Abstract
In this paper, we report an efficient synthetic route for the 23,23-difluoro-25-hydroxyvitamin D3 (5) and its 24-hydroxylated analogues (7,8), which are candidates for the CYP24A1 main metabolites of 5. The key fragments, 23,23-difluoro-CD-ring precursors (9–11), were synthesized starting from Inhoffen-Lythgoe diol (12), and introduction of the C23 difluoro unit to α-ketoester (19) was achieved using N,N-diethylaminosulfur trifluoride (DAST). Preliminary biological evaluation revealed that 23,23-F2-25(OH)D3 (5) showed approximately eight times higher resistance to CYP24A1 metabolism and 12 times lower VDR-binding affinity than its nonfluorinated counterpart 25(OH)D3 (1).
1. Introduction
The introduction of fluorine atom(s) into biologically active compounds has been widely used in the development of pharmaceuticals, with the expected effects of increasing metabolic stability and improving binding affinity to target proteins [1,2,3,4]. Vitamin D3 is no exception. Fluorinated vitamin D3 analogues have been synthesized to extend the biological half-life and modulate the binding affinity to the vitamin D receptor (VDR), and their biological activities have been evaluated [5]. Among them, the side-chain fluorination of vitamin D3 has been vigorously pursued because hydroxylation of the side-chain C23 or C24 position and several subsequent oxidation steps by the metabolic enzyme CYP24A1 are the main deactivation processes of 25-hydroxyvitamin D3 [25(OH)D3] (1) (Scheme 1) [6,7,8]. Falecalcitriol (2), which has been clinically approved for the treatment of secondary hyperparathyroidism [9,10,11], is one such vitamin D3 analogue, and it contains a hexafluoroisopropanol unit in the side chain (Figure 1).
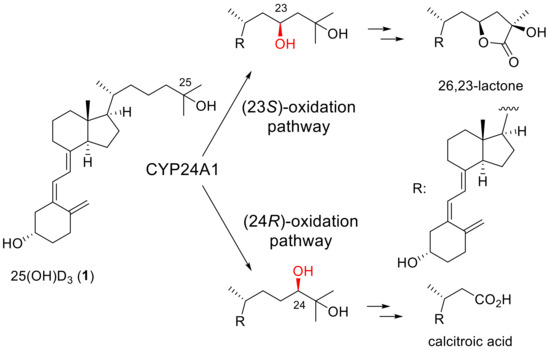
Scheme 1.
Main metabolic pathways of 25(OH)D3 (1) catalyzed by human CYP24A1.
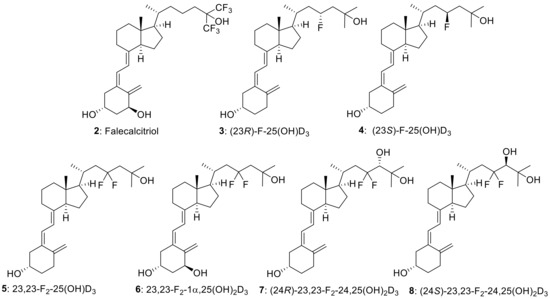
Figure 1.
Structures of Falecalcitriol (2) and C23-fluorinated vitamin D3 analogues (3–8).
In recent years, new aspects of vitamin D3 function have been discovered [12,13,14,15], and syntheses of various vitamin D3 analogues have been carried out [16,17]. As a result, the importance of developing comprehensive and straightforward synthetic methods for fluorinated vitamin D3 analogues has increased. However, the synthetic methods reported so far are limited and mainly use sterol skeletons as starting materials, followed by photochemical transformation and thermal isomerization (Scheme 2) [5]. This strategy leads to a limited number of vitamin D derivatives even after multi-step synthesis with low chemical yields.
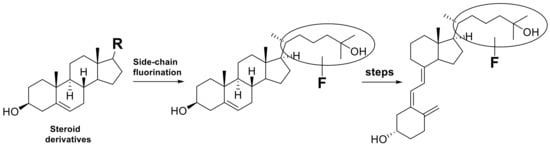
Scheme 2.
Synthetic strategy for side-chain fluorinated vitamin D3 analogues starting from sterols.
During our ongoing vitamin D3 research, we focused on the vitamin D3 side-chain C23 position and explored the efficient synthetic methodology for C23 fluorinated vitamin D3 analogues, in which we achieved the convergent stereoselective synthesis of (23R)-23-fluoro-25-hydroxyvitamin D3 [(23R)-F-25(OH)D3] (3) and its 23S isomer (4) through the corresponding CD-ring parts [18]. This time, we established an efficient synthetic route to 23,23-difluorovitamin D3 analogues. To the best of our knowledge, two 23,23-difluorovitamin D3 analogues: 23,23-difluoro-25-hydroxyvitamin D3 [23,23-F2-25(OH)D3] (5) and 23,23-difluoro-1α,25-dihydroxyvitamin D3 [23,23-F2-1α,25(OH)2D3] (6), were synthesized by Taguchi et al. in 1984 [19] and Nakada et al. in 1985 [20]. The synthetic route to 5 and 6 utilized the same strategy summarized in Scheme 2. They used a sterol-based compound as the starting material. After introducing the 23,23-difluoro unit into the side-chain, the B-ring was opened by photoirradiation, followed by thermal isomerization to afford 5. They also prepared 6 by enzymatic 1α-hydroxylation of 5 [20].
In this study, we herein report an alternative and efficient synthetic route to 23,23-difluorovitamin D3 analogues using a convergent method. First, 23,23-difluoro-CD-ring precursor (9) was prepared as a key intermediate in the synthesis of 5. Next, we synthesized the 23,23-difluoro-24-hydroxy-CD-ring fragments (10,11) as substrates for 23,23-difluoro-24,25(OH)2D3 (7,8) as the possible CYP24A1 metabolites of 23,23-F2-25(OH)D3 (5) (Figure 2).
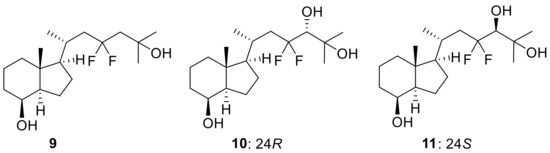
Figure 2.
Structures of key CD-ring fragments (9–11) for the convergent method.
2. Results and Discussion
The retrosynthetic analysis is shown in Scheme 3. The CD-rings (9–11) were synthesized from Inhoffen-Lythgoe diol (12), and the introduction of the difluoro unit to the C23 position was performed using a difluorination reaction of 19 with DAST [21,22,23]. Stereoselective introduction of the hydroxy group to the C24 position was achieved using Sharpless asymmetric dihydroxylation of 28 [24,25]. The A-ring phosphine oxide (14) was prepared from vitamin D3 [26].
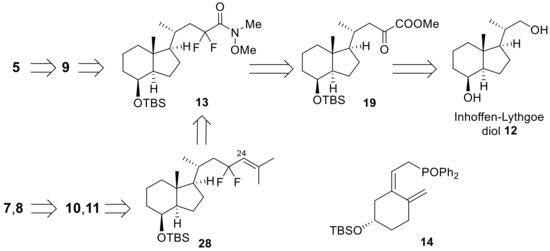
Scheme 3.
Retrosynthetic analysis of C23-fluorinated vitamin D3 analogues (5,7,8) using the convergent method.
In Scheme 4 and Scheme 5, the synthesis of the 23,23-difluoro-CD-ring moiety (9) is described. Selective protection of the C8 secondary hydroxy group of 12 with a TBS group, followed by oxidation at the C22 primary alcohol by TPAP/NMO, afforded aldehyde (16) [27]. The aldehyde was subjected to the Horner-Emmons reagent (17) under basic conditions to create triethylsilyl enol ether (18), which was converted to α-ketoester (19) by selective desilylation of the silyl enol ether unit using TBAF in the presence of acetic acid [23]. The C23-difluoro unit was constructed using DAST toward α-ketoester (19), and the obtained difluoro methyl ester (20) was converted to a Weinreb amide (13) (Scheme 4).
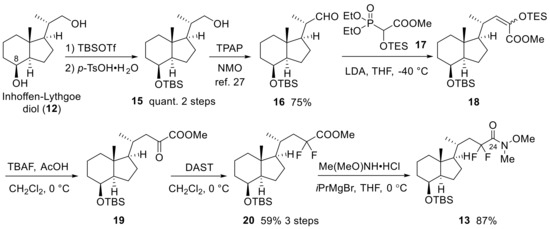
Scheme 4.
Preparation of 23,23-difluoro-24-amide-CD-ring (13) from Inhoffen-Lythgoe diol (12).
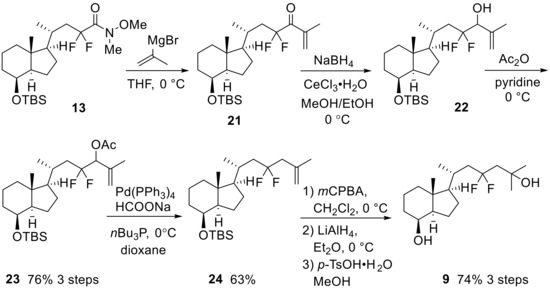
Scheme 5.
Preparation of 23,23-difluoro-CD-ring moiety (9) from 13.
Elongation of the side-chain progressed using isopropenyl Grignard reagent with Weinreb amide (13), and subsequent reduction of the obtained ketone (21) provided alcohol (22). After acetylation of 22, a palladium-catalyzed regio-selective hydride reduction [28] yielded 25,26-alkene (24). Finally, epoxidation of the alkene moiety and reductive opening of the epoxide using LiAlH4, followed by desilylation at C8-OH afforded 9 (Scheme 5).
