
Discovery of Some Heterocyclic Molecules as Bone Morphogenetic Protein 2 (BMP-2)-Inducible Kinase Inhibitors: Virtual Screening, ADME Properties, and Molecular Docking Simulations
 ,
,  ,
,  ,
,  , , , ,
, , , ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
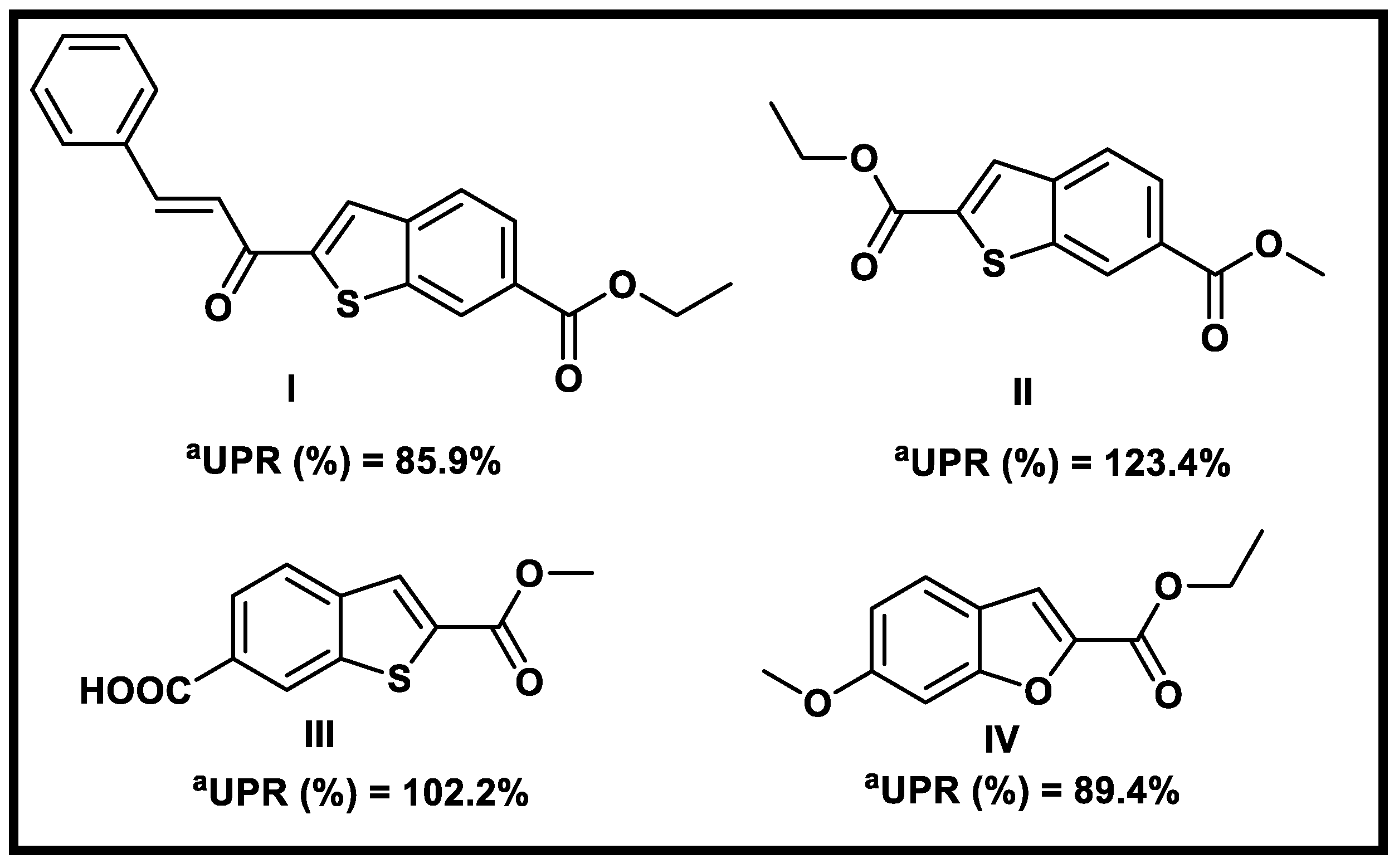
2.1. Virtual Screening
2.2. Computational ADMET Analysis
2.3. Computational Toxicity Studies
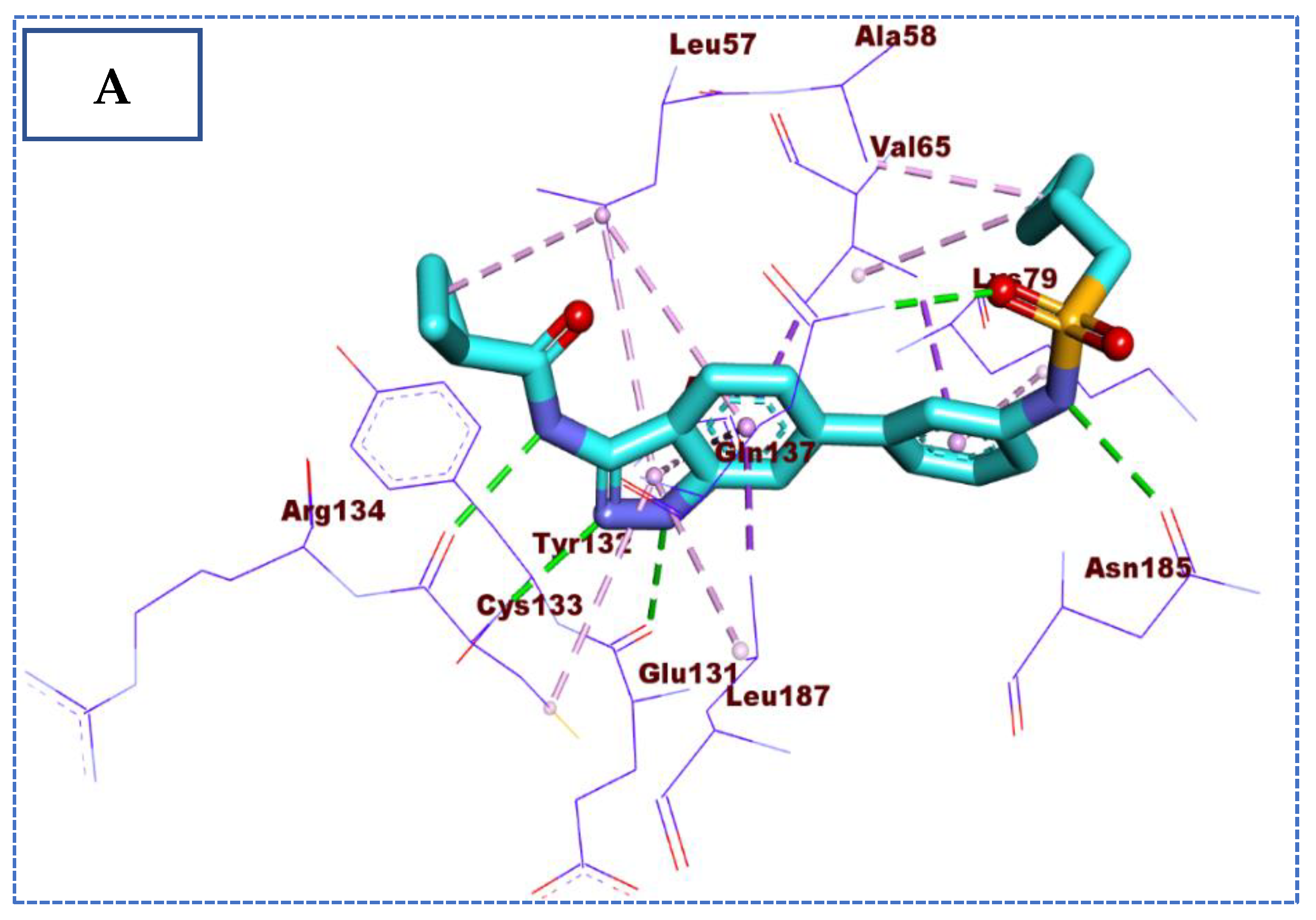
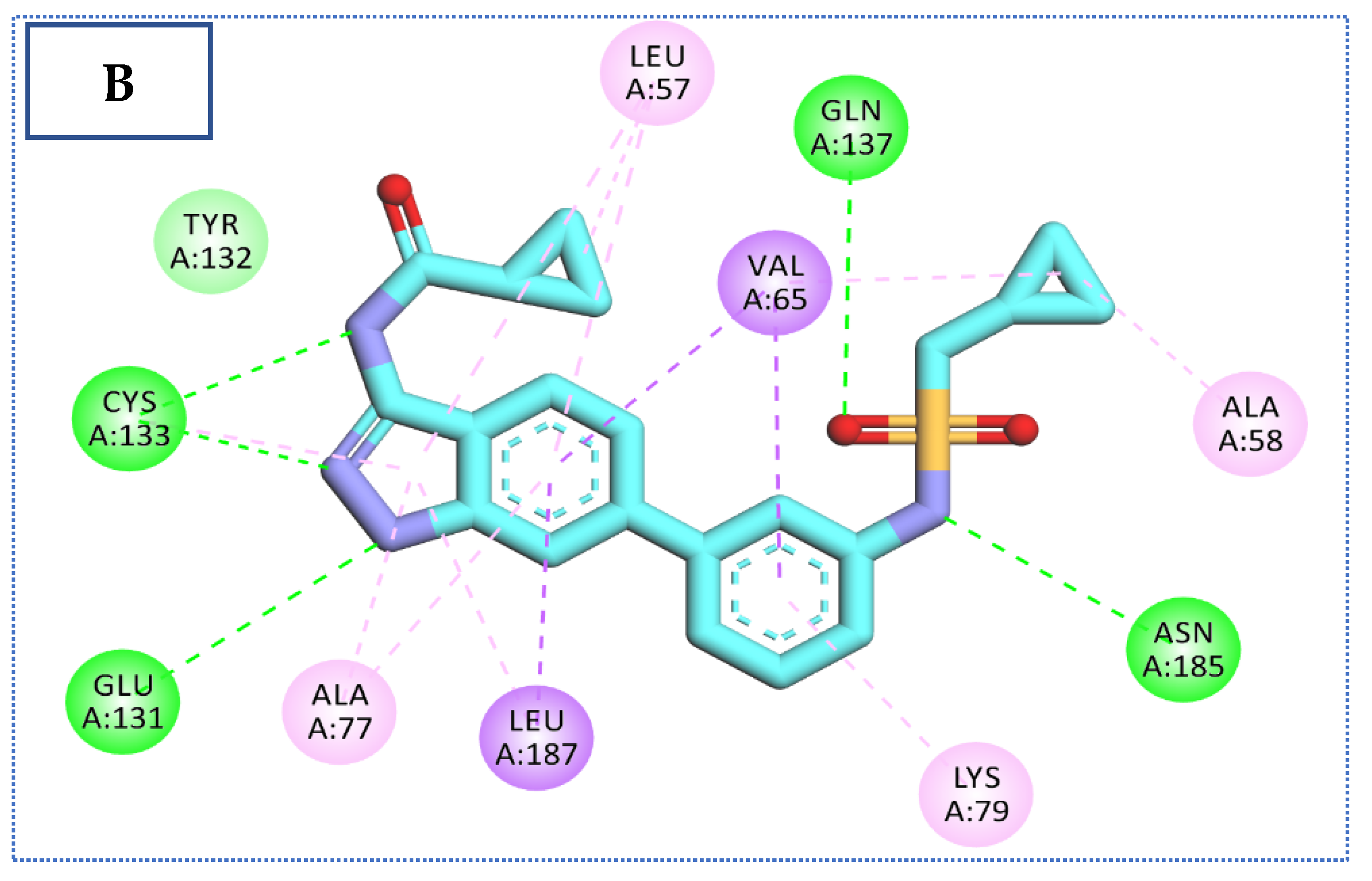
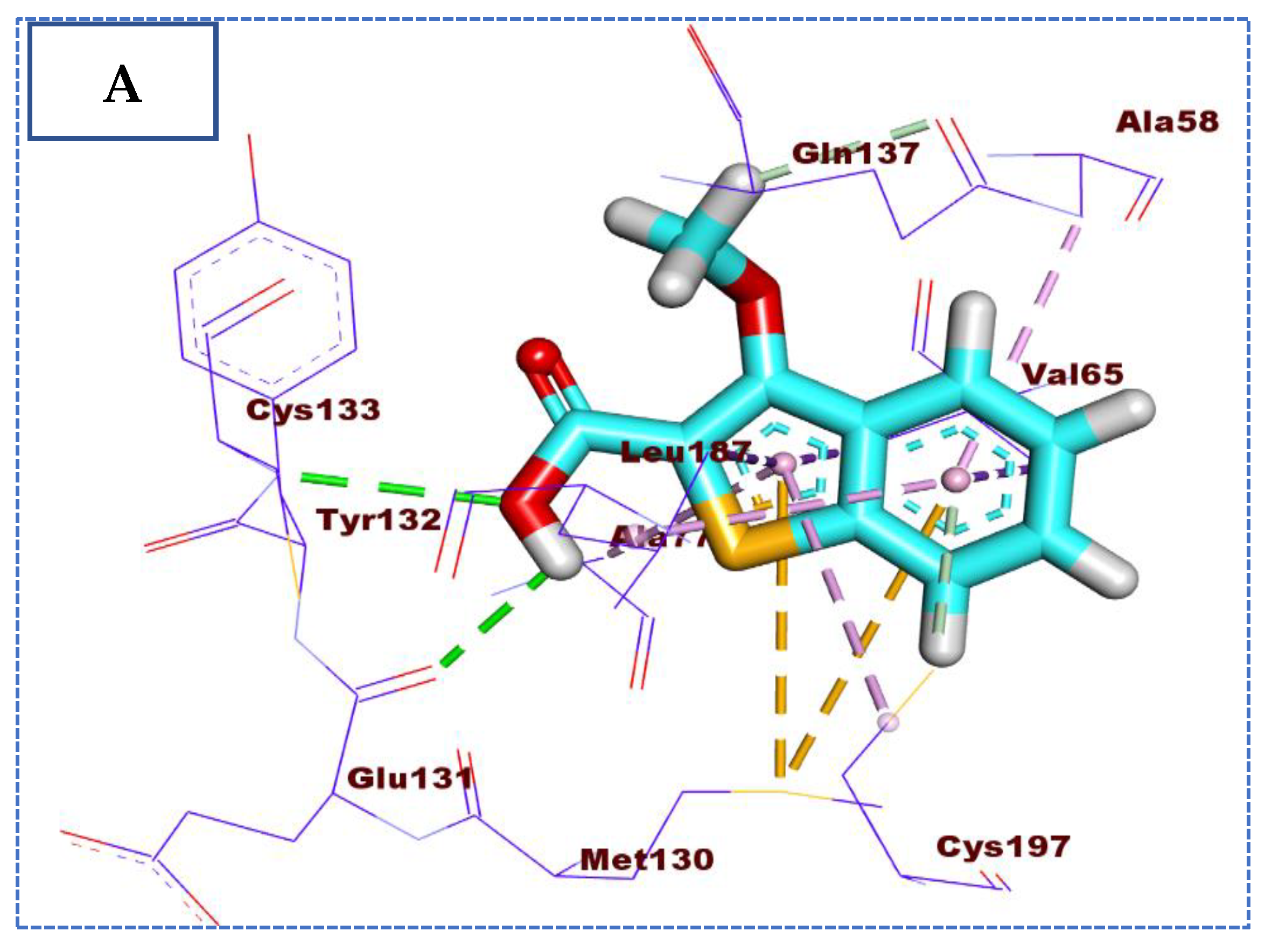
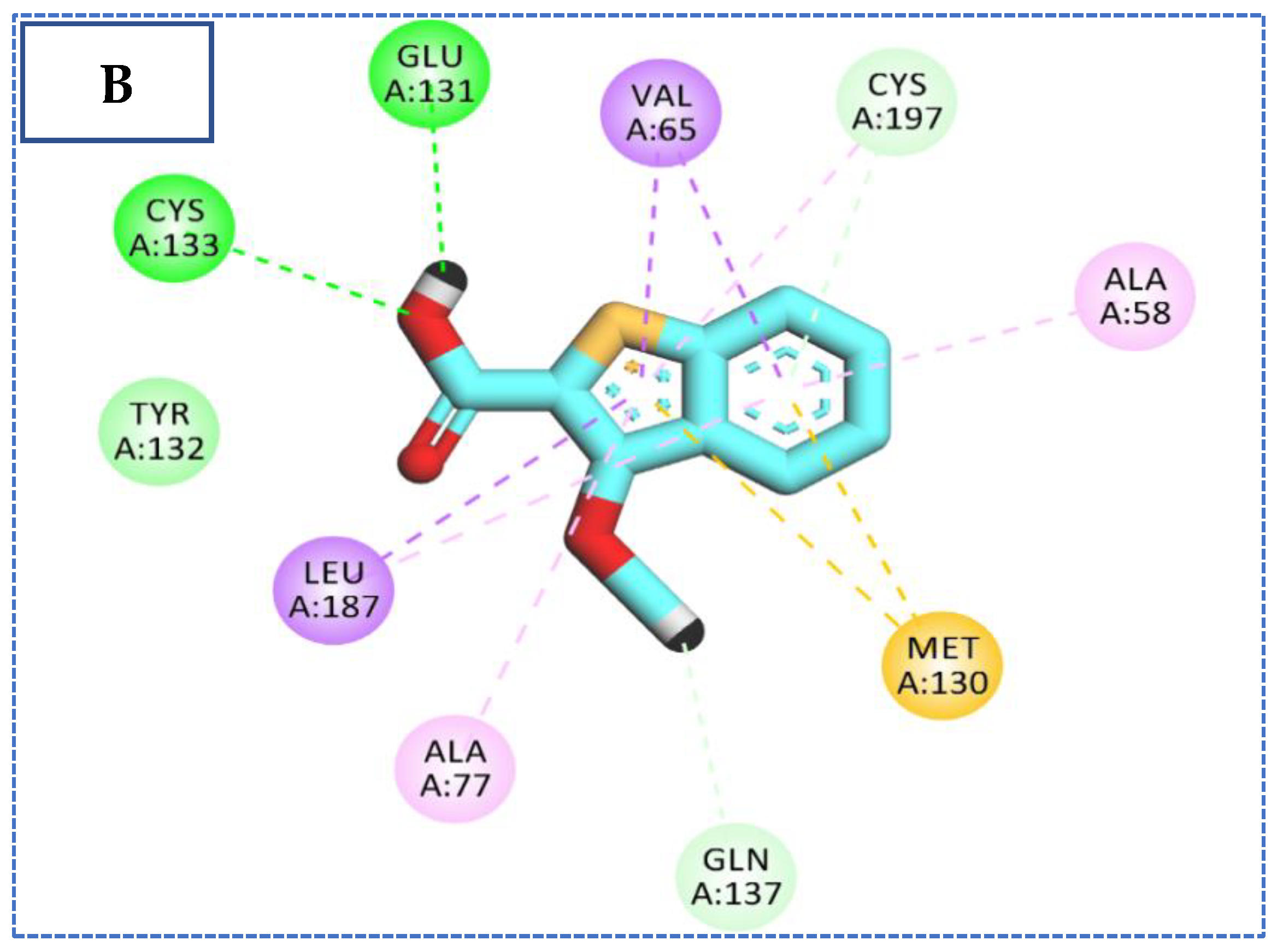
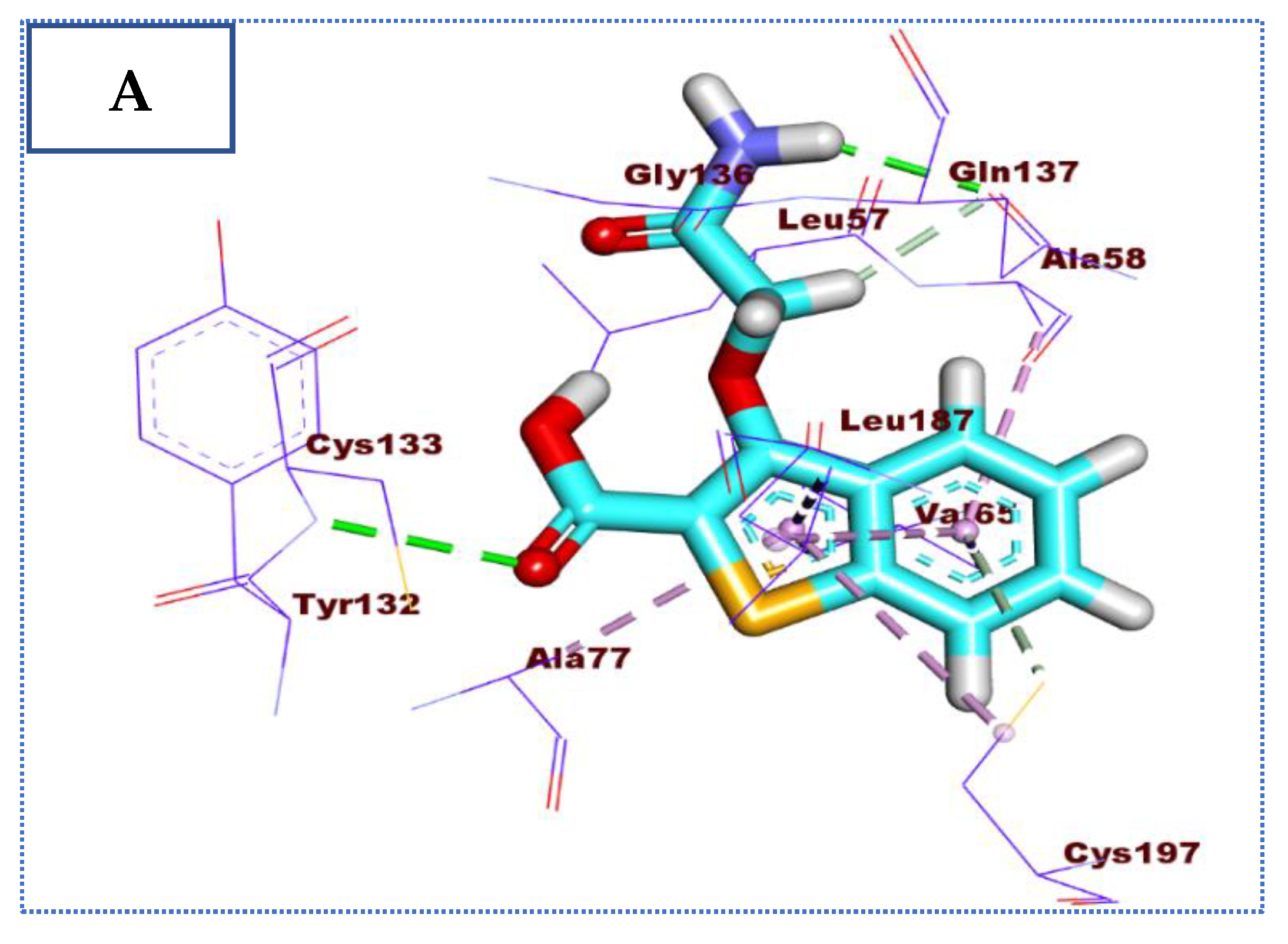
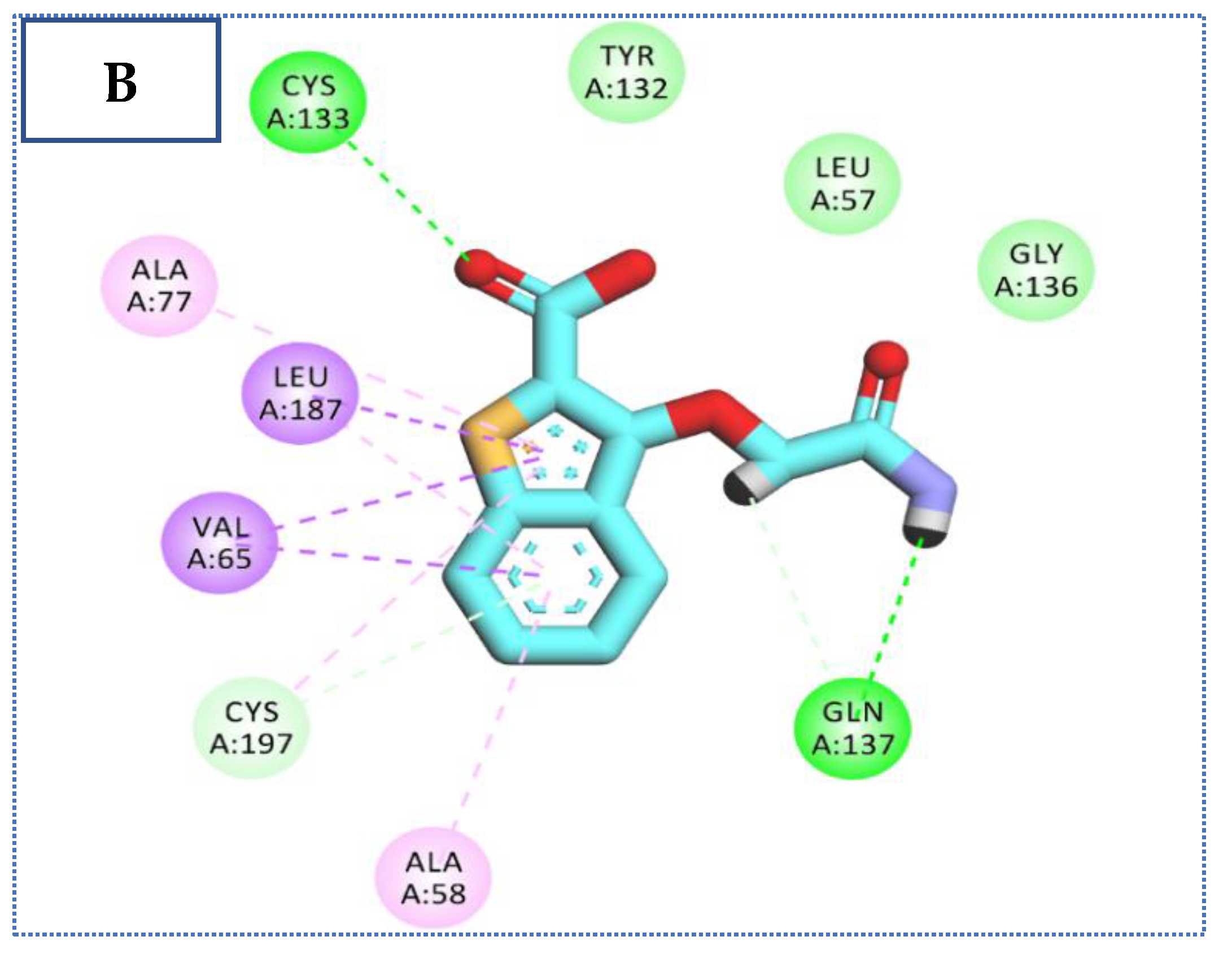
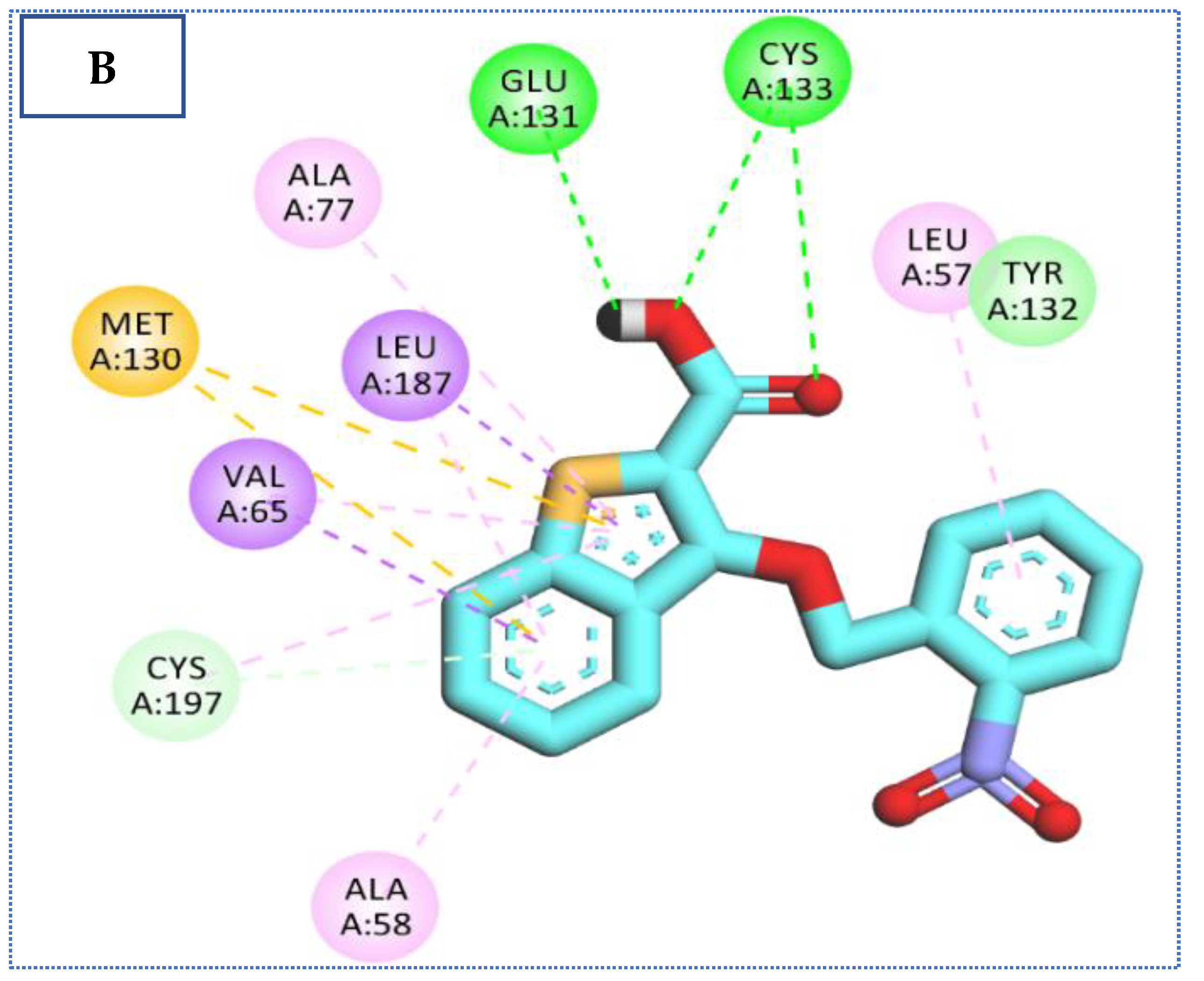
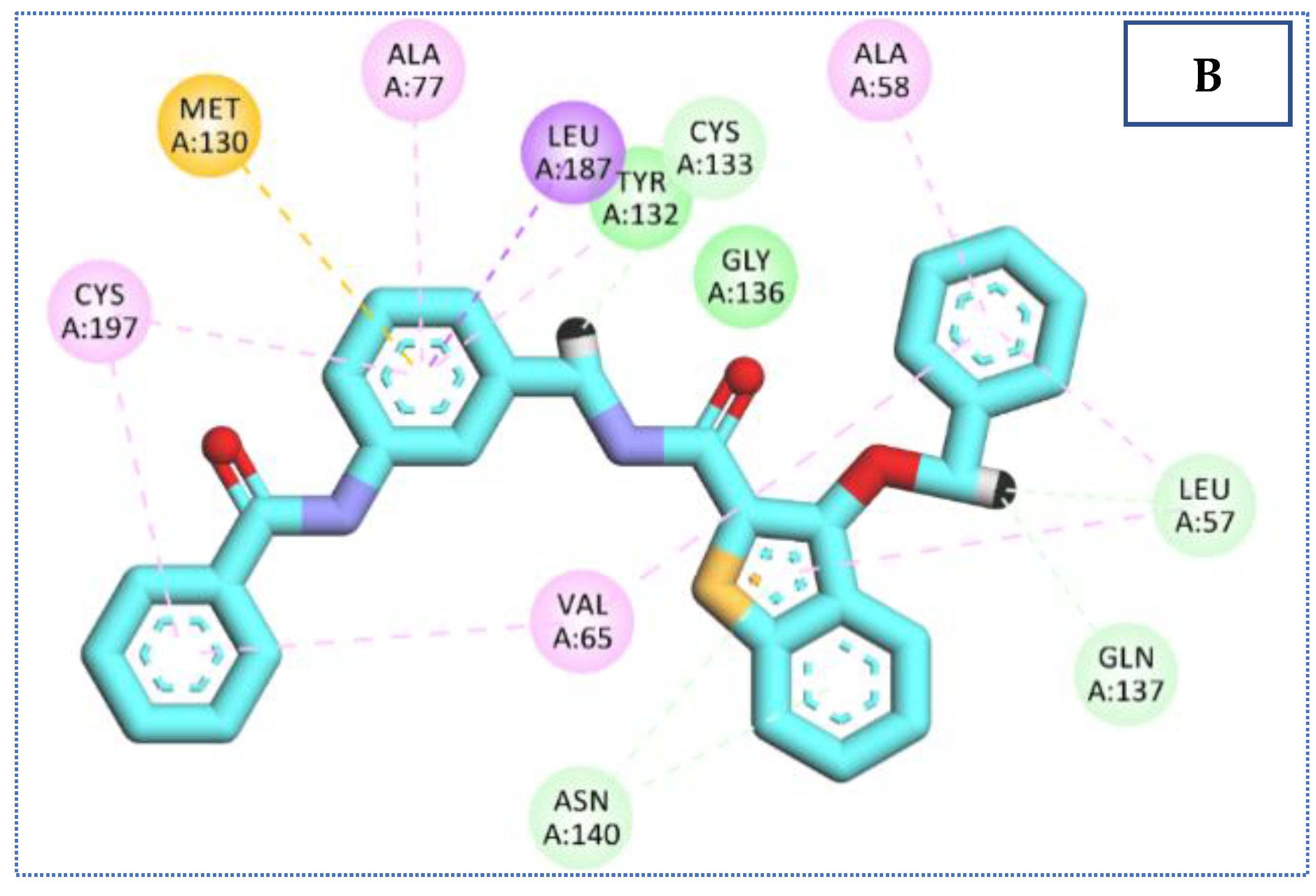
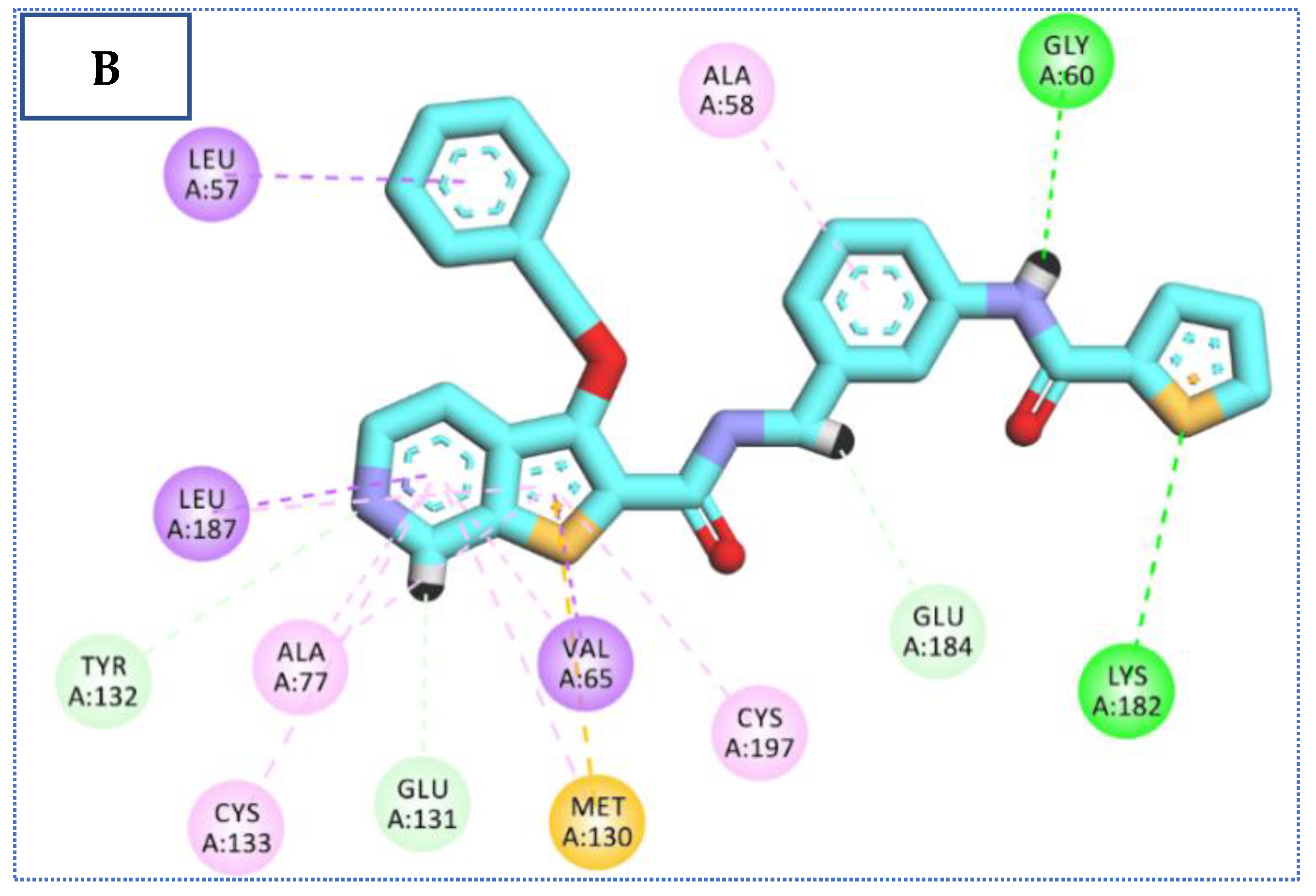
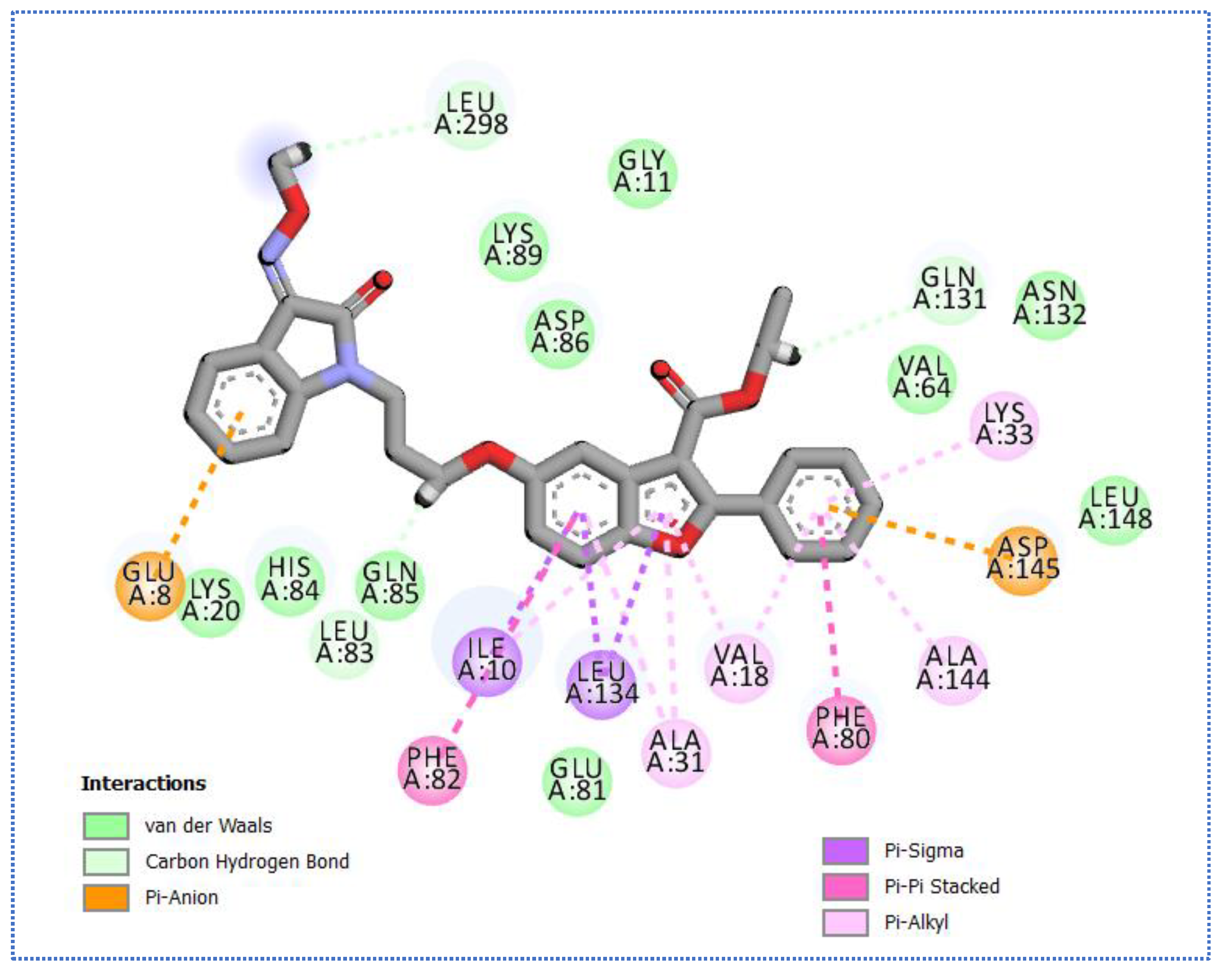
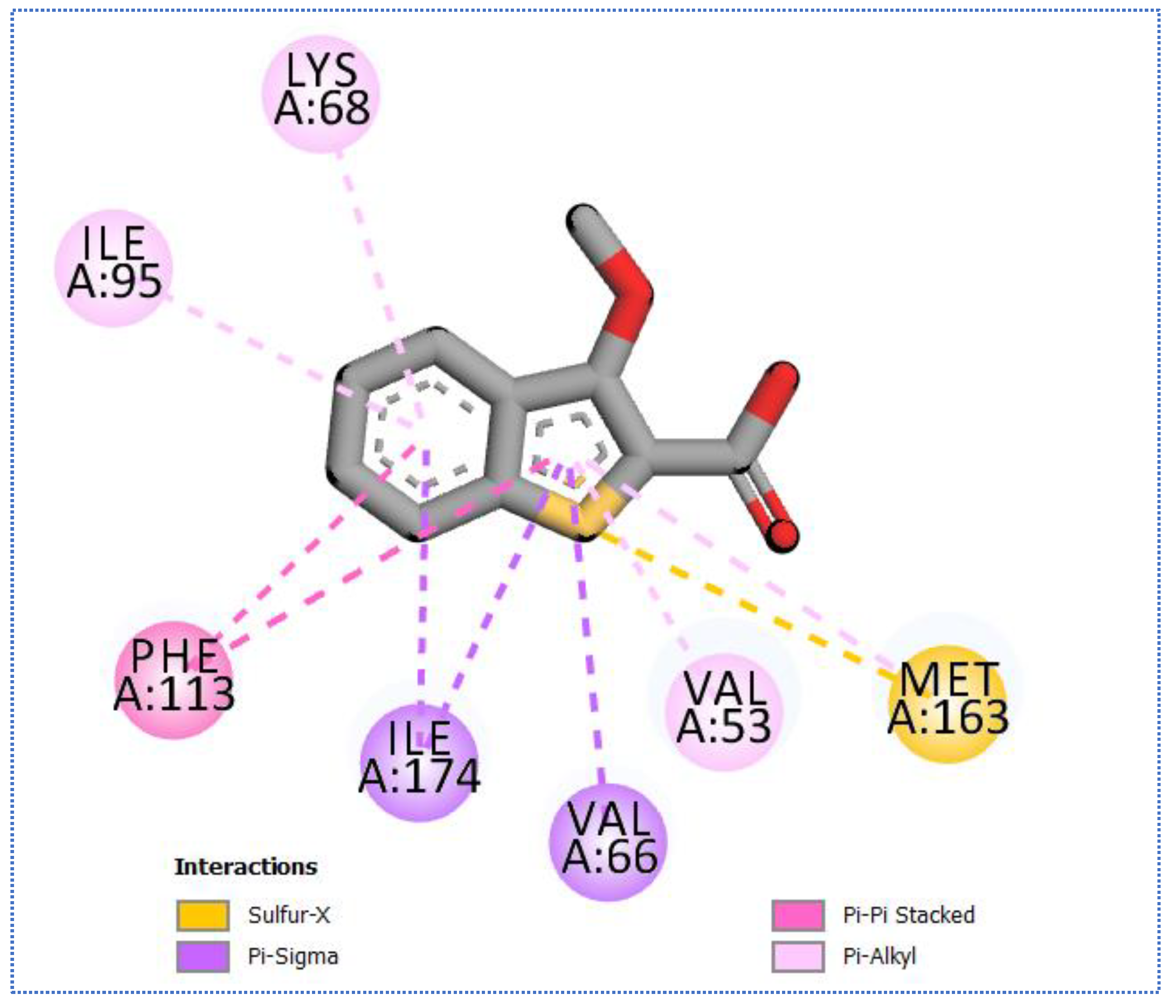
2.4. Molecular Docking Studies
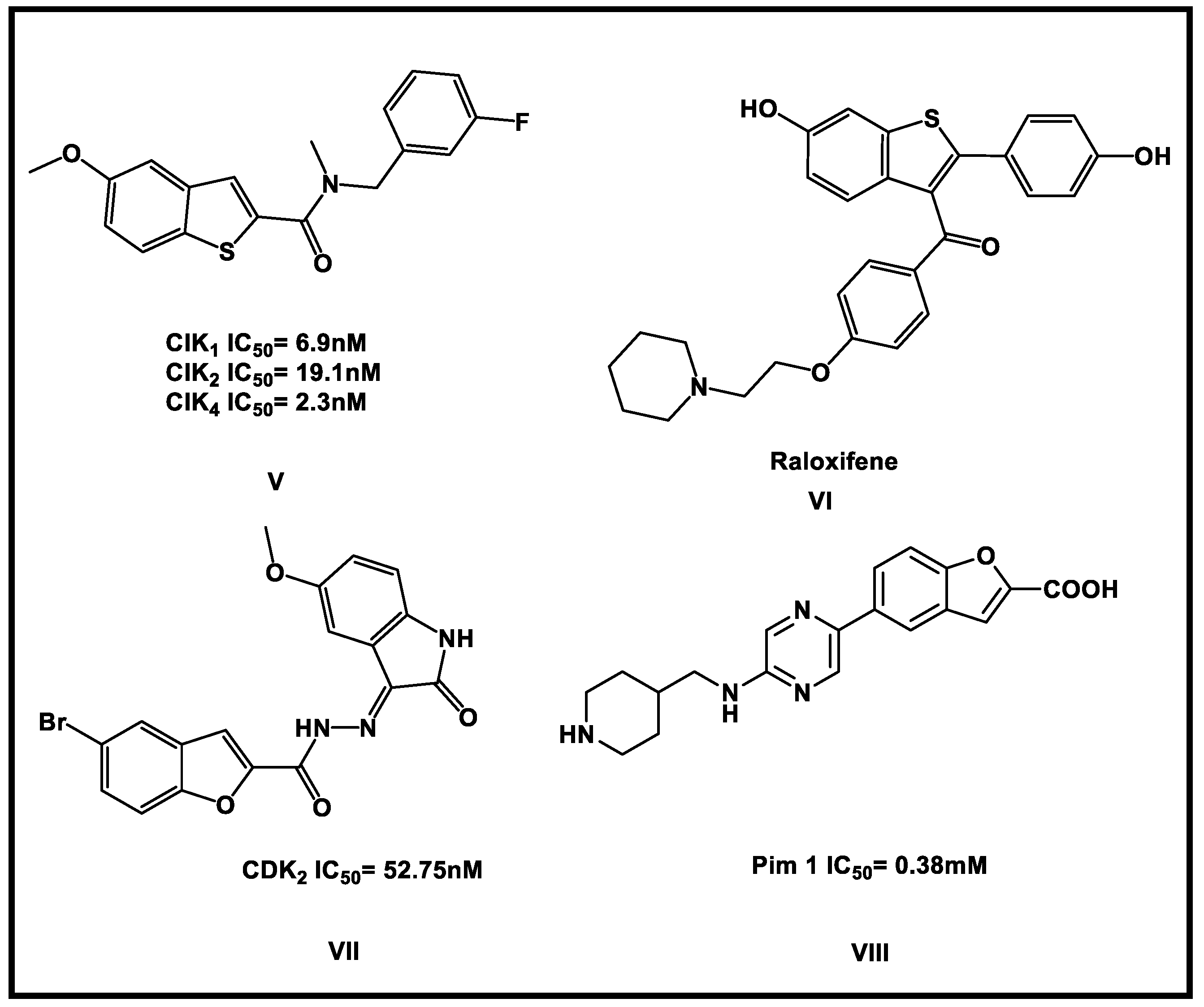
2.5. Further Investigation against Protein Kinases
3. Conclusions
4. Experimental Section
4.1. Screening against Lipinski’s and Veber’s Rules
4.2. Computational ADMET Studies
4.3. Computational Toxicity Studies
4.4. Docking Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiao, Y.T.; Xiang, L.X.; Shao, J.Z. Bone morphogenetic protein. Biochem. Biophys. Res. Commun. 2007, 362, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.H. Bone Morphogenetic Proteins: From Basic Science to Clinical Applications. J. Bone Jt. Surg. 2001, 83, S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A. Cell Biology and Biochemistry of Endochondral Bone Development. Collagen Relat. Res. 1981, 1, 209–226. [Google Scholar] [CrossRef]
- Jain, A.P.; Pundir, S.; Sharma, A. Bone morphogenetic proteins: The anomalous molecules. J. Indian Soc. Periodontol. 2013, 17, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.; Raja, E.; Miyazono, K.; Tsubakihara, Y.; Moustakas, A. Mechanisms of action of bone morphogenetic proteins in cancer. Cytokine Growth Factor Rev. 2015, 27, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Ehata, S.; Yokoyama, Y.; Takahashi, K.; Miyazono, K. Bi-directional roles of bone morphogenetic proteins in cancer: Another molecular Jekyll and Hyde? Pathol. Int. 2013, 63, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.C.; Kodach, L.; Offerhaus, G.J.; Brink, G.R.V.D. Bone morphogenetic protein signalling in colorectal cancer. Nat. Cancer 2008, 8, 806–812. [Google Scholar] [CrossRef]
