
In Vitro Evaluation of In Silico Screening Approaches in Search for Selective ACE2 Binding Chemical Probes

 , , , ,
, , , ,  , , and
, , and 
Abstract
:1. Introduction
2. Methods
2.1. In Silico Screening
2.1.1. UNISTRA Datasets and Their Preliminary Processing
2.1.2. UNISTRA Docking
2.1.3. UNISTRA Ligand-Based Pharmacophore Model
2.1.4. UNISTRA Structure-Based Pharmacophore Model
2.1.5. UNISTRA Antitarget Categorical QSAR Models
2.1.6. ENAMIN Docking into Multiple Active Site Conformers
2.2. Experimental Methods
3. Results
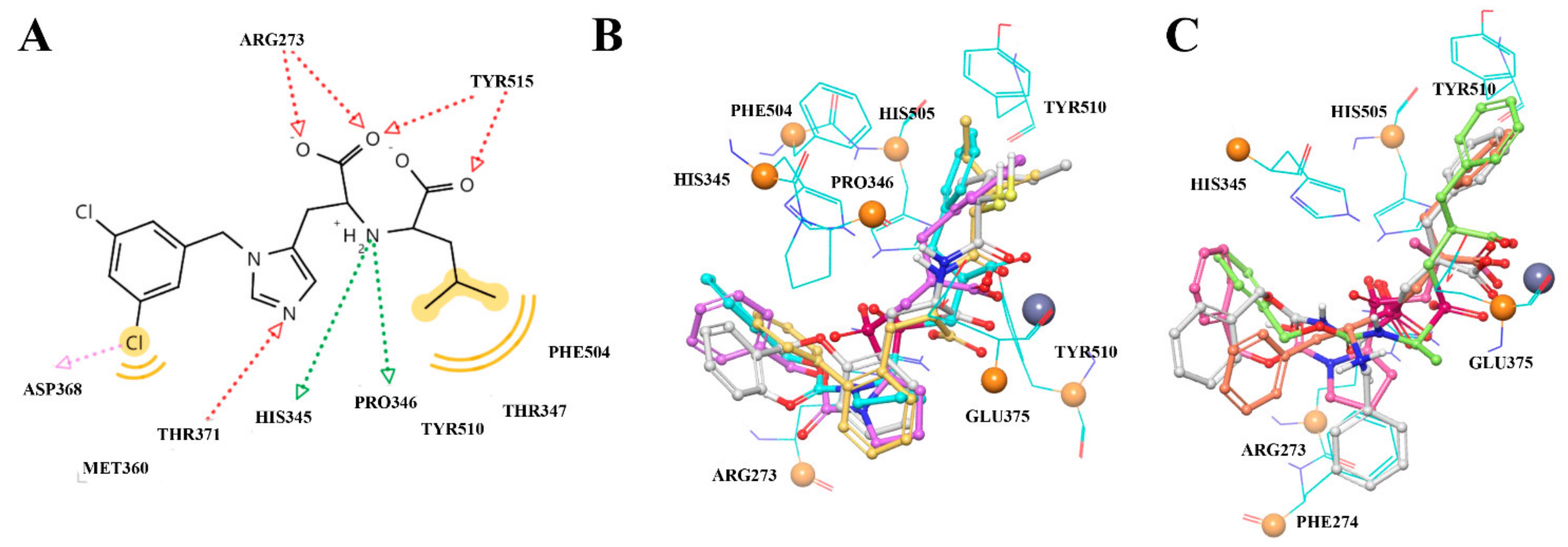
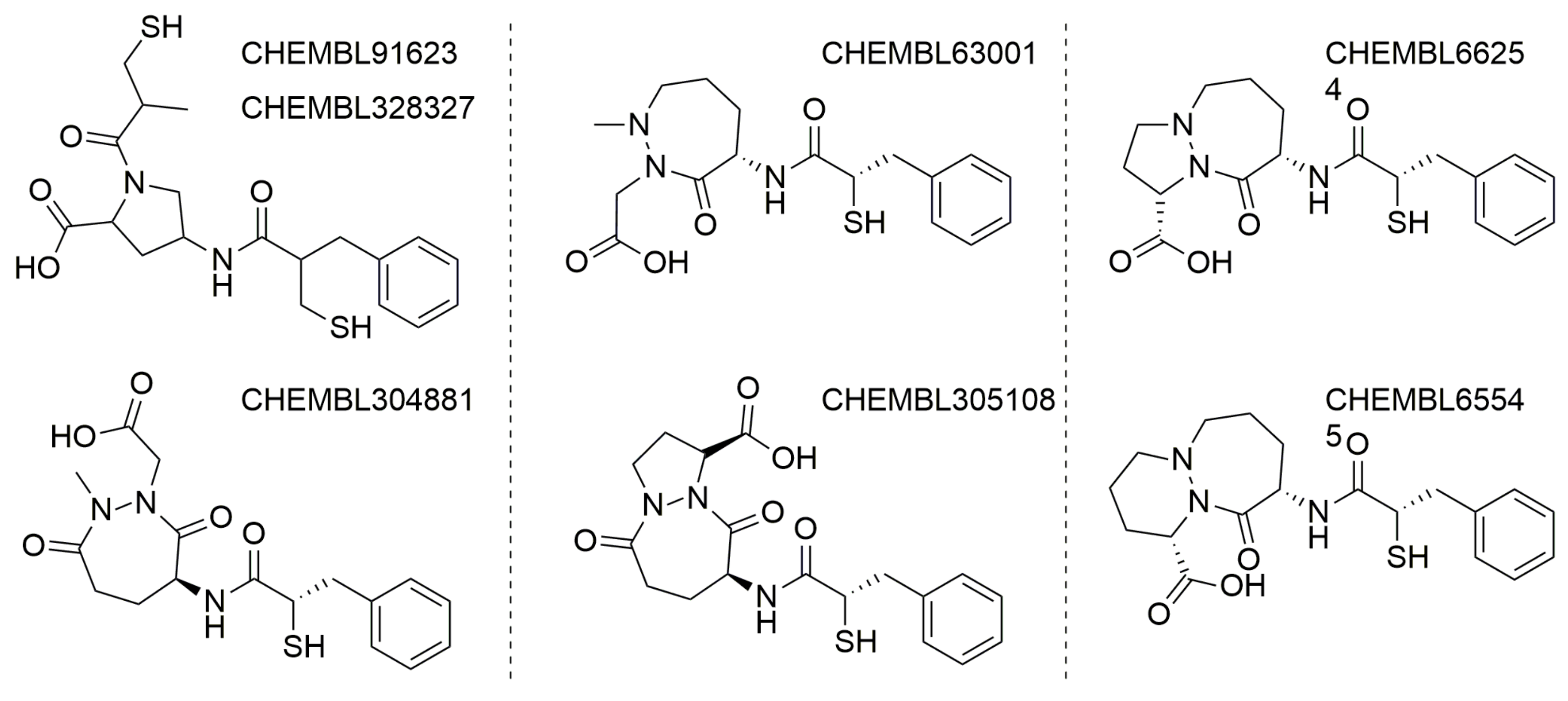
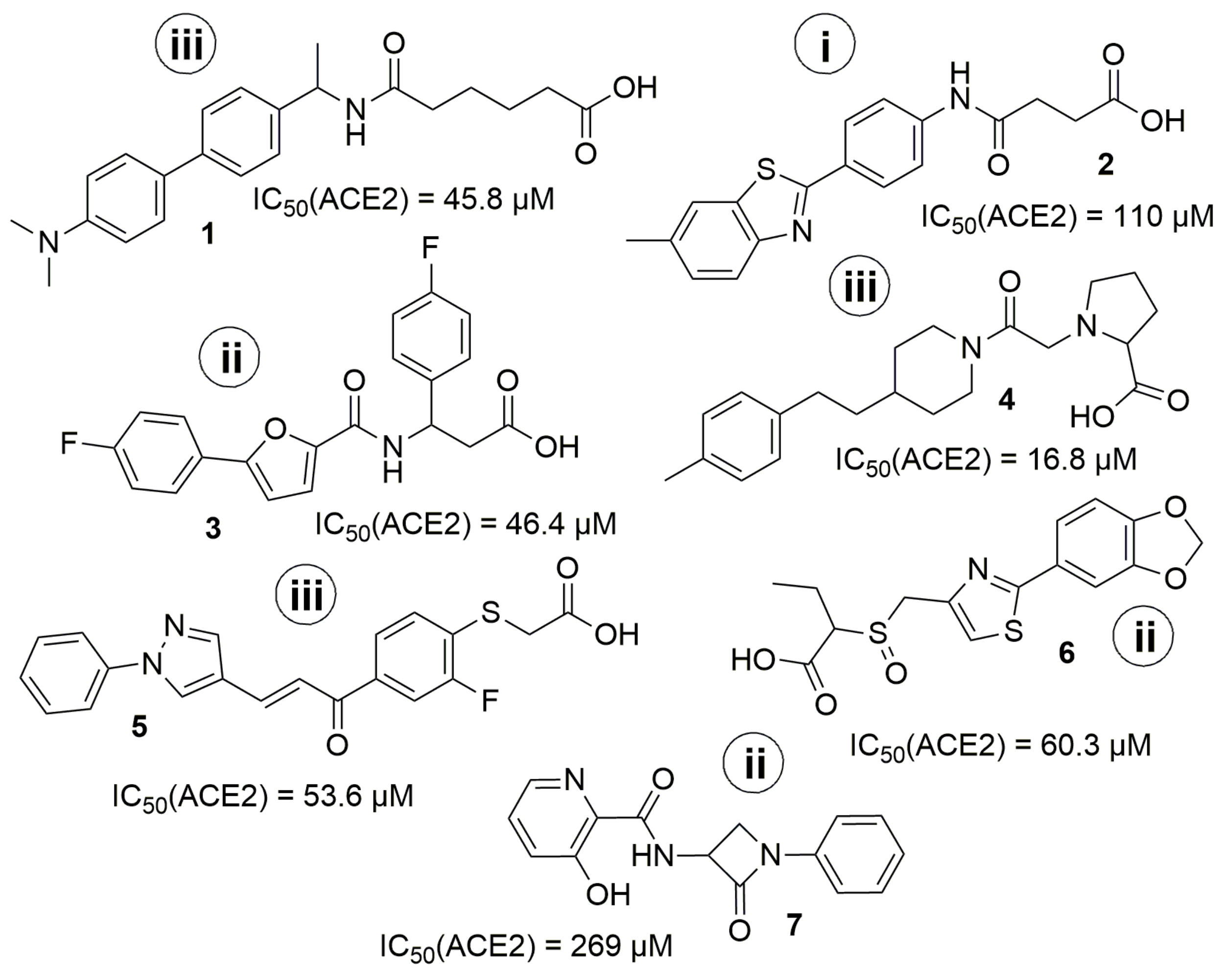
3.1. General Discussion of So-Far Known ACE2 Inhibitors
3.2. UNISTRA Pharmacophore-Based Screening
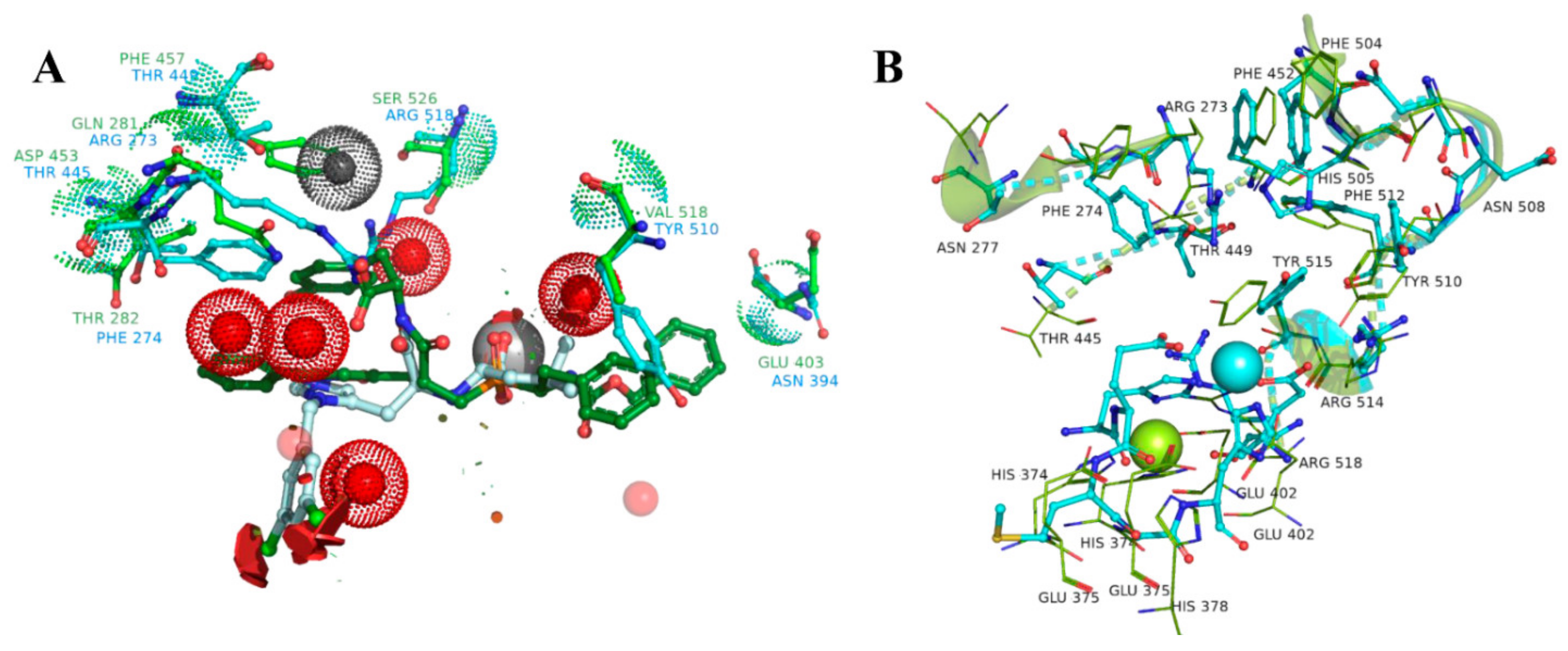
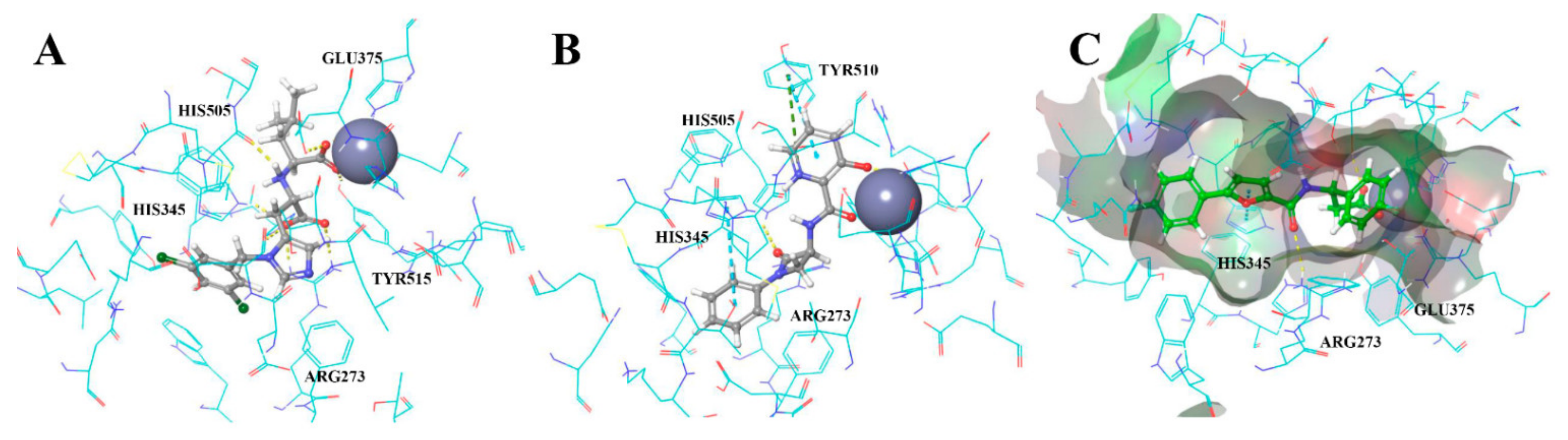
3.3. UNISTRA Docking Results
3.4. UNISTRA QSAR Modeling Results
3.5. ENAMINE MD-Based Docking Results
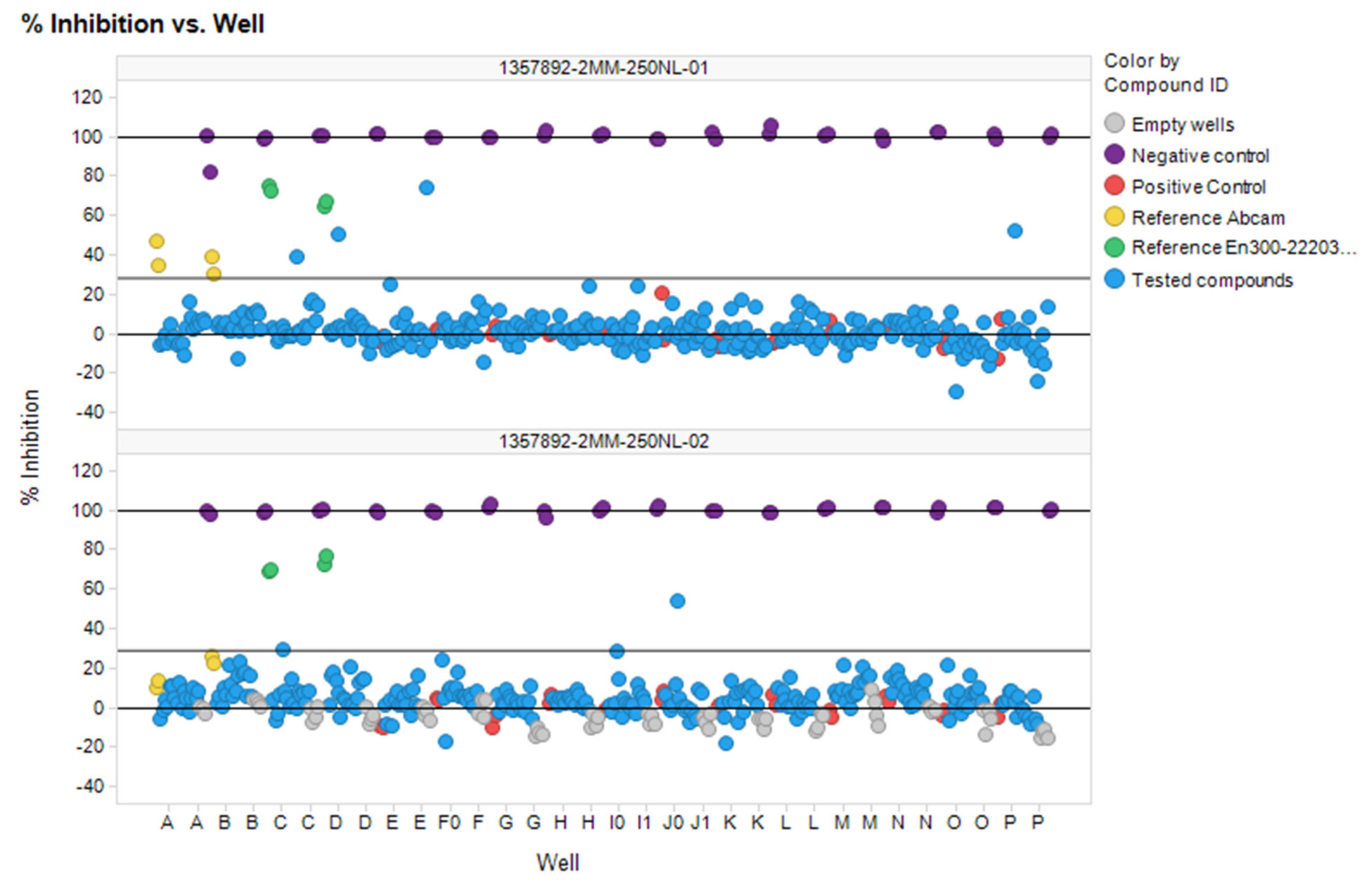
3.6. In Vitro ACE2 Screening
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Deaton, D.N.; Gao, E.N.; Graham, K.P.; Gross, J.W.; Miller, A.B.; Strelow, J.M. Thiol-based angiotensin-converting enzyme 2 inhibitors: P1 modifications for the exploration of the S1 subsite. Bioorg. Med. Chem. Lett. 2008, 18, 732–737. [Google Scholar] [CrossRef]