Next, we developed the synthetic route to two 23,23-difluoro-24-hydroxy-CD-ring moieties (10,11). As shown in Scheme 6, the synthesis started from Weinreb amide 13, which was reacted with isopropyl magnesium bromide to provide ketone (25), followed by reduction with NaBH4 to afford alcohol (26). After trifluoromethanesulfonation of the alcohol, an E2 reaction under basic conditions afforded 24,25-olefin (28). Stereoselective hydroxylation was performed using Sharpless asymmetric dihydroxylation [24,25], followed by desilylation at C8-OH to develop the desired 8,24,25-trihydroxy-CD-rings (10,11). Later, the stereochemistry at the C24 position of 10 was determined to be the same as in compound 7 by X-ray crystallography (Figure 3).
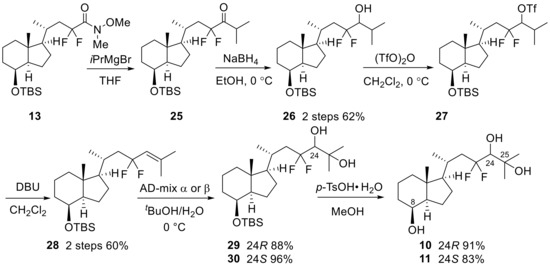
Scheme 6.
Stereoselective synthesis of 23,23-difluoro-24-hydroxy-CD-ring moieties (10,11).
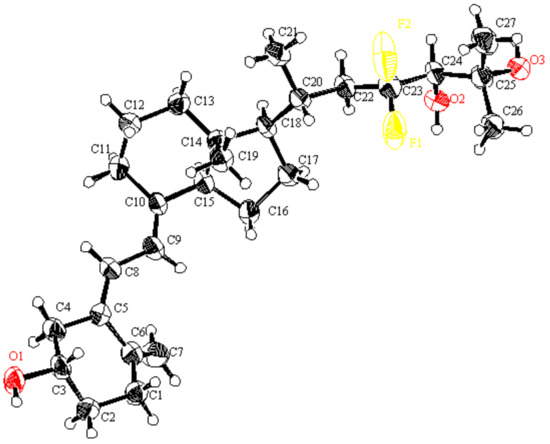
Figure 3.
ORTEP drawings of 7; Ellipsoid contour probability level = 50% (CCDC 2202393).
The triene structures were constructed using 8-keto-CD-rings (32,35,36) and the A-ring (14). The 8-keto-CD-ring (32) was synthesized via the oxidation of 9 and subsequent protection of the C25-OH group with TES. For the synthesis of the 8-keto-CD-rings (35,36), both C24 and C25 hydroxy groups of 10 and 11 were protected with p-methoxybenzylidene acetal that provided inseparable diastereomeric mixtures, respectively, followed by oxidation of the C8-OH group using TPAP/NMO to generate 35 and 36 (Scheme 7). The coupling reaction between the 8-keto-CD-rings (32,35,36) and A-ring phosphine oxide (14) was performed using the Wittig–Horner reaction, followed by deprotection to yield the desired products (5,7,8) (Scheme 8).
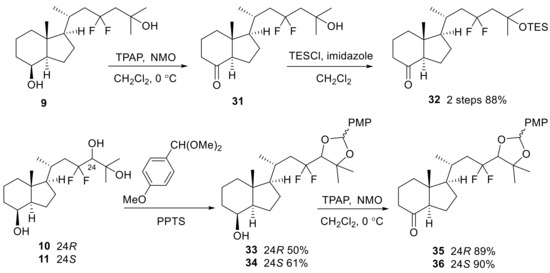
Scheme 7.
Synthesis of 23,23-difluoro-8-keto-CD-rings (32,35,36).
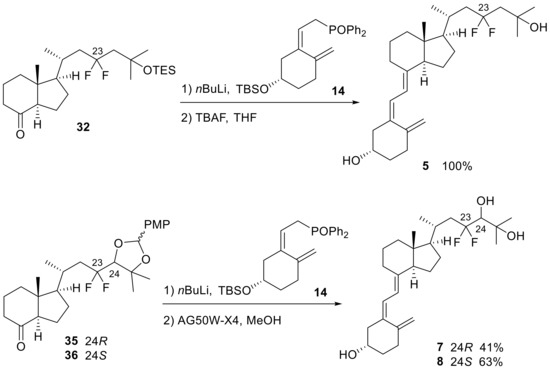
Scheme 8.
The Wittig−Horner coupling reaction between 8-keto-CD-rings (32,35,36) and the A-ring (14) and subsequent deprotection steps for 5, 7, and 8.
The structure of 7, including stereochemistry at the C24 position, was clarified with X-ray crystallography (Figure 3).
Biological Evaluation
The metabolism of 23,23-F2-25(OH)D3 (5) by hCYP24A1 and the binding affinity for hVDR were evaluated (Table 1 and Table 2). 23,23-F2-25(OH)D3 (5) showed eight times higher resistance to CYP24A1 metabolism than its nonfluorinated counterpart 25(OH)D3 (1). We previously reported that 24,24-difluoro-25-hydroxyvitamin D3 showed nearly the same metabolic resistance toward CYP24A1 [23,25].

Table 1.
Hydroxylation activities of human CYP24A1 toward 25(OH)D3 (1) and 23,23-F2-25(OH)D3 (5).
Table 2.
Relative hVDR binding affinity of 23,23-F2-25(OH)D3 (5).
For hVDR binding affinity, Ikekawa and coworkers reported that 23,23-F2-1α,25(OH)2D3 (6) was seven times less active than the natural hormone 1α,25(OH)2D3 [20]. In our experiments, 23,23-F2-25(OH)D3 (5) possessed approximately 12 times lower binding affinity than 25(OH)D3 (1) (Table 2). On the contrary, 24,24-difluoro-25-hydroxyvitamin D3 showed 1.8-times higher binding affinity than 25(OH)D3 (1) [23,25]. These results suggest that the fluorine atoms at the C23 position markedly impair the binding affinity to hVDR.
3. Conclusions
In summary, we developed a novel and efficient synthetic route to 23,23-difluoro-25-hydroxyvitamin D3 analogues (5,7,8) via the key fragments 23,23-difluoro-CD-rings (9–11). The 23,23-difluoro unit was constructed using DAST toward α-ketoester (19). Preliminary biological evaluation revealed that 23,23-F2-25(OH)D3 (5) exhibits higher resistance against CYP24A1 metabolism and lower binding affinity for hVDR than 25(OH)D3 (1).
4. Experimental Section
1H and 13C-NMR spectra were recorded on JEOL AL-400 NMR (400 MHz) and ECP-600 NMR (600 MHz) spectrometers (Tokyo, Japan). 1H-NMR spectra were referenced with (CH3)4Si (δ 0.00 ppm) or CHCl3 (δ 7.26 ppm) as an internal standard. 13C-NMR spectra were referenced with deuterated solvent (δ 77.0 ppm for CDCl3). IR spectra were recorded on a JASCO FT-IR-800 Fourier transform infrared spectrophotometer (Tokyo, Japan). High-resolution mass spectra were obtained on a SHIMADZU LCMS-IT-TOF mass spectrometer (Kyoto, Japan) with an electrospray ionization (ESI) method or atmospheric pressure chemical ionization (APCI). Optical rotations were measured on a JASCO DIP-370 digital polarimeter (Tokyo, Japan). Column chromatography was performed on silica gel 60N (Kanto Chemical Co., Inc., 40–50 μm, Tokyo, Japan) or silica gel 60 (Merck, 0.040–0.063 mm, Tokyo, Japan). All experiments were performed under anhydrous conditions under an atmosphere of argon unless otherwise stated. The supporting information of 1H and 13C NMR spectra of all new compounds: 5, 7–11, 13, 20, 23, 24, 26, 28–30, and 32–36, as well as 19F NMR spectra of compounds: 5, 7, and 8 is available at the link in Supplementary Materials.
- Methyl (R)-4-{(1R,3aR,4S,7aR)-4-[(tert-butyldimethylsilyl)oxy]-7a-methyloctahydro-1H-inden-1-yl}-2,2-difluoropentanoate (20)
To the solution of Horner–Emmons reagent (17) (2.35 g, 7.24 mmol) in THF (3.5 mL), LDA (lithium diisopropylamide) (3.5 mL, 2 M THF/heptane/ethylbenzene solution, 1.75 mmol) was added at −40 °C; the mixture was stirred at the same temperature for 20 min, and a solution of 16 [27] (977.0 mg, 3.01 mmol) in THF (3.5 mL) was added. The reaction mixture was stirred at 0 °C for 20 min. After the reaction was quenched with H2O at 0 °C, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude residue was roughly purified by flash column chromatography on silica gel (hexanen:EtOAc = 3:1, 1% Et3N) to obtain the crude coupling product 18, and it was used for the next reaction without further purification.
To the above coupling product 18 in CH2Cl2 (7 mL), AcOH (1.03 mL) and tetrabutylammonium fluoride (4.8 mL, 1 M THF solution, 4.8 mmol) were added at 0 °C under air, and the mixture was stirred at room temperature for 15 min. After the reaction was quenched with H2O at room temperature, the mixture was extracted with CH2Cl2 three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was partially purified by flash column chromatography on silica gel (hexane:EtOAc = 10:1) to obtain a crude residue of 19 (1.10 g).
To the solution of the above crude residue of 19 (1.10 g), CH2Cl2 (6.5 mL) was slowly added to N,N-diethylaminosulfur trifluoride (DAST) (2.8 mL, 3.4 g, 21.1 mmol) at 0 °C, and the mixture was stirred at room temperature for 16 h. The mixture was cooled to 0 °C, and MeOH and H2O were slowly added. The mixture was extracted with CH2Cl2 three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was partially purified by flash column chromatography on silica gel (hexane:EtOAc = 10:1) to obtain 20 (747.0 mg, 59%, three steps) as a colorless oil.