- Jaeger, E.; Leedham, S.J.; Lewis, A.; Segditsas, S.; Becker, M.; Cuadrado, P.R.; Davis, H.; Kaur, K.; Heinimann, K.; Howarth, K.; et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat. Genet. 2012, 44, 699–703. [Google Scholar] [CrossRef]
- Skovrlj, B.; Koehler, S.M.; Anderson, P.A.; Qureshi, S.A.; Hecht, A.C.; Iatridis, J.C.; Cho, S.K. Association Between BMP-2 and Carcinogenicity. Spine 2015, 40, 1862–1871. [Google Scholar] [CrossRef]
- James, A.W.; Lachaud, G.; Shen, J.; Asatrian, G.; Nguyen, V.; Zhang, X.; Ting, K.; Soo, C. A Review of the Clinical Side Effects of Bone Morphogenetic Protein-2. Tissue Eng. Part B Rev. 2016, 22, 284–297. [Google Scholar] [CrossRef]
- Ehata, S.; Miyazono, K. Bone Morphogenetic Protein Signaling in Cancer; Some Topics in the Recent 10 Years. Front. Cell Dev. Biol. 2022, 10, 883523. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ma, L.; Guo, Q.; Zhang, S. Expression of bone morphogenetic protein-2 and its receptors in epithelial ovarian cancer and their influence on the prognosis of ovarian cancer patients. J. Exp. Clin. Cancer Res. 2010, 29, 85–86. [Google Scholar] [CrossRef]
- Raida, M.; Clement, J.H.; Ameri, K.; Han, C.; Leek, R.D.; Harris, A.L. Expression of bone morphogenetic protein 2 in breast cancer cells inhibits hypoxic cell death. Int. J. Oncol. 2005, 26, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Kim, J.S.; Seo, J.E.; Oh, S.C.; Yoo, Y.A. BMP2 accelerates the motility and invasiveness of gastric cancer cells via activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway. Exp. Cell Res. 2010, 316, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Radadiya, A.; Shah, A. Bioactive benzofuran derivatives: An insight on lead developments, radioligands and advances of the last decade. Eur. J. Med. Chem. 2015, 5, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Chand, K.; Budagumpi, S.; Somappa, S.B.; Patil, S.A.; Nagaraja, B.M. An overview of benzo [b] thiophene-based medicinal chemistry. Eur. J. Med. Chem. 2017, 29, 1002–1033. [Google Scholar] [CrossRef]
- Farhat, J.; Alzyoud, L.; Alwahsh, M.; Al-Omari, B. Structure–Activity Relationship of Benzofuran Derivatives with Potential Anticancer Activity. Cancers 2022, 14, 2196. [Google Scholar] [CrossRef]
