
Density Functional Theory Study on NiNx (x = 1, 2, 3, 4) Catalytic Hydrogenation of Acetylene

Abstract
:
1. Introduction
2. Calculation Methods
3. Results and Discussion
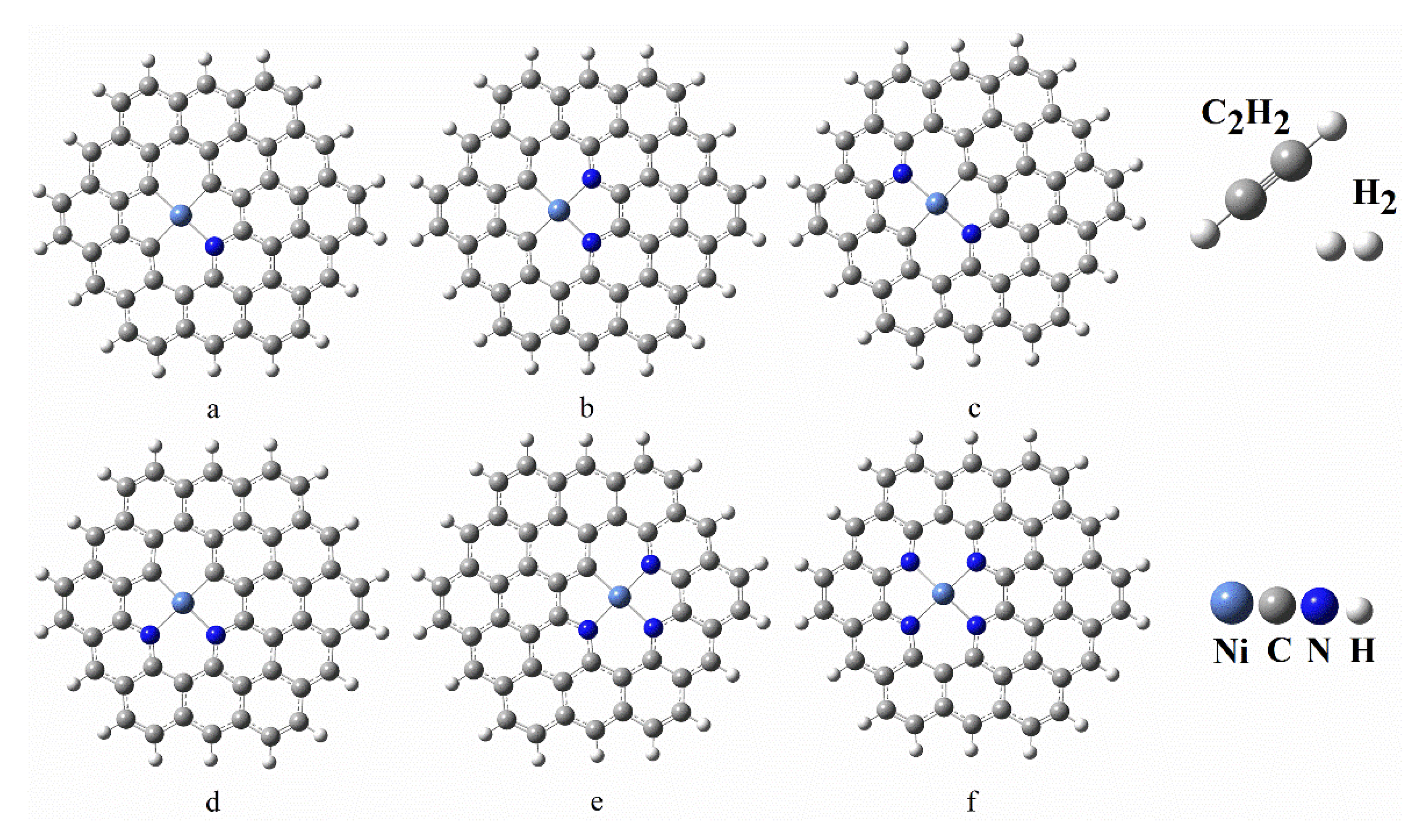
3.1. Geometries of Reactants
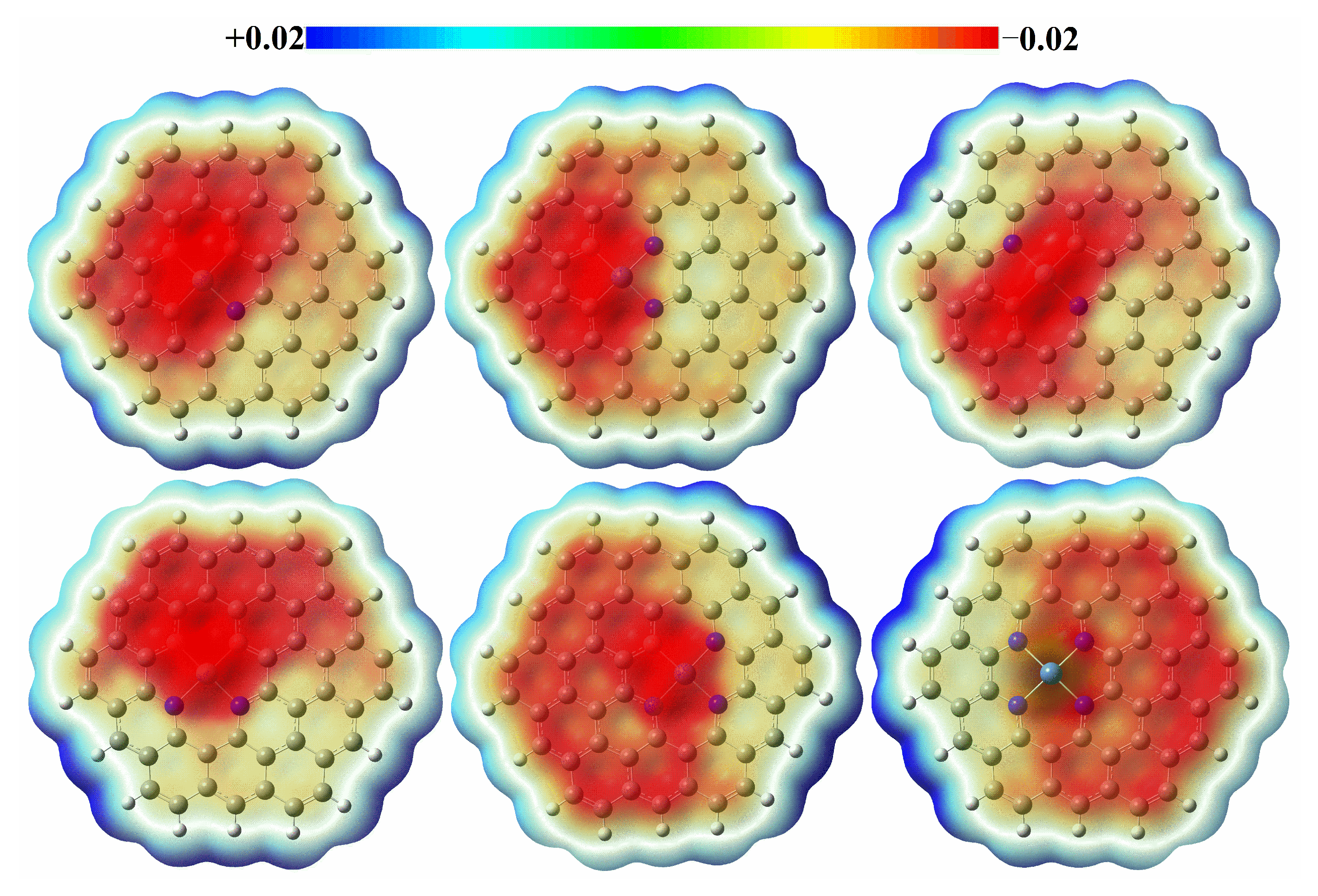
3.2. Adsorption of Reactants
3.3. Reaction Mechanism of Acetylene Hydrogenation Catalyzed Using Graphene-NiNx Catalysts (x = 1, 2, 3, 4)
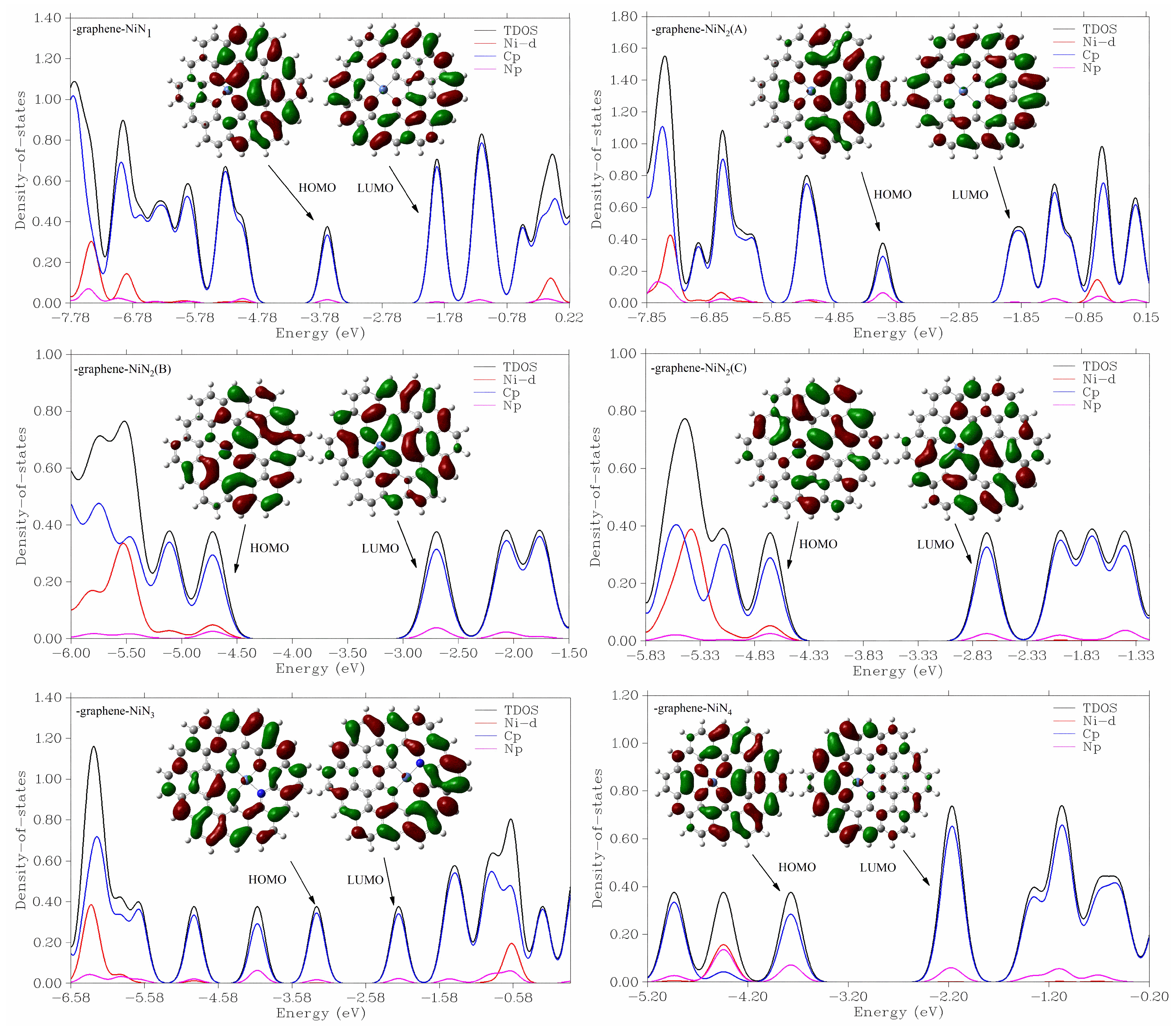
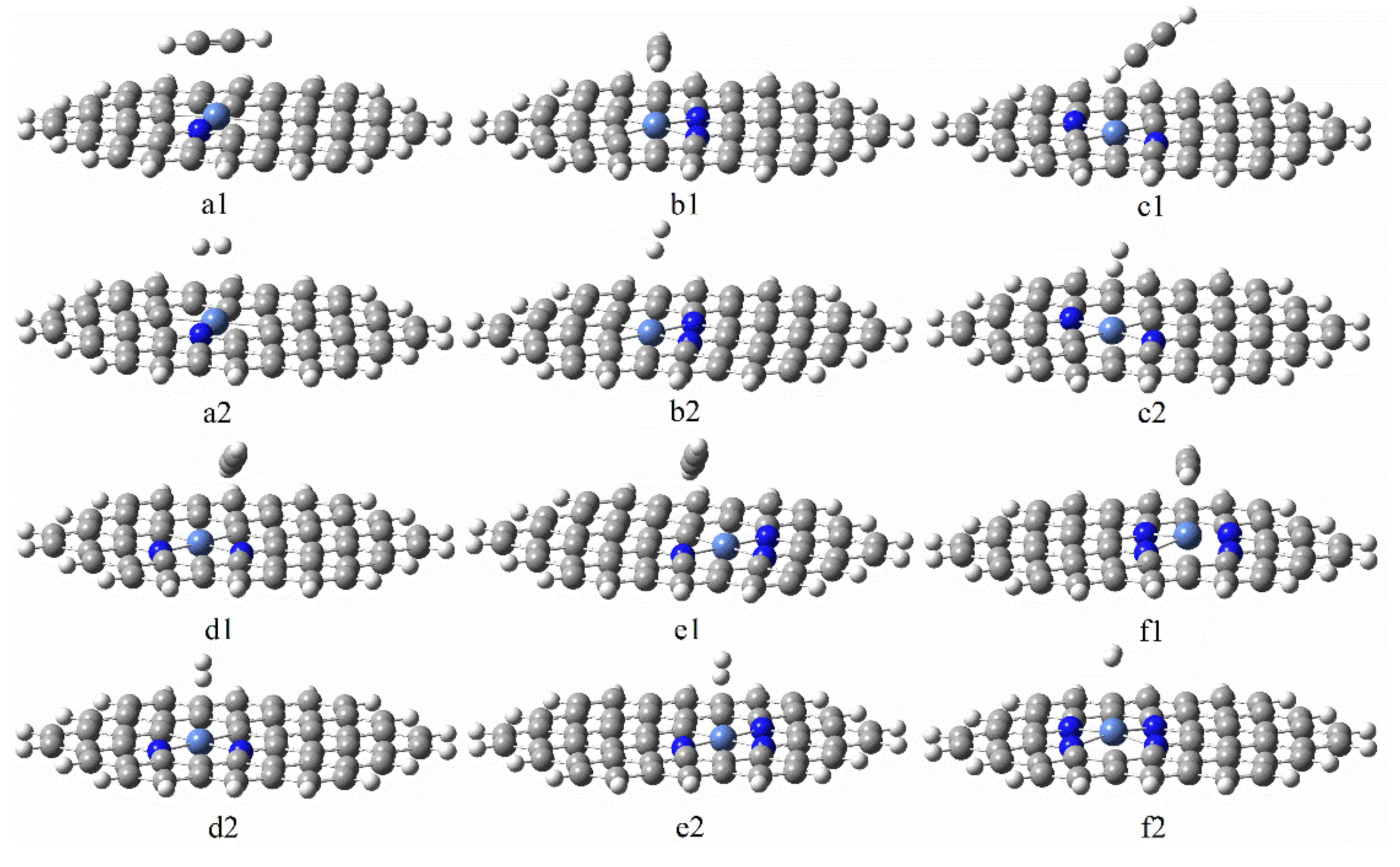
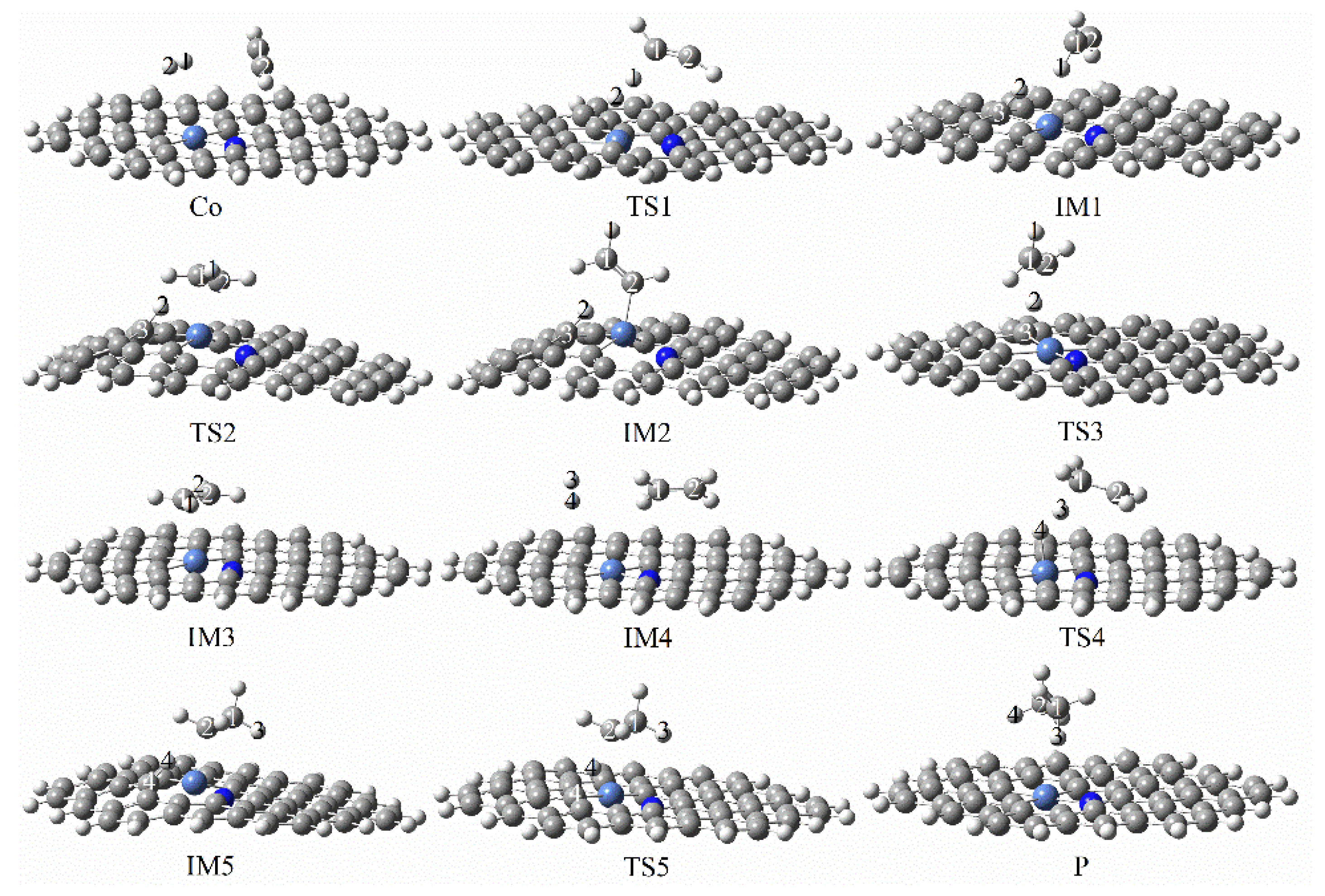
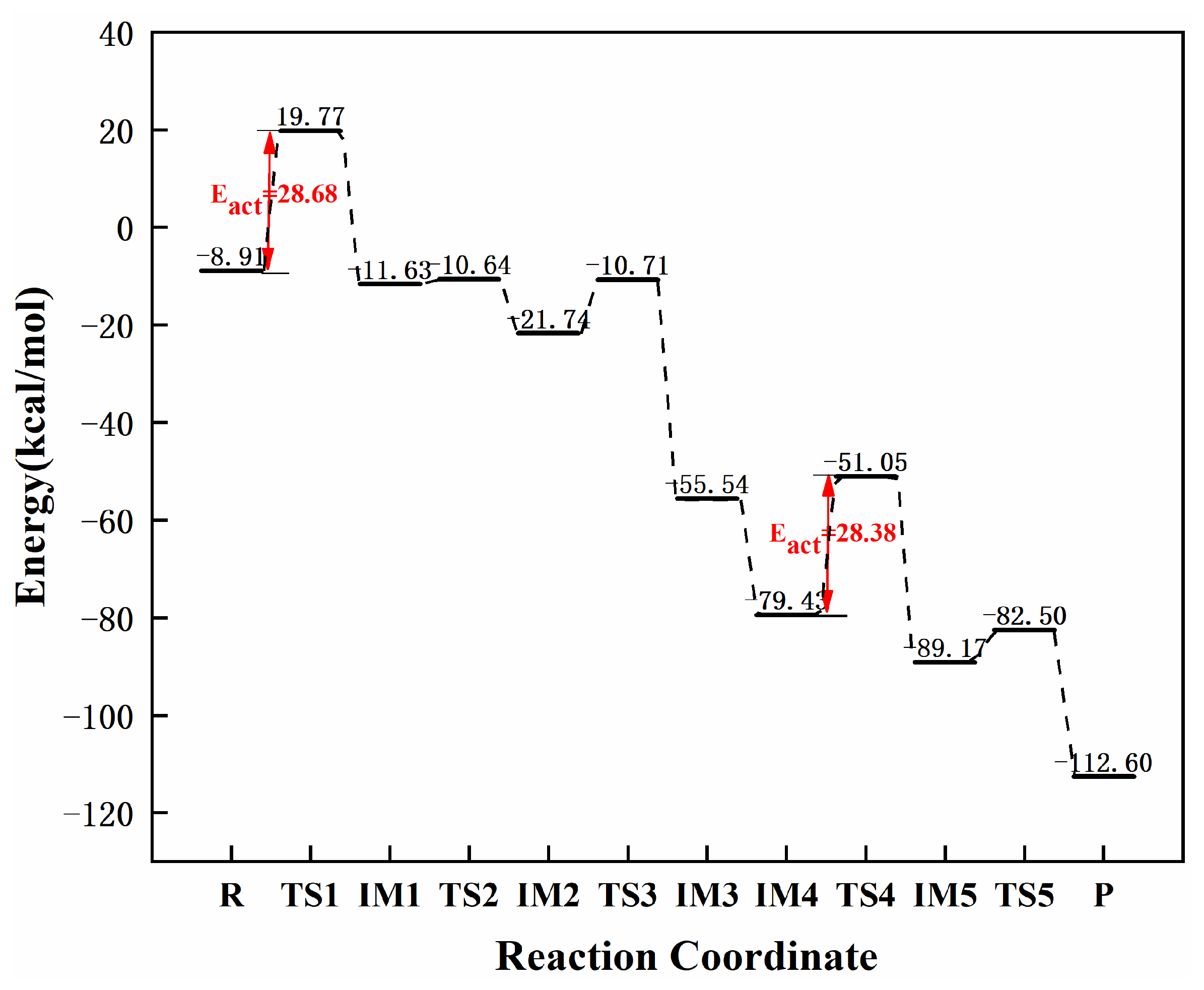
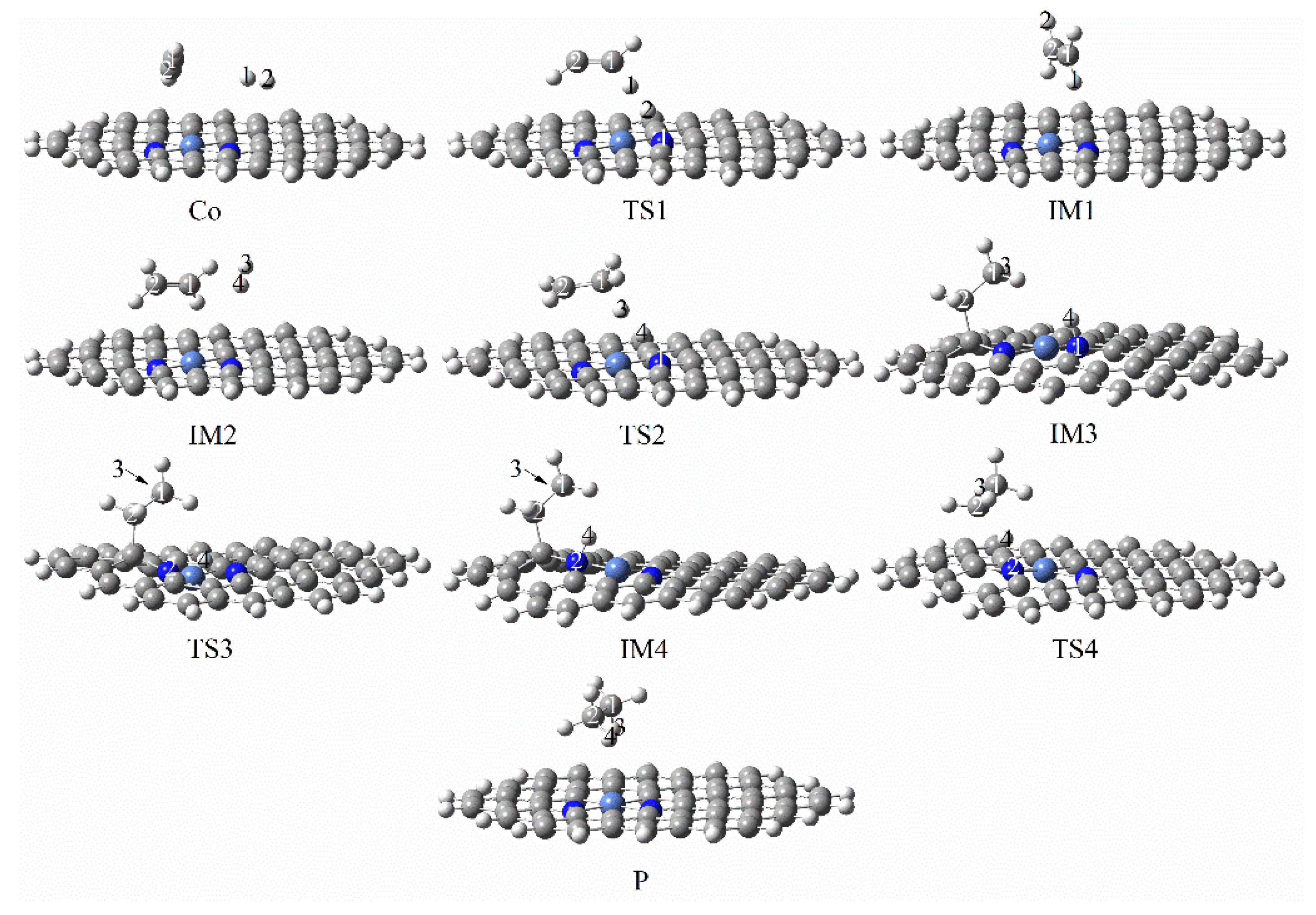
3.3.1. Reaction Mechanism Utilizing Graphene-NiN1
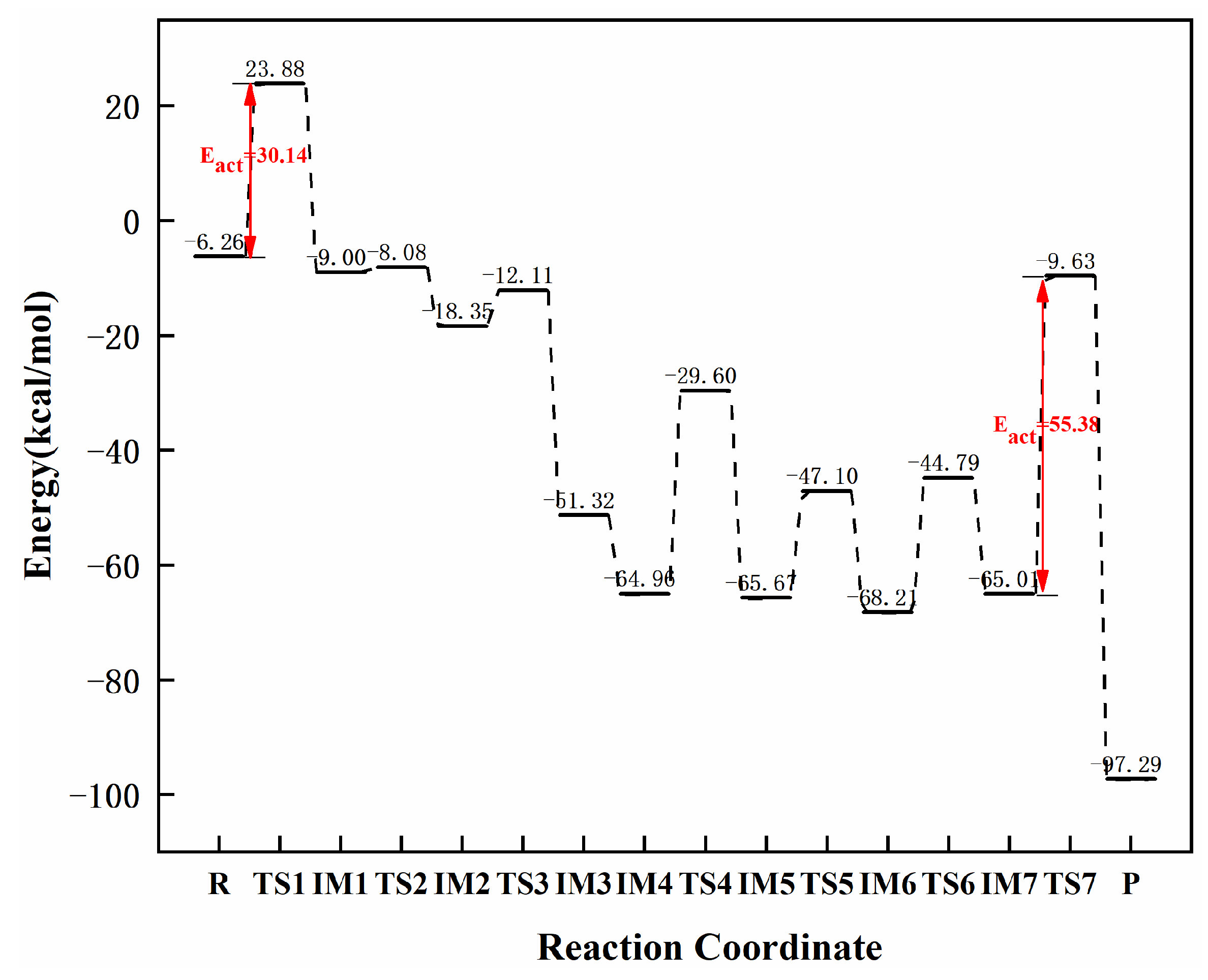
3.3.2. Reaction Mechanism Utilizing Graphene-NiN2 (A)
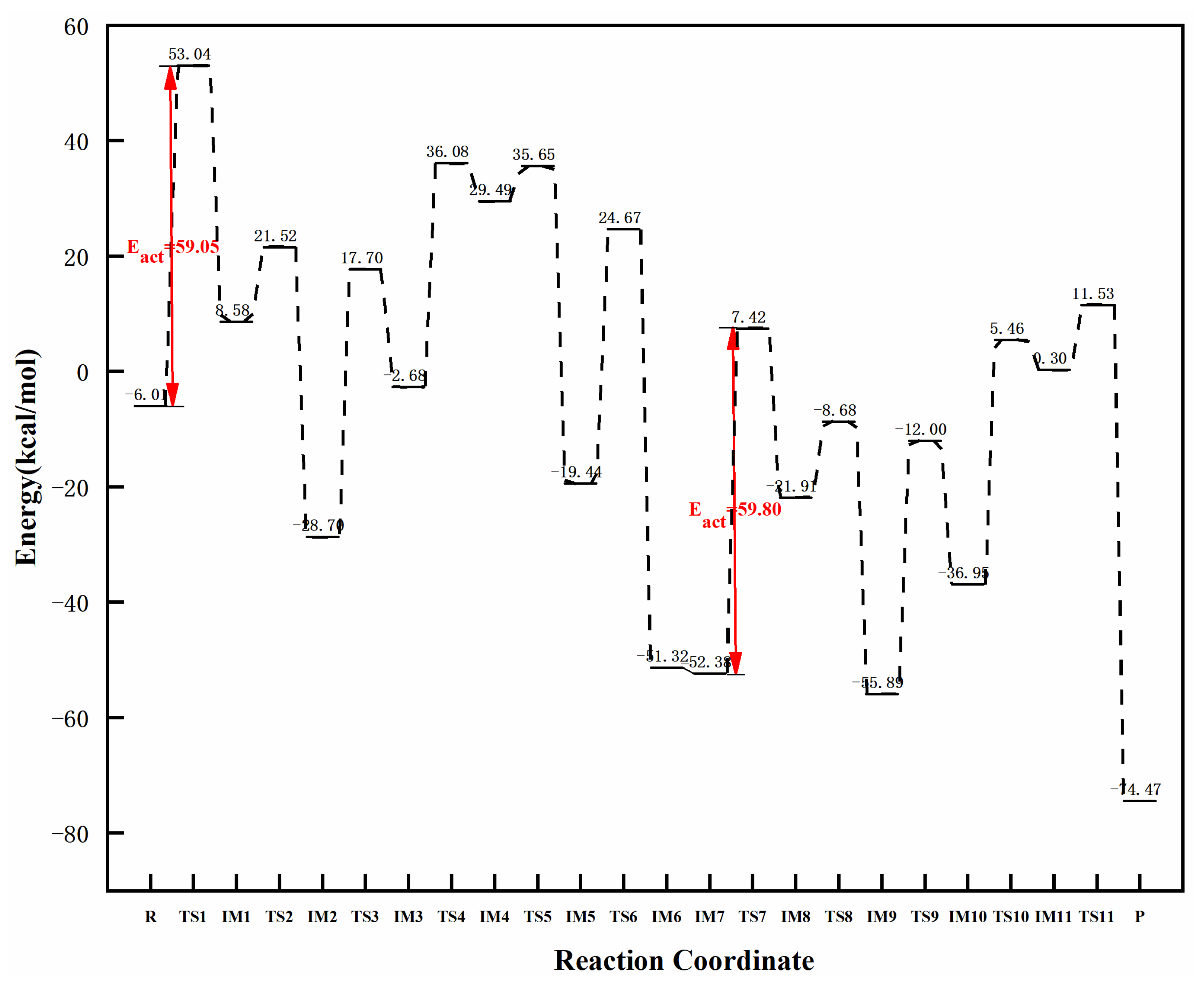
3.3.3. Reaction Mechanism Utilizing Graphene-NiN2 (B)
3.3.4. Reaction Mechanism Utilizing Graphene-NiN2 (C)
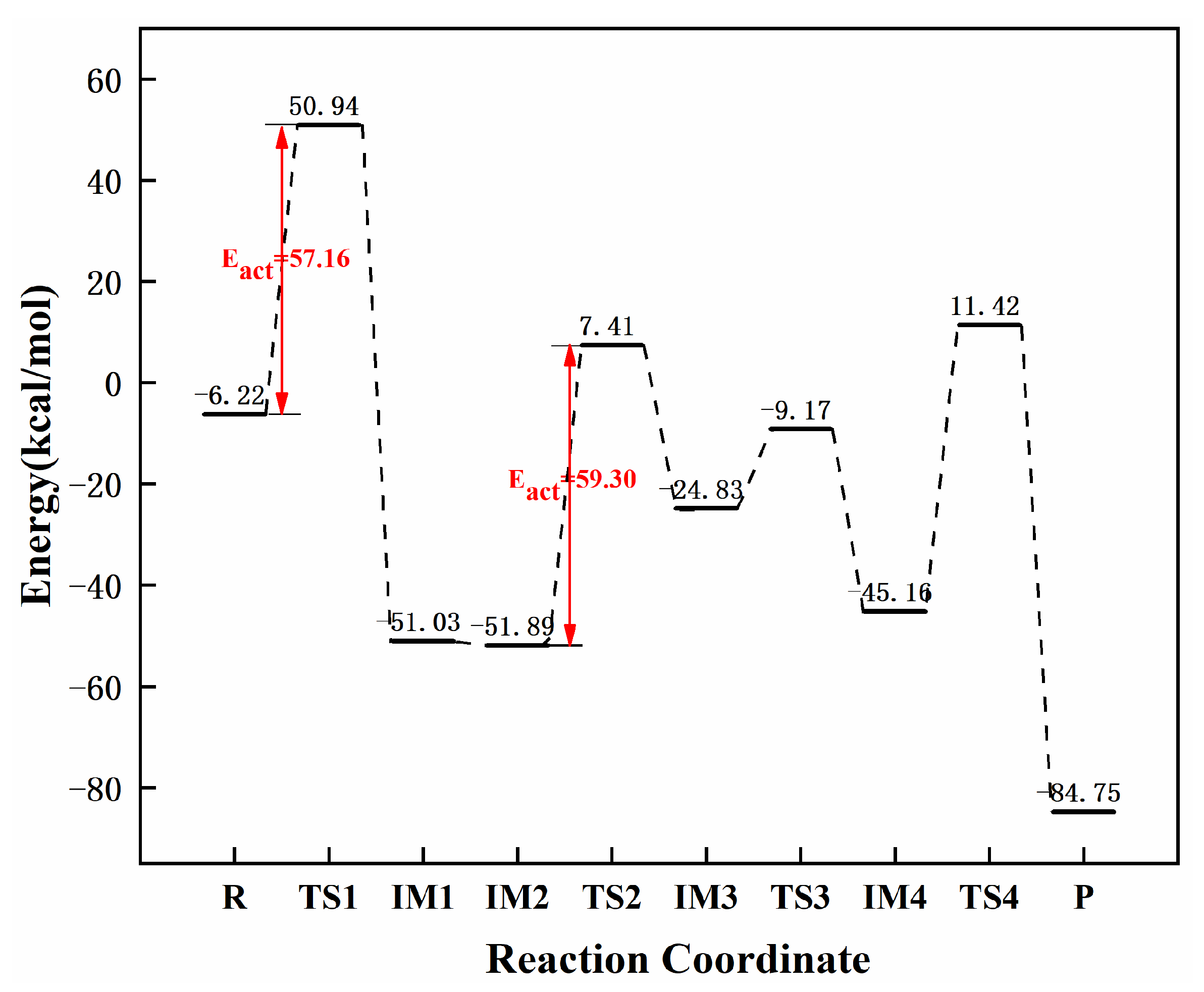
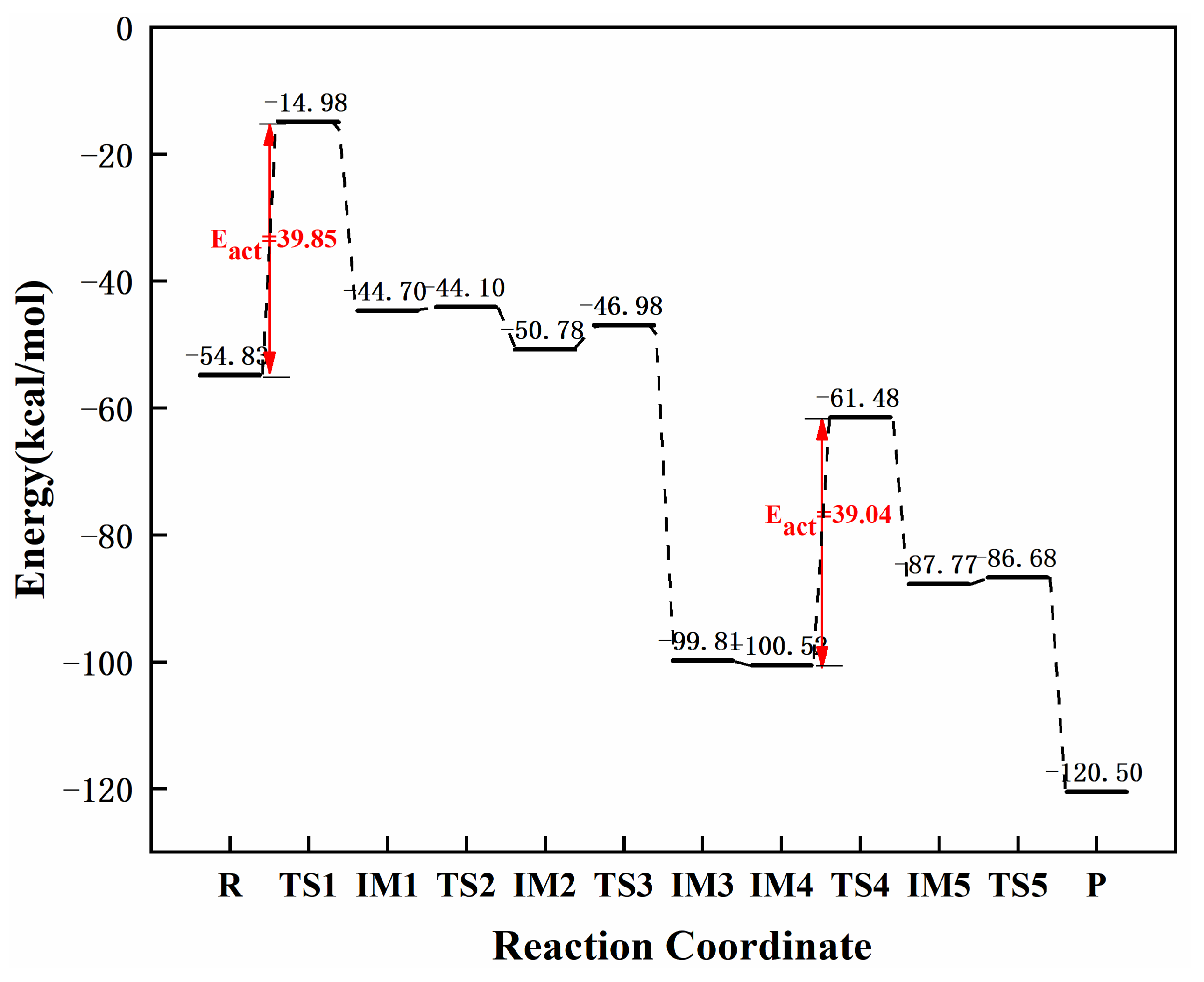
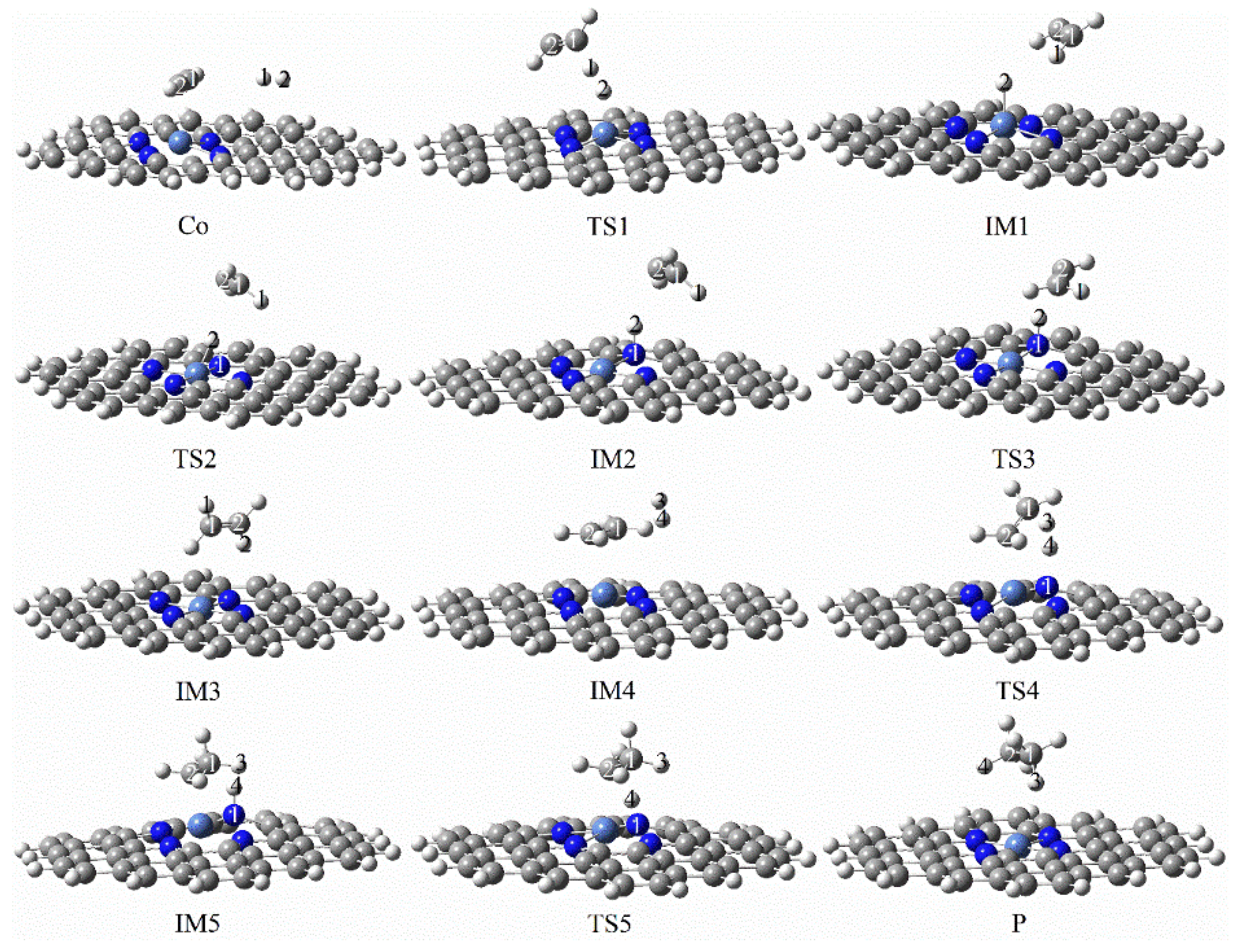
3.3.5. Reaction Mechanism Utilizing Graphene-NiN3
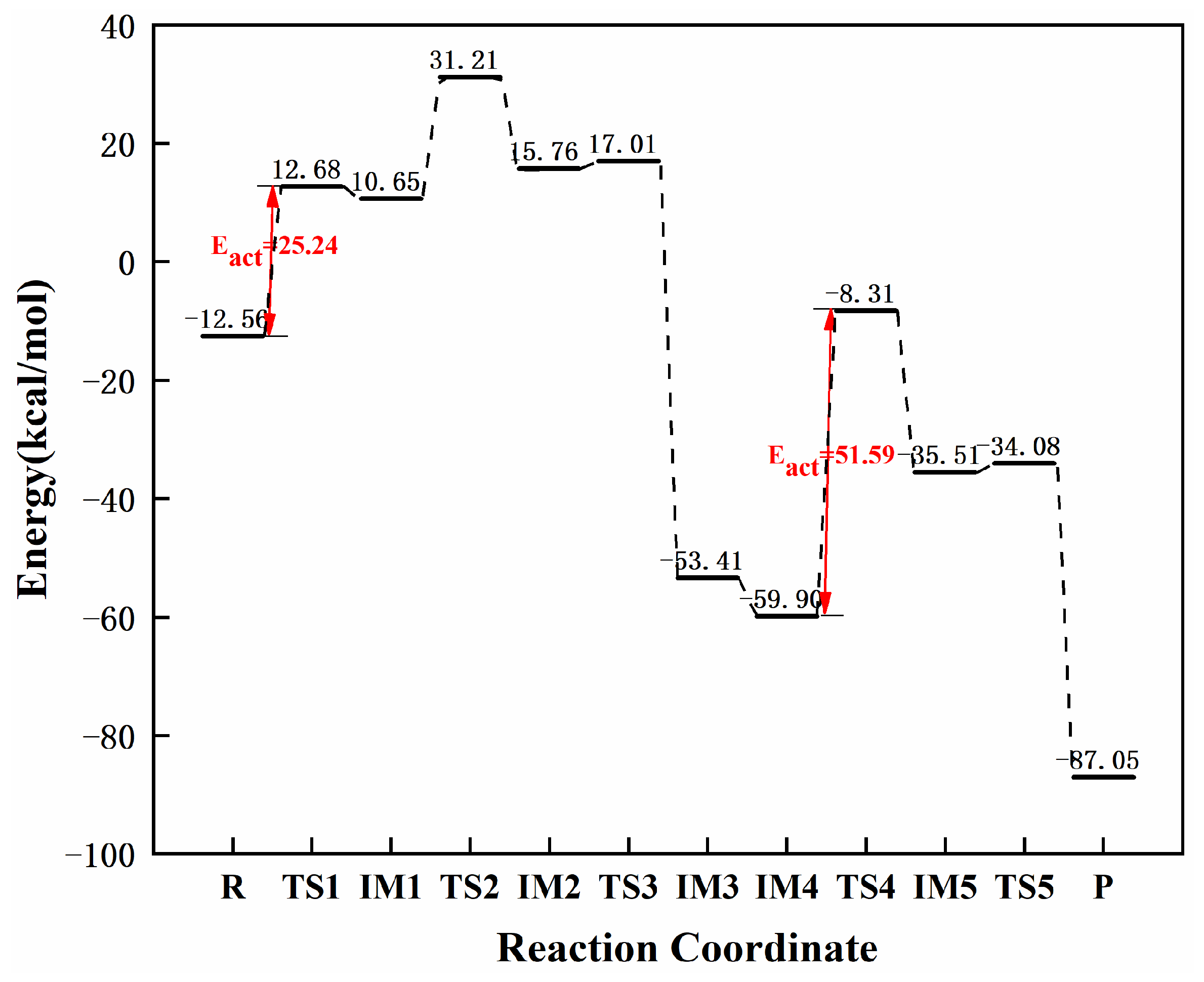
3.3.6. Reaction Mechanism Utilizing Graphene-NiN4
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Glyzdova, D.V.; Smirnova, N.S.; Shlyapin, D.A.; Tsyrul’nikov, P.G. Gas-Phase and Liquid-Phase Hydrogenation of Acetylene in Lean and Enriched Mixtures over Supported Modified Palladium Catalysts. Russ. J. Gen. Chem. 2020, 90, 1120–1140. [Google Scholar] [CrossRef]