20: +36.5 (c 1.90, CHCl3); IR (neat) 1774, 1471, 1253, 1097, 1077, 1029, 838 cm−1; 1H-NMR (600 MHz, CDCl3) δ −0.01 (s, 3H), 0.01 (s, 3H), 0.88 (s, 9H), 0.90 (d, J = 5.4 Hz, 3H), 0.91 (s, 3H), 1.03 (q, J = 9.6 Hz, 1H), 1.10 (td, J = 3.6, 13.2 Hz, 1H), 1.17–1.27 (m, 2H), 1.31–1.38 (m, 2H), 1.43–1.49 (m, 1H), 1.51–1.59 (m, 2H), 1.65–1.67 (m, 1H), 1.74–1.84 (m, 2H), 1.88–1.98 (m, 2H), 2.04–2.16 (m, 1H), 3.86 (s, 3H), 3.98–4.00 (m, 1H); 13C-NMR (150 MHz, CDCl3) δ −5.2, −4.8, 13.7, 17.6, 18.0, 18.3, 23.0, 25.8, 27.0, 27.1, 31.1 (t, J = 22.3 Hz), 34.4, 34.5, 40.6, 42.1, 53.0, 53.2, 56.0, 69.4, 116.8 (t, J = 248.5 Hz), 165.0 (t, J = 33.8 Hz); HRMS (ESI−) calcd for C22H39O3F2Si [M − H]− 417.2642, found 417.2662.
- (R)-4-{(1R,3aR,4S,7aR)-4-[(tert-Butyldimethylsilyl)oxy]-7a-methyloctahydro-1H-inden-1-yl}-2,2-difluoro-N-methoxy-N-methylpentanamide (13)
To the solution of compound 20 (1.01 g, 2.41 mmol) and Me(MeO)NH·HCl (934.3 mg, 9.58 mmol) in THF (20 mL), isopropyl magnesium chloride (19.0 mL, 1 M in THF, 19.0 mmol) was added at 0 °C, and the mixture was stirred at the same temperature for 23 h. After the reaction was quenched with water and aqueous saturated NH4Cl, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 6:1) to obtain 13 (933.9 mg, 87%) as a colorless oil.
13: +39.9 (c 3.47, CHCl3); IR (neat) 1685, 1464, 1379, 1252, 1085, 1039, 984, 837 cm−1; 1H-NMR (600 MHz, CDCl3) δ −0.02 (s, 3H), 0.00 (s, 3H), 0.88 (s, 9H), 0.92 (s, 3H), 1.02 (d, J = 6.6 Hz, 3H), 1.06–1.13 (m, 2H), 1.21–1.26 (m, 2H), 1.30–1.37 (m, 3H), 1.51–1.59 (m, 1H), 1.64–1.66 (m, 1H), 1.74–1.87 (m, 4H), 1.94–1.96 (m, 1H), 2.22–2.32 (m, 1H) 3.24 (brs, 3H), 3.72 (s, 3H), 3.98–3.99 (m, 1H); 13C-NMR (150 MHz, CDCl3) δ −5.2, −4.8, 13.5, 17.6, 18.0, 20.2, 23.0, 25.8, 27.4, 30.6, 33.1, 34.3, 40.1 (t, J = 20.8 Hz), 40.6, 42.2, 53.1, 56.9, 61.9, 69.4, 118.6 (t, J = 249.2 Hz), 164.7 (t, J = 27.9 Hz); HRMS (ESI+) calcd for C23H43NO3F2SiNa [M + Na]+ 470.2872, found 470.2856.
- (6R)-6-{(1R,3aR,4S,7aR)-4-[(tert-Butyldimethylsilyl)oxy]-7a-methyloctahydro-1H-inden-1-yl}-4,4-difluoro-2-methylhept-1-en-3-yl acetate (23)
To the solution of compound 13 (251.5 mg, 0.56 mmol) in THF (20 mL), isopropenyl magnesium bromide (2.25 mL, 0.5 M in THF, 1.12 mmol) was added at 0 °C, and the mixture was stirred at room temperature for 1 h. After the reaction was quenched with water, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 20:1) to obtain the crude product 21.
NaBH4 (45.1 mg, 1.19 mmol) was added to the solution of the above crude 21 and CeCl3·6H2O in EtOH (3 mL) and MeOH (3 mL) at 0 °C, and the mixture was stirred at the same temperature for 5 min. After the reaction was quenched with water, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude 22 was used for the next reaction without further purification.
N,N-Dimethyl-4-aminopyridine (464.5 mg, 3.68 mmol) and Ac2O (1.85 g, 2 mL, 18.1 mmol) were added to the solution of the crude 22 in pyridine (4 mL) at 0 °C, and the mixture was stirred at room temperature for 10 min. After the reaction was quenched with water at room temperature, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 20:1) to obtain 23 (mixture of diastereomers) (201.8 mg, 76%, three steps) as a colorless oil.
23: IR (neat) 1755, 1468, 1375, 1235, 1085, 1039, 841, 775 cm−1; 1H-NMR (600 MHz, CDCl3) δ −0.01 (s, 3H), 0.01 (s, 3H), 0.88 (s, 9H), 0.94 (s, 3H), 1.04–1.15 (m, 5H), 1.17–1.27 (m, 2H), 1.30–1.38 (m, 3H), 1.46–1.60 (m, 2H), 1.65–1.67 (m, 1H), 1.71–2.03 (m, 7H), 2.13–2.14 (m, 3H), 3.98–4.00 (m, 1H), 5.10–5.11 (m, 2H), 5.25–5.32 (m, 1H); 13C-NMR (150 MHz, CDCl3) δ −5.2, −4.8, 13.5, 13.5, 17.6, 18.0, 19.4, 19.7, 20.4, 20.5, 20.8, 23.0, 25.8, 27.5, 27.5, 30.3, 30.4, 34.3, 38.5 (t, J = 22.3 Hz), 38.7 (t, J = 22.3 Hz), 40.7, 42.2, 53.1, 57.0, 57.0, 69.4, 76.3 (t, J = 28.7 Hz), 76.8 (t, J = 27.3 Hz), 117.0, 117.4, 122.9 (t, J = 247.1 Hz), 122.9 (t, J = 246.4 Hz), 138.4, 138.5, 169.3, 169.4; HRMS (ESI+) calcd for C26H50O3NF2SiNa [M + Na]+ 490.3523, found 490.3543.
- tert-Butyl({(1R,3aR,4S,7aR)-1-[(R)-4,4-difluoro-6-methylhept-6-en-2-yl]-7a-methyloctahydro-1H-inden-4-yl}oxy)dimethylsilane (24)
To a solution of sodium formate (155.8 mg, 2.29 mmol) and Pd(PPh3)4 (423.2 mg, 0.37 mmol) in dioxane (1.5 mL), nBu3P (345.5 mg, 427.0 μL, 1.77 mmol) was added at room temperature, and the mixture was stirred at 90 °C for 10 min. Acetate 23 (201.8 mg, 0.43 mmol) was dissolved in dioxane (1.5 mL), and the solution was added to the mixture. After being stirred at the same temperature for 17 h, the reaction mixture was quenched with H2O at room temperature. The mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was partially purified by flash column chromatography on silica gel (hexane:EtOAc = 100:1), and repurification by flash column chromatography on silica gel (hexane only) provided 24 (111.9 mg, 63%) as a colorless oil.
24: +36.0 (c 0.69, CHCl3); IR (neat) 1471, 1379, 1255, 1166, 1085, 1023, 837, 775 cm−1; 1H-NMR (600 MHz, CDCl3) δ 0.00 (s, 3H), 0.01 (s, 3H), 0.89 (s, 9H), 0.95 (s, 3H), 1.04 (d, J = 7.2 Hz, 3H), 1.07 (q, J = 9.6 Hz, 1H), 1.12 (td, J = 3.6, 13.2 Hz, 1H), 1.21–1.27 (m, 2H), 1.31–1.38 (m, 3H), 1.46–1.61 (m, 2H), 1.66–1.68 (m, 1H), 1.75–1.86 (m, 6H), 1.89–2.00 (m, 2H), 2.48–2.59 (m, 2H), 3.99–4.00 (m, 1H), 4.84 (brs, 1H), 4.59 (brs, 1H); 13C-NMR (150 MHz, CDCl3) δ −5.2, −4.8, 13.5, 17.6, 18.0, 20.3, 23.0, 23.3, 25.8, 27.5, 30.9, 34.4, 40.7, 41.7 (t, J = 23.0 Hz), 42.2, 45.6 (t, J = 25.9 Hz), 53.1, 57.1, 69.4, 116.3, 125.2 (t, J = 242.0 Hz), 138.7; HRMS (ESI+) calcd for C24H44OF2SiNa [M + Na]+ 437.3022, found 437.3033.
- (1R,3aR,4S,7aR)-1-[(2R)-4,4-Difluoro-6-hydroxy-6-methylheptan-2-yl]-7a-methyloctahydro-1H-inden-4-ol (9)
mCPBA (286.1 mg, 1.66 mmol) was added to the mixture of 24 (111.9 mg, 0.27 mmol) and NaHCO3 (131.9 mg, 1.57 mmol) in CH2Cl2 (2 mL) at 0 °C, and the mixture was stirred at the same temperature for 100 min under air. After the reaction was quenched with H2O and saturated aqueous NaHCO3 at room temperature, the mixture was extracted with CH2Cl2 three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 20:1) to obtain the crude epoxide.
