

Drugs That Changed Society: Microtubule-Targeting Agents Belonging to Taxanoids, Macrolides and Non-Ribosomal Peptides

Abstract
:1. Introduction
2. The Taxus Diterpenes
2.1. The Genus Taxus
2.2. Taxanes Isolated from the Taxus Genus
2.3. Other Sources for Paclitaxel
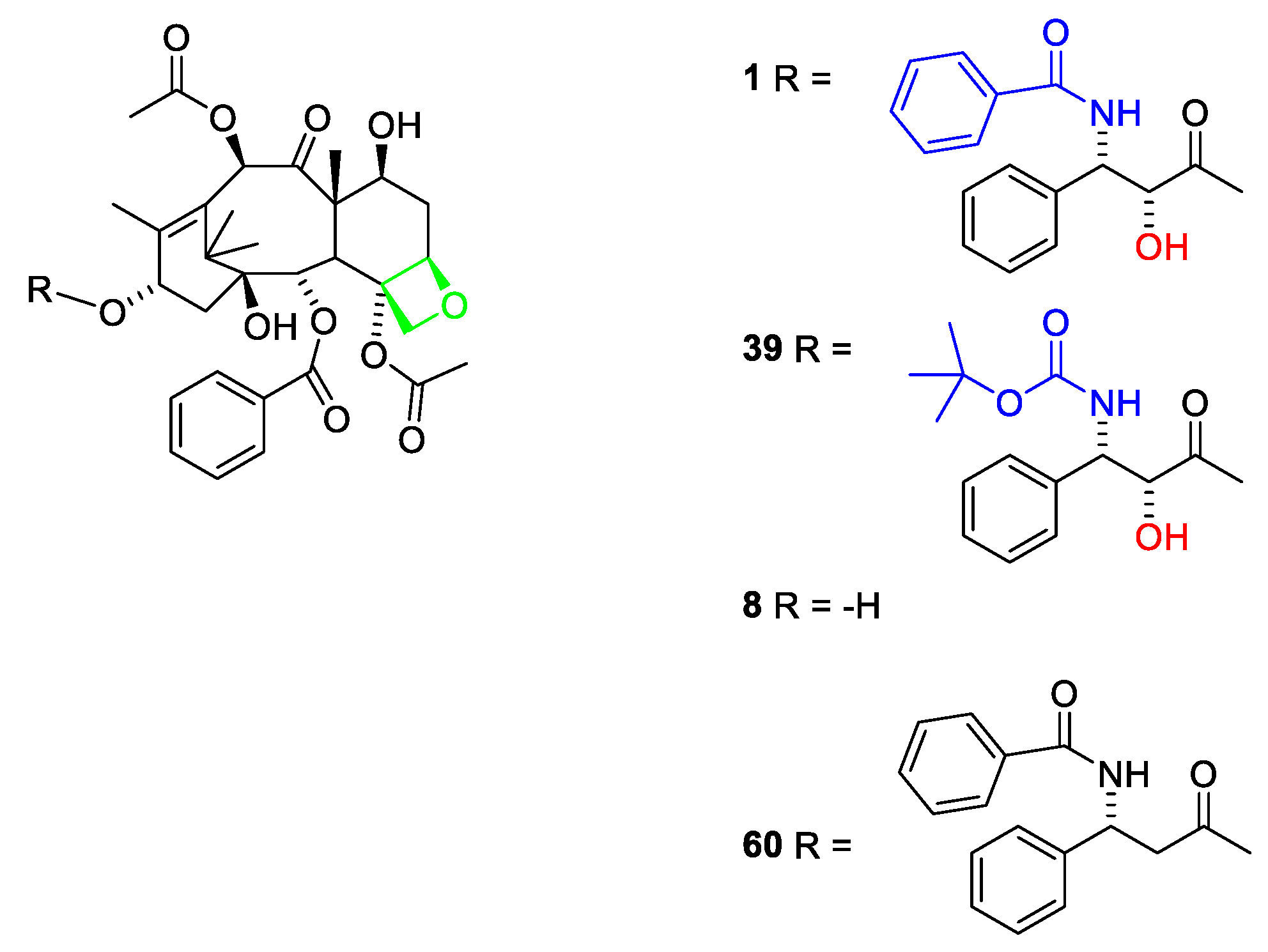
2.4. Isolation and Structure