- Khan, N.A.; Shaikhutdinov, S.; Freund, H.J. Acetylene and ethylene hydrogenation on alumina supported Pd-Ag model catalysts. Catal. Lett. 2006, 108, 159–164. [Google Scholar] [CrossRef]
- Borodzinki, A. Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts. Part 1. Effect of changes to the catalyst during reaction. Catal. Rev.-Sci. Eng. 2006, 48, 91–144. [Google Scholar] [CrossRef]
- Borodzinski, A.; Bond, G.C. Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts, Part 2: Steady-state kinetics and effects of palladium particle size, carbon monoxide, and promoters. Catal. Rev. Sci. Eng. 2008, 50, 379–469. [Google Scholar] [CrossRef]
- Ball, M.R.; Rivera-Dones, K.R.; Gilcher, E.B.; Ausman, S.F.; Hullfish, C.W.; Lebron, E.A.; Dumesic, J.A. AgPd and CuPd Catalysts for Selective Hydrogenation of Acetylene. Acs Catal. 2020, 10, 8567–8581. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Sivadinarayana, C.; Datye, A.K.; Kumar, D.; Goodman, D.W. Acetylene hydrogenation on Au-based catalysts. Catal. Lett. 2003, 86, 1–8. [Google Scholar] [CrossRef]
- Zhuo, H.-Y.; Yu, X.; Yu, Q.; Xiao, H.; Zhang, X.; Li, J. Selective hydrogenation of acetylene on graphene-supported non-noble metal single-atom catalysts. Sci. China-Mater. 2020, 63, 1741–1749. [Google Scholar] [CrossRef]
