

Small Molecule Inhibitors for Hepatocellular Carcinoma: Advances and Challenges
 , ,
, ,  and
and
Abstract
:Highlights:
- Multi tyrosine kinase inhibitors licensed for HCC treatment.
- Multi kinase inhibitors not licensed for HCC treatment.
- Inhibitors of Growth Factor Receptors.
- Small molecules acting as immunomodulators.
- Small molecules inhibiting crucial HCC pathways.
- Small molecules targeting various molecular targets.
Simple Summary
Abstract
1. Introduction
2. Methods
3. Results
4. Tyrosine Kinases Inhibitors
4.1. Multi-Kinase Inhibitors Currently Licensed for HCC
4.1.1. Sorafenib (BAY 43-9006)
Interferon-Lambda 3
Pregnenolone, Lomustine, Carisoprodol, Prestwick-1100, Chlorambucil, and Bretylium Tosilate
MLN4924
Luteolin
SC-2001
Wogonin
419S1 and 420S1
Doxorubicin
Erlotinib
Pravastatin
Celastrol
PI-103
4.1.2. Rogerafenib (BAY 73-4506)
4.1.3. Cabozantinib (XL184, BMS-907351)
4.1.4. Lenvatinib (E7080)
4.2. Multi-Kinase Inhibitors Not Currently Licensed for HCC
4.2.1. Sunitinib (SU11248)
4.2.2. Erlotinib (CP-358774, OSI-774)
4.2.3. Brivanib (BMS-540215)
4.2.4. Cediranib (AZD2171)
4.2.5. Linifanib (ABT-869)
4.2.6. Nintedanib (BIBF1120)
4.2.7. Refametinib (RDEA119/BAY 869766)
4.2.8. Vatalanib (PTK787/ZK222584)
4.2.9. Vandetanib (ZD6474)
4.2.10. Pazopanib (GW786034)
4.2.11. Tivantinib (ARQ 197)
4.2.12. Apatinib (YN968D1)
4.2.13. Dasatinib (BMS-354825)
4.2.14. Imatinib
4.2.15. Gefitinib (E1203)
4.2.16. Lapatinib
4.2.17. Linsitinib (OSI-906/DB06075)
4.2.18. Orantinib (TSU-68)
4.2.19. Axitinib
4.2.20. Donafenib
4.2.21. Anlotinib
4.2.22. Dovitinib (TKI258)
4.2.23. PD0325901
4.2.24. R1498
4.2.25. SGX523
4.2.26. PHA665752
4.2.27. Tepotinib (EMD 1214063)
4.2.28. BLU-9931
4.2.29. FGF401

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Study Design | Sample Size | Results/Primary Endpoint | Secondary Endpoints, Efficacy and Safety | Ref. |
|---|---|---|---|---|---|---|
| Sorafenib vs. Placebo(SHARP) | EGFRs, KIT, PDGFRs, and RAF | phase III; First line; Randomized; Multicenter; Double-blind NCT00105443 | n = 602 | OS Sorafenib: 10.7, Placebo: 7.9 months | TTP (months): Sorafenib: 5.5; Placebo: 2.8 ORR: 2% DCR: 43% TEAEs: 80%; AEs: 52% | [12] |
| Regorafenib vs. Placebo(RESORCE) | EGFR1–3, PDGFR-β, FGFR1, KIT, RET and B-RAF | Phase III; Second line; Randomized; International; Double-blind NCT01774344 | n = 573 | OS Regorafenib: 10.6 Placebo: 7.8 months | TTP (months): Regorafenib: 3.2; Placebo: 1.5 PFS (months): Regorafenib: 3.1; Placebo: 1.5, DCR: 65% ORR: 11% TEAEs: 100%; Grade 3/4 TEAEs: 67%; SAEs: 44 | [69,70] |
| Cabozantinib vs. Placebo (CELESTIAL) | VEGFR1–3, MET, RET and KIT | Phase III; Second line; Randomized; Double-blind NCT01908426 | n = 707 | OS Cabozantinib: 10.2 Placebo: 8 months | PFS (months): Cabozantinib: 5.2; Placebo: 1.9 DCR: 64%, ORR: 4%; Grade 3/4 AEs: 68%; AEs:50% | [76] |
| Lenvatinib vs. Sorafenib (REFLECT) | EGFR1–3, FGFR1–4, PDGFRα, RET, and KIT | Phase III; First line; Multicenter; Non inferiority; Open label NCT01761266 | n = 954 | OS Lenvatinib: 13.6 Sorafenib: 12.3 months | TTP (months): Lenvatinib: 8.9; Sorafenib: 3.7 PFS (months): Lenvatinib: 7.4; Sorafenib: 3.7 DCR:75.5% ORR: 24.1 TEAEs: 99%; Grade ≥ 3 TEAEs: 75%; TEAEs: 43% | [78] |
| Sunitinib vs. Sorafenib | PDGFRα/β, VEGFR-1, VEGFR-2, RET, c-Kit and FLT-3 | Open-label, phase III trial NCT00699374 | n = 1073 | OS 7.9 versus 10.2 months for sunitinib and sorafenib | PFS: Sunitinib; 3.6, sorafenib 3.0 months TTP sunitinib; 4.1, Sorafenib 3.8 months AEs: grade 3/4 | [84] |
| Erlotinib | EGFR | phase II and phase III clinical trials | n = 1020 |
OS 6.25–15.65 months | DCR: 42.5–79.6% (PFS) of 6.5–9.0 months, AEs: 3/4 grade toxicities (fatigue, diarrhea, rash) | [87] |
| Brivanib vs. placebo and best supportive care (sorafenib) | VEGFR-2 and FGFR1 | multinational, randomized, double-blind, phase III trial | n = 1150 | OS 9.9 months for sorafenib and 9.5 months for brivanib | TTP, ORR, and DCR were similar between the study arms. Most frequent grade 3/4 adverse events for sorafenib and brivanib were similar | [93] |
| Cediranib | VEGFR-2 | single-arm phase II study | n = 17 | OS 11.7 (7.5–13.6) months | PFS rate of 77% (60%, 99%). Median PFS was 5.3 (3.5, 9.7) months, stable disease (29%), Grade 3 toxicities: hypertension (29%), hyponatremia (29%), hyperbilirubinemia (18%) | [95] |
| Linifanib vs. sorafenib | VEGF and PDGF | open-label phase III trial NCT01009593 | n = 1035 | OS 9.1 months on the linifanib arm and 9.8 months on the sorafenib arm | TTP was 5.4 months (linifanib) and 4.0 months on sorafenib. Best response rate was 13.0% on the linifanib arm vs. 6.9% on the sorafenib arm. Grade 3/4 (AEs); serious AEs; and AEs leading to discontinuation, dose interruption, and reduction were more frequent with linifanib | [98] |
| Nintedanib vs. sorafenib | VEGFR-1, VEGFR-2 and VEGFR-3, FGFR, PDGFR and Src | randomized, multicenter, open-label, phase I/II study | n = 95 | For nintedanib and sorafenib, median OS 10.2 vs. 10.7 months | For nintedanib and sorafenib, respectively, the median CIR TTP was 2.8 vs. 3.7 months Nintedanib-treated patients had fewer grade 3 or higher AEs (56 vs. 84%), serious AEs (46 vs. 56%), and AEs leading to dose reduction (19 vs. 59%) and drug discontinuation (24 vs. 34%). | [101] |
| Refametinib vs. sorafenib | MEK1/2 | phase II study NCT01204177 | n = 95 |
OS 290 days (n = 70) |
DCR was 44.8% (primary efficacy analysis; n = 58). TTP was 122 days grade 3 AEs | [104] |
| Vatalanib in combination with doxorubicin | VEGFRs, c-Kit, PDGFRβ and c-Fms | phase I/II study | n = 27 | OS: 7.3 months (range, 0.8–23.6 months) |
ORR was 26.0% PFS was 5.4 months (range, 0.27–23.6 months) The commonest grade 3 or 4 non-hematological AEs | [109] |
| Vandetanib vs. placebo | VEGFR-2 and EGFR | a phase II, randomized, double-blind, placebo-controlled study | n = 67 | OS improvement was noticed but statistically insignificant | improved PFS and OS after vandetanib treatment were found, they were statistically insignificant but tumor stabilization rate significant | [186] |
| Pazopanib | VEGFR-1, -2 and -3, PDGFRα/β and c-Kit | phase I dose-escalating study NCT00370513 | n = 28 |
19 patients (73%) had either partial response or stable disease. Diarrhea, skin hypopigmentation, and AST elevation were the most reported AEs | [118] | |
| Tivantinib vs. placebo | c-Met | a phase 3, randomized, placebo-controlled study NCT01755767 | n = 340 |
OS 8·4 months in the tivantinib group and 9·1 months (7·3–10·4) in the placebo group | Grade 3 or worse AEs (ascites, anemia, abdominal pain, and neutropenia) occurred in 56% compared with 55% of patients who received tivantinib and placebo, respectively | [123] |
| Apatinib | VEGFR-2, c-Kit, PDGFRβ and c-Src | single-arm, open-label phase II clinical trial NCT03046979 | n = 23 | The median OS 13.8 months | ORR and DCR were 30.4% and 65.2%, respectively. The median PFS: 8.7 months. The most common treatment-related adverse events were proteinuria (39.1%), hypertension (34.8%), and hand-foot-skin reaction (34.8%). | [126] |
| Imatinib | AKT, p62 and LC3 | phase II clinical trial | n = 17 | Grade 3/4 AEs. There was no objective response, and 5 (33%) patients had stable disease. Median time to treatment failure was 1.8 months | [187] | |
| Gefitinib | EGFR | single arm phase II study | n = 31 | OS 6.5 months | PFS = 2.8 months, Med OS = 6.5 months. Selected grade 3 AEs: neutropenia; rash; diarrhea. There was only 1 grade 4 AE (neutropenia). | [131] |
| Lapatinib | EGFR and HER-2/NEU | A multi-institutional phase II study | n = 25 |
OS 12.6 months | Most common toxicities were diarrhea (73%), nausea (54%), and rash (42%). Ten (40%) patients had stable disease. PFS was 1.9 months | [135] |
| Linsitinib | IGF-1R | Phase II clinical trials | Not completed due to safety issues observed | Not safe | [138] | |
| Orantinib | VEGFR-2, FGFR and PDGFR | a phase I/II clinical trial in patients with unresectable or metastatic HCC NCT00784290 | n = 35 |
OS 13.1 months | TTP was 2.1 months. Common AEs were hypoalbuminemia, diarrhea, anorexia, abdominal pain, malaise, and edema | [139] |
| Axitinib | VEGFR-1, 2, 3 | Multicenter phase II study | n = 45 |
OS 10.1 months |
DCR was 62.2%, and the RR was 6.7%, (PFS): 2.2 months Axitinib has moderate activity and acceptable toxicity for patients with advanced HCC | [144] |
| Donafenib vs. sorafenib | VEGFR, PDGFR, and Raf | A Randomized, Open-Label, Parallel-Controlled Phase II-III Trial | n = 688 | OS was significantly longer with donafenib (12.1) than sorafenib (10.3) months |
PFS: 3.7 vs. 3.6 months. The ORR was 4.6% vs. 2.7% and the disease control rate was 30.8% vs. 28.7%. Drug-related grade ≥ 3 AEs occurred less in donafenib | [149] |
| Anlotinib | VEGFR 1–3, FGF Receptor 1–4, PDGFR α/β, and c-kit | open-label phase II study (ALTER-0802 study) | n = 50 |
PFS rate was 80.8% and (TTP) was 5.9 months. Cohort 2, the 1 PFS rate and median TTP was 72.5% and 4.6 months. The most common grade 3–5 AEs were hypertension (8%), diarrhea (8%) and hand-foot syndrome (6%). | [152] | |
| Dovitinib vs. sorafenib | VEGFR-1, 2, 3, FGFR1, 2, 3, and PDGFR-β | Randomized, open-label phase II study | n = 165 |
The median OS was 8.0 (6.6–9.1) months for dovitinib and 8.4 (5.4–11.3) months for sorafenib | The median TTP per investigator assessment was 4.1 (2.8–4.2) months and 4.1 (2.8–4.3) months for dovitinib and sorafenib, respectively. | [157] |
| Tepotinib | MET | Phase Ib/II trials | n = 121 | Tepotinib induced significant tumor regression in 2 high-level MET amp HCC PDX models (mean tumor volume reduction: 97% and 96%, respectively). | High-level MET amp may be an oncogenic driver in HCC that sensitizes tumors to MET inhibition with tepotinib. Compared with MET overexpression, high-level MET amp could be a better predictive biomarker for MET inhibitors in this setting | [174] |
| Dasatinib combination with irinotecan | Src kinase, SFK/FAK and PI3K/PTEN/Akt | In-vitro study/nine different cell lines | Dasatinib inhibits the proliferation, adhesion, and metastasis of HCC cells in-vitro. | Dasatinib can reinforce the anti-HCC efficacy of irinotecan/SN38 by downregulation of PLK1 synthesis | [129] | |
| PD0325901 | MEK1 and MEK2 | HepG2 and Hep3B human HCC cell lines in-vitro and in Hep3B flank tumors in-vivo | PD0325901 suppressed MEK activity and tumor growth in-vitro in TAMH cells, taken from the livers of TGF-α transgenic mice. | Additionally, it considerably decreased MEK activity in-vivo in athymic mice bearing TAMH flank tumors. | [165] | |
| R1498 vs. sorafenib | VEGFR2 | In-vivo on a panel of GC and HCC xenografts, | R1498 resulted in 80% inhibition of tumor growth and tumor regression in some xenografts. | R1498 anti-tumor efficacy was compared to that of sorafenib in-vivo on a panel of HCC xenograft mouse models. Results reported superior profile of both efficacy and toxicity relative to sorafenib in all the models. | [166] | |
| SGX523 | MET | In-vitro on 2 HCC cell lines: HCC2321 and HCC2309. | Partial inhibition of tumor growth was presented by SGX523 monotherapy at 60 mg/kg and at 10 mg/kg sorafenib monotherapy | SGX523 (60 mg/kg)-sorafenib (10 mg/kg) combination gave no major progress in efficacy | [170] | |
| PHA665752 | c-Met | MHCC97-L and MHCC97-H in xenograft models and cell lines as Huh7 and Hep3B cells (in-vitro or in-vivo) | Inhibition of proliferation and apoptosis was induced in c-Met positive MHCC97-L and MHCC97-H cells by PHA665752. | In accordance with these results, PHA665752 considerably inhibited c-Met positive MHCC97-L and MHCC97-H in xenograft models while c-Met negative cell lines as Huh7 and Hep3B cells were not affected in-vitro or in-vivo | [173] | |
| BLU9931 | FGFR4 | Hep3B cell line | initiation of caspase 3/7 activity, apoptosis, and inhibition of downstream signaling of FGFR4. | BLU9931 is efficacious in tumors with an intact FGFR4 signaling pathway that includes FGF19, FGFR4, and KLB. BLU9931 is the first FGFR4-selective molecule for the treatment of patients with HCC with aberrant FGFR4 signaling. | [175] | |
| FGF401 | FGFR4 | Huh7, SNU878 and Hep3B cell lines and xenografts in-vivo | FGF401 induced tumor stasis at a dose of 10 mg per kg twice a day, as well as tumor regression at these doses: 30 and 100 mg per kg twice a day. These doses were safe and well tolerated. | FGF401 anti-tumor effect was superior in Huh7 xenografts relative to once per day 30 mg/kg sorafenib | [185] |
5. Inhibitors of Growth Factors Receptors
5.1. Galunisertib (LY2157299)
5.2. Vactosertib (EW-7197/TEW-7197)
6. Immunomodulating Small Molecules
CS2164
7. Small Molecules Inhibiting HCC Pathways
7.1. Wnt/β-Catenin Signaling
7.1.1. YC-1
7.1.2. FH535
7.1.3. Mangiferin
7.1.4. IC-2 and PN-3-13
7.2. RAS-RAF-ERK Signaling
Rigosertib (ON-01910)
7.3. JAK/STAT3 Signaling
7.3.1. 2-Ethoxystypandrone
7.3.2. FLLL32
7.3.3. XZH-5
7.4. PI3K/Akt/mTOR Pathway
SC66
8. Small Molecules Targeting Various Molecular Targets
8.1. CMO
8.2. APG-1387 (Apoptosis Inhibitor)
8.3. AC-73 (AN-465/42834501) (CD147)
8.4. VO-OHpic (PTEN Inhibitor)
8.5. Rubone (miR34a)
8.6. FQI1
8.7. AUY922 (Luminespib)
8.8. Compound 81 (CXCR6)
8.9. Cambinol (SIRT-1)
8.10. BI 2536
8.11. THZ1
8.12. IPA-3
8.13. Alisertib (AURKA Inhibitor)
9. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Elghazaly, H.; Gaballah, A.; Eldin, N.B. Clinic-Pathological Pattern of Hepatocellular Carcinoma (Hcc) in Egypt. Ann. Oncol. 2018, 29, v5–v6. [Google Scholar] [CrossRef]
- Jiang, K.; Centeno, B.A. Primary Liver Cancers, Part 2: Progression Pathways and Carcinogenesis. Cancer Control 2018, 25, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of Hepatocellular Carcinoma: Epidemiology, Etiology, and Carcinogenesis. Carcinogenesis 2017, 16, 1. [Google Scholar]
- Savitha, G.; Vishnupriya, V.; Krishnamohan, S. Hepatocellular Carcinoma—A Review. J. Pharm. Sci. Res. 2017, 9, 1276–1280. [Google Scholar] [CrossRef]
- Systemic Treatment for Advanced Hepatocellular Carcinoma—UpToDate. Available online: https://www.uptodate.com/contents/systemic-treatment-for-advanced-hepatocellular-carcinoma (accessed on 23 November 2021).