- Ma, L.L.; Liu, H.M.; Liu, X.M.; Yuan, X.Y.; Xu, C.; Wang, F.; Lin, J.Z.; Xu, R.C.; Zhang, D.K. Screening S protein - ACE2 blockers from natural products: Strategies and advances in the discovery of potential inhibitors of COVID-19. Eur J. Med. Chem 2021, 226, 113857. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, L.; Mo, M.; Liu, T.; Wu, C.; Gong, C.; Lu, K.; Gong, L.; Zhu, W.; Xu, Z. SARS-CoV-2 Omicron RBD shows weaker binding affinity than the currently dominant Delta variant to human ACE2. Signal Transduct. Target. Ther. 2022, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Sepehrinezhad, A.; Shahbazi, A.; Negah, S.S. COVID-19 virus may have neuroinvasive potential and cause neurological complications: A perspective review. J. Neurovirol. 2020, 26, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, V.; Blish, C.A.; South, A.M. A historical perspective on ACE2 in the COVID-19 era. J. Hum. Hypertens. 2021, 35, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L.; Kotz, J.D.; Li, M.; Aube, J.; Austin, C.P.; Reed, J.C.; Rosen, H.; White, E.L.; Sklar, L.A.; Lindsley, C.W.; et al. Advancing Biological Understanding and Therapeutics Discovery with Small-Molecule Probes. Cell 2015, 161, 1252–1265. [Google Scholar] [CrossRef] [Green Version]