Elucidation of Taxol and Other Taxanes
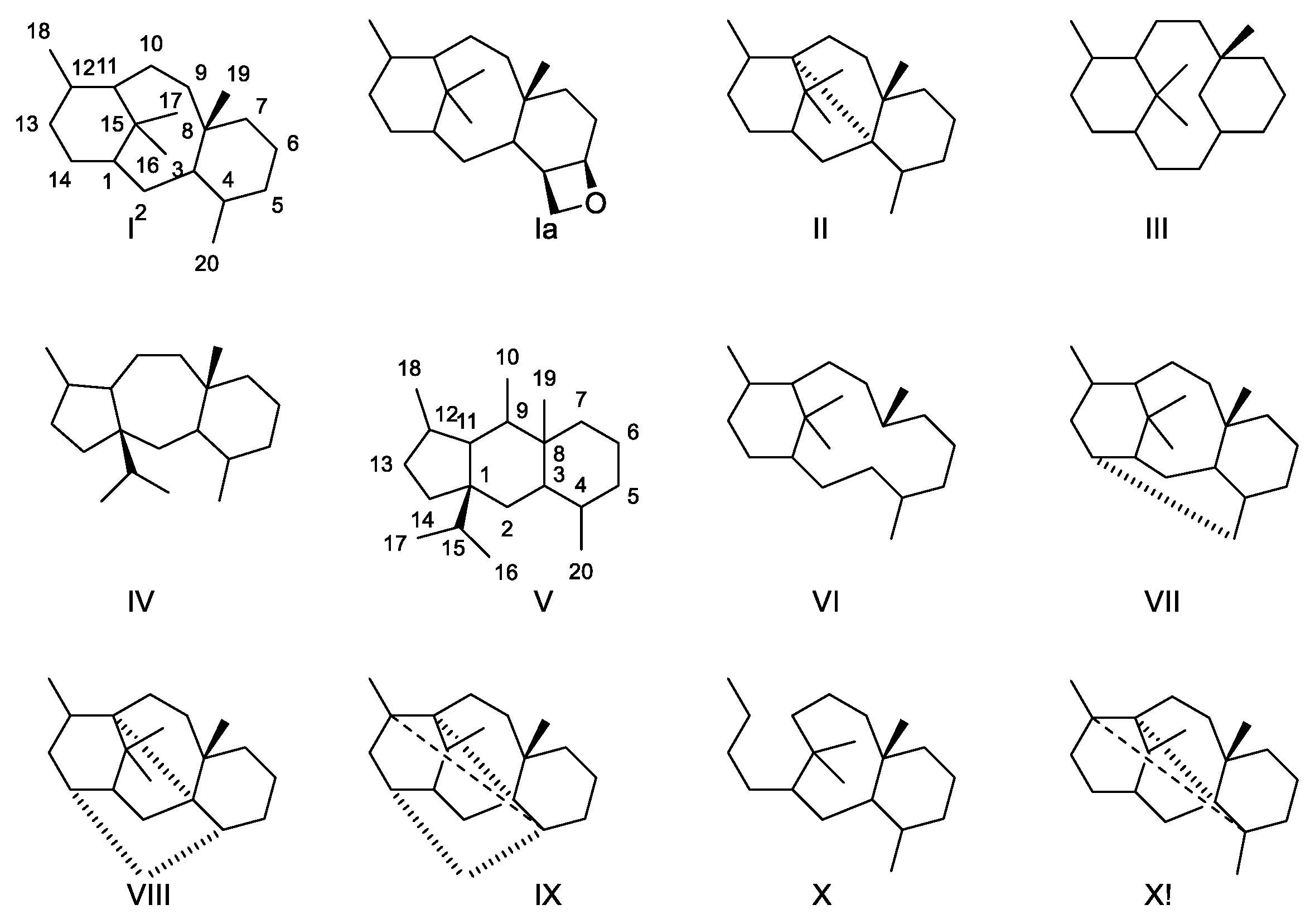
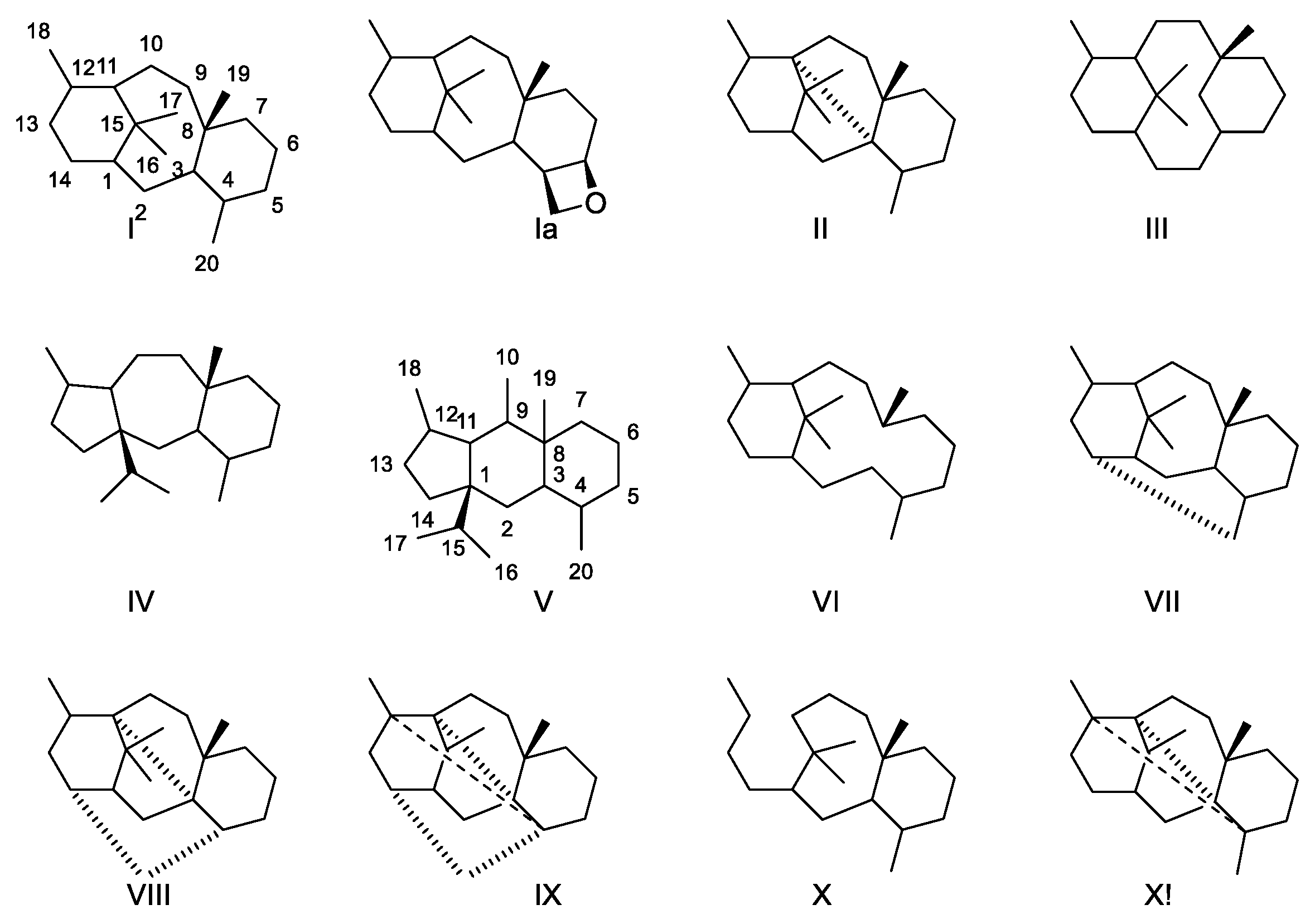
