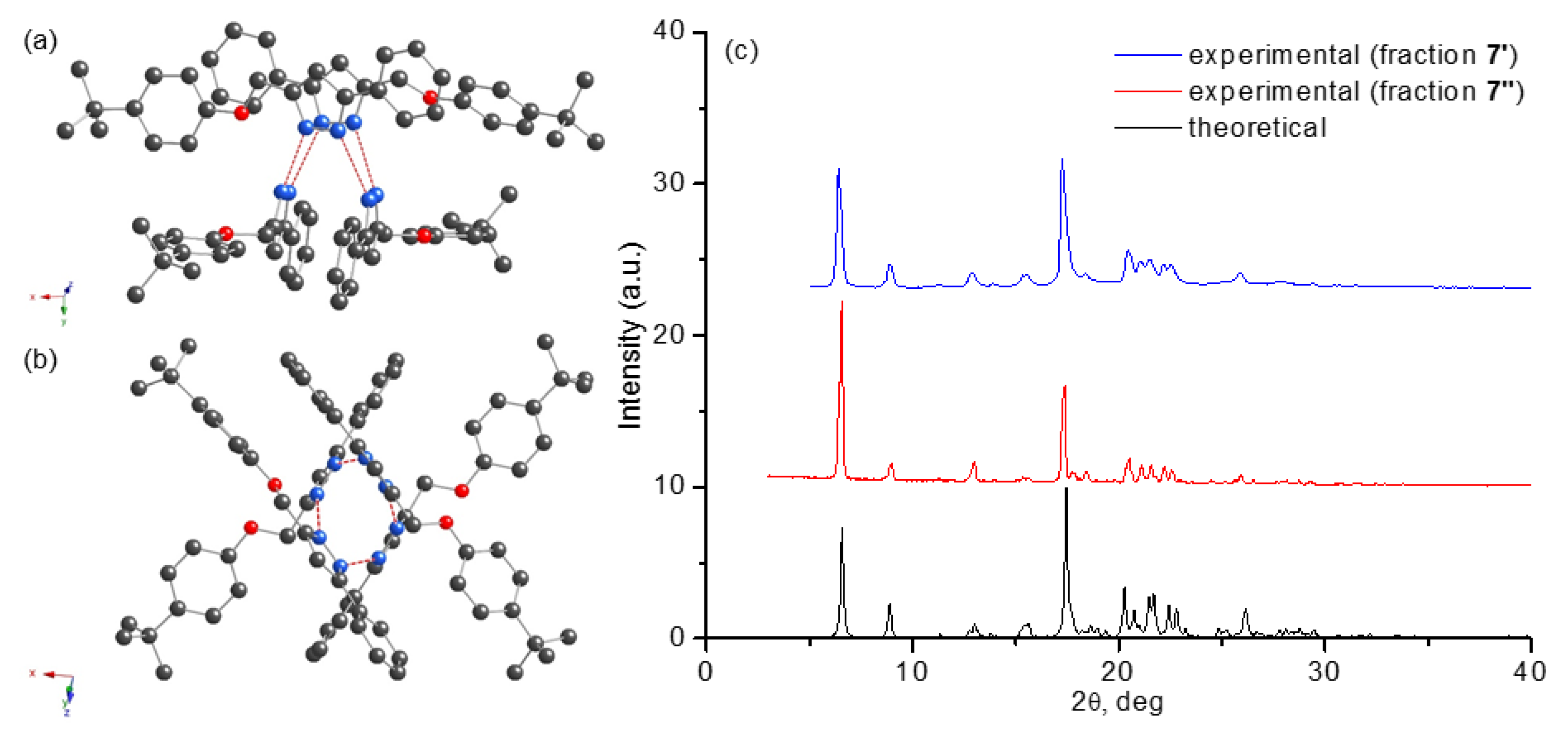
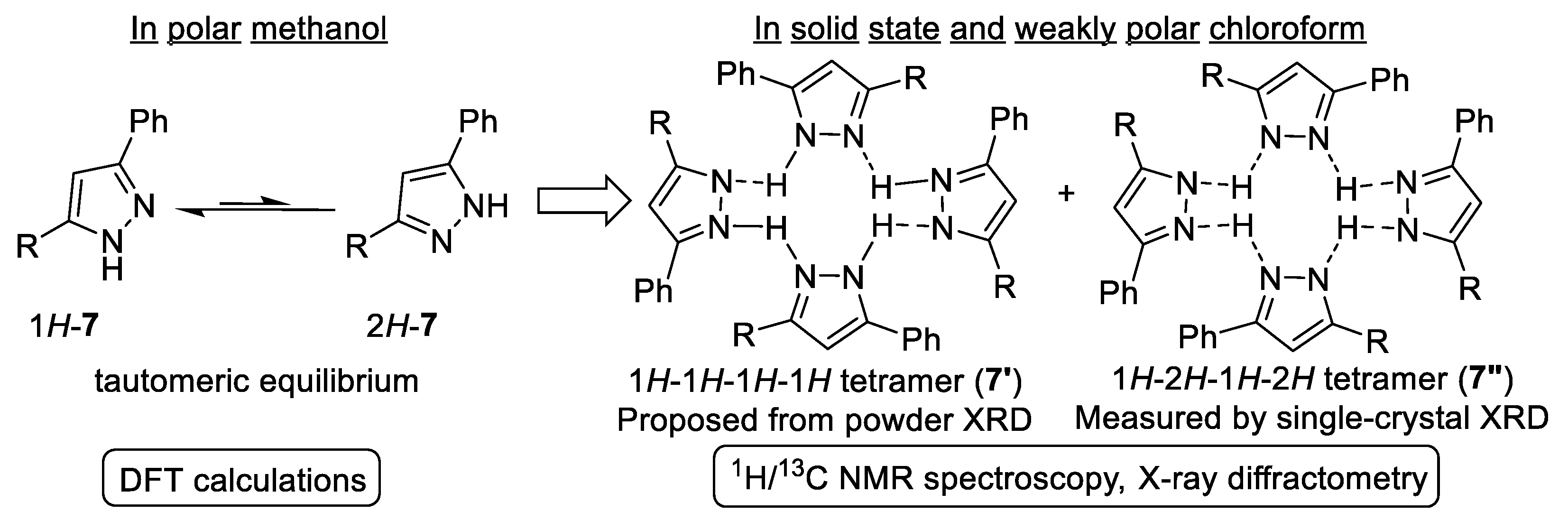
2.2. Structural Characterization of Pyrazole 7
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Johnston, T.; Van Tyne, D.; Chen, R.F.; Fawzi, N.L.; Kwon, B.; Kelso, M.J.; Gilmore, M.S.; Mylonakis, E. Propyl-5-hydroxy-3-methyl-1-phenyl-1H-pyrazole-4-carbodithioate (HMPC): A new bacteriostatic agent against methicillin-resistant Staphylococcus aureus. Sci. Rep. 2018, 8, 7062. [Google Scholar] [CrossRef]
- Frearson, J.A.; Brand, S.; McElroy, S.P.; Cleghorn, L.A.T.; Smid, O.; Stojanovski, L.; Price, H.P.; Guther, M.L.S.; Torrie, L.S.; Robinson, D.A.; et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Mykhailiuk, P.K. Fluorinated Pyrazoles: From Synthesis to Applications. Chem. Rev. 2021, 121, 1670–1715. [Google Scholar] [CrossRef] [PubMed]
- Becerra, D.; Abonia, R.; Castillo, J.-C. Recent Applications of the Multicomponent Synthesis for Bioactive Pyrazole Derivatives. Molecules 2022, 27, 4723. [Google Scholar] [CrossRef]
- Tuong Ly, K.; Chen-Cheng, R.-W.; Lin, H.-W.; Shiau, Y.-J.; Liu, S.-H.; Chou, P.-T.; Tsao, C.-S.; Huang, Y.-C.; Chi, Y. Near-infrared organic light-emitting diodes with very high external quantum efficiency and radiance. Nat. Photonics 2017, 11, 63–68. [Google Scholar] [CrossRef]
- Tigreros, A.; Portilla, J. Recent progress in chemosensors based on pyrazole derivatives. RSC Adv. 2020, 10, 19693–19712. [Google Scholar] [CrossRef]
- Budagumpi, S.; Kulkarni, N.V.; Kurdekar, G.S.; Sathisha, M.P.; Revankar, V.K. Synthesis and spectroscopy of CoII, NiII, CuII and ZnII complexes derived from 3,5-disubstituted-1H-pyrazole derivative: A special emphasis on DNA binding and cleavage studies. Eur. J. Med. Chem. 2010, 45, 455–462. [Google Scholar] [CrossRef]
- Koch, N.; Rosien, J.-R.; Mazik, M. Synthesis of compounds based on a dimesitylmethane scaffold and representative binding studies showing di- vs. monosaccharide preference. Tetrahedron 2014, 70, 8758–8767. [Google Scholar] [CrossRef]