- Siddique, O.; Yoo, E.R.; Perumpail, R.B.; Perumpail, B.J.; Liu, A.; Cholankeril, G.; Ahmed, A. The Importance of a Multidisciplinary Approach to Hepatocellular Carcinoma. J. Multidiscip. Healthc. 2017, 10, 95–100. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A Global View of Hepatocellular Carcinoma: Trends, Risk, Prevention and Management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.Y.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Chang, Y.S.; Adnane, J.; Trail, P.A.; Levy, J.; Henderson, A.; Xue, D.; Bortolon, E.; Ichetovkin, M.; Chen, C.; McNabola, A.; et al. Sorafenib (BAY 43-9006) Inhibits Tumor Growth and Vascularization and Induces Tumor Apoptosis and Hypoxia in RCC Xenograft Models. Cancer Chemother. Pharmacol. 2007, 59, 561–574. [Google Scholar] [CrossRef]
- Carlomagno, F.; Anaganti, S.; Guida, T.; Salvatore, G.; Troncone, G.; Wilhelm, S.M.; Santoro, M. BAY 43-9006 Inhibition of Oncogenic RET Mutants. J. Natl. Cancer Inst. 2006, 98, 326–334. [Google Scholar] [CrossRef]
- Tovoli, F.; Granito, A.; De Lorenzo, S.; Bolondi, L. Regorafenib for the Treatment of Hepatocellular Carcinoma. Drugs Today 2018, 54, 5–13. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and Safety of Sorafenib in Patients in the Asia-Pacific Region with Advanced Hepatocellular Carcinoma: A Phase III Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, L.; He, J.; Liu, P.; Lv, X.; Zhang, Y.; Xu, X.; Zhang, L.; Zhang, Y. Synergy with Interferon-Lambda 3 and Sorafenib Suppresses Hepatocellular Carcinoma Proliferation. Biomed. Pharmacother. 2017, 88, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhu, B.; Zhang, L.; Xie, Q.; Zhuo, W. Detection and Screening of Small Molecule Agents for Overcoming Sorafenib Resistance of Hepatocellular Carcinoma: A Bioinformatics Study. Int. J. Clin. Exp. Med. 2015, 8, 2317. [Google Scholar] [PubMed]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front. Pharmacol. 2018, 9, 1080. [Google Scholar] [CrossRef]
- Cui, D.; Xiong, X.; Zhao, Y. Cullin-RING Ligases in Regulation of Autophagy. Cell Div. 2016, 11, 8. [Google Scholar] [CrossRef]
- Maghames, C.M.; Lobato-Gil, S.; Perrin, A.; Trauchessec, H.; Rodriguez, M.S.; Urbach, S.; Marin, P.; Xirodimas, D.P. NEDDylation Promotes Nuclear Protein Aggregation and Protects the Ubiquitin Proteasome System upon Proteotoxic Stress. Nat. Commun. 2018, 9, 4376. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, J.; Lin, X.; Wu, D.; Li, G.; Zhong, C.; Fang, L.; Jiang, P.; Yin, L.; Zhang, L.; et al. Inhibition of Neddylation Modification by MLN4924 Sensitizes Hepatocellular Carcinoma Cells to Sorafenib. Oncol. Rep. 2019, 41, 3257. [Google Scholar] [CrossRef]
- Huang, Y.T.; Hwang, J.J.; Lee, P.P.; Ke, F.C.; Huang, J.H.; Huang, C.J.; Kandaswami, C.; Middleton, J.; Lee, M.T. Effects of Luteolin and Quercetin, Inhibitors of Tyrosine Kinase, on Cell Growth and Metastasis-Associated Properties in A431 Cells Overexpressing Epidermal Growth Factor Receptor. Br. J. Pharmacol. 1999, 128, 999. [Google Scholar] [CrossRef]
- Leung, H.W.C.; Wu, C.H.; Lin, C.H.; Lee, H.Z. Luteolin Induced DNA Damage Leading to Human Lung Squamous Carcinoma CH27 Cell Apoptosis. Eur. J. Pharmacol. 2005, 508, 77–83. [Google Scholar] [CrossRef]
- Bagli, E.; Stefaniotou, M.; Morbidelli, L.; Ziche, M.; Psillas, K.; Murphy, C.; Fotsis, T. Luteolin Inhibits Vascular Endothelial Growth Factor-Induced Angiogenesis; Inhibition of Endothelial Cell Survival and Proliferation by Targeting Phosphatidylinositol 3′-Kinase Activity. Cancer Res. 2004, 64, 7936–7946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.-M. Luteolin, a Flavonoid with Potentials for Cancer Prevention and Therapy. Curr. Cancer Drug Targets 2008, 8, 634. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Wang, Q.; Zheng, X.; Sun, H.; Zhou, Y.; Li, D.; Lin, Y.; Wang, X. Luteolin Enhances TNF-Related Apoptosis-Inducing Ligand’s Anticancer Activity in a Lung Cancer Xenograft Mouse Model. Biochem. Biophys. Res. Commun. 2012, 417, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Reipas, K.M.; Law, J.H.; Couto, N.; Islam, S.; Li, Y.; Li, H.; Cherkasov, A.; Jung, K.; Cheema, A.S.; Jones, S.J.M.; et al. Luteolin Is a Novel P90 Ribosomal S6 Kinase (RSK) Inhibitor That Suppresses Notch4 Signaling by Blocking the Activation of Y-Box Binding Protein-1 (YB-1). Oncotarget 2013, 4, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Y.; Wang, C.J.; Chen, N.F.; Ho, W.H.; Lu, F.J.; Tseng, T.H. Luteolin Enhances Paclitaxel-Induced Apoptosis in Human Breast Cancer MDA-MB-231 Cells by Blocking STAT3. Chem. Biol. Interact. 2014, 213, 60–68. [Google Scholar] [CrossRef]
- Feng, X.Q.; Rong, L.W.; Wang, R.X.; Zheng, X.L.; Zhang, L.; Zhang, L.; Lin, Y.; Wang, X.; Li, Z.P. Luteolin and Sorafenib Combination Kills Human Hepatocellular Carcinoma Cells through Apoptosis Potentiation and JNK Activation. Oncol. Lett. 2018, 16, 648. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Lin, J.P.; Shiau, C.W.; Tai, W.T.; Liu, C.Y.; Yu, H.C.; Chen, P.J.; Cheng, A.L. Inhibition of Bcl-2 Improves Effect of LCL161, a SMAC Mimetic, in Hepatocellular Carcinoma Cells. Biochem. Pharmacol. 2012, 84, 268–277. [Google Scholar] [CrossRef]
- Chen, K.F.; Su, J.C.; Liu, C.Y.; Huang, J.W.; Chen, K.C.; Chen, W.L.; Tai, W.T.; Shiau, C.W. A Novel Obatoclax Derivative, SC-2001, Induces Apoptosis in Hepatocellular Carcinoma Cells through SHP-1-Dependent STAT3 Inactivation. Cancer Lett. 2012, 321, 27–35. [Google Scholar] [CrossRef]
- Ji, T.; Gong, D.; Han, Z.; Wei, X.; Yan, Y.; Ye, F.; Ding, W.; Wang, J.; Xia, X.; Li, F.; et al. Abrogation of Constitutive Stat3 Activity Circumvents Cisplatin Resistant Ovarian Cancer. Cancer Lett. 2013, 341, 231–239. [Google Scholar] [CrossRef]
- Duan, Z.; Foster, R.; Bell, D.A.; Mahoney, J.; Wolak, K.; Vaidya, A.; Hampel, C.; Lee, H.; Seiden, M.V. Signal Transducers and Activators of Transcription 3 Pathway Activation in Drug-Resistant Ovarian Cancer. Clin. Cancer Res. 2006, 12, 5055–5063. [Google Scholar] [CrossRef]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early Responses of the STAT3 Pathway to Platinum Drugs Are with Cisplatin Resistance in Epithelial Ovarian Cancer. Braz. J. Med. Biol. Res. 2013, 46, 650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colomiere, M.; Findlay, J.; Ackland, L.; Ahmed, N. Epidermal Growth Factor-Induced Ovarian Carcinoma Cell Migration Is Associated with JAK2/STAT3 Signals and Changes in the Abundance and Localization of Alpha6beta1 Integrin. Int. J. Biochem. Cell Biol. 2009, 41, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Sun, X.Y. Mechanisms of Resistance to Sorafenib and the Corresponding Strategies in Hepatocellular Carcinoma. World J. Hepatol. 2013, 5, 345–352. [Google Scholar] [CrossRef]
- Amin, S.; Kumar, A.; Nilchi, L.; Wright, K.; Kozlowski, M. Breast Cancer Cells Proliferation Is Regulated by Tyrosine Phosphatase SHP1 through C-Jun N-Terminal Kinase and Cooperative Induction of RFX-1 and AP-4 Transcription Factors. Mol. Cancer Res. 2011, 9, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Su, J.C.; Tseng, P.H.; Wu, S.H.; Hsu, C.Y.; Tai, W.T.; Li, Y.S.; Chen, I.T.; Liu, C.Y.; Chen, K.F.; Shiau, C.W. SC-2001 Overcomes STAT3-Mediated Sorafenib Resistance through RFX-1/SHP-1 Activation in Hepatocellular Carcinoma. Neoplasia 2014, 16, 595–605. [Google Scholar] [CrossRef]
- Polier, G.; Ding, J.; Konkimalla, B.V.; Eick, D.; Ribeiro, N.; Köhler, R.; Giaisi, M.; Efferth, T.; Desaubry, L.; Krammer, P.H.; et al. Wogonin and Related Natural Flavones Are Inhibitors of CDK9 That Induce Apoptosis in Cancer Cells by Transcriptional Suppression of Mcl-1. Cell Death Dis. 2011, 2, e182. [Google Scholar] [CrossRef]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of Autophagy Potentiates the Antitumor Effect of the Multikinase Inhibitor Sorafenib in Hepatocellular Carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef]
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Hui, B.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; Dai, Z.; Peng, Y.F.; Gu, C.Y.; et al. Targeting Autophagy Enhances Sorafenib Lethality for Hepatocellular Carcinoma via ER Stress-Related Apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef]
- Tai, W.T.; Shiau, C.W.; Chen, H.L.; Liu, C.Y.; Lin, C.S.; Cheng, A.L.; Chen, P.J.; Chen, K.F. Mcl-1-Dependent Activation of Beclin 1 Mediates Autophagic Cell Death Induced by Sorafenib and SC-59 in Hepatocellular Carcinoma Cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef]
- Lin, C.I.; Whang, E.E.; Lorch, J.H.; Ruan, D.T. Autophagic Activation Potentiates the Antiproliferative Effects of Tyrosine Kinase Inhibitors in Medullary Thyroid Cancer. Surgery 2012, 152, 1142–1149. [Google Scholar] [CrossRef]
- Bareford, M.D.; Park, M.A.; Yacoub, A.; Hamed, H.A.; Tang, Y.; Cruickshanks, N.; Eulitt, P.; Hubbard, N.; Tye, G.; Burow, M.E.; et al. Sorafenib Enhances Pemetrexed Cytotoxicity through an Autophagy-Dependent Mechanism in Cancer Cells. Cancer Res. 2011, 71, 4955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajithlal, G.B.; Hamed, H.A.; Cruickshanks, N.; Booth, L.; Tavallai, S.; Syed, J.; Grant, S.; Poklepovic, A.; Dent, P. Sorafenib/Regorafenib and Phosphatidyl Inositol 3 Kinase/Thymoma Viral Proto-Oncogene Inhibition Interact to Kill Tumor Cells. Mol. Pharmacol. 2013, 84, 562–571. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and Everolimus for Patients with Unresectable High-Grade Osteosarcoma Progressing after Standard Treatment: A Non-Randomised Phase 2 Clinical Trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Lee, S.J.; Zhao, F.; Schuchter, L.M.; Flaherty, L.; Kefford, R.; Atkins, M.B.; Leming, P.; Kirkwood, J.M. Phase III Trial of Carboplatin and Paclitaxel with or without Sorafenib in Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 373–379. [Google Scholar] [CrossRef]