LiAlH4 (14.0 mg, 0.37 mmol) was added to the solution of the above crude epoxide in Et2O (3 mL) at 0 °C, and the mixture was stirred at the same temperature for 15 min, and then at room temperature for 20 min. After the reaction was quenched with MeOH, water, and saturated aqueous potassium sodium tartrate, the mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude alcohol was used for the next reaction without further purification.
p-Toluenesulfonic acid monohydrate (981.0 mg, 5.16 mmol) was added to the solution of the above crude alcohol in MeOH (20 mL), and the mixture was stirred at room temperature for 25 h under air. After the reaction was quenched with H2O and saturated aqueous NaHCO3 at room temperature, the mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 2:1) to obtain 9 (63.8 mg, 74%, three steps) as a colorless oil.
9: +26.9 (c 2.20, CHCl3); IR (neat) 3412, 1471, 1375, 1170, 987, 864 cm−1; 1H-NMR (600 MHz, CDCl3) δ 0.97 (s, 3H), 1.04 (d, J = 6.6 Hz, 3H), 1.08–1.18 (m, 2H), 1.23–1.38 (m, 8H), 1.39–1.49 (m, 3H), 1.54–1.88 (m, 8H), 1.92–2.12 (m, 4H), 4.06–4.08 (m, 1H); 13C-NMR (150 MHz, CDCl3) δ 13.3, 17.4, 20.3, 22.4, 27.5, 30.2, 30.4, 31.0, 33.5, 40.3, 41.9, 44.1 (t, J = 23.0 Hz), 48.6 (t, J = 23.0 Hz), 52.7, 56.8, 69.3, 69.9, 126.6 (t, J = 241.3 Hz); HRMS (ESI+) calcd for C18H32O2F2Na [M + Na]+ 341.2263 found 341.2249.
- (6R)-6-{(1R,3aR,4S,7aR)-4-[(tert-Butyldimethylsilyl)oxy]-7a-methyloctahydro-1H-inden-1-yl}-4,4-difluoro-2-methylheptan-3-ol (26)
To the solution of compound 13 (933.9 mg, 2.09 mmol) in THF (10 mL), isopropyl magnesium chloride (6.3 mL, 1 M in THF, 6.26 mmol) was added at 0 °C, and the mixture was stirred at room temperature for 1 h. After the reaction was quenched with water and aqueous saturated NH4Cl at 0 °C, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 10:1) to obtain the crude product 25.
NaBH4 (45.1 mg, 1.19 mmol) was added to the solution of the above crude 25 in EtOH (3 mL) at 0 °C, and the mixture was stirred at the same temperature for 5 min. After the reaction was quenched with water, the mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 10:1) to obtain 26 (mixture of diastereomers) (563.4 mg, 62%, two steps) as a colorless oil.
26: IR (neat) 3442, 1471, 1367, 1255, 1166, 1085, 1023, 837, 775 cm−1; 1H-NMR (600 MHz, CDCl3) δ −0.01 (s, 3H), 0.01 (s, 3H), 0.89 (s, 9H), 0.95–1.02 (m, 8H), 1.04–1.15 (m, 4H), 1.23–1.38 (m, 6H), 1.49–1.67 (m, 3H), 1.77–2.12 (m, 7H), 3.38–3.45 (m, 1H), 3.99–4.00 (m, 1H); 13C-NMR (150 MHz, CDCl3) δ −5.2, −4.8, 13.5, 13.6, 17.0, 17.5, 17.6, 18.0, 20.2, 20.3, 20.5, 20.7, 23.0, 25.8, 27.6, 27.6, 28.5, 28.6, 30.3, 30.5, 34.4, 38.4 (t, J = 21.5 Hz), 40.7, 40.7, 42.2, 53.1, 57.1, 69.4, 77.4 (t, J = 27.3 Hz), 77.6 (t, J = 28.9 Hz), 125.3 (t, J = 244.9 Hz), 125.5 (t, J = 244.9 Hz); HRMS (ESI+) calcd for C24H47O2F2Si [M + H]+ 433.3308, found 433.3290.
- tert-Butyl({(1R,3aR,4S,7aR)-1-[(R)-4,4-difluoro-6-methylhept-5-en-2-yl]-7a-methyloctahydro-1H-inden-4-yl}oxy)dimethylsilane (28)
Trifluoromethanesulfonic anhydride (366.8 mg, 213 μL, 1.30 mmol) was added to the solution of 26 (467.3 mg, 1.08 mmol) and 2,6-di-tert-butylpyridine (464.5 mg, 3.68 mmol) in CH2Cl2 (4 mL) at 0 °C, and the mixture was stirred at the same temperature for 75 min. After the reaction was quenched with water and aqueous saturated NH4Cl at room temperature, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude 27 was used for the next reaction without further purification.
1,8-Diazabicyclo [5.4.0]undec-7-ene (DBU) (600 μL) was added to the solution of the above crude 27 in CH2Cl2 (4 mL) at room temperature and the mixture was stirred at the same temperature for 15 h. After the reaction was quenched with water and aqueous saturated NH4Cl at room temperature, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane only) to obtain 28 (269.8 mg, 60%, two steps) as a colorless oil.
28: +59.4 (c 0.79, CHCl3); IR (neat) 1468, 1375, 1255, 1166, 1085, 1027, 984, 837, 771 cm−1; 1H-NMR (600 MHz, CDCl3) δ −0.01 (s, 3H), 0.01 (s, 3H), 0.89 (s, 9H), 0.94 (s, 3H), 1.03 (d, J = 6.6 Hz, 3H), 1.07 (q, J = 9.6 Hz, 1H), 1.10 (td, J = 3.6, 13.2 Hz, 1H), 1.21–1.27 (m, 2H), 1.31–1.38 (m, 3H), 1.53–1.84 (m, 12H), 1.95–2.06 (m, 2H), 3.99–4.00 (m, 1H), 5.32 (t, J = 8.1 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ −5.2, −4.8, 13.5, 17.6, 18.0, 19.1, 20.4, 23.0, 25.8, 26.5, 27.6, 31.2, 34.4, 40.7, 42.2, 44.2 (t, J = 25.1 Hz), 53.1, 57.0, 69.4, 121.6 (t, J = 26.5 Hz), 123.2 (t, J = 238.5 Hz), 141.3 (t, J = 5.8 Hz); HRMS (ESI+) calcd for C24H44OF2SiNa [M + Na]+ 437.3022, found 437.3004.
- (3R,6R)-6-{(1R,3aR,4S,7aR)-4-[(tert-Butyldimethylsilyl)oxy]-7a-methyloctahydro-1H-inden-1-yl}-4,4-difluoro-2-methylheptane-2,3-diol (29)
The mixture of AD-mix α (605.3 mg) in tBuOH (4 mL) and H2O (4 mL) was stirred at 0 °C for 20 min, and 28 (55.9 mg, 0.13 mmol) was added to the mixture at 0 °C with stirring at the same temperature for 4 h, and then at 4 °C for 20 h under air. After the reaction was quenched with water, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 1:1) to obtain 29 (53.4 mg, 88%) as a colorless oil.
29: +35.0 (c 2.71, CHCl3); IR (neat) 3408, 1471, 1375, 1255, 1166, 1085, 1023, 833, 775 cm−1; 1H-NMR (600 MHz, CDCl3) δ −0.01 (s, 3H), 0.01 (s, 3H), 0.88 (s, 9H), 0.95 (s, 3H), 1.06 (d, J = 7.2 Hz, 3H), 1.09–1.16 (m, 2H), 1.22–1.38 (m, 11H), 1.53–1.67 (m, 3H), 1.67–1.88 (m, 3H), 1.95–2.00 (m, 2H), 2.26 (ddd, J = 7.8, 13.8, 34.2 Hz, 1H), 2.79 (d, J = 6.6 Hz, 1H), 3.41 (ddd, J = 5.4, 7.8, 19.2 Hz, 1H), 3.99–3.99 (m, 1H); 13C-NMR (150 MHz, CDCl3) δ −5.2, −4.8, 13.5, 17.6, 18.0, 21.1, 23.0, 25.6, 25.8, 27.6, 28.2, 30.1, 34.4, 39.5 (t, J = 22.3 Hz), 40.7, 42.2, 53.2, 57.1, 69.4, 72.1, 77.5 (t, J = 27.3 Hz), 126.3 (t, J = 245.6 Hz); HRMS (ESI−) calcd for C25H47O5F2Si [M + HCOO]− 439.3166, found 439.3170.
- (3S,6R)-6-{(1R,3aR,4S,7aR)-4-[(tert-Butyldimethylsilyl)oxy]-7a-methyloctahydro-1H-inden-1-yl}-4,4-difluoro-2-methylheptane-2,3-diol (30)
The mixture of AD-mix β (704.8 mg) in tBuOH (4 mL) and H2O (4 mL) was stirred at 0 °C for 30 min, and 28 (51.8 mg, 0.13 mmol) was added to the mixture at 0 °C with stirring at the same temperature for 5 h, and then at 4 °C for 19 h under air. After the reaction was quenched with water, the mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 2:1) to obtain 30 (53.8 mg, 96%) as a white powder.
30: +44.7 (c 0.30, CHCl3); IR (neat) 3431, 1471, 1375, 1255, 1162, 1081, 1019, 837, 771 cm−1; 1H-NMR (400 MHz, CDCl3)
δ −0.01 (s, 3H), 0.00 (s, 3H), 0.88 (s, 9H), 0.96 (s, 3H), 1.03 (d, J = 6.4 Hz, 3H), 1.07–1.16 (m, 2H), 1.21–1.39 (m, 11H), 1.52–1.86 (m, 6H), 1.95–2.19 (m, 3H), 2.84 (d, J = 7.6 Hz, 1H), 3.42 (ddd, J = 3.6, 8.0, 21.5 Hz, 1H), 3.99–3.99 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ −5.2, −4.8, 13.6, 17.6, 18.0, 20.2, 23.0, 25.7, 25.8, 27.5, 28.2, 30.6, 34.4, 39.9 (t, J = 21.9 Hz), 40.7, 42.2, 53.1, 57.1, 69.4, 72.3, 76.6 (t, J = 28.2 Hz), 126.8 (t, J = 246.0 Hz); HRMS (ESI−) calcd for C25H47O5F2Si [M + HCOO]− 439.3166, found 439.3164.