- Guo, H.-F.; Shao, H.-Y.; Yang, Z.-Y.; Xue, S.-T.; Li, X.; Liu, Z.-Y.; He, X.-B.; Jiang, J.-D.; Zhang, Y.-Q.; Si, S.-Y.; et al. Substituted Benzothiophene or Benzofuran Derivatives as a Novel Class of Bone Morphogenetic Protein-2 Up-Regulators: Synthesis, Structure−Activity Relationships, and Preventive Bone Loss Efficacies in Senescence Accelerated Mice (SAMP6) and Ovariectomized Rats. J. Med. Chem. 2010, 53, 1819–1829. [Google Scholar] [CrossRef]
- Xue, S.-T.; Guo, H.-F.; Liu, M.-J.; Jin, J.; Ju, D.-H.; Liu, Z.-Y.; Li, Z.-R. Synthesis of a novel class of substituted benzothiophene or benzofuran derivatives as BMP-2 up-regulators and evaluation of the BMP-2-up-regulating effects in vitro and the effects on glucocorticoid-induced osteoporosis in rats. Eur. J. Med. Chem. 2015, 96, 151–161. [Google Scholar] [CrossRef]
- ElHady, A.K.; El-Gamil, D.S.; Chen, P.J.; Hwang, T.L.; Abadi, A.H.; Abdel-Halim, M.; Engel, M. 5-Methoxybenzothiophene-2-Carboxamides as Inhibitors of Clk1/4: Optimization of Selectivity and Cellular Potency. Molecules 2021, 26, 1001. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Maklad, R.M.; Almahli, H.; Al-Warhi, T.; Elkaeed, E.B.; Abourehab, M.A.S.; Abdel-Aziz, H.A.; El Kerdawy, A.M. Identification of 3-(piperazinylmethyl)benzofuran derivatives as novel type II CDK2 inhibitors: Design, synthesis, biological evaluation, and in silico insights. J. Enzym. Inhib. Med. Chem. 2022, 37, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Wadood, A.; Jamal, S.B.; Riaz, M.; Mir, A. Computational analysis of benzofuran-2-carboxlic acids as potent Pim-1 kinase inhibitors. Pharm. Biol. 2014, 52, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Wang, T.; Gao, M.; Zhang, X.; Liu, Z.; Zhao, S.-J.; Lv, Z.-S.; Xiao, J. Benzofuran-isatin-imine hybrids tethered via different length alkyl linkers: Design, synthesis and in vitro evaluation of anti-tubercular and anti-bacterial activities as well as cytotoxicity. Eur. J. Med. Chem. 2019, 165, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Y.; Zhang, J.; Ullah, S.; Kang, N.; Zhao, Y.; Zhou, H. Benzothiophene-2-carboxamide derivatives as SENPs inhibitors with selectivity within SENPs family. Eur. J. Med. Chem. 2020, 204, 112553. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorganic Chem. 2021, 112, 104947. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Dahab, M.A.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Mahdy, H.A.; Elkady, H. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: Design, molecular modeling, and synthesis. Bioorg. Chem. 2021, 110, 104807. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Elkady, H.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Alanazi, M.M.; Alharbi, M.A.; Eissa, I.H.; Dahab, M.A. New quinoxaline-based VEGFR-2 inhibitors: Design, synthesis, and antiproliferative evaluation with in silico docking, ADMET, toxicity, and DFT studies. RSC Adv. 2021, 11, 30315–30328. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Alesawy, M.S.; Eissa, S.I.; El-Fakharany, E.M.; Kalaba, M.H.; Sharaf, M.H.; Shama, N.M.A.; Mahmoud, S.H.; Mostafa, A.; Al-Karmalawy, A.A.; et al. Design and synthesis of new 4-(2-nitrophenoxy)benzamide derivatives as potential antiviral agents: Molecular modeling and in vitro antiviral screening. New J. Chem. 2021, 45, 16557–16571. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Eissa, I.H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.F.; Albassam, H.; Elkady, H.; Elwan, A. Design, synthesis, docking, ADMET studies, and anticancer evaluation of new 3-methylquinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1760–1782. [Google Scholar] [CrossRef] [PubMed]
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Elwan, A.; Elkady, H. Discovery of new VEGFR-2 inhibitors based on bis ([1, 2, 4] triazolo)[4, 3-a: 3’, 4’-c] quinoxaline derivatives as anticancer agents and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1093–1114. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Maliski, E.G.; Gallant, A.P.; Rogers, D. Classification of Kinase Inhibitors Using a Bayesian Model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef]
- BIOVIA, QSAR, ADMET and Predictive Toxicology. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html.
- Molecular Operating Environment (MOE), 2014.09, Chemical Computing Group Inc., Montréal, Canada. Available online: https://www.chemcomp.com.
- Mehany, A.B.M.; Belal, A.; Mohamed, A.F.; Shaaban, S.; Abdelhamid, G. Apoptotic and anti-angiogenic effects of propolis against human bladder cancer: Molecular docking and in vitro screening. Biomarkers 2022, 27, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Belal, A. 3D-Pharmacophore Modeling, Molecular Docking, and Virtual Screening for Discovery of Novel CDK4/6 Selective Inhibitors. Russ. J. Bioorg. Chem. 2021, 47, 317–333. [Google Scholar] [CrossRef]