- Riley, C.; De La Riva, A.; Zhou, S.; Wan, Q.; Peterson, E.; Artyushkova, K.; Farahani, M.D.; Friedrich, H.B.; Burkemper, L.; Atudorei, N.-V.; et al. Synthesis of Nickel-Doped Ceria Catalysts for Selective Acetylene Hydrogenation. Chemcatchem 2019, 11, 1526–1533. [Google Scholar] [CrossRef]
- Abdollahi, T.; Farmanzadeh, D. Graphene-supported Cu-11 nanocluster as a candidate catalyst for the selective hydrogenation of acetylene: A density functional study. J. Alloys Compd. 2018, 735, 117–130. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.-E.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Kalavakunda, V.; Hosmane, N.S. Graphene and its analogues. Nanotechnol. Rev. 2016, 5, 369–376. [Google Scholar] [CrossRef]
- Wei, Z.; Hou, Y.; Yang, Y.; Liu, Y. The progress on graphene-based catalysis. Curr. Org. Chem. 2016, 20, 2055–2082. [Google Scholar] [CrossRef]
- Fauzi, F.; Rianjanu, A.; Santoso, I.; Triyana, K. Gas and humidity sensing with quartz crystal microbalance (QCM) coated with graphene-based materials–A mini review. Sens. Actuators A Phys. 2021, 330, 112837. [Google Scholar] [CrossRef]
- Cai, X.; Lai, L.; Shen, Z.; Lin, J. Graphene and graphene-based composites as Li-ion battery electrode materials and their application in full cells. J. Mater. Chem. A 2017, 5, 15423–15446. [Google Scholar] [CrossRef]
- Xinmin, H.; Ting, Z.; Fei, C.; Jun, J. Applications of graphene in composite thermoelectric materials. Prog. Chem. 2018, 30, 439. [Google Scholar]