- Rong, L.W.; Wang, R.X.; Zheng, X.L.; Feng, X.Q.; Zhang, L.; Zhang, L.; Lin, Y.; Li, Z.P.; Wang, X. Combination of Wogonin and Sorafenib Effectively Kills Human Hepatocellular Carcinoma Cells through Apoptosis Potentiation and Autophagy Inhibition. Oncol. Lett. 2017, 13, 5028. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Emelyanov, A.; Koh, C.H.V.; Spitsbergen, J.M.; Lam, S.H.; Mathavan, S.; Parinov, S.; Gong, Z. A High Level of Liver-Specific Expression of Oncogenic Kras(V12) Drives Robust Liver Tumorigenesis in Transgenic Zebrafish. Dis. Model. Mech. 2011, 4, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.W.; Hsia, Y.; Tu, H.C.; Hsiao, Y.C.; Yang, W.Y.; Wang, H.D.; Yuh, C.H. Liver Development and Cancer Formation in Zebrafish. Birth Defects Res. C Embryo Today 2011, 93, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Siew, H.L.; Yi, L.W.; Vega, V.B.; Miller, L.D.; Spitsbergen, J.; Tong, Y.; Zhan, H.; Govindarajan, K.R.; Lee, S.; Mathavan, S.; et al. Conservation of Gene Expression Signatures between Zebrafish and Human Liver Tumors and Tumor Progression. Nat. Biotechnol. 2006, 24, 73–75. [Google Scholar] [CrossRef]
- Goessling, W.; North, T.E.; Zon, L.I. Ultrasound Biomicroscopy Permits in Vivo Characterization of Zebrafish Liver Tumors. Nat. Methods 2007, 4, 551–553. [Google Scholar] [CrossRef]
- Lin, H.S.; Huang, Y.L.; Wang, Y.R.; Hsiao, E.; Hsu, T.A.; Shiao, H.Y.; Jiaang, W.T.; Sampurna, B.P.; Lin, K.H.; Wu, M.S.; et al. Identification of Novel Anti-Liver Cancer Small Molecules with Better Therapeutic Index than Sorafenib via Zebrafish Drug Screening Platform. Cancers 2019, 11, 739. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Shi, Q.; Knox, J.J.; Kaubisch, A.; Niedzwiecki, D.; Posey, J.; Tan, B.R.; Kavan, P.; Goel, R.; Lammers, P.E.; et al. Assessment of Treatment With Sorafenib Plus Doxorubicin vs. Sorafenib Alone in Patients With Advanced Hepatocellular Carcinoma: Phase 3 CALGB 80802 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1582. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.; Bharthuar, A. A Review of Erlotinib--an Oral, Selective Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. Expert Opin. Pharmacother. 2010, 11, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Bassullu, N.; Turkmen, I.; Dayangac, M.; Korkmaz, P.Y.; Yasar, R.; Akyildiz, M.; Yaprak, O.; Tokat, Y.; Yuzer, Y.; Dogusoy, G.B. The Predictive and Prognostic Significance of C-Erb-B2, EGFR, PTEN, MTOR, PI3K, P27, and ERCC1 Expression in Hepatocellular Carcinoma. Hepat. Mon. 2012, 12, e7492. [Google Scholar] [CrossRef] [PubMed]
- Ezzoukhry, Z.; Louandre, C.; Trécherel, E.; Godin, C.; Chauffert, B.; Dupont, S.; Diouf, M.; Barbare, J.C.; Mazière, J.C.; Galmiche, A. EGFR Activation Is a Potential Determinant of Primary Resistance of Hepatocellular Carcinoma Cells to Sorafenib. Int. J. Cancer 2012, 131, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Duran, I.; Hotté, S.J.; Hirte, H.; Chen, E.X.; MacLean, M.; Turner, S.; Duan, L.; Pond, G.R.; Lathia, C.; Walsh, S.; et al. Phase I Targeted Combination Trial of Sorafenib and Erlotinib in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2007, 13, 4849–4857. [Google Scholar] [CrossRef]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.J.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix, J.; Qin, S.; Thuluvath, P.J.; Llovet, J.M.; et al. SEARCH: A Phase III, Randomized, Double-Blind, Placebo-Controlled Trial of Sorafenib plus Erlotinib in Patients with Advanced Hepatocellular Carcinoma. J. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef]
- Kawata, S.; Nagase, T.; Yamasaki, E.; Ishiguro, H.; Matsuzawa, Y. Modulation of the Mevalonate Pathway and Cell Growth by Pravastatin and D-Limonene in a Human Hepatoma Cell Line (Hep G2). Br. J. Cancer 1994, 69, 1015–1020. [Google Scholar] [CrossRef]
- Sutter, A.P.; Maaser, K.; Höpfner, M.; Huether, A.; Schuppan, D.; Scherübl, H. Cell Cycle Arrest and Apoptosis Induction in Hepatocellular Carcinoma Cells by HMG-CoA Reductase Inhibitors. Synergistic Antiproliferative Action with Ligands of the Peripheral Benzodiazepine Receptor. J. Hepatol. 2005, 43, 808–816. [Google Scholar] [CrossRef]
- Kawata, S.; Takaishi, K.; Nagase, T.; Ito, N.; Matsuda, Y.; Tannini, S.; Mat Su/Awa, Y.; Tarui, S. Increase in the Active Form of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase in Human Hepatocellular Carcinoma: Possible Mechanism for Alteration of Cholesterol Biosynthesis. Cancer Res. 1990, 50, 3270–3273. [Google Scholar]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basañez, G.; Antonsson, B.; Prieto, J.; García-Ruiz, C.; Colell, A.; et al. Mitochondrial Cholesterol Contributes to Chemotherapy Resistance in Hepatocellular Carcinoma. Cancer Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef]
- Juneja, M.; Kobelt, D.; Walther, W.; Voss, C.; Smith, J.; Specker, E.; Neuenschwander, M.; Gohlke, B.O.; Dahlmann, M.; Radetzki, S.; et al. Statin and Rottlerin Small-Molecule Inhibitors Restrict Colon Cancer Progression and Metastasis via MACC1. PLOS Biol. 2017, 15, e2000784. [Google Scholar] [CrossRef]
- Jouve, J.L.; Lecomte, T.; Bouché, O.; Barbier, E.; Khemissa Akouz, F.; Riachi, G.; Nguyen Khac, E.; Ollivier-Hourmand, I.; Debette-Gratien, M.; Faroux, R.; et al. Pravastatin Combination with Sorafenib Does Not Improve Survival in Advanced Hepatocellular Carcinoma. J. Hepatol. 2019, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, D.; Sharma, A.; Tuli, H.S.; Sak, K.; Mukherjee, T.; Bishayee, A. Molecular Targets of Celastrol in Cancer: Recent Trends and Advancements. Crit. Rev. Oncol. Hematol. 2018, 128, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Chen, H.L.; Tai, W.T.; Feng, W.C.; Hsu, C.H.; Chen, P.J.; Cheng, A.L. Activation of Phosphatidylinositol 3-Kinase/Akt Signaling Pathway Mediates Acquired Resistance to Sorafenib in Hepatocellular Carcinoma Cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, Z.; Wu, S.S.; Xu, J.; Kong, L.C.; Wei, P. Celastrol Enhances the Anti-Liver Cancer Activity of Sorafenib. Med. Sci. Monit. 2019, 25, 4068. [Google Scholar] [CrossRef]
- Gedaly, R.; Angulo, P.; Hundley, J.; Daily, M.F.; Chen, C.; Koch, A.; Mark Evers, B. PI-103 and Sorafenib Inhibit Hepatocellular Carcinoma Cell Proliferation by Blocking Ras/Raf/MAPK and PI3K/AKT/MTOR Pathways. Anticancer Res. 2010, 30, 4951. [Google Scholar]
- Gryziak, M.; Woźniak, K.; Kraj, L.; Stec, R. Milestones in the Treatment of Hepatocellular Carcinoma: A Systematic Review. Crit. Rev. Oncol. Hematol. 2021, 157, 103179. [Google Scholar] [CrossRef]
- Regorafenib|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/regorafenib (accessed on 18 July 2022).
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for Patients with Hepatocellular Carcinoma Who Progressed on Sorafenib Treatment (RESORCE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Bruix, J.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; Gerolami, R.; Masi, G.; et al. Efficacy, Safety, and Health-Related Quality of Life (HRQoL) of Regorafenib in Patients with Hepatocellular Carcinoma (HCC) Progressing on Sorafenib: Results of the International, Double-Blind Phase 3 RESORCE Trial. Ann. Oncol. 2016, 27, vi564. [Google Scholar] [CrossRef]
- Shlomai, A.; Leshno, M.; Goldstein, D.A. Regorafenib Treatment for Patients with Hepatocellular Carcinoma Who Progressed on Sorafenib-A Cost-Effectiveness Analysis. PLoS ONE 2018, 13, e0207132. [Google Scholar] [CrossRef]
- Parikh, N.D.; Singal, A.G.; Hutton, D.W. Cost Effectiveness of Regorafenib as Second-Line Therapy for Patients with Advanced Hepatocellular Carcinoma. Cancer 2017, 123, 3725–3731. [Google Scholar] [CrossRef] [PubMed]
- Klank-Sokołowska, E.; Kucharewicz, M.; Wojtukiewicz, M.Z. Cabozantinib in the Treatment of Advanced Hepatocellular Carcinoma Patients. Oncol. Clin. Pract. 2019, 15, 195–201. [Google Scholar] [CrossRef]
- FDA Approves Cabozantinib for Hepatocellular Carcinoma|FDA. Available online: https://www.fda.gov/drugs/fda-approves-cabozantinib-hepatocellular-carcinoma (accessed on 18 July 2022).
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Nair, A.; Reece, K.; Donoghue, M.B.; Yuan, W.; Rodriguez, L.; Keegan, P.; Pazdur, R. FDA Supplemental Approval Summary: Lenvatinib for the Treatment of Unresectable Hepatocellular Carcinoma. Oncologist 2021, 26, e484–e491. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus Sorafenib in First-Line Treatment of Patients with Unresectable Hepatocellular Carcinoma: A Randomised Phase 3 Non-Inferiority Trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kudo, M.; Izumi, N.; Kaneko, S.; Azuma, M.; Copher, R.; Meier, G.; Pan, J.; Ishii, M.; Ikeda, S. Cost-Effectiveness Analysis of Lenvatinib Treatment for Patients with Unresectable Hepatocellular Carcinoma (UHCC) Compared with Sorafenib in Japan. J. Gastroenterol. 2019, 54, 558–570. [Google Scholar] [CrossRef]
- Crunkhorn, S. Kinase Inhibitor Combination Combats Liver Cancer. Nat. Rev. Drug Discov. 2021, 20, 668. [Google Scholar] [CrossRef]
- Faivre, S.; Demetri, G.; Sargent, W.; Raymond, E. Molecular Basis for Sunitinib Efficacy and Future Clinical Development. Nat. Rev. Drug Discov. 2007, 6, 734–745. [Google Scholar] [CrossRef]
- Mendel, D.B.; Douglas Laird, A.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In Vivo Antitumor Activity of SU11248, a Novel Tyrosine Kinase Inhibitor Targeting Vascular Endothelial Growth Factor and Platelet-Derived Growth Factor Receptors: Determination of a Pharmacokinetic/Pharmacodynamic Relationship. Clin. Canger Res. 2003, 9, 327–337. [Google Scholar]
- Farrell, A.O.; Foran, J.M.; Fiedler, W.; Serve, H.; Paquette, R.L.; Cooper, M.A.; Yuen, H.A.; Louie, S.G.; Kim, H.; Nicholas, S.; et al. An Innovative Phase I Clinical Study Demonstrates Inhibition of FLT3 Phosphorylation by SU11248 in Acute Myeloid Leukemia Patients. Clin. Cancer Res. 2003, 9, 5465–5476. [Google Scholar]
- Cheng, A.L.; Kang, Y.K.; Lin, D.Y.; Park, J.W.; Kudo, M.; Qin, S.; Chung, H.C.; Song, X.; Xu, J.; Poggi, G.; et al. Sunitinib versus Sorafenib in Advanced Hepatocellular Cancer: Results of a Randomized Phase III Trial. J. Clin. Oncol. 2013, 31, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Induction of Apoptosis and Cell Cycle Arrest by CP-358,774, an Inhibitor of Epidermal Growth Factor Receptor Tyrosine Kinase—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/9354447/ (accessed on 18 July 2022).