- (3R,6R)-4,4-Difluoro-6-[(1R,3aR,4S,7aR)-4-hydroxy-7a-methyloctahydro-1H-inden-1-yl]-2-methylheptane-2,3-diol (10)
To the solution of 29 (53.4 mg, 0.12 mmol) in MeOH (10 mL), p-toluenesulfonic acid monohydrate (222.8 mg, 1.17 mmol) was added, and the mixture was stirred at room temperature for 39 h under air. After the reaction was quenched with H2O and saturated aqueous NaHCO3 at room temperature, the mixture was extracted with CH2Cl2 three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 1:2) to obtain 10 (36.1 mg, 91%, in two steps) as a white powder.
10: +28.7 (c 2.34, CHCl3); IR (neat) 3415, 1471, 1371, 1267, 1162, 1073, 991, 741 cm−1; 1H-NMR (600 MHz, CDCl3) δ 0.96 (s, 3H), 1.05 (d, J = 7.2 Hz, 3H), 1.11–1.17 (m, 2H), 1.27–1.66 (m, 14H), 1.77–1.91 (m, 4H), 1.99–2.02 (m, 1H), 2.16 (brs, 1H), 2.21–2.30 (m, 1H), 3.03 (d, J = 7.2 Hz, 1H), 3.41 (dt, J = 6.0, 18.6 Hz, 1H), 4.06–4.07 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ 13.6, 17.4, 20.1, 22.4, 25.6 (t, J = 3.8 Hz), 27.4, 28.2, 30.6, 33.5, 39.8 (t, J = 21.9 Hz), 40.3, 41.9, 52.6, 56.9, 69.3, 72.3, 76.7 (t, J = 30.5 Hz), 126.7 (t, J = 246.0 Hz); HRMS (ESI−) calcd for C18H32O3F2Cl [M + Cl]− 369.2014, found 369.1989.
- (3S,6R)-4,4-Difluoro-6-[(1R,3aR,4S,7aR)-4-hydroxy-7a-methyloctahydro-1H-inden-1-yl]-2-methylheptane-2,3-diol (11)
To the solution of 30 (48.9 mg, 0.11 mmol) in MeOH (5 mL) and CH2Cl2 (5 mL), p-toluenesulfonic acid monohydrate (204.1 mg, 1.07 mmol) was added, and the mixture was stirred at room temperature for 66 h under air. After the reaction was quenched with H2O and saturated aqueous NaHCO3 at room temperature, the mixture was extracted with CH2Cl2 three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 1:2) to obtain 11 (30.4 mg, 83%, in two steps) as a white powder.
11: +22.6 (c 2.78, CHCl3); IR (neat) 3396, 1468, 1379, 1275, 1162, 1073, 987, 741 cm−1; 1H-NMR (600 MHz, CDCl3) δ 0.97 (s, 3H), 1.04 (d, J = 6.0 Hz, 3H), 1.08–1.20 (m, 2H), 1.28–1.91 (m, 18H), 2.00–2.19 (m, 3H), 2.93 (d, J = 7.8 Hz, 1H), 3.42 (ddd, J = 4.1, 7.8, 21.1 Hz, 1H), 4.06–4.08 (m, 1H); 13C-NMR (150 MHz, CDCl3) δ 13.3, 17.3, 21.0 (d, J = 3.0 Hz), 22.4, 25.5, 27.4, 28.2, 30.1 (d, J = 2.9 Hz), 33.5, 39.4 (t, J = 22.3 Hz), 40.3, 41.9, 52.7, 56.9, 69.3, 72.0, 77.5 (t, J = 25.0 Hz), 126.1 (t, J = 245.6 Hz); HRMS (ESI−) calcd for C18H32O3F2Cl [M + Cl]− 369.2014, found 369.1983.
- (1R,3aR,7aR)-1-{(2R)-4,4-Difluoro-6-methyl-6[(triethylsilyl)oxy] heptan-2-yl}-7a-methyloctahydro-4H-inden-4-one (32)
4-Methylmorpholine N-oxide (36.6 mg, 0.31 mmol) was added to the solution of 9 (63.8 mg, 0.20 mmol) in CH2Cl2 (2 mL), and the mixture was cooled to 0 °C. TPAP (37.8 mg, 0.11 mmol) was added to the mixture, and the mixture was stirred at 0 °C for 1 h. The reaction was diluted with an excess amount of Et2O. The mixture was directly purified by flash column chromatography on silica gel (Et2O only) to obtain the crude ketone (31), and this was used for the next reaction without further purification.
TESCl (211.0 mg, 234 μL, 1.4 mmol) was added to the 0 °C cooled solution of the above crude ketone (31) and imidazole (130.6 mg, 1.92 mmol) in CH2Cl2 (5 mL), and the mixture was stirred at room temperature for 28 h. After the reaction was quenched with H2O at 0 °C, the mixture was extracted with CH2Cl2 three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 20:1–10:1) to obtain 32 (76.1 mg, 88%, in two steps) as a colorless oil.
32: +6.4 (c 2.36, CHCl3); IR (neat) 1715, 1464, 1387, 1371, 1239, 1177, 1042, 748 cm−1; 1H-NMR (600 MHz, CDCl3) δ 0.58 (q, J = 7.8 Hz, 6H), 0.68 (s, 3H), 0.95 (t, J = 7.8 Hz, 9H), 1.10 (d, J = 6.0 Hz, 3H), 1.31–1.37 (m, 7H), 1.46 (q, J = 9.6 Hz, 1H), 1.50–1.78 (m, 4H), 1.82–2.06 (m, 7H), 2.12–2.16 (m, 1H), 2.19–2.30 (m, 2H), 2.46 (dd, J = 7.8, 12.0 Hz, 1H); 13C-NMR (150 MHz, CDCl3) δ 6.7, 7.0, 12.3, 19.1, 21.0, 23.9, 27.7, 30.7, 30.9, 38.9, 40.9, 43.1 (t, J = 23.0 Hz), 49.8, 51.2 (t, J = 23.7 Hz), 57.0, 62.0, 72.1, 125.3 (t, J = 240.6 Hz), 211.8; HRMS (ESI+) calcd for C24H44O2F2SiNa [M + Na]+ 453.2971 found 453.2953.
- (1R,3aR,4S,7aR)-1-{(2R)-4,4-Difluoro-4-[(4R)-2-(4-methoxyphenyl)-5,5-dimethyl-1,3-dioxolan-4-yl]butan-2-yl}-7a-methyloctahydro-1H-inden-4-ol (33)
Pyridinium p-toluenesulfonate (PPTS) (19.3 mg, 0.08 mmol) was added to a solution of 10 (45.7 mg, 0.14 mmol) in anisaldehyde dimethyl acetal (1.5 mL) at room temperature, and the mixture was stirred at the same temperature for 18 h. After the reaction was quenched with water and saturated aqueous NaHCO3, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was partially purified by flash column chromatography on silica gel (hexane:EtOAc = 4:1–2:1), and repurification by flash column chromatography on silica gel (hexane:EtOAc = 3:1) provided 33 (mixture of diastereomers) (31.3 mg, 50%) as a colorless oil.
33: IR (neat) 3481, 1523, 1460, 1379, 1252, 1170, 1096, 991, 829 cm−1; 1H-NMR (600 MHz, CDCl3) δ 0.96–0.96 (m, 3H), 1.06–1.18 (m, 5H), 1.25–1.35 (m, 3H), 1.41–1.62 (m, 12H), 1.79–1.93 (m, 4H), 1.99–2.20 (m, 1H), 2.17–2.28 (m, 1H), 3.79–3.83 (m, 4H), 4.06 (brs, 1H), 5.87 (s, 0.57H), 6.03 (s, 0.45H), 6.89–6.91 (m, 2H), 7.38–7.44 (m, 2H); 13C-NMR (150 MHz, CDCl3) δ 13.3, 17.4, 20.7, 20.8, 21.4, 21.4, 22.4, 23.4, 27.1, 27.3, 27.4, 28.0, 30.1, 30.2, 30.3, 30.3, 33.5, 40.3, 40.3, 40.5 (t, J = 23.0 Hz), 41.3 (t, J = 22.3 Hz), 41.9, 41.9, 52.7, 55.3, 56.8, 56.9, 69.3, 80.9, 81.6, 83.3, 83.5, 83.6, 83.6, 83.7, 83.8, 83.9, 84.0, 101.8, 102.1, 113.7, 113.7, 123.5 (dd, J = 241.2, 251.3 Hz), 123.7 (dd, J = 241.4, 251.4 Hz), 127.9, 128.4, 129.3, 130.5, 160.4, 160.5; HRMS (ESI+) calcd for C26H38O4F2Na [M + Na]+ 475.2630, found 475.2636.
- (1R,3aR,4S,7aR)-1-{(2R)-4,4-Difluoro-4-[(4S)-2-(4-methoxyphenyl)-5,5-dimethyl-1,3-dioxolan-4-yl]butan-2-yl}-7a-methyloctahydro-1H-inden-4-ol (34)
Pyridinium p-toluenesulfonate (PPTS) (158.6 mg, 0.63 mmol) was added to a solution of 11 (36.1 mg, 0.11 mmol) in anisaldehyde dimethyl acetal (3 mL) at room temperature, and the mixture was stirred at the same temperature for 18 h. After the reaction was quenched with water and saturated aqueous NaHCO3, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was partially purified by flash column chromatography on silica gel (hexane only–hexane:EtOAc = 4:1–2:1), and repurification by flash column chromatography on silica gel (hexane:EtOAc = 4:1) provided 34 (mixture of diastereomers) (22.9 mg, 61%) as a colorless oil.