- Liu, Y.; Ge, X.; Li, J. Graphene lubrication. Appl. Mater. Today 2020, 20, 100662. [Google Scholar] [CrossRef]
- Yang, F.; Song, P.; Liu, X.; Mei, B.; Xing, W.; Jiang, Z.; Gu, L.; Xu, W. Highly efficient CO2 electroreduction on ZnN4-based single-atom catalyst. Angew. Chem. Int. Ed. 2018, 57, 12303–12307. [Google Scholar] [CrossRef]
- Wagner, S.; Auerbach, H.; Tait, C.E.; Martinaiou, I.; Kumar, S.C.; Kübel, C.; Sergeev, I.; Wille, H.C.; Behrends, J.; Wolny, J.A. Elucidating the Structural Composition of an Fe–N–C Catalyst by Nuclear-and Electron-Resonance Techniques. Angew. Chem. 2019, 131, 10596–10602. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, X.; Wu, D.; Han, X.; Zhang, L.; Wang, H.; Xu, F.; Gao, Z.; Jiang, K. Ni and nitrogen-codoped ultrathin carbon nanosheets with strong bonding sites for efficient CO2 electrochemical reduction. J. Colloid Interface Sci. 2020, 570, 31–40. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Yang, Z. CO2 reduction on metal-and nitrogen-codoped graphene: Balancing activity and selectivity via coordination engineering. ACS Sustain. Chem. Eng. 2020, 8, 6134–6141. [Google Scholar] [CrossRef]
- Xu, L.; Deng, D.; Tian, Y.; Li, H.; Qian, J.; Wu, J.; Li, H. Dual-active-sites design of CoNx anchored on zinc-coordinated nitrogen-codoped porous carbon with efficient oxygen catalysis for high-stable rechargeable zinc-air batteries. Chem. Eng. J. 2021, 408, 127321. [Google Scholar] [CrossRef]