- Pollack, V.A.; Savage, D.M.; Baker, D.A.; Tsaparikos, K.E.; Sloan, D.E.; Moyer, J.D.; Barbacci, E.G.; Pustilnik, L.R.; Smolarek, T.A.; Davis, J.A.; et al. Inhibition of Epidermal Growth Factor Receptor-Associated Tyrosine Phosphorylation in Human Carcinomas with CP-358,774: Dynamics of Receptor Inhibition In Situ and Antitumor Effects in Athymic Mice 1. J. Pharmacol. Exp. Ther. 1999, 291, 739–748. [Google Scholar] [PubMed]
- Zhang, J.; Zong, Y.; Xu, G.Z.; Xing, K. Erlotinib for Advanced Hepatocellular Carcinoma. A Systematic Review of Phase II/III Clinical Trials. Saudi Med. J. 2016, 37, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Bhide, R.S.; Cai, Z.W.; Zhang, Y.Z.; Qian, L.; Wei, D.; Barbosa, S.; Lombarde, L.J.; Borzilleri, R.M.; Zheng, X.; Wu, L.I.; et al. Discovery and Preclinical Studies of (R)-1-(4-(4-Fluoro-2-Methyl-1H-Indol-5-Yloxy)-5-Methylpyrrolo[2,1-f][1,2,4]Triazin-6-Yloxy)Propan-2-Ol (BMS-540215), an in Vivo Active Potent VEGFR-2 Inhibitor. J. Med. Chem. 2006, 49, 2143–2146. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.W.; Zhang, Y.; Borzilleri, R.M.; Qian, L.; Barbosa, S.; Wei, D.; Zheng, X.; Wu, L.; Fan, J.; Shi, Z.; et al. Discovery of Brivanib Alaninate ((S)-((R)-1-(4-(4-Fluoro-2-Methyl-1H-Indol-5-Yloxy)-5-Methylpyrrolo[2,1-f][1,2,4]Triazin-6-Yloxy)Propan-2-Yl)2-Aminopropanoate), a Novel Prodrug of Dual Vascular Endothelial Growth Factor Receptor-2 and Fibroblast Growth Fa. J. Med. Chem. 2008, 51, 1976–1980. [Google Scholar] [CrossRef]
- Huynh, H.; Ngo, V.C.; Fargnoli, J.; Ayers, M.; Khee, C.S.; Heng, N.K.; Choon, H.T.; Hock, S.O.; Chung, A.; Chow, P.; et al. Brivanib Alaninate, a Dual Inhibitor of Vascular Endothelial Growth Factor Receptor and Fibroblast Growth Factor Receptor Tyrosine Kinases, Induces Growth Inhibition in Mouse Models of Human Hepatocellular Carcinoma. Clin. Cancer Res. 2008, 14, 6146–6153. [Google Scholar] [CrossRef]
- Bhide, R.S.; Lombardo, L.J.; Hunt, J.T.; Cai, Z.W.; Barrish, J.C.; Galbraith, S.; Jeyaseelan, R.; Mortillo, S.; Wautlet, B.S.; Krishnan, B.; et al. The Antiangiogenic Activity in Xenograft Models of Brivanib, a Dual Inhibitor of Vascular Endothelial Growth Factor Receptor-2 and Fibroblast Growth Factor Receptor-1 Kinases. Mol. Cancer Ther. 2010, 9, 369–378. [Google Scholar] [CrossRef]
- Ogasawara, S.; Yano, H.; Iemura, A.; Hisaka, T.; Kojiro, M. Expressions of Basic Fibroblast Growth Factor and Its Receptors and Their Relationship to Proliferation of Human Hepatocellular Carcinoma Cell Lines. Hepatology 1996, 24, 198–205. [Google Scholar] [CrossRef]
- Bolos, D.; Finn, R.S. Systemic Therapy in HCC: Lessons from Brivanib. J. Hepatol. 2014, 61, 947–950. [Google Scholar] [CrossRef]
- Wedge, S.R.; Kendrew, J.; Hennequin, L.F.; Valentine, P.J.; Barry, S.T.; Brave, S.R.; Smith, N.R.; James, N.H.; Dukes, M.; Curwen, J.O.; et al. AZD2171: A Highly Potent, Orally Bioavailable, Vascular Endothelial Growth Factor Receptor-2 Tyrosine Kinase Inhibitor for the Treatment of Cancer. Cancer Res. 2005, 65, 4389–4400. [Google Scholar] [CrossRef]
- Zhu, A.X.; Ancukiewicz, M.; Supko, J.G.; Sahani, D.V.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Abrams, T.A.; McCleary, N.J.; Bhargava, P.; Muzikansky, A.; et al. Efficacy, Safety, Pharmacokinetics, and Biomarkers of Cediranib Monotherapy in Advanced Hepatocellular Carcinoma: A Phase II Study. Clin. Cancer Res. 2013, 19, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Albert, D.H.; Tapang, P.; Magoc, T.J.; Pease, L.J.; Reuter, D.R.; Wei, R.Q.; Li, J.; Guo, J.; Bousquet, P.F.; Ghoreishi-Haack, N.S.; et al. Preclinical Activity of ABT-869, a Multitargeted Receptor Tyrosine Kinase Inhibitor. Mol. Cancer Ther. 2006, 5, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Jasinghe, V.J.; Xie, Z.; Zhou, J.; Khng, J.; Poon, L.F.; Senthilnathan, P.; Glaser, K.B.; Albert, D.H.; Davidsen, S.K.; Chen, C.S. ABT-869, a Multi-Targeted Tyrosine Kinase Inhibitor, in Combination with Rapamycin Is Effective for Subcutaneous Hepatocellular Carcinoma Xenograft. J. Hepatol. 2008, 49, 985–997. [Google Scholar] [CrossRef]
- Cainap, C.; Qin, S.; Huang, W.T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.K.; Chen, P.J.; Toh, H.C.; et al. Linifanib Versus Sorafenib in Patients With Advanced Hepatocellular Carcinoma: Results of a Randomized Phase III Trial. J. Clin. Oncol. 2015, 33, 172. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. Design, Synthesis, and Evaluation of Indolinones as Triple Angiokinase Inhibitors and the Discovery of a Highly Specific 6-Methoxycarbonyl-Substituted Indolinone (BIBF 1120). J. Med. Chem. 2009, 52, 4466–4480. [Google Scholar] [CrossRef]
- Tai, W.T.; Shiau, C.W.; Li, Y.S.; Chang, C.W.; Huang, J.W.; Hsueh, T.T.; Yu, H.C.; Chen, K.F. Nintedanib (BIBF-1120) Inhibits Hepatocellular Carcinoma Growth Independent of Angiokinase Activity. J. Hepatol. 2014, 61, 89–97. [Google Scholar] [CrossRef]
- Yen, C.J.; Kim, T.Y.; Feng, Y.H.; Chao, Y.; Lin, D.Y.; Ryoo, B.Y.; Huang, D.C.L.; Schnell, D.; Hocke, J.; Loembé, A.B.; et al. A Phase I/Randomized Phase II Study to Evaluate the Safety, Pharmacokinetics, and Efficacy of Nintedanib versus Sorafenib in Asian Patients with Advanced Hepatocellular Carcinoma. Liver Cancer 2018, 7, 165–178. [Google Scholar] [CrossRef]
- Iverson, C.; Larson, G.; Lai, C.; Yeh, L.T.; Dadson, C.; Weingarten, P.; Appleby, T.; Vo, T.; Maderna, A.; Vernier, J.M.; et al. RDEA119/BAY 869766: A Potent, Selective, Allosteric Inhibitor of MEK1/2 for the Treatment of Cancer. Cancer Res. 2009, 69, 6839–6847. [Google Scholar] [CrossRef]
- Schmieder, R.; Puehler, F.; Neuhaus, R.; Kissel, M.; Adjei, A.A.; Miner, J.N.; Mumberg, D.; Ziegelbauer, K.; Scholz, A. Allosteric MEK1/2 Inhibitor Refametinib (BAY 86-9766) in Combination with Sorafenib Exhibits Antitumor Activity in Preclinical Murine and Rat Models of Hepatocellular Carcinoma. Neoplasia 2013, 15, 1161–1171. [Google Scholar] [CrossRef]
- Lim, H.Y.; Heo, J.; Choi, H.J.; Lin, C.Y.; Yoon, J.H.; Hsu, C.; Rau, K.M.; Poon, R.T.P.; Yeo, W.; Park, J.W.; et al. A Phase II Study of the Efficacy and Safety of the Combination Therapy of the MEK Inhibitor Refametinib (BAY 86-9766) plus Sorafenib for Asian Patients with Unresectable Hepatocellular Carcinoma. Clin. Cancer Res. 2014, 20, 5976–5985. [Google Scholar] [CrossRef]
- Wood, J.M.; Bold, G.; Buchdunger, E.; Cozens, R.; Ferrari, S.; Frei, J.; Hofmann, F.; Rgen Mestan, J.; Mett, H.; O’reilly, T.; et al. PTK787/ZK 222584, a Novel and Potent Inhibitor of Vascular Endothelial Growth Factor Receptor Tyrosine Kinases, Impairs Vascular Endothelial Growth Factor-Induced Responses and Tumor Growth after Oral Administration. Cancer Res. 2000, 60, 2178–2189. [Google Scholar] [PubMed]
- Liu, Y.; Poon, R.T.; Li, Q.; Kok, T.W.; Lau, C.; Fan, S.T. Both Antiangiogenesis- and Angiogenesis-Independent Effects Are Responsible for Hepatocellular Carcinoma Growth Arrest by Tyrosine Kinase Inhibitor PTK787/ZK222584. Cancer Res. 2005, 65, 3691–3699. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kobayashi, S.; Marubashi, S.; Tomimaru, Y.; Noda, T.; Wada, H.; Eguchi, H.; Takeda, Y.; Tanemura, M.; Umeshita, K.; et al. Tyrosine Kinase Inhibitor PTK/ZK Enhances the Antitumor Effects of Interferon-α/5-Fluorouracil Therapy for Hepatocellular Carcinoma Cells. Ann. Surg. Oncol. 2011, 18, 589–596. [Google Scholar] [CrossRef]
- Katsura, Y.; Wada, H.; Murakami, M.; Akita, H.; Hama, N.; Kawamoto, K.; Kobayashi, S.; Marubashi, S.; Eguchi, H.; Tanemura, M.; et al. PTK787/ZK222584 Combined with Interferon Alpha and 5-Fluorouracil Synergistically Inhibits VEGF Signaling Pathway in Hepatocellular Carcinoma. Ann. Surg. Oncol. 2013, 20, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Chan, P.; Pang, R.; Ng, K.; Fan, S.T.; Poon, R.T. Phase 1-2 Trial of PTK787/ZK222584 Combined with Intravenous Doxorubicin for Treatment of Patients with Advanced Hepatocellular Carcinoma: Implication for Antiangiogenic Approach to Hepatocellular Carcinoma. Cancer 2010, 116, 5022–5029. [Google Scholar] [CrossRef]
- Hennequin, L.F.; Stokes, E.S.E.; Thomas, A.P.; Johnstone, C.; Plé, P.A.; Ogilvie, D.J.; Dukes, M.; Wedge, S.R.; Kendrew, J.; Curwen, J.O. Novel 4-Anilinoquinazolines with C-7 Basic Side Chains: Design and Structure Activity Relationship of a Series of Potent, Orally Active, VEGF Receptor Tyrosine Kinase Inhibitors. J. Med. Chem. 2002, 45, 1300–1312. [Google Scholar] [CrossRef]
- Ciardiello, F.; Caputo, R.; Damiano, V.; Caputo, R.; Troiani, T.; Vitagliano, D.; Carlomagno, F.; Veneziani, B.M.; Fontanini, G.; Bianco, A.R.; et al. Antitumor Effects of ZD6474, a Small Molecule Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitor, with Additional Activity against Epidermal Growth Factor Receptor Tyrosine Kinase. Clin. Cancer Res. 2003, 9, 1546–1556. [Google Scholar]
- Giannelli, G.; Azzariti, A.; Sgarra, C.; Porcelli, L.; Antonaci, S.; Paradiso, A. ZD6474 Inhibits Proliferation and Invasion of Human Hepatocellular Carcinoma Cells. Biochem. Pharmacol. 2006, 71, 479–485. [Google Scholar] [CrossRef]
- Inoue, K.; Torimura, T.; Nakamura, T.; Iwamoto, H.; Masuda, H.; Abe, M.; Hashimoto, O.; Koga, H.; Ueno, T.; Yano, H.; et al. Vandetanib, an Inhibitor of VEGF Receptor-2 and EGF Receptor, Suppresses Tumor Development and Improves Prognosis of Liver Cancer in Mice. Clin. Cancer Res. 2012, 18, 3924–3933. [Google Scholar] [CrossRef]
- Kim, S.T.; Jang, H.L.; Lee, S.J.; Lee, J.; Choi, Y.L.; Kim, K.M.; Cho, J.; Park, S.H.; Park, Y.S.; Lim, H.Y.; et al. Pazopanib, a Novel Multitargeted Kinase Inhibitor, Shows Potent in Vitro Antitumor Activity in Gastric Cancer Cell Lines with FGFR2 Amplification. Mol. Cancer Ther. 2014, 13, 2527–2536. [Google Scholar] [CrossRef]
- Kumar, R.; Knick, V.B.; Rudolph, S.K.; Johnson, J.H.; Crosby, R.M.; Crouthamel, M.C.; Hopper, T.M.; Miller, C.G.; Harrington, L.E.; Onori, J.A.; et al. Pharmacokinetic-Pharmacodynamic Correlation from Mouse to Human with Pazopanib, a Multikinase Angiogenesis Inhibitor with Potent Antitumor and Antiangiogenic Activity. Mol. Cancer Ther. 2007, 6, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Tonon, G.; Sattler, M.; Tai, Y.T.; LeGouill, S.; Yasui, H.; Ishitsuka, K.; Kumar, S.; Kumar, R.; Pandite, L.N.; et al. The Small-Molecule VEGF Receptor Inhibitor Pazopanib (GW786034B) Targets Both Tumor and Endothelial Cells in Multiple Myeloma. Proc. Natl. Acad. Sci. USA 2006, 103, 19478–19483. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.D.; Zhang, J.B.; Fan, P.L.; Xiong, Y.Q.; Zhuang, P.Y.; Zhang, W.; Xu, H.X.; Gao, D.M.; Kong, L.Q.; Wang, L.; et al. Antiangiogenic Effects of Pazopanib in Xenograft Hepatocellular Carcinoma Models: Evaluation by Quantitative Contrast-Enhanced Ultrasonography. BMC Cancer 2011, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Chen, P.J.; Chan, P.; Curtis, C.M.; Murphy, P.S.; Suttle, A.B.; Gauvin, J.; Hodge, J.P.; Dar, M.M.; Poon, R.T. Phase I Dose-Finding Study of Pazopanib in Hepatocellular Carcinoma: Evaluation of Early Efficacy, Pharmacokinetics, and Pharmacodynamics. Clin. Cancer Res. 2011, 17, 6914–6923. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.; Jeay, S.; Li, Y.; Chen, C.R.; France, D.S.; Ashwell, M.A.; Hill, J.; Moussa, M.M.; Leggett, D.S.; Li, C.J. ARQ 197, a Novel and Selective Inhibitor of the Human c-Met Receptor Tyrosine Kinase with Antitumor Activity. Mol. Cancer Ther. 2010, 9, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Abstract #820: Combination Studies of Tyrosine Kinase Inhibitors (TKIs): Assessment of Potential Cytotoxic Synergy of ARQ 197 with Sorafenib or Sunitinib|Cancer Research|American Association for Cancer Research. Available online: https://aacrjournals.org/cancerres/article/69/9_Supplement/820/558578/Abstract-820-Combination-studies-of-tyrosine (accessed on 18 July 2022).