- Koch, N.; Seichter, W.; Mazik, M. Compounds Based on a Triethyl- or Trimethoxybenzene Scaffold Bearing Pyrazole, Pyridine, and Pyrimidine Groups: Syntheses and Representative Binding Studies towards Carbohydrates. Synthesis 2016, 48, 2757–2767. [Google Scholar] [CrossRef]
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to Mid-2010: A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-T.; Kuo, S.-C.; Lin, H.-C. Solvent- and transition metal catalyst-dependent regioselectivity in the [3+2] cyclocondensation of trifluoromethyl-α,β-ynones with hydrazines: Switchable access to 3- and 5-trifluoromethylpyrazoles. Adv. Synth. Catal. 2015, 357, 683–689. [Google Scholar] [CrossRef]
- Muzalevskiy, V.M.; Rulev, A.Y.; Romanov, A.R.; Kondrashov, E.V.; Ushakov, I.A.; Chertkov, V.A.; Nenajdenko, V.G. Selective, metal-free approach to 3- or 5-CF3-pyrazoles: Solvent switchable reaction of CF3-ynones with hydrazines. J. Org. Chem. 2017, 82, 7200–7214. [Google Scholar] [CrossRef] [PubMed]
- Topchiy, M.A.; Zharkova, D.A.; Asachenko, A.F.; Muzalevskiy, V.M.; Chertkov, V.A.; Nenajdenko, V.G.; Nechaev, M.S. Mild and regioselective synthesis of 3-CF3-pyrazoles by the AgOTf-catalyzed reaction of CF3-ynones with hydrazines. Eur. J. Org. Chem. 2018, 2018, 3750–3755. [Google Scholar] [CrossRef]
- Ahmed, M.S.M.; Kobayashi, K.; Mori, A. One-Pot Construction of Pyrazoles and Isoxazoles with Palladium-Catalyzed Four-Component Coupling. Org. Lett. 2005, 7, 4487–4489. [Google Scholar] [CrossRef]
- Niedballa, J.; Müller, T.J.J. Heterocycles by consecutive multicomponent syntheses via catalytically generated alkynoyl intermediates. Catalysts 2022, 12, 90. [Google Scholar] [CrossRef]
- Kobychev, V.B.; Vitkovskaya, N.M.; Klyba, N.S.; Trofimov, B.A. Acetylene-allene rearrangement of propargyl systems X—CH2—C≡CH (X = H, Me, NMe2, OMe, F, SMe): An ab initio study. Russ. Chem. Bull. 2002, 51, 774–782. [Google Scholar] [CrossRef]
- Lee, K.Y.; Lee, M.J.; Kim, J.N. Facile synthesis of α,β-acetylenic ketones and 2,5-disubstituted furans: Consecutive activation of triple and double bond with ZnBr2 toward the synthesis of furan ring. Tetrahedron 2005, 61, 8705–8710. [Google Scholar] [CrossRef]