- Chen, Z.; Mou, K.; Yao, S.; Liu, L. Zinc-coordinated nitrogen-codoped graphene as an efficient catalyst for selective electrochemical reduction of CO2 to CO. ChemSusChem 2018, 11, 2944–2952. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Kang, L. A DFT study of graphene-FeNx (x = 4, 3, 2, 1) catalysts for acetylene hydrochlorination. Colloids Surf. A Physicochem. Eng. Asp. 2021, 618, 126495. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Zheng, L.; Shang, J.; Wan, X.; Hu, R.; Guo, X.; Hong, S.; Shui, J. Preparation of Fe–N–C catalysts with FeNx (x= 1, 3, 4) active sites and comparison of their activities for the oxygen reduction reaction and performances in proton exchange membrane fuel cells. J. Mater. Chem. A 2019, 7, 26147–26153. [Google Scholar] [CrossRef]
- Lin, L.; Li, H.; Wang, Y.; Li, H.; Wei, P.; Nan, B.; Si, R.; Wang, G.; Bao, X. Temperature-Dependent CO2 Electroreduction over Fe-N-C and Ni-N-C Single-Atom Catalysts. Angew. Chem. 2021, 133, 26786–26790. [Google Scholar] [CrossRef]
- Li, Y.; Adli, N.M.; Shan, W.; Wang, M.; Zachman, M.J.; Hwang, S.; Tabassum, H.; Karakalos, S.; Feng, Z.; Wang, G. Atomically dispersed single Ni site catalysts for high-efficiency CO2 electroreduction at industrial-level current densities. Energy Environ. Sci. 2022, 15, 2108–2119. [Google Scholar] [CrossRef]