- Workman, P.; Collins, I. Probing the probes: Fitness factors for small molecule tools. Chem. Biol. 2010, 17, 561–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrowsmith, C.H.; Audia, J.E.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.E.; Brown, P.J.; Bunnage, M.E.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Kravets, I.O.; Dudenko, D.V.; Pashenko, A.E.; Borisova, T.A.; Tolstanova, G.M.; Ryabukhin, S.V.; Volochnyuk, D.M. Virtual Screening in Search for a Chemical Probe for Angiotensin-Converting Enzyme 2 (ACE2). Molecules 2021, 26, 7584. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [Green Version]
- Varnek, A.; Fourches, D.; Horvath, D.; Klimchuk, O.; Gaudin, C.; Vayer, P.; Solov’ev, V.; Hoonakker, F.; Tetko, I.V.; Marcou, G. ISIDA - Platform for virtual screening based on fragment and pharmacophoric descriptors. Curr. Comput.-Aided Drug Des. 2008, 4, 191–198. [Google Scholar] [CrossRef]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA—An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput.-Aided Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef]
- Sidorov, P.; Gaspar, H.; Marcou, G.; Varnek, A.; Horvath, D. Mappability of drug-like space: Towards a polypharmacologically competent map of drug-relevant compounds. J. Comput.-Aided Mol. Des. 2015, 29, 1087–1108. [Google Scholar] [CrossRef]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Horvath, D.; Brown, J.; Marcou, G.; Varnek, A. An evolutionary optimizer of libsvm models. Challenges 2014, 5, 450–472. [Google Scholar] [CrossRef]