- Zhaorigetu, I.M.F.; Belal, A.; al Badawi, M.H.; Abdelhady, A.A.; Galala, F.M.A.; El-Sharkawy, A.; El-Dahshan, A.A.; Mehany, A.B. Antiproliferative, Apoptotic Effects and Suppression of Oxidative Stress of Quercetin against Induced Toxicity in Lung Cancer Cells of Rats: In vitro and In vivo Study. J. Cancer 2021, 12, 5249. [Google Scholar] [CrossRef]
- Belal, A. Pyrrolizines as Potential Anticancer Agents: Design, Synthesis, Caspase-3 activation and Micronucleus (MN) Induction. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem.-Anti-Cancer Agents) 2018, 18, 2124–2130. [Google Scholar] [CrossRef]
- Belal, A. Synthesis, molecular docking and antitumor activity of novel pyrrolizines with potential as EGFR-TK inhibitors. Bioorg. Chem. 2015, 59, 124–129. [Google Scholar] [CrossRef]
- El-Helby, A.-G.A.; Ayyad, R.R.; El-Adl, K.; Elkady, H. Phthalazine-1, 4-dione derivatives as non-competitive AMPA receptor antagonists: Design, synthesis, anticonvulsant evaluation, ADMET profile and molecular docking. Mol. Divers. 2019, 23, 283–298. [Google Scholar] [CrossRef]
- El-Zahabi, M.A.; Elbendary, E.R.; Bamanie, F.H.; Radwan, M.F.; Ghareib, S.A.; Eissa, I.H. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of phthalimide-sulfonylurea hybrids as PPARγ and SUR agonists. Bioorg. Chem. 2019, 91, 103115. [Google Scholar] [CrossRef]
- Ibrahim, M.K.; Eissa, I.H.; Alesawy, M.S.; Metwaly, A.M.; Radwan, M.M.; ElSohly, M.A. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4 (3H)-one derivatives as potential PPARγ and SUR agonists. Bioorg. Med. Chem. 2017, 25, 4723–4744. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, A.; Metwaly, A.M.; Hassan, A.; El-Aziz, A.; Mohamed, T.; Elkaeed, E.B.; Eissa, I.H.; Arafa, R.K.; Stockand, J.D. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021, 11, 460. [Google Scholar] [CrossRef] [PubMed]
- El-Gamal, K.M.; El-Morsy, A.M.; Saad, A.M.; Eissa, I.H.; Alswah, M. Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J. Mol. Struct. 2018, 1166, 15–33. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Mahdy, H.A.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Dahab, M.A.; Eissa, I.H. New bis ([1, 2, 4] triazolo)[4, 3-a: 3′, 4′-c] quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and anticancer evaluation. Bioorg. Chem. 2021, 112, 104949. [Google Scholar] [CrossRef]