- Wu, X.; Chen, J.; Wang, M.; Li, X.; Yang, L.; Li, G.; Shan, L.; Li, X.; Lin, Y.; Jiang, J. High-curvature carbon-supported Ni single atoms with charge polarization for highly efficient CO2 reduction. Chem. Commun. 2022, 58, 2914–2917. [Google Scholar] [CrossRef]
- Wang, Y.; You, L.; Zhou, K. Origin of the N-coordinated single-atom Ni sites in heterogeneous electrocatalysts for CO2 reduction reaction. Chem. Sci. 2021, 12, 14065–14073. [Google Scholar] [CrossRef]
- Ma, Y.; Fan, H.; Wu, C.; Zhang, M.; Yu, J.; Song, L.; Li, K.; He, J. An efficient dual-metal single-atom catalyst for bifunctional catalysis in zinc-air batteries. Carbon 2021, 185, 526–535. [Google Scholar] [CrossRef]
- Hu, X.; Yao, S.; Chen, L.; Zhang, X.; Jiao, M.; Lu, Z.; Zhou, Z. Understanding the role of axial O in CO2 electroreduction on NiN4 single-atom catalysts via simulations in realistic electrochemical environment. J. Mater. Chem. A 2021, 9, 23515–23521. [Google Scholar] [CrossRef]
- Maulana, A.L.; Saputro, A.G.; Prasetyo, Y.; Mahyuddin, M.H.; Iqbal, M.; Yudistira, H.T.; Wenten, I.G.; Dipojono, H.K. Two-Electron Electrochemical Reduction of CO2 on B-Doped Ni–N–C Catalysts: A First-Principles Study. J. Phys. Chem. C 2021, 125, 19247–19258. [Google Scholar] [CrossRef]