- Horvath, D.; Marcou, G.; Varnek, A. Predicting the Predictability: A Unified Approach to the Applicability Domain Problem of QSAR Models. J. Chem. Inf. Model. 2009, 49, 1762–1776. [Google Scholar]
- James, T.; Hsieh, M.L.; Knipling, L.; Hinton, D. Determining the Architecture of a Protein-DNA Complex by Combining FeBABE Cleavage Analyses, 3-D Printed Structures, and the ICM Molsoft Program. Methods Mol. Biol. 2015, 1334, 29–40. [Google Scholar] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Perez, A.; Marchan, I.; Svozil, D.; Sponer, J.; Cheatham, T.E., 3rd; Laughton, C.A.; Orozco, M. Refinement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Gready, J.E. Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. J. Comput. Chem. 2011, 32, 2031–2040. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM? A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Fan, Y.; Zhang, X.; Lan, H.; Yang, T.; Shao, M.; Li, S. Rapid Discrimination for Traditional Complex Herbal Medicines from Different Parts, Collection Time, and Origins Using High-Performance Liquid Chromatography and Near-Infrared Spectral Fingerprints with Aid of Pattern Recognition Methods. J. Anal. Methods Chem. 2015, 2015, 727589. [Google Scholar] [CrossRef] [Green Version]