- Eissa, I.H.; El-Helby, A.G.A.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; et al. Discovery of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorg. Chem. 2020, 105, 104380. [Google Scholar] [CrossRef]
- Eissa, I.H.; Dahab, M.A.; Ibrahim, M.K.; Alsaif, N.A.; Alanazi, A.; Eissa, S.I.; Belal, A.; Beauchemin, A.M. Design and discovery of new antiproliferative 1, 2, 4-triazin-3 (2H)-ones as tubulin polymerization inhibitors targeting colchicine binding site. Bioorg. Chem. 2021, 112, 104965. [Google Scholar] [CrossRef]
- Eissa, I.H.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; Abdelhady, A.A.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Mahdy, H.A.J.B.C. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorg. Chem. 2021, 107, 104532. [Google Scholar] [CrossRef] [PubMed]
- Mehany, A.B.M.; Belal, A.; Santali, E.Y.; Shaaban, S.; Abourehab, M.A.S.; El-Feky, O.A.; Diab, M.; Galala, F.M.A.A.; Elkaeed, E.B.; Abdelhamid, G. Biological Effect of Quercetin in Repairing Brain Damage and Cerebral Changes in Rats: Molecular Docking and In Vivo Studies. BioMed Res. Int. 2022, 2022, 1–12. [Google Scholar] [CrossRef]
- Belal, A.; Elanany, M.A.; Raafat, M.; Hamza, H.T.; Mehany, A.B.M. Calendula officinalis Phytochemicals for the Treatment of Wounds Through Matrix Metalloproteinases-8 and 9 (MMP-8 and MMP-9): In Silico Approach. Nat. Prod. Commun. 2022, 17, 1934578X221098848. [Google Scholar] [CrossRef]
- Sun, X.; Belal, A.; Elanany, M.A.; Alsantali, R.I.; Alrooqi, M.M.; Mohamed, A.R.; Hasabelnaby, S. Identification of Some Promising Heterocycles Useful in Treatment of Allergic Rhinitis: Virtual Screening, Pharmacophore Mapping, Molecular Docking, and Molecular Dynamics. Russ. J. Bioorg. Chem. 2022, 48, 438–456. [Google Scholar]