- Chen, J.; Li, H.; Fan, C.; Meng, Q.; Tang, Y.; Qiu, X.; Fu, G.; Ma, T. Dual single-atomic Ni-N4 and Fe-N4 sites constructing Janus hollow graphene for selective oxygen electrocatalysis. Adv. Mater. 2020, 32, 2003134. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, J.; Wei, S.; Bi, S.; Xia, Y.; Chen, M.; Hou, Y.; Qiu, M.; Yuan, C.; Su, Y. Atomic Ni anchored covalent triazine framework as high efficient electrocatalyst for carbon dioxide conversion. Adv. Funct. Mater. 2019, 29, 1806884. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, T.-S.; Hong, S.; Fan, Q.; Soo, Y.-L.; Masa, J.; Qiu, J.; Sun, Z. Efficient electrochemical reduction of CO2 by Ni–N catalysts with tunable performance. ACS Sustain. Chem. Eng. 2019, 7, 15030–15035. [Google Scholar] [CrossRef]
- Yang, F.; Wang, M.; Liu, W.; Yang, B.; Wang, Y.; Luo, J.; Tang, Y.; Hou, L.; Li, Y.; Li, Z. Atomically dispersed Ni as the active site towards selective hydrogenation of nitroarenes. Green Chem. 2019, 21, 704–711. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision D, 1st ed.; Gaussian, Inc.: Wallingford, CT, USA, 2009; Available online: http://www.gaussian.com (accessed on 15 November 2021).
- Lavine, M.S. A comparison of DFT methods. Science 2016, 351, 1411–1413. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| d (Ni–N) | d (Ni–C) | |
|---|---|---|
| graphene-NiN1 | 1.951 | 1.880 |
| graphene-NiN2 (A) | 1.922 | 1.871 |
| graphene-NiN2 (B) | 1.932 | 1.872 |
| graphene-NiN2 (C) | 1.951 | 1.863 |
| graphene-NiN3 | 1.909 | 1.862 |
| graphene-NiN4 | 1.961 | -- |