34: IR (neat) 3516, 1519, 1460, 1375, 1252, 1166, 1093, 987, 829 cm−1; 1H-NMR (600 MHz, CDCl3) δ 0.96 (s, 3H), 1.05–1.35 (m, 8H), 1.40–1.88 (m, 15H), 1.96–2.07 (m, 2H), 3.81–3.84 (m, 4H), 4.05–4.07 (m, 1H), 5.55 (s, 0.56H), 6.04 (s, 0.44H), 6.90 (d, J = 7.8 Hz, 2H), 7.39–7.45 (m, 2H); 13C-NMR (150 MHz, CDCl3) δ 13.3, 13.3, 17.3, 21.5, 21.5, 22.4, 23.3 (t, J = 4.4 Hz), 27.1, 27.3, 27.4, 28.1, 30.6, 33.5, 40.3, 40.4 (t, J = 21.5 Hz), 41.2 (t, J = 23.0 Hz), 41.9, 52.6, 55.3, 56.8, 56.8, 69.3, 80.9, 81.6, 82.8, 82.9, 83.0, 83.0, 83.2, 83.2, 83.3, 83.5, 101.9, 102.2, 113.7, 113.8, 123.7 (dd, J = 239.9, 251.4 Hz), 123.9 (dd, J = 241.2, 251.3 Hz), 127.8, 128.4, 129.4, 130.5, 160.4, 160.5; HRMS (ESI+) calcd for C26H38O4F2Na [M + Na]+ 475.2630, found 475.2604.
- (1R,3aR,7aR)-1-{(2R)-4,4-Difluoro-4-[(4R)-2-(4-methoxyphenyl)-5,5-dimethyl-1,3-dioxolan-4-yl]butan-2-yl}-7a-methyloctahydro-4H-inden-4-one (35)
4-Methylmorpholine N-oxide (14.4 mg, 0.12 mmol) was added to the solution of 33 (29.9 mg, 0.07 mmol) in CH2Cl2 (3 mL), and the mixture was cooled to 0 °C. TPAP (14.2 mg, 0.04 mmol) was added to the mixture, and the mixture was stirred at 0 °C for 40 min. The reaction was diluted with an excess amount of Et2O. The mixture was directly purified by flash column chromatography on silica gel (Et2O only) followed by purification on flash column chromatography on silica gel (hexane:EtOAc = 4:1) to obtain 35 (mixture of diastereomers) (26.5 mg, 89%) as a colorless oil.
35: IR (neat) 1712, 1615, 1519, 1468, 1387, 1252, 1170, 1096, 1031, 833, 736 cm−1; 1H-NMR (600 MHz, CDCl3) δ 0.65–0.67 (m, 3H), 1.12 (t, J = 6.9 Hz, 3H), 1.22–1.38 (m, 1H), 1.46–1.77 (m, 11H), 1.82–1.94 (m, 3H), 1.98–2.03 (m, 1H), 2.10–2.30 (m, 4H), 2.42–2.46 (m, 1H), 3.78–3.82 (m, 4H), 5.87 (s, 0.55H), 6.03 (s, 0.44H), 6.88–6.91 (m, 2H), 7.38–7.44 (m, 2H); 13C-NMR (150 MHz, CDCl3) δ 12.3, 12.3, 19.0, 20.9, 21.0, 21.4, 21.4, 23.3, 23.9, 27.1, 27.5, 27.6, 28.0, 30.2, 30.2, 30.4, 30.4, 38.8, 38.9, 40.4 (t, J = 22.3 Hz), 40.9, 41.3 (t, J = 23.0 Hz), 49.8, 55.3, 56.8, 56.8, 62.0, 80.9, 81.6, 83.3, 83.4, 83.5, 83.7, 83.8, 84.0, 101.9, 102.2, 113.7, 113.8, 121.8, 121.9, 123.4, 123.5, 123.6, 125.0, 125.2, 127.9, 128.4, 129.2, 130.3, 160.4, 160.6, 211.7; HRMS (ESI+) calcd for C26H36O4F2Na [M + Na]+ 473.2474, found 473.2492.
- (1R,3aR,7aR)-1-{(2R)-4,4-Difluoro-4-[(4S)-2-(4-methoxyphenyl)-5,5-dimethyl-1,3-dioxolan-4-yl]butan-2-yl}-7a-methyloctahydro-4H-inden-4-one (36)
4-Methylmorpholine N-oxide (13.0 mg, 0.11 mmol) was added to the solution of 34 (31.3 mg, 0.07 mmol) in CH2Cl2 (3 mL), and the mixture was cooled to 0 °C. TPAP (13.1 mg, 0.04 mmol) was added to the mixture, and the mixture was stirred at 0 °C for 35 min. The reaction was diluted with an excess amount of Et2O. The mixture was directly purified by flash column chromatography on silica gel (Et2O only) followed by purification on flash column chromatography on silica gel (hexane:EtOAc = 4:1) to obtain 36 (mixture of diastereomers) (28.2 mg, 90%) as a colorless oil.
36: IR (neat) 1712, 1611, 1519, 1464, 1383, 1248, 1174, 1096, 1035, 837 cm−1; 1H-NMR (600 MHz, CDCl3) δ 0.66 (s, 3H), 1.10–1.12 (m, 3H), 1.24–1.36 (m, 1H), 1.40–1.61 (m, 10H), 1.66–2.04 (m, 7H), 2.11–2.14 (m, 1H), 2.18–2.24 (m, 1H), 2.27–2.30 (m, 1H), 2.41–2.46 (m, 1H), 3.79–3.83 (m, 4H), 5.88 (s, 0.53H), 6.04 (s, 0.44H), 6.89–6.91 (m, 2H), 7.38–7.44 (m, 2H); 13C-NMR (150 MHz, CDCl3) δ 12.4, 19.0, 20.5, 20.6, 21.4, 21.5, 23.2, 23.9, 27.1, 27.5, 27.6, 28.1, 30.7, 38.9, 40.4 (t, J = 22.3 Hz), 41.0, 41.1 (t, J = 23.0 Hz), 49.8, 55.3, 55.3, 56.7, 56.8, 62.0, 80.4, 81.6, 82.8, 82.9, 83.0, 83.0, 83.2, 83.2, 83.3, 83.5, 101.9, 102.2, 113.7, 113.8, 122.0, 122.1, 123.6, 123.6, 123.7, 123.8, 125.2, 125.4, 127.8, 128.4, 129.3, 130.4, 160.4, 160.6, 211.7, 211.7; HRMS (ESI+) calcd for C26H36O4F2Na [M + Na]+ 473.2474, found 473.2499.
- (3R,5Z,7E)-23,23-Difluoro-9,10-seco-5,7,10(19)-cholestatriene-3,25-diol: 23,23-Difluoro-25-hydroxyvitamin D3 (5)
nBuLi (183 μL, 1.55 M hexane solution, 0.29 mmol) was added to a solution of A-ring phosphine oxide 14 (129.1 mg, 0.29 mmol) in THF (1 mL) at −78 °C. After stirring for 20 min, a solution of 32 (30.7 mg, 0.07 mmol) in THF (1.5 mL) was added to the reaction mixture, and the mixture was stirred at −78 °C for 1 h. After the reaction was quenched with H2O at the same temperature, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 20:1) to obtain the crude coupling product (52.0 mg), and it was used for the next reaction without further purification.
Tetrabutylammonium fluoride (710 μL, 1 M THF solution, 0.71 mmol) was added to the solution of the above crude coupling product (52.0 mg) in THF (4 mL), and the mixture was stirred at room temperature for 16 h. After the reaction was quenched with H2O and aqueous saturated NH4Cl at room temperature, the mixture was extracted with EtOAc three times. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 1:1) to afford 5 (30.8 mg, 100%, in two steps) as a white powder.
5: +53.4 (c 2.37, EtOH); IR (neat) 3381, 1441, 1379, 1220, 1174, 1054, 895, 860, 741 cm−1; 1H-NMR (600 MHz, CD3OD) δ 0.63 (s, 3H), 1.12 (d, J = 7.2 Hz, 3H), 1.33–1.76 (m, 16H), 1.83–2.18 (m, 9H), 2.23 (dd, J = 9.6, 12.0 Hz, 1H), 2.44 (dt, J = 5.1, 13.2 Hz, 1H), 2.57 (dd, J = 3.9, 12.9 Hz, 1H), 2.88–2.91 (m, 1H), 3.78–3.82 (m, 1H), 4.78 (d, J = 1.2 Hz, 1H), 5.08 (brs, 1H), 6.08 (d, J = 10.8 Hz, 1H), 6.26 (d, J = 10.8 Hz, 1H); 13C-NMR (150 MHz, CD3OD) δ 12.6, 21.7, 23.5, 24.8, 29.2, 30.2, 30.6, 30.8, 33.1, 33.9, 36.9, 42.1, 45.2 (t, J = 23.0 Hz), 47.2, 47.3, 50.6 (t, J = 23.7 Hz), 57.9, 58.5, 70.4, 70.9, 113.0, 119.4, 122.9, 127.0 (t, J = 240.5 Hz), 137.8, 142.6, 147.3; 19F NMR (565 MHz, CD3OD) δ −94.3 (d, J = 224.7 Hz), −92.7 (d, J = 243.4 Hz); HRMS (ESI+) calcd for C27H42O4F2Na [M + Na]+ 459.3045, found 459.3033.