- Kel’in, A.V.; Gevorgyan, V. Efficient synthesis of 2-mono- and 2,5-disubstituted furans via the CuI-catalyzed cycloisomerization of alkynyl ketones. J. Org. Chem. 2002, 67, 95–98. [Google Scholar] [CrossRef]
- Dudnik, A.S.; Sromek, A.W.; Rubina, M.; Kim, J.T.; Kel’i, A.V.; Gevorgyan, V. Metal-Catalyzed 1,2-Shift of Diverse Migrating Groups in Allenyl Systems as a New Paradigm toward Densely Functionalized Heterocycles. J. Am. Chem. Soc. 2008, 130, 1440–1452. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Van, S.; Combs, D.; Lev, D.A.; Schneider, C.; Rideout, M.; Meyer, C.; Hernandez, G.; Mejorado, L. New Flexible Synthesis of Pyrazoles with Different, Functionalized Substituents at C3 and C5. J. Org. Chem. 2002, 67, 9200–9209. [Google Scholar] [CrossRef] [PubMed]
- Muravev, A.A.; Solovieva, S.E.; Galieva, F.B.; Bazanova, O.B.; Rizvanov, I.K.; Ivshin, K.A.; Kataeva, O.N.; Matthews, S.E.; Antipin, I.S. Calixarene alpha-ketoacetylenes: Versatile platforms for reaction with hydrazine nucleophile. RSC Adv. 2018, 8, 32765–32769. [Google Scholar] [CrossRef] [PubMed]
- Solovieva, S.; Muravev, A.; Galieva, F.; Agarkov, A.; Sapunova, A.; Voloshina, A.; Matthews, S.; Antipin, I. Calix[4]arene stereoisomers with pyrazole pharmacophoric groups: Potential cervical carcinoma therapy. Eur. J. Clin. Investig. 2019, 49, 169. [Google Scholar]
- Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A.S.K. An Industrial Perspective on Counter Anions in Gold Catalysis: Underestimated with Respect to “Ligand Effects”. Adv. Synth. Catal. 2018, 360, 2493–2502. [Google Scholar] [CrossRef]
- Wu, Y.; Pan, M.; Dai, Y.; Liu, B.; Cui, J.; Shi, W.; Qiu, Q.; Huang, W.; Qian, H. Design, synthesis and biological evaluation of LBM-A5 derivatives as potent P-glycoprotein-mediated multidrug resistance inhibitors. Bioorg. Med. Chem. 2016, 24, 2287–2297. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Z.-Q.; Zhang, Y.-P.; Liang, T.; Xu, Y. Crystal structure of di-(μ-iodo)bis[(triphenylphosphine) iodopalladium(II)], Pd2I4(PC18H15)2. Z. Kristallogr. 2010, 225, 273–274. [Google Scholar] [CrossRef]