- Belal, A.; Gawad, N.M.A.; Mehany, A.B.M.; Abourehab, M.A.S.; Elkady, H.; Al-Karmalawy, A.A.; Ismael, A.S. Design, synthesis and molecular docking of new fused 1H-pyrroles, pyrrolo[3,2-d]pyrimidines and pyrrolo[3,2-e][1, 4]diazepine de-rivatives as potent EGFR/CDK2 inhibitors. J. Enzym. Inhib. Med. Chem. 2022, 37, 1884–1902. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Elkady, H.; Belal, A.; Alsfouk, B.A.; Ibrahim, T.H.; Abdelmoaty, M.; Arafa, R.K.; Metwaly, A.M.; Eissa, H.I. Mul-ti-Phase In Silico Discovery of Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors among 3009 Clinical and FDA-Approved Related Drugs. Processes 2022, 10, 530. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Eissa, I.H.; Belal, A.; El-Sayed, A.A. Design, eco-friendly synthesis, molecular modeling and anticancer eval-uation of thiazol-5 (4 H)-ones as potential tubulin polymerization inhibitors targeting the colchicine binding site. RSC Adv. 2020, 10, 2791–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Lipinski’s Rule | Veber’s Rule | ||||
|---|---|---|---|---|---|---|
| Num HD | Num HA | M Wt | AlogP | Num Rotatable Bonds | TPSA | |
| 11 | 0 | 8 | 498.527 | 4.994 | 10 | 90.57 |
| 26 | 1 | 3 | 208.234 | 2.596 | 2 | 74.77 |
| 27 | 3 | 5 | 251.258 | 1.45 | 4 | 117.86 |
| 28 | 1 | 3 | 284.33 | 4.18 | 4 | 74.77 |
| 29 | 1 | 6 | 329.327 | 4.074 | 5 | 120.59 |
| 30 | 1 | 6 | 329.327 | 4.074 | 5 | 120.59 |
| 31 | 1 | 6 | 329.327 | 4.074 | 5 | 120.59 |
| 32 | 1 | 3 | 298.356 | 4.666 | 4 | 74.77 |
| 33 | 2 | 5 | 416.492 | 4.543 | 6 | 95.67 |
| 34 | 4 | 7 | 459.517 | 3.396 | 8 | 138.76 |
| 35 | 2 | 5 | 492.588 | 6.126 | 8 | 95.67 |
| 40 | 3 | 4 | 388.482 | 4.595 | 6 | 92.59 |
| 61 | 3 | 6 | 481.566 | 5.511 | 8 | 111.46 |
| 62 | 2 | 6 | 482.55 | 5.521 | 8 | 108.81 |
| 63 | 2 | 5 | 498.616 | 6.08 | 8 | 123.91 |
| 65 | 2 | 6 | 493.576 | 4.976 | 8 | 108.56 |
| 68 | 2 | 5 | 430.519 | 4.462 | 7 | 95.67 |
| 69 | 2 | 5 | 444.545 | 5.129 | 8 | 95.67 |
| 70 | 2 | 5 | 458.572 | 5.585 | 9 | 95.67 |
| 71 | 2 | 5 | 458.572 | 5.591 | 8 | 95.67 |
| 72 | 2 | 6 | 499.604 | 4.929 | 8 | 136.8 |
| Comp. | BBB Level a | Solubility Level b | Absorption Level c | CYP2D6 Prediction d | PPB Prediction e |
|---|---|---|---|---|---|
| 11 | 4 | 2 | 1 | false | True |
| 26 | 2 | 3 | 0 | false | True |
| 27 | 3 | 3 | 0 | false | True |
| 28 | 1 | 2 | 0 | false | True |
| 29 | 2 | 2 | 0 | false | True |
| 30 | 2 | 2 | 0 | false | true |
| 31 | 2 | 2 | 0 | false | True |
| 32 | 1 | 2 | 0 | false | True |
| 33 | 1 | 2 | 0 | false | True |
| 34 | 4 | 2 | 0 | false | True |
| 35 | 4 | 1 | 1 | false | True |
| 40 | 1 | 2 | 0 | false | True |
| 61 | 4 | 1 | 1 | false | True |