- (3R,24R,5Z,7E)-23,23-Difluoro-9,10-seco-5,7,10(19)-cholestatriene-3,24,25-triol: (24R)-23,23-Difluoro-24,25-dihydroxyvitamin D3 (7)
nBuLi (152 μL, 1.55 M hexane solution, 0.24 mmol) was added to a solution of A-ring phosphine oxide 14 (112.9 mg, 0.25 mmol) in THF (1 mL) at −78 °C. After stirring for 20 min, a solution of 35 (26.5 mg, 0.06 mmol) in THF (1 mL) was added to the reaction mixture, and the mixture was stirred at −78 °C for 2 h. After the reaction was quenched with H2O at the same temperature, the mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 5:1) to obtain the crude coupling product (25.5 mg), and it was used for the next reaction without further purification.
The above crude residue was dissolved in MeOH (5 mL), and AG 50W-X4 resin H+ form (183.1 mg) was added. The mixture was stirred for 3 h, insoluble materials were filtered off, and the solution was diluted with H2O and saturated aqueous NaHCO3. The mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (hexane:EtOAc = 1:1) to afford 7 (10.8 mg, 41%, two steps) as a white solid. Crystal of 7 was obtained by dissolving 7 in EtOH and allowing the solvent to slowly evaporate at room temperature (colorless needles).
7: +90.0 (c 0.83, EtOH); IR (neat) 3388, 1433, 1375, 1158, 1058, 1000, 957, 895 cm−1; 1H-NMR (600 MHz, CD3OD) δ 0.63 (s, 3H), 1.13 (d, J = 7.2 Hz, 3H), 1.29–1.63 (m, 13H), 1.70–1.79 (m, 3H), 1.88–2.33 (m, 9H), 2.44 (dt, J = 5.4, 13.8 Hz, 1H), 2.57 (dd, J = 3.6, 12.6 Hz, 1H), 2.88–2.91 (m, 1H), 3.44 (dd, J = 9.0, 13.8 Hz, 1H), 3.78–3.82 (m, 1H), 4.78 (d, J = 1.2 Hz, 1H), 5.08 (brs, 1H), 6.07 (d, J = 11.4 Hz, 1H), 6.26 (d, J = 10.8 Hz, 1H); 13C-NMR (150 MHz, CD3OD) δ 12.5, 22.2, 23.5, 24.8, 27.0 (d, J = 4.4 Hz), 27.8, 29.2, 30.2, 32.4, 33.9, 36.9, 41.1 (t, J = 22.3 Hz), 42.1, 47.2, 47.3, 57.9, 58.7, 70.9, 73.0, 79.5 (t, J = 26.6 Hz), 113.0, 119.4, 122.9, 127.1 (t, J = 244.1 Hz), 137.7, 142.6, 147.3; 19F-NMR (565 MHz, CD3OD) δ −107.1 (d, J = 251.8 Hz), −101.8 (d, J = 251.2 Hz); HRMS (ESI−) calcd for C27H42O3F2Cl [M + Cl]− 487.2796, found 487.2778.
4.1. Crystal Data of 7 (CCDC 2202393)
C27H42F2O3: Mr = 452.63, CuKα (λ = 1.54187 Å), orthorhombic, Pbca, colorless block 0.200 × 0.150 × 0.040 mm, crystal dimensions a = 6.74568(15) Å, b = 10.5822(3) Å, c = 36.4192(7) Å, α = 90°, β = 90°, γ = 90°, T = 173 K, Z = 4, V = 32599.74(10) Å3, Dcalc = 1.156 g/cm3, μCuKα = 6.723 cm−1, F000 = 984.00, GOF = 1.162, Rint = 0.0686, R1 = 0.0614, wR2 = 0.1337.
All measurements were taken on a Rigaku Raxis Rapid imaging plate area detector with graphite monochromated Cu-Kα radiation. The data were collected at a temperature of –100 °C. The structure was solved by direct-method SIR97 and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. All calculations were performed using the Crystal Structure (Crystal Structure 4.2.2) crystallographic software package except for refinement, which was performed using SHELXL97.
4.2. Accession Codes of Compound 7
CCDC 2202393 contains the supplementary crystallographic data for this study. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif (accessed on 21 August 2022), by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223-336033.
- (3R,24S,5Z,7E)-23,23-Difluoro-9,10-seco-5,7,10(19)-cholestatriene-3,24,25-triol: (24S)-23,23-Difluoro-24,25-dihydroxyvitamin D3 (8)
nBuLi (161 μL, 1.55 M hexane solution, 0.25 mmol) was added to a solution of A-ring phosphine oxide 14 (115.4 mg, 0.25 mmol) in THF (1 mL) at −78 °C. After stirring for 20 min, a solution of 36 (28.2 mg, 0.06 mmol) in THF (1 mL) was added to the reaction mixture, and the mixture was stirred at −78 °C for 100 min. After the reaction was quenched with H2O at the same temperature, the mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (hexane:EtOAc = 4:1) to obtain the crude coupling product (41.6 mg), and it was used for the next reaction without further purification.
The above crude residue was dissolved in MeOH (5 mL), and AG 50W-X4 resin H+ form (188.3 mg) was added. The mixture was stirred for 3 h 10 min, insoluble materials were filtered off, and the solution was diluted with H2O and saturated aqueous NaHCO3. The mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (hexane:EtOAc = 1:1) to afford 8 (19.6 mg, 63%, two steps) as a white powder.
8: +12.0 (c 0.25, CHCl3); IR (neat) 3388, 1437, 1375, 1263, 1162, 1066, 996, 891, 741 cm−1; 1H-NMR (600 MHz, CD3OD) δ 0.64 (s, 3H), 1.12 (d, J = 7.2 Hz, 3H), 1.30–1.42 (m, 10H), 1.47–1.63 (m, 4H), 1.68–1.79 (m, 3H), 1.87–2.31 (m, 9H), 2.44 (dt, J = 5.1, 13.2 Hz, 1H), 2.57 (dd, J = 3.6, 12.6 Hz, 1H), 2.88–2.91 (m, 1H), 3.46 (dd, J = 7.2, 16.8 Hz, 1H), 3.78–3.82 (m, 1H), 4.79 (d, J = 2.4 Hz, 1H), 5.08 (brs, 1H), 6.08 (d, J = 10.8 Hz, 1H), 6.26 (d, J = 10.8 Hz, 1H); 13C-NMR (150 MHz, CD3OD) δ 12.6, 21.6, 23.6, 24.8, 26.7 (d, J = 4.2 Hz), 27.9, 29.1, 30.2, 32.7, 33.9, 36.9, 41.3 (t, J = 22.2 Hz), 42.2, 47.2, 47.3, 57.9, 58.7, 70.9, 73.1, 79.0 (t, J = 27.3 Hz), 113.0, 119.4, 122.9, 127.3 (t, J = 244.9 Hz), 137.7, 142.6, 147.3; 19F-NMR (565 MHz, CD3OD) δ −105.0 (d, J = 251.5 Hz), −101.6 (d, J = 250.9 Hz); HRMS (ESI−) calcd for C27H42O3F2Cl [M + Cl]− 487.2796, found 487.2794.
4.3. Metabolism of 25(OH)D3 (1) and 5 by Recombinant hCYP24A1
The metabolism of 25(OH)D3 and its analogue 5 by CYP24A1 was analyzed using the membrane fraction prepared from recombinant Escherichia coli cells expressing human CYP24A1, as described in our previous study [29]. Briefly, the reaction mixture containing 0.02 µM of human CYP24A1, 2.0 µM of adrenodoxin (ADX), 0.2 µM of NADPH-adrenodoxin reductase (ADR), 1 mM of EDTA, 1 mM of NADPH, and 5.0 µM of each substrate in 100 mM Tris-HCl (pH 7.4) was incubated at 37 °C for 5 or 15 min. The metabolites were extracted with four volumes of CHCl3-CH3OH (3:1) and analyzed by HPLC under the following conditions: column, CAPCELL PAK C18 UG120 (5 μm) (4.6 × 250 mm) (SHISEIDO, Tokyo, Japan); UV detection, 265 nm; flow-rate, 1.0 mL min−1; column temperature, 40 °C; mobile phase, CH3CN: a linear gradient of 20–100% CH3CN aqueous solution per 25 min and 100% CH3CN for 10 min.
4.4. Measurement of hVDR Binding Affinity of 25(OH)D3 (1) and 5
The binding affinity of each analogue for hVDR was evaluated using the in vitro system based on the split-luciferase technique described in our previous study [30]. Briefly, 50 μL of cell lysate prepared from recombinant E. coli expressing split-luciferase vitamin D biosensor protein was added to each well of a 96-well plate and left for 10 min at room temperature. Then, 50 μL of the Luciferin solution containing 20 mM of MgSO4, 2 mM of D-luciferin, and 4 mM of adenosine triphosphate in 25 mM Tris-HCl (pH 7.4) were injected into each well and incubated for 15 min at room temperature. The luminescence (photon counts) was measured using a luminometer (Infinite 200 Pro 96-microplate luminometer, Tecan). The relative hVDR binding affinity of each analogue was evaluated based on the concentration at which the luminescence showed 50% of the maximum value.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/molecules27165352/s1. 1H and 13C-NMR spectra of all new compounds: 5, 7–11, 13, 20, 23, 24, 26, 28–30, and 32–36, as well as 19F-NMR spectra of compounds: 5, 7, and 8.