- Mo, J.; Choi, W.; Min, J.; Kim, C.-E.; Eom, D.; Kim, S.H.; Lee, P.H. ICl-Mediated Intramolecular Twofold Iodoarylation of Diynes and Diynyl Diethers and Amines: Synthesis of Bis(2H-hydronaphthalene and chromene) and 2H-Quinoline Bearing an Alkenyl Iodide Moiety. J. Org. Chem. 2013, 78, 11382–11388. [Google Scholar] [CrossRef]
- Fakhrutdinov, A.N.; Karlinskii, B.Y.; Minyaev, M.E.; Ananikov, V.P. Unusual effect of impurities on the spectral characterization of 1,2,3-triazoles synthesized by the Cu-catalyzed azide–alkyne click reaction. J. Org. Chem. 2021, 86, 11456–11463. [Google Scholar] [CrossRef]
- Claramunt, R.M.; Cornago, P.; Torres, V.; Pinilla, E.; Torres, M.R.; Samat, A.; Lokshin, V.; Valés, M.; Elguero, J. The structure of pyrazoles in the solid state: A combined CPMAS, NMR, and crystallographic study. J. Org. Chem. 2006, 71, 6881–6891. [Google Scholar] [CrossRef]
- Secrieru, A.; O’Neill, P.M.; Cristiano, M.L.S. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules 2020, 25, 42. [Google Scholar] [CrossRef]
- Moore, F.H.; White, A.H.; Willis, A.C. 3-Methyl-5-phenylpyrazole: A neutron diffraction study. J. Chem. Soc. Perkin Trans. 2 1975, 10, 1068–1071. [Google Scholar] [CrossRef]
- Claramunt, R.M.; Cornago, P.; Santa María, M.D.; Torres, V.; Pinilla, E.; Torres, M.R.; Elguero, J. The Structure of a Non-Symmetric Disordered Tetramer: A Crystallographic and Solid State Multinuclear NMR Study of the Properties of 3(5)-Ethyl-5(3)-Phenyl-1H-Pyrazole. Supramol. Chem. 2006, 18, 349–356. [Google Scholar] [CrossRef]
- Armarego, W.L.F. Purification of Laboratory Chemicals, 8th ed.; Butterworth-Heinemann: Oxford, UK, 2017. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- ChemCraft. Available online: http://www.chemcraftprog.com (accessed on 18 June 2022).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Pd/Cu (mol %) | Solvent | t (h) | TEA (mol %) | Percentage of Unreacted Alkyne and Coupling Products in the Reaction Mixture 1 | |||
|---|---|---|---|---|---|---|---|---|
| Alkyne 2 | Diyne 4 | Furan 5 | Ynone 6 | |||||