| 62 | 4 | 1 | 1 | false | True |
| 63 | 4 | 1 | 1 | false | True |
| 65 | 4 | 2 | 0 | false | True |
| 68 | 1 | 2 | 0 | false | True |
| 69 | 1 | 2 | 0 | false | True |
| 70 | 1 | 1 | 0 | false | True |
| 71 | 1 | 1 | 1 | false | True |
| 72 | 2 | 2 | 0 | false | True |
| IDK | 4 | 2 | 0 | false | True |
| Comp | ∆G Kcal/mol | No. of H Bonds | Distance (Å) | Amino Acid Involved |
|---|---|---|---|---|
| 26 | −22.90 | 4 | 2.59 | Glu131 |
| 2.15 | Cys133 | |||
| 2.92 | Cys197 | |||
| 2.78 | Gln137 | |||
| 27 | −23.34 | 4 | 2.92 | Cys133 |
| 2.39 | Cys197 | |||
| 2.35, 2.44 | Gln137 | |||
| 31 | −22.22 | 4 | 2.24 | Glu131 |
| 3.08, 3.18 | Cys133 | |||
| 2.52 | Cys197 | |||
| 35 | −24.25 | 5 | 2.80, 2.72 | Asn140 |
| 2.33 | Cys 133 | |||
| 2.85 | Gln 137 | |||
| 2.86 | Leu 57 | |||
| 72 | −24.22 | 5 | 2.72 | Glu131 |
| 2.75 | Glu184 | |||
| 2.68 | Lys 182 | |||
| 2.76 | Tyr 132 | |||
| 2.65 | Gly 60 | |||
| Co-crystallized ligand (IDK) | −22.72 | 5 | 3.22 | Glu131 |
| 2.79, 2.83 | Cys133 | |||
| 3.36 | Asn185 | |||
| 2.89 | Gln137 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belal, A.; Elkady, H.; Al-Karmalawy, A.A.; Amin, A.H.; Ghoneim, M.M.; El-Sherbiny, M.; Al-Serwi, R.H.; Abdou, M.A.; Ibrahim, M.H.; Mehany, A.B.M. Discovery of Some Heterocyclic Molecules as Bone Morphogenetic Protein 2 (BMP-2)-Inducible Kinase Inhibitors: Virtual Screening, ADME Properties, and Molecular Docking Simulations. Molecules 2022, 27, 5571. https://doi.org/10.3390/molecules27175571
Belal A, Elkady H, Al-Karmalawy AA, Amin AH, Ghoneim MM, El-Sherbiny M, Al-Serwi RH, Abdou MA, Ibrahim MH, Mehany ABM. Discovery of Some Heterocyclic Molecules as Bone Morphogenetic Protein 2 (BMP-2)-Inducible Kinase Inhibitors: Virtual Screening, ADME Properties, and Molecular Docking Simulations. Molecules. 2022; 27(17):5571. https://doi.org/10.3390/molecules27175571
Chicago/Turabian StyleBelal, Amany, Hazem Elkady, Ahmed A. Al-Karmalawy, Ali H. Amin, Mohammed M. Ghoneim, Mohamed El-Sherbiny, Rasha Hamed Al-Serwi, Mohamed Attia Abdou, Mona H. Ibrahim, and Ahmed B. M. Mehany. 2022. "Discovery of Some Heterocyclic Molecules as Bone Morphogenetic Protein 2 (BMP-2)-Inducible Kinase Inhibitors: Virtual Screening, ADME Properties, and Molecular Docking Simulations" Molecules 27, no. 17: 5571. https://doi.org/10.3390/molecules27175571