2.4.1. Structure Elucidation of Taxanes
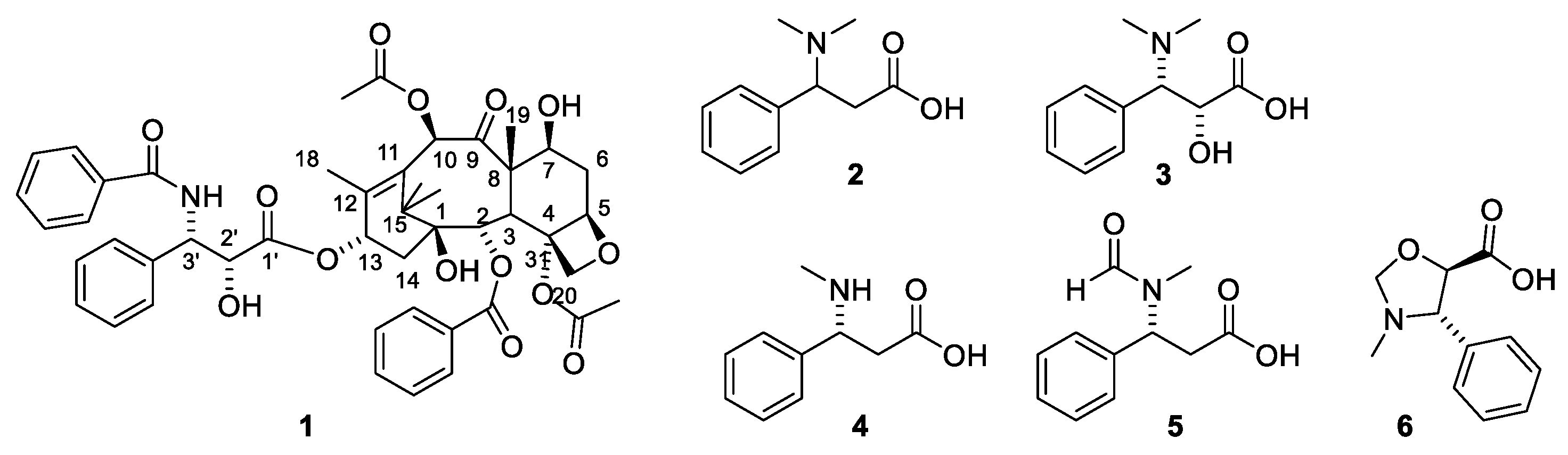
2.4.2. Structure