| 1 | 1%/2% | THF | 12 | 440 | 94 | 6 | – | – |
| 2 | 2%/4% | THF | 12 | 440 | – | 2 | 98 | – |
| 3 | 4%/8% | THF | 12 | 440 | – | 3 | 97 | – |
| 4 | 2%/4% | CH2Cl2 | 12 | 440 | – | 5 | 85 | 10 |
| 5 | 2%/4% | TEA | 12 | 5000 | 14 | 16 | 50 | 20 |
| 6 | 2%/4% | PhCH3 | 12 | 440 | – | 7 | 15 | 78 |
| 7 | 2%/4% | C6H14 | 12 | 440 | – | 3 | 14 | 83 |
| 8 | 2%/4% | C6H14 | 72 | 440 | – | 5 | 95 | – |
| 9 | 2%/4% | C6H14 | 3 | 440 | 36 | 8 | – | 56 |
| 10 | 2%/4% | THF | 3 | 440 | – | 5 | 66 | 29 |
| 11 | 2%/4% | C6H14 | 12 | 120 | 51 | 3 | – | 46 |
| 12 | 2%/4% | THF | 12 | 120 | – | 20 | – | 80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muravev, A.A.; Ovsyannikov, A.S.; Konorov, G.V.; Islamov, D.R.; Usachev, K.S.; Novikov, A.S.; Solovieva, S.E.; Antipin, I.S. Thermodynamic vs. Kinetic Control in Synthesis of O-Donor 2,5-Substituted Furan and 3,5-Substituted Pyrazole from Heteropropargyl Precursor. Molecules 2022, 27, 5178. https://doi.org/10.3390/molecules27165178
Muravev AA, Ovsyannikov AS, Konorov GV, Islamov DR, Usachev KS, Novikov AS, Solovieva SE, Antipin IS. Thermodynamic vs. Kinetic Control in Synthesis of O-Donor 2,5-Substituted Furan and 3,5-Substituted Pyrazole from Heteropropargyl Precursor. Molecules. 2022; 27(16):5178. https://doi.org/10.3390/molecules27165178
Chicago/Turabian StyleMuravev, Anton A., Alexander S. Ovsyannikov, Gennady V. Konorov, Daut R. Islamov, Konstantin S. Usachev, Alexander S. Novikov, Svetlana E. Solovieva, and Igor S. Antipin. 2022. "Thermodynamic vs. Kinetic Control in Synthesis of O-Donor 2,5-Substituted Furan and 3,5-Substituted Pyrazole from Heteropropargyl Precursor" Molecules 27, no. 16: 5178. https://doi.org/10.3390/molecules27165178
APA StyleMuravev, A. A., Ovsyannikov, A. S., Konorov, G. V., Islamov, D. R., Usachev, K. S., Novikov, A. S., Solovieva, S. E., & Antipin, I. S. (2022). Thermodynamic vs. Kinetic Control in Synthesis of O-Donor 2,5-Substituted Furan and 3,5-Substituted Pyrazole from Heteropropargyl Precursor. Molecules, 27(16), 5178. https://doi.org/10.3390/molecules27165178