- Bouattour, M.; Raymond, E.; Qin, S.; Cheng, A.L.; Stammberger, U.; Locatelli, G.; Faivre, S. Recent Developments of C-Met as a Therapeutic Target in Hepatocellular Carcinoma. Hepatology 2018, 67, 1132. [Google Scholar] [CrossRef] [PubMed]
- Remsing Rix, L.L.; Kuenzi, B.M.; Luo, Y.; Remily-Wood, E.; Kinose, F.; Wright, G.; Li, J.; Koomen, J.M.; Haura, E.B.; Lawrence, H.R.; et al. GSK3 Alpha and Beta Are New Functionally Relevant Targets of Tivantinib in Lung Cancer Cells. ACS Chem. Biol. 2014, 9, 353–358. [Google Scholar] [CrossRef]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Rota Caremoli, E.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for Second-Line Treatment of MET-High, Advanced Hepatocellular Carcinoma (METIV-HCC): A Final Analysis of a Phase 3, Randomised, Placebo-Controlled Study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- Tian, S.; Quan, H.; Xie, C.; Guo, H.; Lü, F.; Xu, Y.; Li, J.; Lou, L. YN968D1 Is a Novel and Selective Inhibitor of Vascular Endothelial Growth Factor Receptor-2 Tyrosine Kinase with Potent Activity in Vitro and in Vivo. Cancer Sci. 2011, 102, 1374–1380. [Google Scholar] [CrossRef]
- Yang, C.; Qin, S. Apatinib Targets Both Tumor and Endothelial Cells in Hepatocellular Carcinoma. Cancer Med. 2018, 7, 4570. [Google Scholar] [CrossRef]
- Hou, Z.; Zhu, K.; Yang, X.; Chen, P.; Zhang, W.; Cui, Y.; Zhu, X.; Song, T.; Li, Q.; Li, H.; et al. Apatinib as First-Line Treatment in Patients with Advanced Hepatocellular Carcinoma: A Phase II Clinical Trial. Ann. Transl. Med. 2020, 8, 1047. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.Y.; Wang, M. Molecular Mechanisms of Action and Potential Biomarkers of Growth Inhibition of Dasatinib (BMS-354825) on Hepatocellular Carcinoma Cells. BMC Cancer 2013, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, I.; Helmy, M.W.; El-Abhar, H.S. Inhibition of SRC/FAK Cue: A Novel Pathway for the Synergistic Effect of Rosuvastatin on the Anti-Cancer Effect of Dasatinib in Hepatocellular Carcinoma. Life Sci. 2018, 213, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhu, Y.; Shao, J.; Chen, M.; Yan, H.; Li, G.; Zhu, Y.; Xu, Z.; Yang, B.; Luo, P.; et al. Dasatinib Synergises with Irinotecan to Suppress Hepatocellular Carcinoma via Inhibiting the Protein Synthesis of PLK1. Br. J. Cancer 2017, 116, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.C.; Qian, H.; Huang, C.K.; Zheng, B.N.; Yan, F.Z.; Liu, F.; Zhang, X.; Chen, S.J.; Luo, C.; Xie, W.F. Imatinib Inhibits the Malignancy of Hepatocellular Carcinoma by Suppressing Autophagy. Eur. J. Pharmacol. 2021, 906, 174217. [Google Scholar] [CrossRef]
- O’Dwyer, P.J.; Giantonio, B.J.; Levy, D.E.; Kauh, J.S.; Fitzgerald, D.B.; Benson, A.B., III. Gefitinib in Advanced Unresectable Hepatocellular Carcinoma: Results from the Eastern Cooperative Oncology Group’s Study E1203. J. Clin. Oncol. 2006, 24 (Suppl. 18), 4143. [Google Scholar] [CrossRef]
- Tong, Y.; Wang, M.; Huang, H.; Zhang, J.; Huang, Y.; Chen, Y.; Pan, H. Inhibitory Effects of Genistein in Combination with Gefitinib on the Hepatocellular Carcinoma Hep3B Cell Line. Exp. Ther. Med. 2019, 18, 3793. [Google Scholar] [CrossRef]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Glennon, K.; Knight, W.B.; et al. The Effects of the Novel, Reversible Epidermal Growth Factor Receptor/ErbB-2 Tyrosine Kinase Inhibitor, GW2016, on the Growth of Human Normal and Tumor-Derived Cell Lines in Vitro and in Vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar]
- Shewchuk, L.; Hassell, A.; Wisely, B.; Rocque, W.; Holmes, W.; Veal, J.; Kuyper, L.F. Binding Mode of the 4-Anilinoquinazoline Class of Protein Kinase Inhibitor: X-Ray Crystallographic Studies of 4-Anilinoquinazolines Bound to Cyclin-Dependent Kinase 2 and P38 Kinase. J. Med. Chem. 2000, 43, 133–138. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; Markowitz, J.; Prescott, N.; Sadee, W.; Heerema, N.; Wei, L.; Dai, Z.; Papp, A.; Campbell, A.; Culler, K.; et al. A Multi-Institutional Phase II Study of the Efficacy and Tolerability of Lapatinib in Patients with Advanced Hepatocellular Carcinomas. Clin. Cancer Res. 2009, 15, 5895. [Google Scholar] [CrossRef]
- Yan1, Y.-Y.; Guo2, Y.; Zhang2, W.; Yan, Y.-Y. Celastrol Enhanced the Anticancer Effect of Lapatinib in Human Hepatocellular Carcinoma Cells in Vitro. JBUON 2014, 19, 412. [Google Scholar]
- Wu, J.; Zhu, A.X. Targeting Insulin-like Growth Factor Axis in Hepatocellular Carcinoma. J. Hematol. Oncol. 2011, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ngo, M.H.T.; Jeng, H.Y.; Kuo, Y.C.; Nanda, J.D.; Brahmadhi, A.; Ling, T.Y.; Chang, T.S.; Huang, Y.H. The Role of IGF/IGF-1R Signaling in Hepatocellular Carcinomas: Stemness-Related Properties and Drug Resistance. Int. J. Mol. Sci. 2021, 22, 1931. [Google Scholar] [CrossRef] [PubMed]
- Kanai, F.; Yoshida, H.; Tateishi, R.; Sato, S.; Kawabe, T.; Obi, S.; Kondo, Y.; Taniguchi, M.; Tagawa, K.; Ikeda, M.; et al. A Phase I/II Trial of the Oral Antiangiogenic Agent TSU-68 in Patients with Advanced Hepatocellular Carcinoma. Cancer Chemother. Pharmacol. 2011, 67, 315–324. [Google Scholar] [CrossRef]
- Yamashita, T.; Wang, X.W. Cancer Stem Cells in the Development of Liver Cancer. J Clin Invest 2013, 123, 1911–1918. [Google Scholar] [CrossRef]
- Hidaka, H.; Izumi, N.; Aramaki, T.; Ikeda, M.; Inaba, Y.; Imanaka, K.; Okusaka, T.; Kanazawa, S.; Kaneko, S.; Kora, S.; et al. Subgroup Analysis of Efficacy and Safety of Orantinib in Combination with TACE in Japanese HCC Patients in a Randomized Phase III Trial (ORIENTAL). Med. Oncol. 2019, 36, 52. [Google Scholar] [CrossRef]
- Kang, Y.K.; Yau, T.; Park, J.W.; Lim, H.Y.; Lee, T.Y.; Obi, S.; Chan, S.L.; Qin, S.K.; Kim, R.D.; Casey, M.; et al. Randomized Phase II Study of Axitinib versus Placebo plus Best Supportive Care in Second-Line Treatment of Advanced Hepatocellular Carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 2457–2463. [Google Scholar] [CrossRef]
- Yang, K.L.; Chi, M.S.; Ko, H.L.; Huang, Y.Y.; Huang, S.C.; Lin, Y.M.; Chi, K.H. Axitinib in Combination with Radiotherapy for Advanced Hepatocellular Carcinoma: A Phase I Clinical Trial. Radiat. Oncol. 2021, 16, 18. [Google Scholar] [CrossRef]
- Lin, Z.-Z.; Chen, B.-B.; Hung, Y.-P.; Huang, P.-H.; Shen, Y.-C.; Shao, Y.-Y.; Hsu, C.-H.; Cheng, A.-L.; Lee, R.-C.; Chao, Y.; et al. A Multicenter Phase II Study of Second-Line Axitinib for Patients with Advanced Hepatocellular Carcinoma Failing First-Line Sorafenib Monotherapy. Oncologist 2020, 25, e1280–e1285. [Google Scholar] [CrossRef]
- Rao, S.S.; Thompson, C.; Cheng, J.; Haimovitz-Friedman, A.; Powell, S.N.; Fuks, Z.; Kolesnick, R.N. Axitinib Sensitization of High Single Dose Radiotherapy. Radiother. Oncol. 2014, 111, 88–93. [Google Scholar] [CrossRef]
- Hillman, G.G.; Lonardo, F.; Hoogstra, D.J.; Rakowski, J.; Yunker, C.K.; Joiner, M.C.; Dyson, G.; Gadgeel, S.; Singh-Gupta, V. Axitinib Improves Radiotherapy in Murine Xenograft Lung Tumors. Transl. Oncol. 2014, 7, 400–409. [Google Scholar] [CrossRef]
- Li, X.; Qiu, M.; Wang, S.J.; Zhu, H.; Feng, B.; Zheng, L. A Phase I Dose-Escalation, Pharmacokinetics and Food-Effect Study of Oral Donafenib in Patients with Advanced Solid Tumours. Cancer Chemother. Pharmacol. 2020, 85, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.; Qiu, M.; Chai, X.; Niu, J.; Ding, Y.; Bai, Y.; Wu, L.; Shentu, J.; Hao, P.; Chen, J.; et al. A Multicenter Phase II Study of Donafenib in Patients with Advanced Hepatocellular Carcinoma. J. Clin. Oncol. 2017, 35, e15682. [Google Scholar] [CrossRef]
- Qin, S.; Bi, F.; Gu, S.; Bai, Y.; Chen, Z.; Wang, Z.; Ying, J.; Lu, Y.; Meng, Z.; Pan, H.; et al. Donafenib Versus Sorafenib in First-Line Treatment of Unresectable or Metastatic Hepatocellular Carcinoma: A Randomized, Open-Label, Parallel-Controlled Phase II-III Trial. J. Clin. Oncol. 2021, 39, 3002–3011. [Google Scholar] [CrossRef]
- Shen, G.; Zheng, F.; Ren, D.; Du, F.; Dong, Q.; Wang, Z.; Zhao, F.; Ahmad, R.; Zhao, J. Anlotinib: A Novel Multi-Targeting Tyrosine Kinase Inhibitor in Clinical Development. J. Hematol. Oncol. 2018, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wu, T.; Hao, Y. Anlotinib Induces Hepatocellular Carcinoma Apoptosis and Inhibits Proliferation via Erk and Akt Pathway. Biochem. Biophys. Res. Commun. 2018, 503, 3093–3099. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, A.; Zhang, W.; Jiang, Z.; Chen, B.; Zhao, J.; Li, Z.; Wang, L.; Bi, X.; Zhao, H.; et al. Anlotinib in the Treatment of Advanced Hepatocellular Carcinoma: An Open-Label Phase II Study (ALTER-0802 Study). Hepatol. Int. 2021, 15, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Taeger, J.; Moser, C.; Hellerbrand, C.; Mycielska, M.E.; Glockzin, G.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O.; Lang, S.A. Targeting FGFR/PDGFR/VEGFR Impairs Tumor Growth, Angiogenesis, and Metastasis by Effects on Tumor Cells, Endothelial Cells, and Pericytes in Pancreatic Cancer. Mol. Cancer Ther. 2011, 10, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.B.; Chesney, J.; Robinson, D.; Gardner, H.; Shi, M.M.; Kirkwood, J.M. Phase I/II and Pharmacodynamic Study of Dovitinib (TKI258), an Inhibitor of Fibroblast Growth Factor Receptors and VEGF Receptors, in Patients with Advanced Melanoma. Clin. Cancer Res. 2011, 17, 7451–7461. [Google Scholar] [CrossRef]
- Chen, K.F.; Chen, H.L.; Liu, C.Y.; Tai, W.T.; Ichikawa, K.; Chen, P.J.; Cheng, A.L. Dovitinib Sensitizes Hepatocellular Carcinoma Cells to TRAIL and Tigatuzumab, a Novel Anti-DR5 Antibody, through SHP-1-Dependent Inhibition of STAT3. Biochem. Pharmacol. 2012, 83, 769–777. [Google Scholar] [CrossRef]
- Tai, W.T.; Cheng, A.L.; Shiau, C.W.; Liu, C.Y.; Ko, C.H.; Lin, M.W.; Chen, P.J.; Chen, K.F. Dovitinib Induces Apoptosis and Overcomes Sorafenib Resistance in Hepatocellular Carcinoma through SHP-1-Mediated Inhibition of STAT3. Mol. Cancer Ther. 2012, 11, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Thongprasert, S.; Lim, H.Y.; Sukeepaisarnjaroen, W.; Yang, T.S.; Wu, C.C.; Chao, Y.; Chan, S.L.; Kudo, M.; Ikeda, M.; et al. Randomized, Open-Label Phase 2 Study Comparing Frontline Dovitinib versus Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Hepatology 2016, 64, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Sebolt-Leopold, J.S.; Herrera, R. Targeting the Mitogen-Activated Protein Kinase Cascade to Treat Cancer. Nat. Rev. Cancer 2004, 4, 937–947. [Google Scholar] [CrossRef] [PubMed]