Elucidation of Paclitaxel
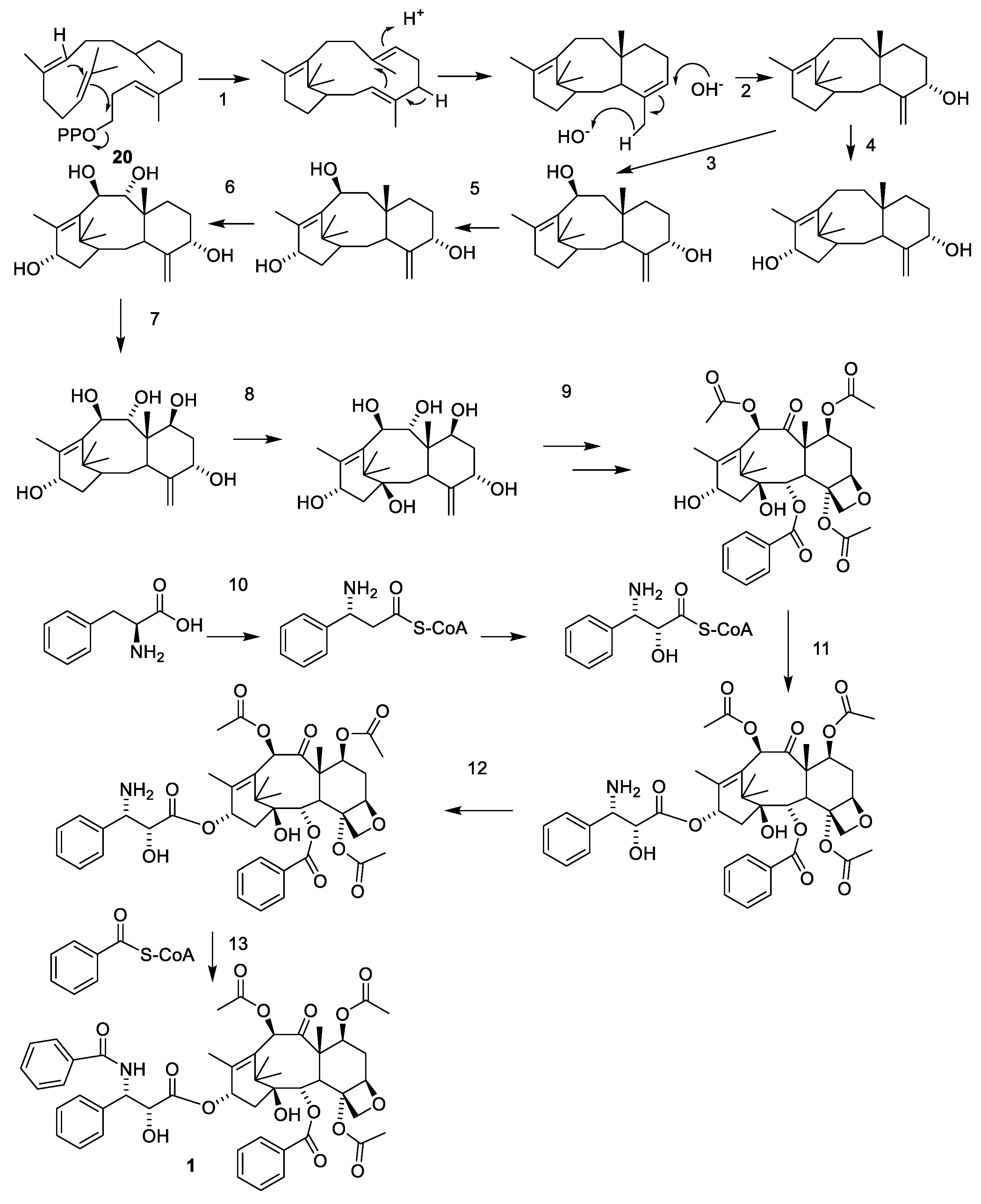
2.4.3. Biosynthesis of Taxanes