- Mores, A.; Matziari, M.; Beau, F.; Cuniasse, P.; Yiotakis, A.; Dive, V. Development of potent and selective phosphinic peptide inhibitors of angiotensin-converting enzyme 2. J. Med. Chem. 2008, 51, 2216–2226. [Google Scholar] [CrossRef]
- Dales, N.A.; Gould, A.E.; Brown, J.A.; Calderwood, E.F.; Guan, B.; Minor, C.A.; Gavin, J.M.; Hales, P.; Kaushik, V.K.; Stewart, M.; et al. Substrate-based design of the first class of angiotensin-converting enzyme-related carboxypeptidase (ACE2) inhibitors. J. Am. Chem. Soc. 2002, 124, 11852–11853. [Google Scholar] [CrossRef] [PubMed]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. Transport of small molecules through the blood-brain barrier: Biology and methodology. Adv. Drug Deliv. Rev. 1995, 15, 5–36. [Google Scholar] [CrossRef]
- Jullien, N.; Makritis, A.; Georgiadis, D.; Beau, F.; Yiotakis, A.; Dive, V. Phosphinic tripeptides as dual angiotensin-converting enzyme C-domain and endothelin-converting enzyme-1 inhibitors. J. Med. Chem. 2010, 53, 208–220. [Google Scholar] [CrossRef]