- McKillop, I.H.; Schmidt, C.M.; Cahill, P.A.; Sitzmann, J.V. Altered Expression of Mitogen-Activated Protein Kinases in a Rat Model of Experimental Hepatocellular Carcinoma. Hepatology 1997, 26, 1484–1491. [Google Scholar] [CrossRef]
- Osada, S.; Kanematsu, M.; Imai, H.; Goshima, S.; Sugiyama, Y. Evaluation of Extracellular Signal Regulated Kinase Expression and Its Relation to Treatment of Hepatocellular Carcinoma. J. Am. Coll. Surg. 2005, 201, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Thorgeirsson, S.S. Molecular Mechanisms of Hepatocarcinogenesis in Transgenic Mouse Models of Liver Cancer. Toxicol. Pathol. 2005, 33, 181–184. [Google Scholar] [CrossRef]
- Chung, Y.-H.; Kim, J.A.; Song, B.-C.; Chan Lee, G.; Soo Koh, M.; Sang Lee, Y.; Gyu Lee, S.; Jin Suh, D.; Author, C.; Chung, Y.-H. Expression of Transforming Growth Factor Alpha MRNA in Livers of Patients with Chronic Viral Hepatitis and Hepatocellular Carcinoma. Korean J. Hepatol. 2000, 6, 33–40. [Google Scholar]
- Prenzel, N.; Fischer, O.M.; Streit, S.; Hart, S.; Ullrich, A. The Epidermal Growth Factor Receptor Family as a Central Element for Cellular Signal Transduction and Diversification. Endocr. Relat. Cancer 2001, 8, 11–31. [Google Scholar] [CrossRef]
- Hege Thoresen, G.; Guren, T.K.; Sandnes, D.; Peak, M.; Agius, L.; Christoffersen, T. Response to Transforming Growth Factor a (TGFa) and Epidermal Growth Factor (EGF) in Hepatocytes: Lower EGF Receptor Affinity of TGFa Is Associated With More Sustained Activation of P42/P44 Mitogen-Activated Protein Kinase and Greater Efficacy in Stimulat. J. Cell. Physiol. 1998, 175, 10–18. [Google Scholar] [CrossRef]
- Hennig, M.; Yip-Schneider, M.T.; Wentz, S.; Wu, H.; Hekmatyar, S.K.; Klein, P.; Bansal, N.; Max Schmidt, C. Targeting Mitogen-Activated Protein Kinase Kinase with the Inhibitor PD0325901 Decreases Hepatocellular Carcinoma Growth in Vitro and in Mouse Model Systems. Hepatology 2010, 51, 1218–1225. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, X.; Zhang, M.; Zhu, L.; Zhao, R.; Xu, D.; Lin, Z.; Liang, C.; Chen, T.; Chen, L.; et al. Small Molecule R1498 as a Well-Tolerated and Orally Active Kinase Inhibitor for Hepatocellular Carcinoma and Gastric Cancer Treatment via Targeting Angiogenesis and Mitosis Pathways. PLoS ONE 2013, 8, e65264. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Bradley, R.; Kang, L.; Koeman, J.; Ascierto, M.L.; Worschech, A.; De Giorgi, V.; Wang, E.; Kefene, L.; Su, Y.; et al. Hepatocyte Growth Factor (HGF) Autocrine Activation Predicts Sensitivity to MET Inhibition in Glioblastoma. Proc. Natl. Acad. Sci. USA 2012, 109, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour Micro-Environment Elicits Innate Resistance to RAF Inhibitors through HGF Secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread Potential for Growth-Factor-Driven Resistance to Anticancer Kinase Inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef]
- Xie, Q.; Su, Y.; Dykema, K.; Johnson, J.; Koeman, J.; De Giorgi, V.; Huang, A.; Schlegel, R.; Essenburg, C.; Kang, L.; et al. Overexpression of HGF Promotes HBV-Induced Hepatocellular Carcinoma Progression and Is an Effective Indicator for Met-Targeting Therapy. Genes Cancer 2013, 4, 247–260. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Ding, W.; You, H.; Dang, H.; LeBlanc, F.; Galicia, V.; Lu, S.C.; Stiles, B.; Rountree, C.B. Epithelial-to-Mesenchymal Transition of Murine Liver Tumor Cells Promotes Invasion. Hepatology 2010, 52, 945–953. [Google Scholar] [CrossRef]
- You, H.; Ding, W.; Dang, H.; Jiang, Y.; Rountree, C.B. C-Met Represents a Potential Therapeutic Target for Personalized Treatment in Hepatocellular Carcinoma. Hepatology 2011, 54, 879–889. [Google Scholar] [CrossRef]
- Faivre, S.J.; Blanc, J.; Pan, H.; Grando, V.; Lim, H.Y.; Gracián, A.C.; Isambert, N.; Johne, A.; Schumacher, K.; Stroh, C.; et al. Activity of Tepotinib in Hepatocellular Carcinoma (HCC) with High-Level MET Amplification (METamp): Preclinical and Clinical Evidence. J. Clin. Oncol. 2021, 39, 329. [Google Scholar] [CrossRef]
- Hagel, M.; Miduturu, C.; Sheets, M.; Rubin, N.; Weng, W.; Stransky, N.; Bifulco, N.; Kim, J.L.; Hodous, B.; Brooijmans, N.; et al. First Selective Small Molecule Inhibitor of FGFR4 for the Treatment of Hepatocellular Carcinomas with an Activated FGFR4 Signaling Pathway. Cancer Discov. 2015, 5, 424–437. [Google Scholar] [CrossRef]
- Wu, A.L.; Coulter, S.; Liddle, C.; Wong, A.; Eastham-Anderson, J.; French, D.M.; Peterson, A.S.; Sonoda, J. FGF19 Regulates Cell Proliferation, Glucose and Bile Acid Metabolism via FGFR4-Dependent and Independent Pathways. PLoS ONE 2011, 6, e17868. [Google Scholar] [CrossRef] [PubMed]
- Desnoyers, L.R.; Pai, R.; Ferrando, R.E.; Hötzel, K.; Le, T.; Ross, J.; Carano, R.; D’Souza, A.; Qing, J.; Mohtashemi, I.; et al. Targeting FGF19 Inhibits Tumor Growth in Colon Cancer Xenograft and FGF19 Transgenic Hepatocellular Carcinoma Models. Oncogene 2008, 27, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Li, W.Y.; Chen, D.; Henry, J.R.; Li, H.Y.; Chen, Z.; Zia-Ebrahimi, M.; Bloem, L.; Zhai, Y.; Huss, K.; et al. A Novel, Selective Inhibitor of Fibroblast Growth Factor Receptors That Shows a Potent Broad Spectrum of Antitumor Activity in Several Tumor Xenograft Models. Mol. Cancer Ther. 2011, 10, 2200–2210. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Wong, M.J.; Moran, L.; Wardwell, S.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Clackson, T.; Rivera, V.M. Ponatinib (AP24534), a Multitargeted Pan-FGFR Inhibitor with Activity in Multiple FGFR-Amplified or Mutated Cancer Models. Mol. Cancer Ther. 2012, 11, 690–699. [Google Scholar] [CrossRef]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-Dichloro-3,5-Dimethoxy-Phenyl)-1-{6-[4-(4-Ethyl-Piperazin-1-Yl)-Phenylamino]-Pyrimidin-4-Yl}-1-Methyl-Urea (NVP-BGJ398), a Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An Orally Bioavailable, Potent, and Selective Inhibitor of the Fibroblast Growth Factor Receptor Tyrosine Kinase Family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Arnedos, M.; Andre, F.; Soria, J.C.; Dieci, M.V. Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.; Adler, F.; Buhles, A.; Stamm, C.; Fairhurst, R.A.; Kiffe, M.; Sterker, D.; Centeleghe, M.; Wartmann, M.; Kinyamu-Akunda, J.; et al. FGF401, A First-In-Class Highly Selective and Potent FGFR4 Inhibitor for the Treatment of FGF19-Driven Hepatocellular Cancer. Mol. Cancer Ther. 2019, 18, 2194–2206. [Google Scholar] [CrossRef]
- Hsu, C.; Yang, T.S.; Huo, T.I.; Hsieh, R.K.; Yu, C.W.; Hwang, W.S.; Hsieh, T.Y.; Huang, W.T.; Chao, Y.; Meng, R.; et al. Vandetanib in Patients with Inoperable Hepatocellular Carcinoma: A Phase II, Randomized, Double-Blind, Placebo-Controlled Study. J. Hepatol. 2012, 56, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Eckel, F.; Von Delius, S.; Mayr, M.; Dobritz, M.; Fend, F.; Hosius, C.; Schleyer, E.; Schulte-Frohlinde, E.; Schmid, R.M.; Lersch, C. Pharmacokinetic and Clinical Phase II Trial of Imatinib in Patients with Impaired Liver Function and Advanced Hepatocellular Carcinoma. Oncology 2005, 69, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.S.; Anderson, B.D.; Beight, D.W.; Campbell, R.M.; Jones, M.L.; Herron, D.K.; Lampe, J.W.; McCowan, J.R.; McMillen, W.T.; Mort, N.; et al. Synthesis and Activity of New Aryl- and Heteroaryl-Substituted Pyrazole Inhibitors of the Transforming Growth Factor-Beta Type I Receptor Kinase Domain. J. Med. Chem. 2003, 46, 3953–3956. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical Development of Galunisertib (LY2157299 Monohydrate), a Small Molecule Inhibitor of Transforming Growth Factor-Beta Signaling Pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef] [PubMed]
- Yingling, J.M.; McMillen, W.T.; Yan, L.; Huang, H.; Sawyer, J.S.; Graff, J.; Clawson, D.K.; Britt, K.S.; Anderson, B.D.; Beight, D.W.; et al. Preclinical Assessment of Galunisertib (LY2157299 Monohydrate), a First-in-Class Transforming Growth Factor-β Receptor Type I Inhibitor. Oncotarget 2017, 9, 6659–6677. [Google Scholar] [CrossRef]
- Rodon, J.; Carducci, M.A.; Sepulveda-Sánchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Braña, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.L.; et al. First-in-Human Dose Study of the Novel Transforming Growth Factor-β Receptor I Kinase Inhibitor LY2157299 Monohydrate in Patients with Advanced Cancer and Glioma. Clin. Cancer Res. 2015, 21, 553–560. [Google Scholar] [CrossRef]
- Giannelli, G.; Bergamini, C.; Fransvea, E.; Sgarra, C.; Antonaci, S. Laminin-5 with Transforming Growth Factor-Beta1 Induces Epithelial to Mesenchymal Transition in Hepatocellular Carcinoma. Gastroenterology 2005, 129, 1375–1383. [Google Scholar] [CrossRef]
- Giannelli, G.; Mazzocca, A.; Fransvea, E.; Lahn, M.; Antonaci, S. Inhibiting TGF-β Signaling in Hepatocellular Carcinoma. Biochim. Biophys. Acta-Rev. Cancer 2011, 1815, 214–223. [Google Scholar] [CrossRef]
- Giannelli, G.; Fransvea, E.; Marinosci, F.; Bergamini, C.; Colucci, S.; Schiraldi, O.; Antonaci, S. Transforming Growth Factor-Beta1 Triggers Hepatocellular Carcinoma Invasiveness via Alpha3beta1 Integrin. Am. J. Pathol. 2002, 161, 183–193. [Google Scholar] [CrossRef]
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the Epithelial-Mesenchymal Transition by TGF-Beta. Future Oncol. 2009, 5, 1145–1168. [Google Scholar] [CrossRef]
- Wang, N.; Wang, S.; Li, M.Y.; Hu, B.G.; Liu, L.P.; Yang, S.L.; Yang, S.; Gong, Z.; Lai, P.B.S.; Chen, G.G. Cancer Stem Cells in Hepatocellular Carcinoma: An Overview andpromising Therapeutic Strategies. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816287. [Google Scholar] [CrossRef] [PubMed]