| Catalyst | C2H2 | H2 | Co-Adsorption |
|---|---|---|---|
| graphene-NiN1 | −8.43 | −0.60 | −8.91 |
| graphene-NiN2 (A) | −4.11 | −1.23 | −6.26 |
| graphene-NiN2 (B) | −5.22 | −1.35 | −6.01 |
| graphene-NiN2 (C) | −5.29 | −1.42 | −6.22 |
| graphene-NiN3 | −53.83 | −50.13 | −54.83 |
| graphene-NiN4 | −11.55 | −1.79 | −12.56 |
| Catalyst | Eethylene barrier | Eethane barrier | Selectivity | Source |
|---|---|---|---|---|
| graphene-NiN1 | 28.68 | 28.38 | 0.30 | this work |
| graphene-NiN2 (A) | 30.14 | 55.38 | 25.24 | this work |
| graphene-NiN2 (B) | 59.05 | 59.80 | 0.75 | this work |
| graphene-NiN2 (C) | 57.16 | 59.30 | 2.14 | this work |
| graphene-NiN3 | 39.85 | 39.04 | 0.81 | this work |
| graphene-NiN4 | 25.24 | 51.59 | 26.35 | this work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, C.; Kang, L.; Zhu, M. Density Functional Theory Study on NiNx (x = 1, 2, 3, 4) Catalytic Hydrogenation of Acetylene. Molecules 2022, 27, 5437. https://doi.org/10.3390/molecules27175437
Hou C, Kang L, Zhu M. Density Functional Theory Study on NiNx (x = 1, 2, 3, 4) Catalytic Hydrogenation of Acetylene. Molecules. 2022; 27(17):5437. https://doi.org/10.3390/molecules27175437
Chicago/Turabian StyleHou, Cuili, Lihua Kang, and Mingyuan Zhu. 2022. "Density Functional Theory Study on NiNx (x = 1, 2, 3, 4) Catalytic Hydrogenation of Acetylene" Molecules 27, no. 17: 5437. https://doi.org/10.3390/molecules27175437