- Almquist, R.G.; Jennings-White, C.; Chao, W.R.; Steeger, T.; Wheeler, K.; Rogers, J.; Mitoma, C. Synthesis and biological activity of pentapeptide analogues of the potent angiotensin converting enzyme inhibitor 5(S)-benzamido-4-oxo-6-phenylhexanoyl-L-proline. J. Med. Chem. 1985, 28, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.M.; Mir, S. Molecular docking: Approaches, types, applications and basic challenges. J. Anal. Bioanal. Tech. 2017, 8, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angiotensin II Converting Enzyme (ACE2) Inhibitor Screening Kit (ab273373). Available online: https://www.abcam.com/angiotensin-ii-converting-enzyme-ace2-inhibitor-screening-kit-ab273373.html (accessed on 10 May 2017).
- Daina, A.; Michielin, , O.; Zoete, , V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | 1:1 | 1:2 | 1:4 | 1:8 | ||||
|---|---|---|---|---|---|---|---|---|
| Enzyme | S/B | Z-Prime | S/B | Z-Prime | S/B | Z-Prime | S/B | Z-Prime |
| 1:1 | 24.29 | 0.92 | 13.14 | 0.87 | 7.26 | 0.80 | 4.44 | 0.82 |
| 1:2 | 13.61 | 0.90 | 7.68 | 0.94 | 4.46 | 0.90 | 2.97 | 0.79 |
| 1:4 | 6.77 | 0.94 | 4.09 | 0.87 | 2.62 | 0.78 | 1.92 | 0.82 |
| 1:8 | 4.56 | 0.89 | 2.87 | 0.80 | 1.96 | 0.73 | 1.56 | 0.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayevsky, A.V.; Poturai, A.S.; Kravets, I.O.; Pashenko, A.E.; Borisova, T.A.; Tolstanova, G.M.; Volochnyuk, D.M.; Borysko, P.O.; Vadzyuk, O.B.; Alieksieieva, D.O.; et al. In Vitro Evaluation of In Silico Screening Approaches in Search for Selective ACE2 Binding Chemical Probes. Molecules 2022, 27, 5400. https://doi.org/10.3390/molecules27175400