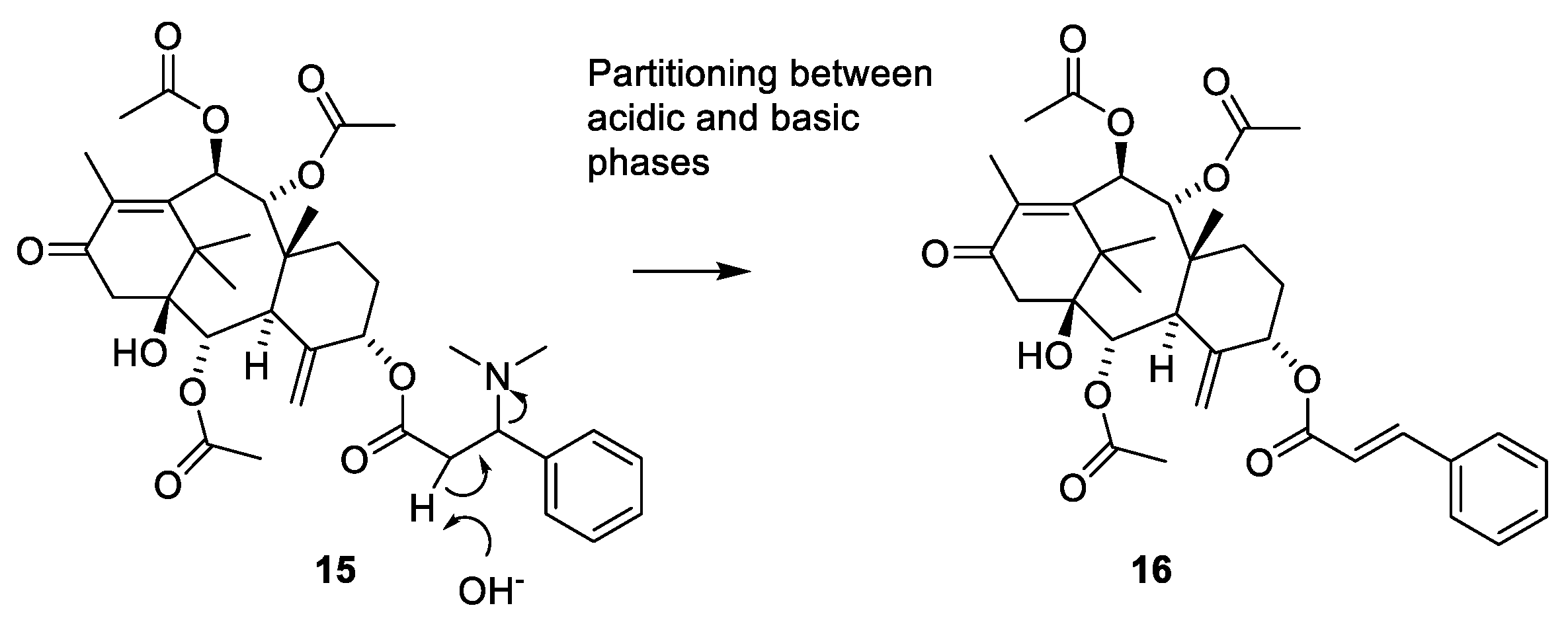
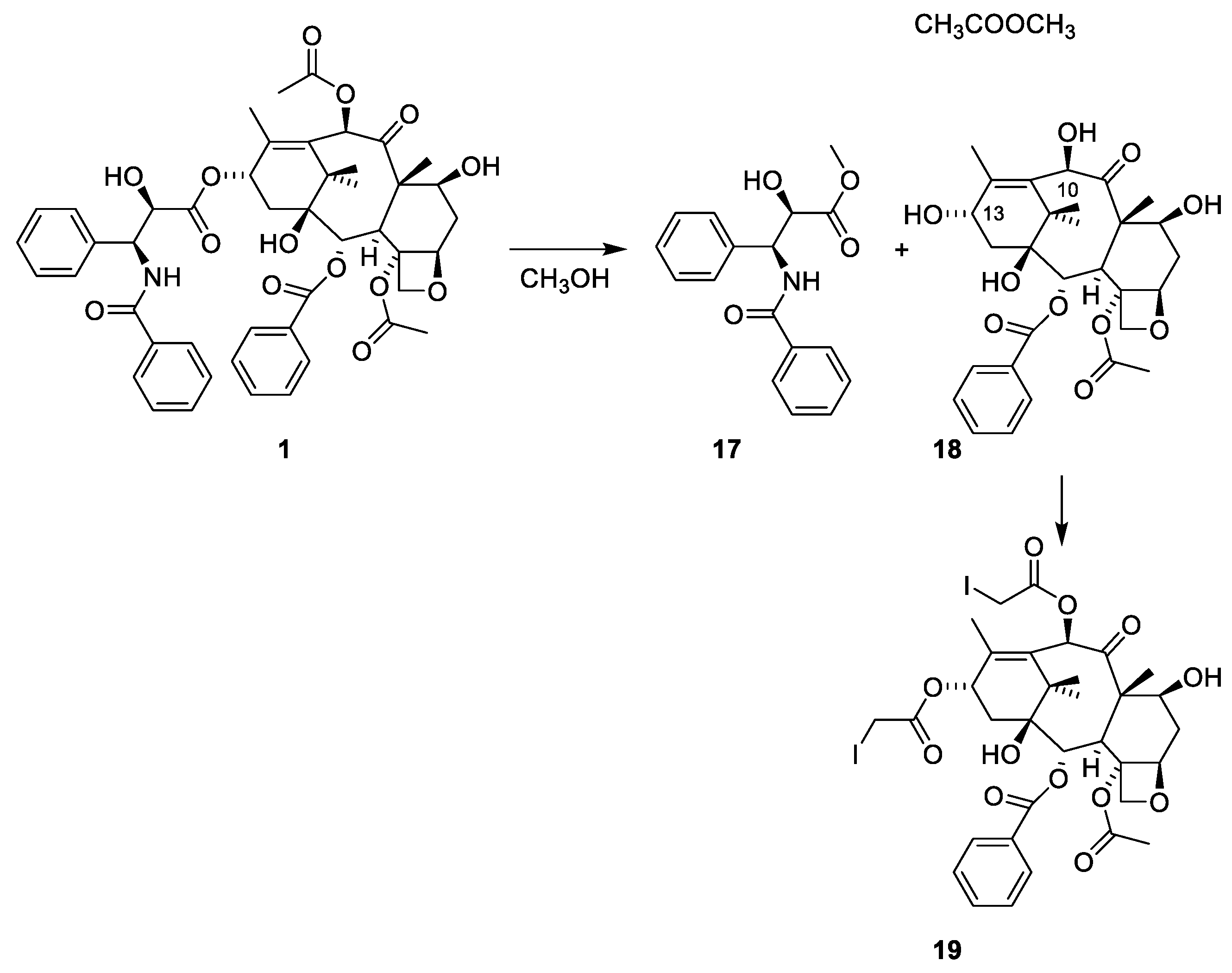
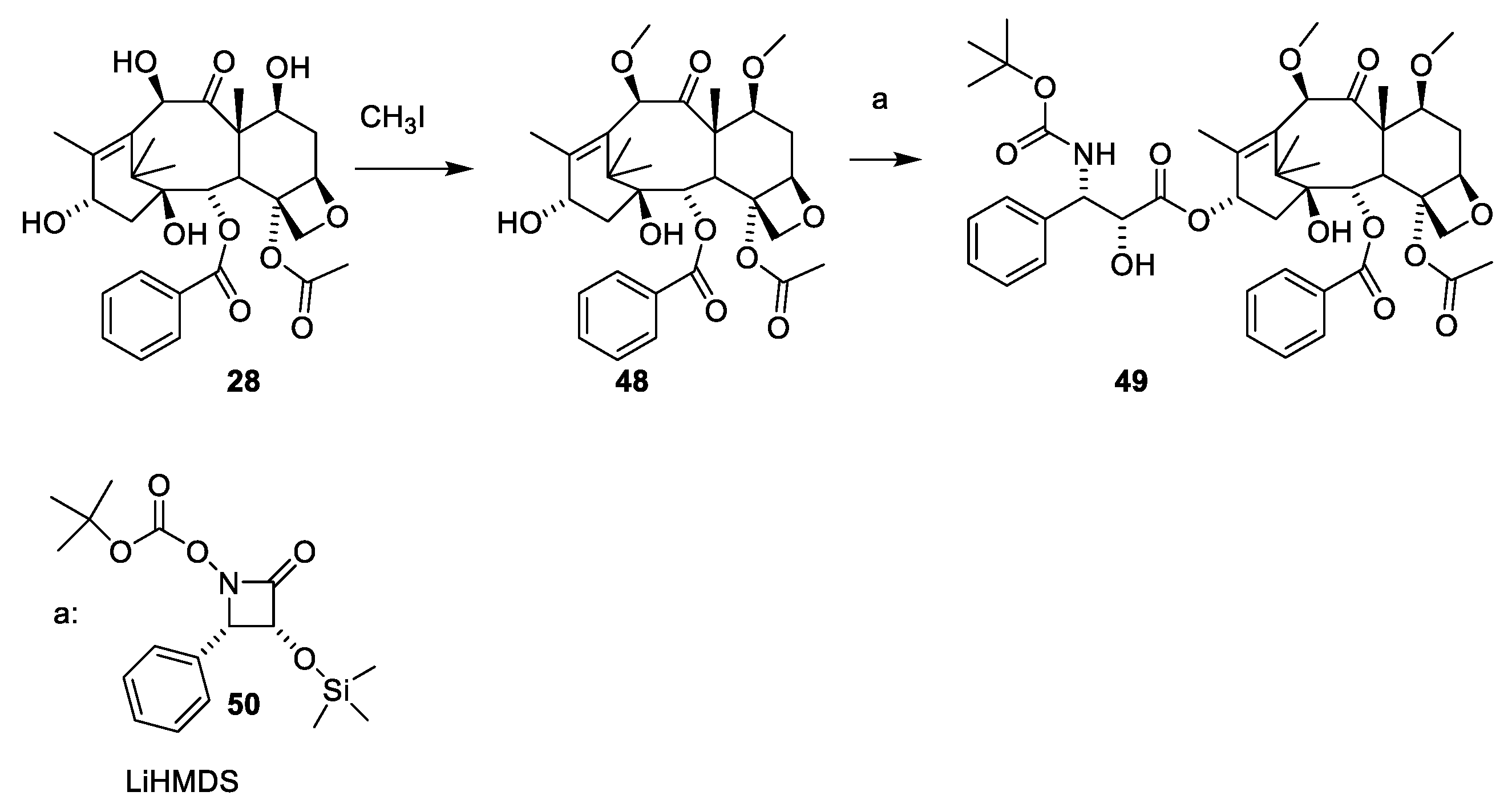