- Raoul, J.L.; Bruix, J.; Greten, T.F.; Sherman, M.; Mazzaferro, V.; Hilgard, P.; Scherubl, H.; Scheulen, M.E.; Germanidis, G.; Dominguez, S.; et al. Relationship between Baseline Hepatic Status and Outcome, and Effect of Sorafenib on Liver Function: SHARP Trial Subanalyses. J. Hepatol. 2012, 56, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Santoro, A.; Kelley, R.K.; Gane, E.; Paradis, V.; Cleverly, A.; Smith, C.; Estrem, S.T.; Man, M.; Wang, S.; et al. Biomarkers and Overall Survival in Patients with Advanced Hepatocellular Carcinoma Treated with TGF-ΒRI Inhibitor Galunisertib. PLoS ONE 2020, 15, e0222259. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Agarwal, R.; Dituri, F.; Lupo, L.; Trerotoli, P.; Mancarella, S.; Winter, P.; Giannelli, G. NGS-Based Transcriptome Profiling Reveals Biomarkers for Companion Diagnostics of the TGF-β Receptor Blocker Galunisertib in HCC. Cell Death Dis. 2017 82 2017, 8, e2634. [Google Scholar] [CrossRef] [PubMed]
- Ungerleider, N.; Han, C.; Zhang, J.; Yao, L.; Wu, T. TGFβ Signaling Confers Sorafenib Resistance via Induction of Multiple RTKs in Hepatocellular Carcinoma Cells. Mol. Carcinog. 2017, 56, 1302–1311. [Google Scholar] [CrossRef]
- Serova, M.; Tijeras-Raballand, A.; Dos Santos, C.; Albuquerque, M.; Paradis, V.; Neuzillet, C.; Benhadji, K.A.; Raymond, E.; Faivre, S.; de Gramont, A. Effects of TGF-Beta Signalling Inhibition with Galunisertib (LY2157299) in Hepatocellular Carcinoma Models and in Ex Vivo Whole Tumor Tissue Samples from Patients. Oncotarget 2015, 6, 21614–21627. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-Β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125. [Google Scholar] [CrossRef]
- Coulouarn, C.; Clément, B. Stellate Cells and the Development of Liver Cancer: Therapeutic Potential of Targeting the Stroma. J. Hepatol. 2014, 60, 1306–1309. [Google Scholar] [CrossRef]
- Yang, J.D.; Nakamura, I.; Roberts, L.R. The Tumor Microenvironment in Hepatocellular Carcinoma: Current Status and Therapeutic Targets. Semin. Cancer Biol. 2011, 21, 35–43. [Google Scholar] [CrossRef]
- Desmoulière, A.; Guyot, C.; Gabbiani, G. The Stroma Reaction Myofibroblast: A Key Player in the Control of Tumor Cell Behavior. Int. J. Dev. Biol. 2004, 48, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.K.; Liu, X.W.; Chirco, R.; Fridman, R.; Kim, H.R.C. Identification of CD63 as a Tissue Inhibitor of Metalloproteinase-1 Interacting Cell Surface Protein. EMBO J. 2006, 25, 3934. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Kim, M.J.; Park, S.Y.; Kim, J.S.; Lim, W.; Nam, J.S.; Yhong Sheen, Y. TIMP-1 Mediates TGF-β-Dependent Crosstalk between Hepatic Stellate and Cancer Cells via FAK Signaling. Sci. Rep. 2015, 5, 16492. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fu, C.; Kong, Y.; Pan, D.; Wang, Y.; Huang, S.; Li, Z.; Ning, Z.; Lu, X.; Shan, S.; et al. Antitumor and Immunomodulatory Effects of a Novel Multitarget Inhibitor, CS2164, in Mouse Hepatocellular Carcinoma Models. Anticancer Drugs 2019, 30, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, A.M.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/β-Catenin Signaling in Hepatocellular Carcinoma, Pathogenesis, and Clinical Significance. J. Hepatocell. Carcinoma 2018, 5, 61. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Gammons, M.; Bienz, M. Multiprotein Complexes Governing Wnt Signal Transduction. Curr. Opin. Cell Biol. 2018, 51, 42–49. [Google Scholar] [CrossRef]
- Tsao, C.M.; Yan, M.D.; Shih, Y.L.; Yu, P.N.; Kuo, C.C.; Lin, W.C.; Li, H.J.; Lin, Y.W. SOX1 Functions as a Tumor Suppressor by Antagonizing the WNT/β-Catenin Signaling Pathway in Hepatocellular Carcinoma. Hepatology 2012, 56, 2277–2287. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/Beta-Catenin Pathway in Cancer: Update on Effectors and Inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Ko, H.R.; Chang, Y.S.; Park, W.S.; Ahn, J.Y. Opposing Roles of the Two Isoforms of ErbB3 Binding Protein 1 in Human Cancer Cells. Int. J. Cancer 2016, 139, 1202–1208. [Google Scholar] [CrossRef]
- Wu, J.Y.; Shih, Y.L.; Lin, S.P.; Hsieh, T.Y.; Lin, Y.W. YC-1 Antagonizes Wnt/β-Catenin Signaling Through the EBP1 P42 Isoform in Hepatocellular Carcinoma. Cancers. 2019, 11, 661. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/Beta-Catenin Pathway Activation Is Enriched in Basal-like Breast Cancers and Predicts Poor Outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Honma, T.; Matsuda, Y.; Suzuki, Y.; Narisawa, R.; Ajioka, Y.; Asakura, H. Nuclear Translocation of Beta-Catenin in Colorectal Cancer. Br. J. Cancer 2000, 82, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Baehrecke, E.H. Autophagy, Cell Death, and Cancer. Mol. Cell. Oncol. 2015, 2, e985913. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and Autophagy in Cancer Cells, from Tumor Initiation to Cancer Therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef]
- Turcios, L.; Chacon, E.; Garcia, C.; Eman, P.; Cornea, V.; Jiang, J.; Spear, B.; Liu, C.; Watt, D.S.; Marti, F.; et al. Autophagic Flux Modulation by Wnt/β-Catenin Pathway Inhibition in Hepatocellular Carcinoma. PLoS ONE 2019, 14, e0212538. [Google Scholar] [CrossRef]
- Núñez Selles, A.J.; Daglia, M.; Rastrelli, L. The Potential Role of Mangiferin in Cancer Treatment through Its Immunomodulatory, Anti-Angiogenic, Apoptopic, and Gene Regulatory Effects. Biofactors 2016, 42, 475–491. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Guo, W.; Man, K.; Cheng, C.S.; Chen, Z.; Feng, Y. Repression of WT1-Mediated LEF1 Transcription by Mangiferin Governs β-Catenin-Independent Wnt Signalling Inactivation in Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2018, 47, 1819–1834. [Google Scholar] [CrossRef]
- Li, H.; Huang, J.; Yang, B.; Xiang, T.; Yin, X.; Peng, W.; Cheng, W.; Wan, J.; Luo, F.; Li, H.; et al. Mangiferin Exerts Antitumor Activity in Breast Cancer Cells by Regulating Matrix Metalloproteinases, Epithelial to Mesenchymal Transition, and β-Catenin Signaling Pathway. Toxicol. Appl. Pharmacol. 2013, 272, 180–190. [Google Scholar] [CrossRef]
- Yang, G.; Shang, X.; Cui, G.; Zhao, L.; Zhao, H.; Wang, N. Mangiferin Attenuated Diethynitrosamine-Induced Hepatocellular Carcinoma in Sprague-Dawley Rats via Alteration of Oxidative Stress and Apoptotic Pathway. J. Environ. Pathol. Toxicol. Oncol. 2019, 38, 1–12. [Google Scholar] [CrossRef]
- Ji, J.; Wang, X.W. Clinical Implications of Cancer Stem Cell Biology in Hepatocellular Carcinoma. Semin. Oncol. 2012, 39, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, H.; Tao, K.; Li, R.; Song, Z.; Zhao, Q.; Zhang, F.; Dou, K. Expression and Clinical Significance of the Stem Cell Marker CD133 in Hepatocellular Carcinoma. Int. J. Clin. Pract. 2008, 62, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.K.; Lam, C.T.; Poon, R.T.P.; Fan, S.T. Significance of CD90+ Cancer Stem Cells in Human Liver Cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt Pathways in Cancer Stem Cells: Clinical Update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Koo, B.S.; Kim, J.M.; Huang, S.; Rho, Y.S.; Bae, W.J.; Kang, H.J.; Kim, Y.S.; Moon, J.H.; Lim, Y.C. Wnt/β-Catenin Signalling Maintains Self-Renewal and Tumourigenicity of Head and Neck Squamous Cell Carcinoma Stem-like Cells by Activating Oct4. J. Pathol. 2014, 234, 99–107. [Google Scholar] [CrossRef]
- Quan, M.F.; Xiao, L.H.; Liu, Z.H.; Guo, H.; Ren, K.Q.; Liu, F.; Cao, J.G.; Deng, X.Y. 8-Bromo-7-Methoxychrysin Inhibits Properties of Liver Cancer Stem Cells via Downregulation of β-Catenin. World J. Gastroenterol. 2013, 19, 7680. [Google Scholar] [CrossRef] [PubMed]
- Seto, K.; Sakabe, T.; Itaba, N.; Azumi, J.; Oka, H.; Morimoto, M.; Umekita, Y.; Shiota, G. A Novel Small-Molecule WNT Inhibitor, IC-2, Has the Potential to Suppress Liver Cancer Stem Cells. Anticancer Res. 2017, 37, 3569–3579. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Freese, K.; Mahli, A.; Thasler, W.E.; Hellerbrand, C.; Bosserhoff, A.K. Combined Effects of PLK1 and RAS in Hepatocellular Carcinoma Reveal Rigosertib as Promising Novel Therapeutic “Dual-Hit” Option. Oncotarget 2018, 9, 3605. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Q.; Chen, K.; Sima, Z.; Liu, J.; Yu, Q.; Liu, J. 2-Ethoxystypandrone, a Novel Small-Molecule STAT3 Signaling Inhibitor from Polygonum Cuspidatum, Inhibits Cell Growth and Induces Apoptosis of HCC Cells and HCC Cancer Stem Cells. BMC Complement. Altern. Med. 2019, 19, 38. [Google Scholar] [CrossRef]
- Liu, Y.; Fuchs, J.; Li, C.; Lin, J. IL-6, a Risk Factor for Hepatocellular Carcinoma: FLLL32 Inhibits IL-6-Induced STAT3 Phosphorylation in Human Hepatocellular Cancer Cells. Cell Cycle 2010, 9, 3423–3427. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, A.; Xu, Z.; Yu, W.; Wang, H.; Li, C.; Lin, J. XZH-5 Inhibits STAT3 Phosphorylation and Causes Apoptosis in Human Hepatocellular Carcinoma Cells. Apoptosis 2011, 16, 502–510. [Google Scholar] [CrossRef]
- Jo, H.; Lo, P.K.; Li, Y.; Loison, F.; Green, S.; Wang, J.; Silberstein, L.E.; Ye, K.; Chen, H.; Luo, H.R. Deactivation of Akt by a Small Molecule Inhibitor Targeting Pleckstrin Homology Domain and Facilitating Akt Ubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 6486–6491. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Del Barrio, M.G.; Portillo, F.; Nieto, M.A. The Transcription Factor Snail Controls Epithelial–Mesenchymal Transitions by Repressing E-Cadherin Expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Cusimano, A.; Puleio, R.; D’Alessandro, N.; Loria, G.R.; McCubrey, J.A.; Montalto, G.; Cervello, M. Cytotoxic Activity of the Novel Small Molecule AKT Inhibitor SC66 in Hepatocellular Carcinoma Cells. Oncotarget 2015, 6, 1707–1722. [Google Scholar] [CrossRef]
- Mohan, C.D.; Anilkumar, N.C.; Rangappa, S.; Shanmugam, M.K.; Mishra, S.; Chinnathambi, A.; Alharbi, S.A.; Bhattacharjee, A.; Sethi, G.; Kumar, A.P.; et al. Novel 1,3,4-Oxadiazole Induces Anticancer Activity by Targeting NF-ΚB in Hepatocellular Carcinoma Cells. Front. Oncol. 2018, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Zhang, H.; Yu, J.; Francisco, R.; Dent, P.; Ebert, M.P.A.; Röcken, C.; Farrell, G. Constitutive Activation of NF-KappaB in Human Hepatocellular Carcinoma: Evidence of a Cytoprotective Role. Hum. Gene Ther. 2006, 17, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Constitutive Activation of Nuclear Factor KappaB in Hepatocellular Carcinoma—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/11147598/ (accessed on 20 July 2022).