Rayevsky AV, Poturai AS, Kravets IO, Pashenko AE, Borisova TA, Tolstanova GM, Volochnyuk DM, Borysko PO, Vadzyuk OB, Alieksieieva DO, et al. In Vitro Evaluation of In Silico Screening Approaches in Search for Selective ACE2 Binding Chemical Probes. Molecules. 2022; 27(17):5400. https://doi.org/10.3390/molecules27175400
Chicago/Turabian StyleRayevsky, Alexey V., Andrii S. Poturai, Iryna O. Kravets, Alexander E. Pashenko, Tatiana A. Borisova, Ganna M. Tolstanova, Dmitriy M. Volochnyuk, Petro O. Borysko, Olga B. Vadzyuk, Diana O. Alieksieieva, and et al. 2022. "In Vitro Evaluation of In Silico Screening Approaches in Search for Selective ACE2 Binding Chemical Probes" Molecules 27, no. 17: 5400. https://doi.org/10.3390/molecules27175400
APA StyleRayevsky, A. V., Poturai, A. S., Kravets, I. O., Pashenko, A. E., Borisova, T. A., Tolstanova, G. M., Volochnyuk, D. M., Borysko, P. O., Vadzyuk, O. B., Alieksieieva, D. O., Zabolotna, Y., Klimchuk, O., Horvath, D., Marcou, G., Ryabukhin, S. V., & Varnek, A. (2022). In Vitro Evaluation of In Silico Screening Approaches in Search for Selective ACE2 Binding Chemical Probes. Molecules, 27(17), 5400. https://doi.org/10.3390/molecules27175400