Author Contributions
Conceptualization, F.K., T.S., and A.K.; investigation, F.K., S.M., H.M., and K.Y.; original draft preparation, F.K. and K.Y.; writing—review and editing, A.K.; supervision, A.K. and T.S.; funding acquisition, F.K., A.K., and T.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by Grants-in-Aid from the Japan Society for the Promotion of Science (No. 22K14688 to F.K., No. 18K06556 to A.K., and No. 19H02889 to T.S.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
Abbreviations
DAST: N:N-diethylaminosulfur trifluoride; VDR: vitamin D receptor; TBS: tert-butyldimethylsilyl; TPAP: tetrapropylammonium perruthenate; NMO: 4-methylmorpholine N-oxide; TBAF: tetra-n-butylammonium fluoride; TES: triethylsilyl; LDA: lithium diisopropylamide; DBU: 1,8-Diazabicyclo [5.4.0]undec-7-ene; PPTS: pyridinium p-toluenesulfonate.
References
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; Del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-containing drugs approved by the FDA in 2018. Chemistry 2019, 25, 11797–11819. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Shu, Y.Z.; Zhuo, X.; Meanwell, N.A. Metabolic and pharmaceutical aspects of fluorinated compounds. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef]
- Kawagoe, F.; Mototani, S.; Kittaka, A. Design and synthesis of fluoro analogues of vitamin D. Int. J. Mol. Sci. 2021, 22, 8191. [Google Scholar] [CrossRef]
- Sakaki, T.; Sawada, N.; Komai, K.; Shiozawa, S.; Yamada, S.; Yamamoto, K.; Ohyama, Y.; Inouye, K. Dual metabolic pathway of 25-hydroxyvitamin D3 catalyzed by human CYP24. Eur. J. Biochem. 2000, 267, 6158–6165. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, T.; Kagawa, N.; Yamamoto, K.; Inouye, K. Metabolism of vitamin D3 by cytochromes P450. Front. Biosci. 2005, 10, 119–134. [Google Scholar]
- Yasuda, K.; Nishikawa, M.; Okamoto, K.; Horibe, K.; Mano, H.; Yamaguchi, M.; Okon, R.; Nakagawa, K.; Tsugawa, N.; Okano, T.; et al. Elucidation of metabolic pathways of 25-hydroxyvitamin D3 mediated by Cyp24A1 and Cyp3A using Cyp24a1 knockout rats generated by CRISPR/Cas9 system. J. Biol. Chem. 2021, 296, 100668. [Google Scholar] [CrossRef]
- Kawagoe, F.; Sugiyama, T.; Uesugi, M.; Kittaka, A. Recent developments for introducing a hexafluoroisopropanol unit into the vitamin D side chain. J. Steroid Biochem. Mol. Biol. 2018, 177, 250–254. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Taguchi, T.; Mitsuhashi, S.; Eguchi, T.; Ohshima, E.; Ikekawa, N. Studies on organic fluorine compounds. XXXIX. Studies on steroids. LXXIX. Synthesis of 1α, 25-dihydroxy-26,26,26,27,27,27-hexafluorovitamin D3. Chem. Pharm. Bull. 1982, 30, 4297–4304. [Google Scholar] [CrossRef]
- Tanaka, Y.; DeLuca, H.F.; Kobayashi, Y.; Ikekawa, N. 26,26,26,27,27,27-Hexafluoro-1,25-dihydroxyvitamin D3: A highly potent, long-lasting analog of 1,25-dihydroxyvitamin D3. Arch. Biochem. Biophys. 1984, 229, 348–354. [Google Scholar] [CrossRef]
- Asano, L.; Watanabe, M.; Ryoden, Y.; Usuda, K.; Yamaguchi, T.; Khambu, B.; Takashima, M.; Sato, S.; Sakai, J.; Nagasawa, K.; et al. Vitamin D metabolite, 25-hydroxyvitamin D, regulates lipid metabolism by inducing degradation of SREBP/SCAP. Cell Chem. Biol. 2017, 24, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, F.; Mendoza, A.; Hayata, Y.; Asano, L.; Kotake, K.; Mototani, S.; Kawamura, S.; Kurosaki, S.; Akagi, Y.; Takemoto, Y.; et al. Discovery of a vitamin D receptor-silent vitamin D derivative that impairs sterol regulatory element-binding protein in vivo. J. Med. Chem. 2021, 64, 5689–5709. [Google Scholar] [CrossRef] [PubMed]
- Nagata, A.; Akagi, Y.; Asano, L.; Kotake, K.; Kawagoe, F.; Mendoza, A.; Masoud, S.S.; Usuda, K.; Yasui, K.; Takemoto, Y.; et al. Synthetic chemical probes that dissect vitamin D activities. ACS Chem. Biol. 2019, 14, 2851–2858. [Google Scholar] [CrossRef]
- Mendoza, A.; Takemoto, Y.; Cruzado, K.T.; Masoud, S.S.; Nagata, A.; Tantipanjaporn, A.; Okuda, S.; Kawagoe, F.; Sakamoto, R.; Odagi, M.; et al. Controlled lipid β-oxidation and carnitine biosynthesis by a vitamin D metabolite. Cell Chem. Biol. 2022, 29, 660–669. [Google Scholar] [CrossRef]
- Maestro, M.A.; Molnár, F.; Carlberg, C. Vitamin D and its synthetic analogs. J. Med. Chem. 2019, 62, 6854–6875. [Google Scholar] [CrossRef]
- Kawagoe, F.; Mototani, S.; Kittaka, A. The synthesis and biological evaluation of D-ring-modified vitamin D analogues. Biomolecules 2021, 11, 1639. [Google Scholar] [CrossRef]
- Kawagoe, F.; Yasuda, K.; Mototani, S.; Sugiyama, T.; Uesugi, M.; Sakaki, T.; Kittaka, A. Synthesis and CYP24A1-dependent metabolism of 23-fluorinated vitamin D3 analogues. ACS Omega 2019, 4, 11332–11337. [Google Scholar] [CrossRef]
- Taguchi, T.; Mitsuhashi, S.; Yamanouchi, A.; Kobayashi, Y. Synthesis of 23,23-difluoro-25-hydroxyvitamin D3. Tetrahedron Lett. 1984, 25, 4933–4936. [Google Scholar] [CrossRef]
- Nakada, M.; Tanaka, Y.; DeLuca, H.F.; Kobayashi, Y.; Ikekawa, N. Biological activities and binding properties of 23,23-difluoro-25-hydroxyvitamin D3 and its 1α-hydroxy derivative. Arch. Biochem. Biophys. 1985, 241, 173–178. [Google Scholar] [CrossRef]
- Nakamura, E. New acyl anion equivalent. A short route to the enol lactam intermediate in Cytochalasin synthesis. Tetrahedron Lett. 1981, 22, 663–666. [Google Scholar] [CrossRef]
- Ando, K.; Kondo, F.; Koike, F.; Takayama, H. An improved synthesis of 24,24-difluoro-1α,25-dihydroxyvitamin D3 from vitamin D2. Chem. Pharm. Bull. 1992, 40, 1662–1664. [Google Scholar] [CrossRef][Green Version]
- Kawagoe, F.; Mototani, S.; Yasuda, K.; Nagasawa, K.; Uesugi, M.; Sakaki, T.; Kittaka, A. Introduction of fluorine atoms to vitamin D3 side-chain and synthesis of 24,24-difluoro-25-hydroxyvitamin D3. J. Steroid Biochem. Mol. Biol. 2019, 195, 105477. [Google Scholar] [CrossRef]
- Ono, Y.; Watanabe, H.; Taira, I.; Takahashi, K.; Ishihara, J.; Hatakeyama, S.; Kubodera, N. Synthesis of putative metabolites of 1α,25-dihydroxy-2β-(3-hydroxypropoxy)vitamin D3 (ED-71). Steroids 2006, 71, 529–540. [Google Scholar] [CrossRef]
- Kawagoe, F.; Yasuda, K.; Mototani, S.; Mano, H.; Sakaki, T.; Kittaka, A. Stereoselective synthesis of 24-fluoro-25-hydroxyvitamin D3 analogues and their stability to hCYP24A1-dependent catabolism. Int. J. Mol. Sci. 2021, 22, 11863. [Google Scholar] [CrossRef]
- Toh, H.T.; Okamura, W.H. Studies on a convergent route to side-chain analogues of vitamin D: 25-hydroxy-23-oxavitamin D3. J. Org. Chem. 1983, 48, 1414–1417. [Google Scholar] [CrossRef]
- Grzywacz, P.; Plum, L.A.; Sicinski, R.R.; Clagett-Dame, M.; DeLuca, H.F. Methyl substitution of the 25-hydroxy group on 2-methylene-19-nor-1α,25-dihydroxyvitamin D3 (2MD) reduces potency but allows bone selectivity. Arch. Biochem. Biophys. 2007, 460, 274–284. [Google Scholar] [CrossRef]
- Sawada, N.; Kusudo, T.; Sakaki, T.; Hatakeyama, S.; Hanada, M.; Abe, D.; Kamao, M.; Okano, T.; Ohta, M.; Inouye, K. Novel metabolism of 1α,25-dihydroxyvitamin D3 with C24–C25 bond cleavage catalyzed by human CYP24A1. Biochemistry 2004, 43, 4530–4537. [Google Scholar] [CrossRef]
- Kusudo, T.; Sakaki, T.; Abe, D.; Fujishima, T.; Kittaka, A.; Takayama, H.; Hatakeyama, S.; Ohta, M.; Inouye, K. Metabolism of A-ring diastereomers of 1α,25-dihydroxyvitamin D3 by CYP24A1. Biochem. Biophys. Res. Commun. 2004, 321, 774–782. [Google Scholar] [CrossRef]
- Mano, H.; Ikushiro, S.; Saito, N.; Kittaka, A.; Sakaki, T. Development of a highly sensitive in vitro system to detect and discriminate between vitamin D receptor agonists and antagonists based on split-luciferase technique. J. Steroid Biochem. Mol. Biol. 2018, 178, 55–59. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).