- Baburajeev, C.P.; Mohan, C.D.; Rangappa, S.; Mason, D.J.; Fuchs, J.E.; Bender, A.; Barash, U.; Vlodavsky, I.; Basappa; Rangappa, K.S. Identification of Novel Class of Triazolo-Thiadiazoles as Potent Inhibitors of Human Heparanase and Their Anticancer Activity. BMC Cancer 2017, 17, 1–14. [Google Scholar] [CrossRef]
- Roopashree, R.; Mohan, C.D.; Swaroop, T.R.; Jagadish, S.; Raghava, B.; Balaji, K.S.; Jayarama, S.; Basappa; Rangappa, K.S. Novel Synthetic Bisbenzimidazole That Targets Angiogenesis in Ehrlich Ascites Carcinoma Bearing Mice. Bioorg. Med. Chem. Lett. 2015, 25, 2589–2593. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a Novel Azaspirane That Targets the Janus Kinase-Signal Transducer and Activator of Transcription (STAT) Pathway in Hepatocellular Carcinoma in Vitro and in Vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef] [Green Version]
- Traenckner, E.B.M.; Pahl, H.L.; Henkel, T.; Schmidt, K.N.; Wilk, S.; Baeuerle, P.A. Phosphorylation of Human I Kappa B-Alpha on Serines 32 and 36 Controls I Kappa B-Alpha Proteolysis and NF-Kappa B Activation in Response to Diverse Stimuli. EMBO J. 1995, 14, 2876–2883. [Google Scholar] [CrossRef]
- Ebert, G.; Allison, C.; Preston, S.; Cooney, J.; Toe, J.G.; Stutz, M.D.; Ojaimi, S.; Baschuk, N.; Nachbur, U.; Torresi, J.; et al. Eliminating Hepatitis B by Antagonizing Cellular Inhibitors of Apoptosis. Proc. Natl. Acad. Sci. USA 2015, 112, 5803–5808. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Darding, M.; Miasari, M.; Santoro, M.M.; Zender, L.; Xue, W.; Tenev, T.; da Fonseca, P.C.A.; Zvelebil, M.; Bujnicki, J.M.; et al. IAPs Contain an Evolutionarily Conserved Ubiquitin-Binding Domain That Regulates NF-KappaB as Well as Cell Survival and Oncogenesis. Nat. Cell Biol. 2008, 10, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Dougan, M.; Dougan, S.; Slisz, J.; Firestone, B.; Vanneman, M.; Draganov, D.; Goyal, G.; Li, W.; Neuberg, D.; Blumberg, R.; et al. IAP Inhibitors Enhance Co-Stimulation to Promote Tumor Immunity. J. Exp. Med. 2010, 207, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Mirza, N.N.; Garbitt, V.M.; Sharik, M.E.; Dueck, A.C.; Asmann, Y.W.; Akhmetzyanova, I.; Kosiorek, H.E.; Calcinotto, A.; Riggs, D.L.; et al. IAP Antagonists Induce Anti-Tumor Immunity in Multiple Myeloma. Nat. Med. 2016, 22, 1411–1420. [Google Scholar] [CrossRef]
- Pan, W.; Luo, Q.; Yan, X.; Yuan, L.; Yi, H.; Zhang, L.; Li, B.; Zhang, Y.; Sun, J.; Qiu, M.Z.; et al. A Novel SMAC Mimetic APG-1387 Exhibits Dual Antitumor Effect on HBV-Positive Hepatocellular Carcinoma with High Expression of CIAP2 by Inducing Apoptosis and Enhancing Innate Anti-Tumor Immunity. Biochem. Pharmacol. 2018, 154, 127–135. [Google Scholar] [CrossRef]
- Fu, Z.G.; Wang, L.; Cuiv, H.Y.; Peng, J.L.; Wangc, S.J.; Gengc, J.J.; Liu, J.D.; Feng, F.; Song, F.; Li, L.; et al. A Novel Small-Molecule Compound Targeting CD147 Inhibits the Motility and Invasion of Hepatocellular Carcinoma Cells. Oncotarget 2016, 7, 9429–9447. [Google Scholar] [CrossRef]
- Augello, G.; Puleio, R.; Emma, M.R.; Cusimano, A.; Loria, G.R.; McCubrey, J.A.; Montalto, G.; Cervello, M. A PTEN Inhibitor Displays Preclinical Activity against Hepatocarcinoma Cells. Cell Cycle 2016, 15, 573. [Google Scholar] [CrossRef]
- Xiao, Z.; Han Li, C.; Chan, S.L.; Xu, F.; Feng, L.; Wang, Y.; Jiang, J.D.; Sung, J.J.Y.; Cheng, C.H.K.; Chen, Y. A Small-Molecule Modulator of the Tumor-Suppressor MiR34a Inhibits the Growth of Hepatocellular Carcinoma. Cancer Res. 2014, 74, 6236–6247. [Google Scholar] [CrossRef]
- Grant, T.J.; Bishop, J.A.; Christadore, B.M.; Barot, G.; Chin, H.G.; Woodson, S.; Kavouris, J.; Siddiq, A.; Gredler, R.; Shen, X.N.; et al. Antiproliferative Small-Molecule Inhibitors of Transcription Factor LSF Reveal Oncogene Addiction to LSF in Hepatocellular Carcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 4503–4508. [Google Scholar] [CrossRef] [Green Version]
- Augello, G.; Emma, M.R.; Cusimano, A.; Azzolina, A.; Mongiovì, S.; Puleio, R.; Cassata, G.; Gulino, A.; Belmonte, B.; Gramignoli, R.; et al. Targeting HSP90 with the Small Molecule Inhibitor AUY922 (Luminespib) as a Treatment Strategy against Hepatocellular Carcinoma. Int. J. Cancer 2019, 144, 2613–2624. [Google Scholar] [CrossRef]
- Peddibhotla, S.; Hershberger, P.M.; Jason Kirby, R.; Sugarman, E.; Maloney, P.R.; Hampton Sessions, E.; Divlianska, D.; Morfa, C.J.; Terry, D.; Pinkerton, A.B.; et al. Discovery of Small Molecule Antagonists of Chemokine Receptor CXCR6 That Arrest Tumor Growth in SK-HEP-1 Mouse Xenografts as a Model of Hepatocellular Carcinoma. Bioorg. Med. Chem. Lett. 2020, 30, 126899. [Google Scholar] [CrossRef] [PubMed]
- Portmann, S.; Fahrner, R.; Lechleiter, A.; Keogh, A.; Overney, S.; Laemmle, A.; Mikami, K.; Montani, M.; Tschan, M.P.; Candinas, D.; et al. Antitumor Effect of SIRT1 Inhibition in Human HCC Tumor Models in Vitro and in Vivo. Mol. Cancer Ther. 2013, 12, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Haupenthal, J.; Bihrer, V.; Korkusuz, H.; Kollmar, O.; Schmithals, C.; Kriener, S.; Engels, K.; Pleli, T.; Benz, A.; Canamero, M.; et al. Reduced Efficacy of the Plk1 Inhibitor BI 2536 on the Progression of Hepatocellular Carcinoma Due to Low Intratumoral Drug Levels. Neoplasia 2012, 14, 410. [Google Scholar] [CrossRef] [PubMed]
- Tsang, F.H.C.; Law, C.T.; Tang, T.C.C.; Cheng, C.L.H.; Chin, D.W.C.; Tam, W.S.V.; Wei, L.; Wong, C.C.L.; Ng, I.O.L.; Wong, C.M. Aberrant Super-Enhancer Landscape in Human Hepatocellular Carcinoma. Hepatology 2019, 69, 2502–2517. [Google Scholar] [CrossRef] [PubMed]
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.E.; Myers, C.; Chernoff, J.; Peterson, J.R. An Isoform-Selective, Small-Molecule Inhibitor Targets the Autoregulatory Mechanism of P21-Activated Kinase. Chem. Biol. 2008, 15, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Viaud, J.; Peterson, J.R. An Allosteric Kinase Inhibitor Binds the P21-Activated Kinase (Pak) Autoregulatory Domain Covalently. Mol. Cancer Ther. 2009, 8, 2559. [Google Scholar] [CrossRef]
- Fan, S.; Gao, M.; Meng, Q.; Laterra, J.J.; Symons, M.H.; Coniglio, S.; Pestell, R.G.; Goldberg, I.D.; Rosen, E.M. Role of NF-KappaB Signaling in Hepatocyte Growth Factor/Scatter Factor-Mediated Cell Protection. Oncogene 2005, 24, 1749–1766. [Google Scholar] [CrossRef]
- Frost, J.A.; Swantek, J.L.; Stippec, S.; Yin, M.J.; Gaynor, R.; Cobb, M.H. Stimulation of NFkappa B Activity by Multiple Signaling Pathways Requires PAK1. J. Biol. Chem. 2000, 275, 19693–19699. [Google Scholar] [CrossRef]
- Wong, L.L.Y.; Lam, I.P.Y.; Wong, T.Y.N.; Lai, W.L.; Liu, H.F.; Yeung, L.L.; Ching, Y.P. IPA-3 Inhibits the Growth of Liver Cancer Cells By Suppressing PAK1 and NF-ΚB Activation. PLoS One 2013, 8, e68843. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, W.; Kang, W.; Wong, S.H.; Wang, M.; Zhou, Y.; Fang, X.; Zhang, X.; Yang, H.; Wong, C.H.; et al. Genomic Analysis of Liver Cancer Unveils Novel Driver Genes and Distinct Prognostic Features. Theranostics 2018, 8, 1740–1751. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022. [Google Scholar] [CrossRef]
- Bukowska, B.; Gajek, A.; Marczak, A. Two Drugs Are Better than One. A Short History of Combined Therapy of Ovarian Cancer. Contemp. Oncol. Onkol. 2014, 19, 350–353. [Google Scholar] [CrossRef]
- Palmer, A.C.; Chidley, C.; Sorger, P.K. A Curative Combination Cancer Therapy Achieves High Fractional Cell Killing through Low Cross Resistance and Drug Additivity. eLife 2019, 8, e50036. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A. Oet al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Ismail, M.; Khan, S.; Khan, F.; Noor, S.; Sajid, H.; Yar, S.; Rasheed, I. Prevalence and Significance of Potential Drug-Drug Interactions among Cancer Patients Receiving Chemotherapy. BMC Cancer 2020, 20, 1–9. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Mayer, L.D. Improving Combination Cancer Therapy: The CombiPlex® Development Platform. Future Oncol. 2018, 14, 1317–1332. [Google Scholar] [CrossRef]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Narayan, R.S.; Molenaar, P.; Teng, J.; Cornelissen, F.M.G.; Roelofs, I.; Menezes, R.; Dik, R.; Lagerweij, T.; Broersma, Y.; Petersen, N.; et al. A Cancer Drug Atlas Enables Synergistic Targeting of Independent Drug Vulnerabilities. Nat. Commun. 2020, 11, 2935. [Google Scholar] [CrossRef]
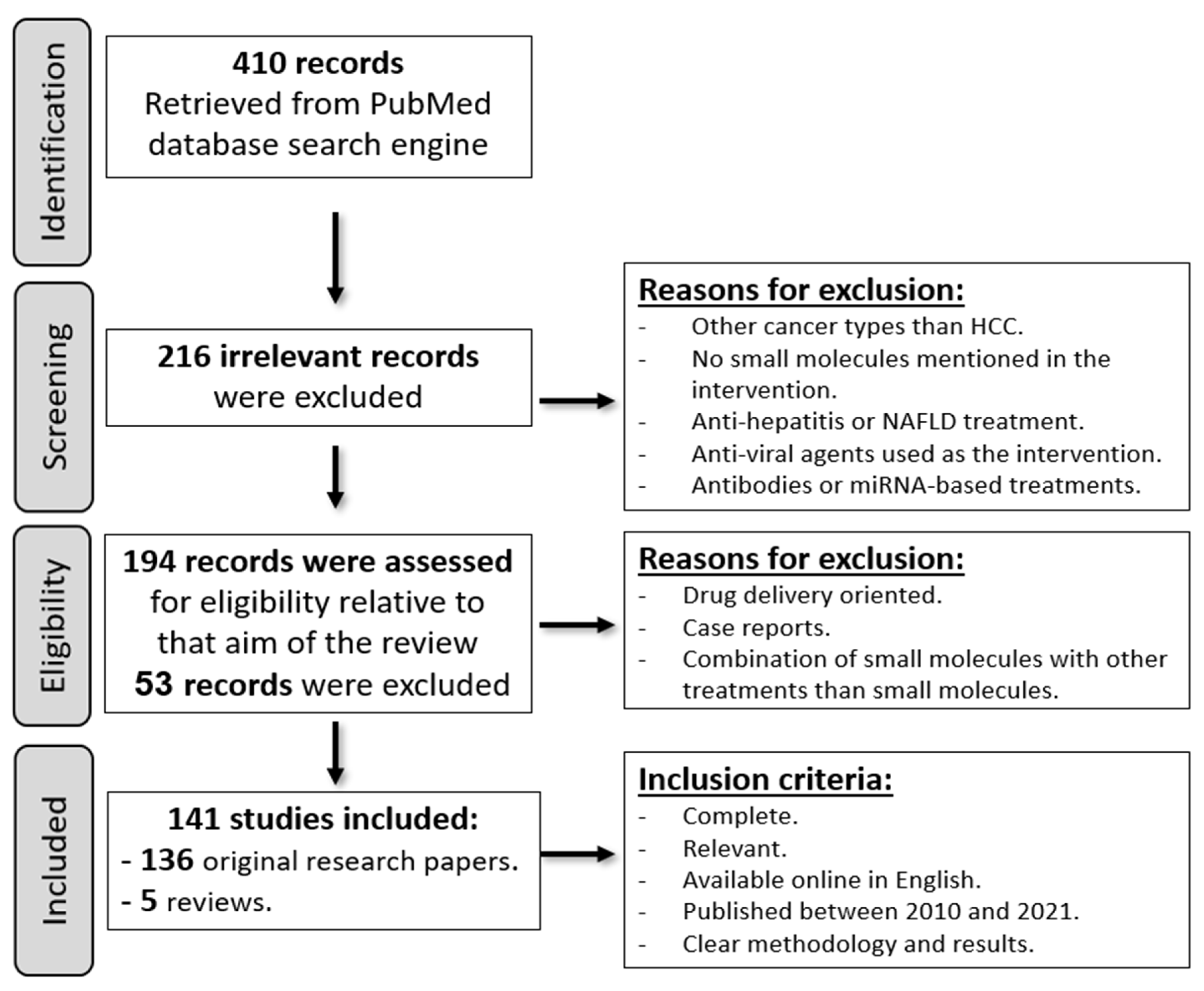





| Small Molecule Inhibitor | Target |
|---|---|
| CMO | P65 protein |
| APG-1387 | Inhibitor of apoptosis proteins (IAPs) |
| AC-73 | CD147 |
| VO-OHpic | Phosphatase and tensin homolog (PTEN) |
| Rubone | miR34a |
| FQI1 | Transcription factor LSF |
| AUY922 (luminespib) | Heat shock protein 90 (HSP-90) |
| Compound 81 | Chemokine receptor 6 (CXCR6) |
| Cambinol | Sirtuin 1 (SIRT-1) |
| BI 2536 | Polo-like kinase 1 (plk-1) |
| THZ1 | cyclin dependent kinase 7 (CDK7) |
| IPA-3 | p21-activated kinase 1 (PAK1) |
| Alisertib | AURKA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamal, M.A.; Mandour, Y.M.; Abd El-Aziz, M.K.; Stein, U.; El Tayebi, H.M. Small Molecule Inhibitors for Hepatocellular Carcinoma: Advances and Challenges. Molecules 2022, 27, 5537. https://doi.org/10.3390/molecules27175537
Kamal MA, Mandour YM, Abd El-Aziz MK, Stein U, El Tayebi HM. Small Molecule Inhibitors for Hepatocellular Carcinoma: Advances and Challenges. Molecules. 2022; 27(17):5537. https://doi.org/10.3390/molecules27175537
Chicago/Turabian StyleKamal, Monica A., Yasmine M. Mandour, Mostafa K. Abd El-Aziz, Ulrike Stein, and Hend M. El Tayebi. 2022. "Small Molecule Inhibitors for Hepatocellular Carcinoma: Advances and Challenges" Molecules 27, no. 17: 5537. https://doi.org/10.3390/molecules27175537
APA StyleKamal, M. A., Mandour, Y. M., Abd El-Aziz, M. K., Stein, U., & El Tayebi, H. M. (2022). Small Molecule Inhibitors for Hepatocellular Carcinoma: Advances and Challenges. Molecules, 27(17), 5537. https://doi.org/10.3390/molecules27175537