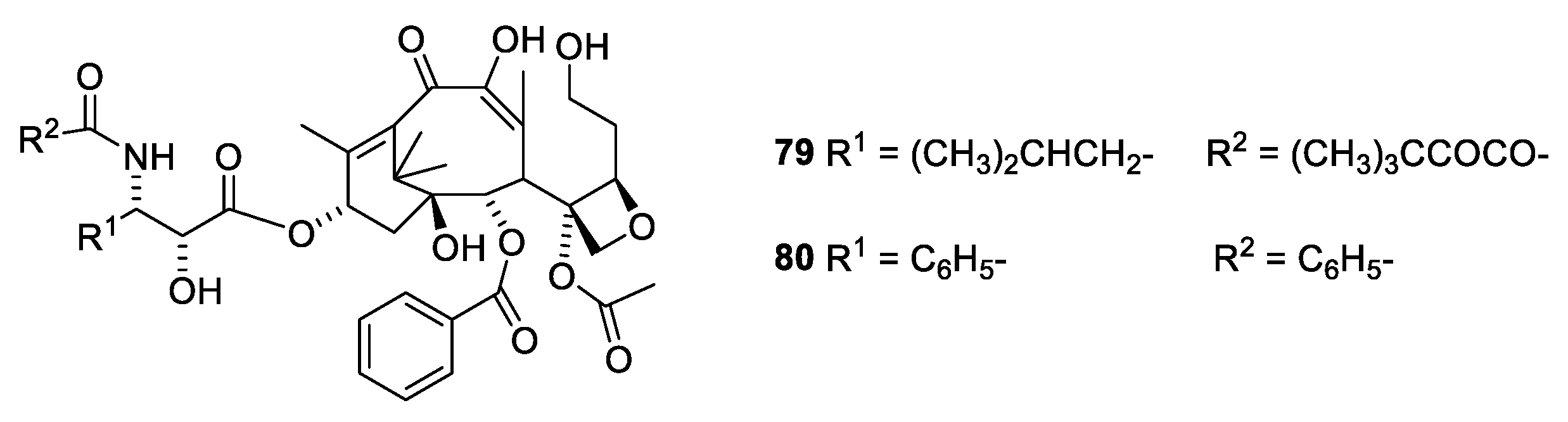
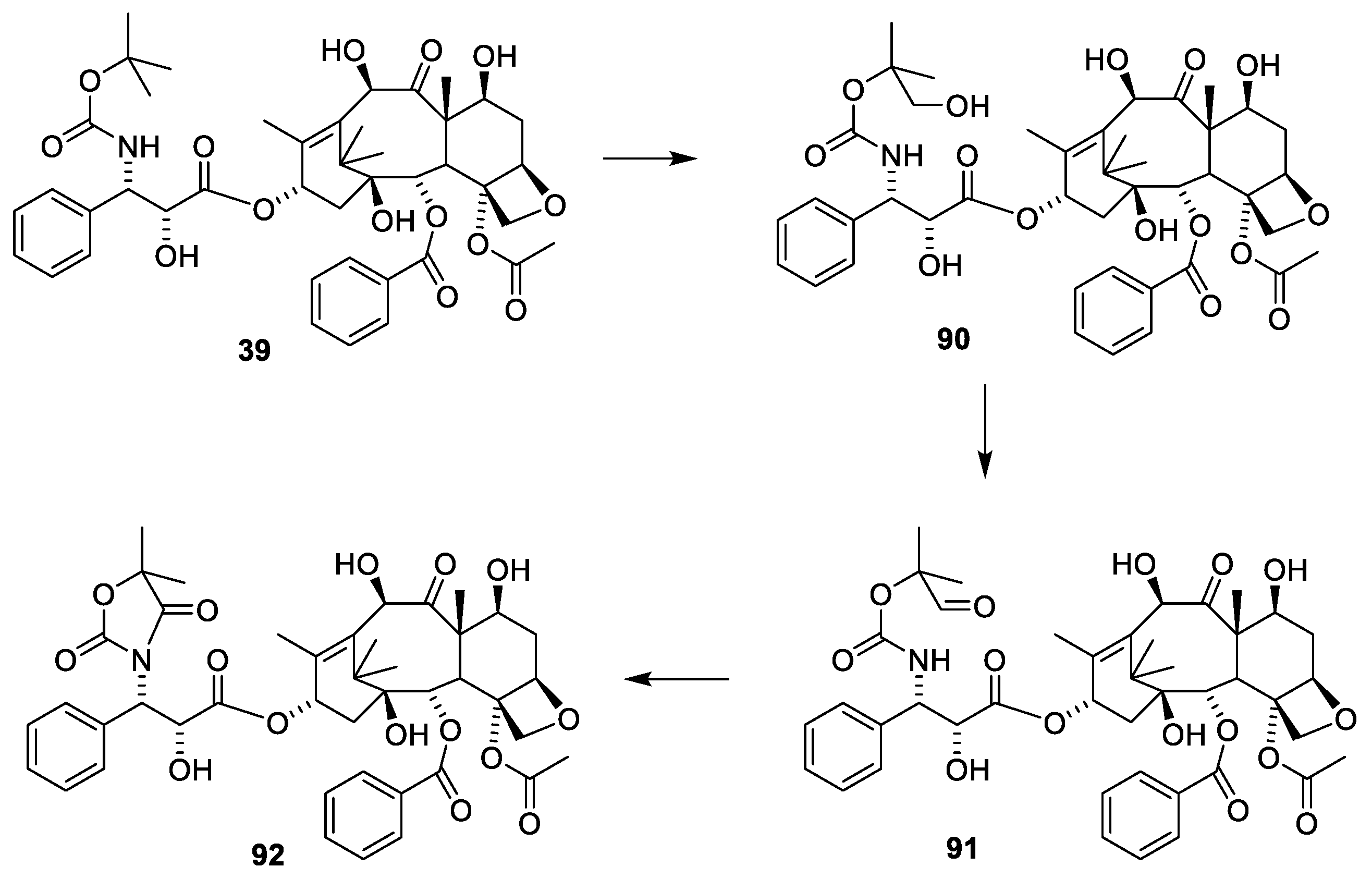
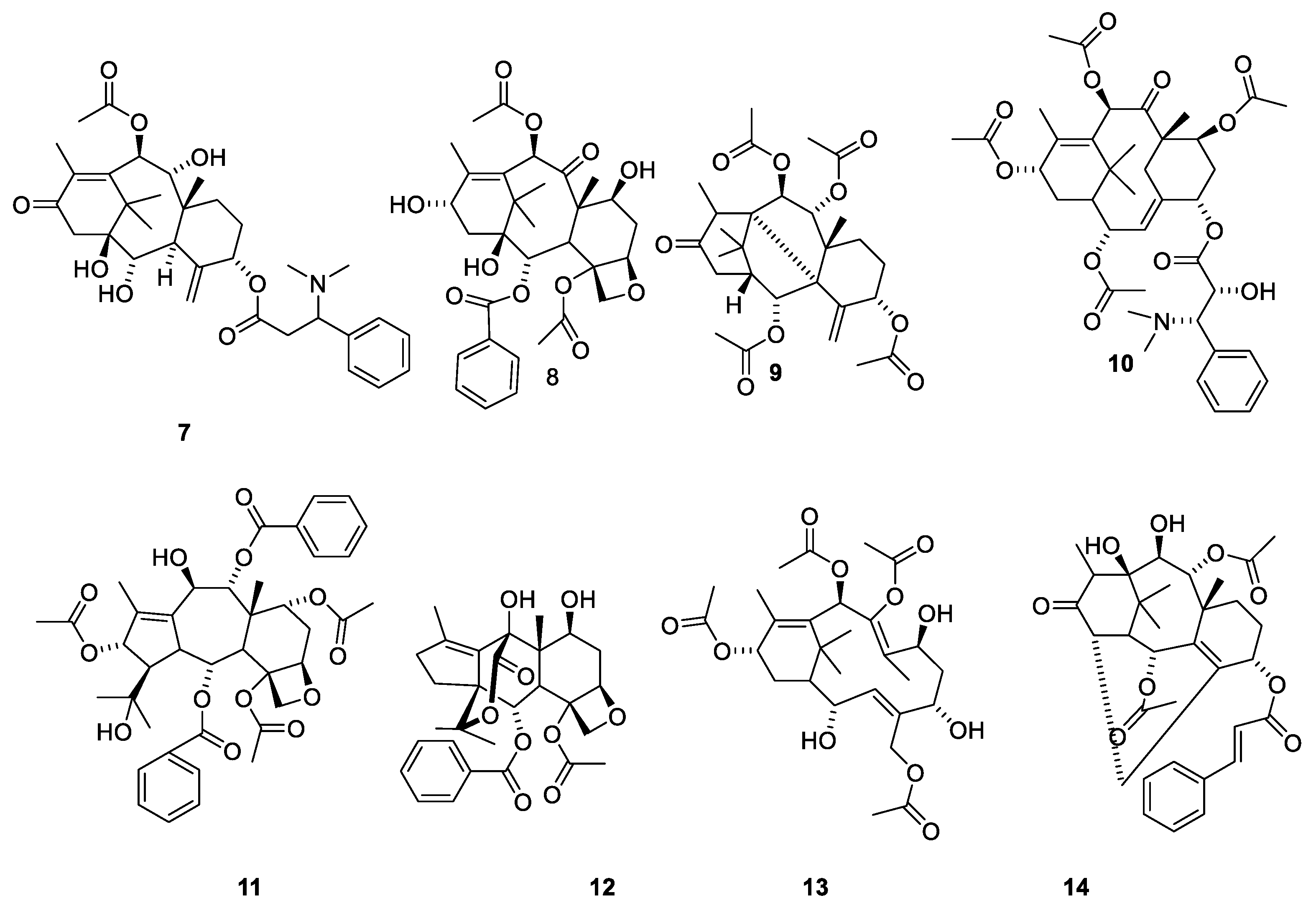
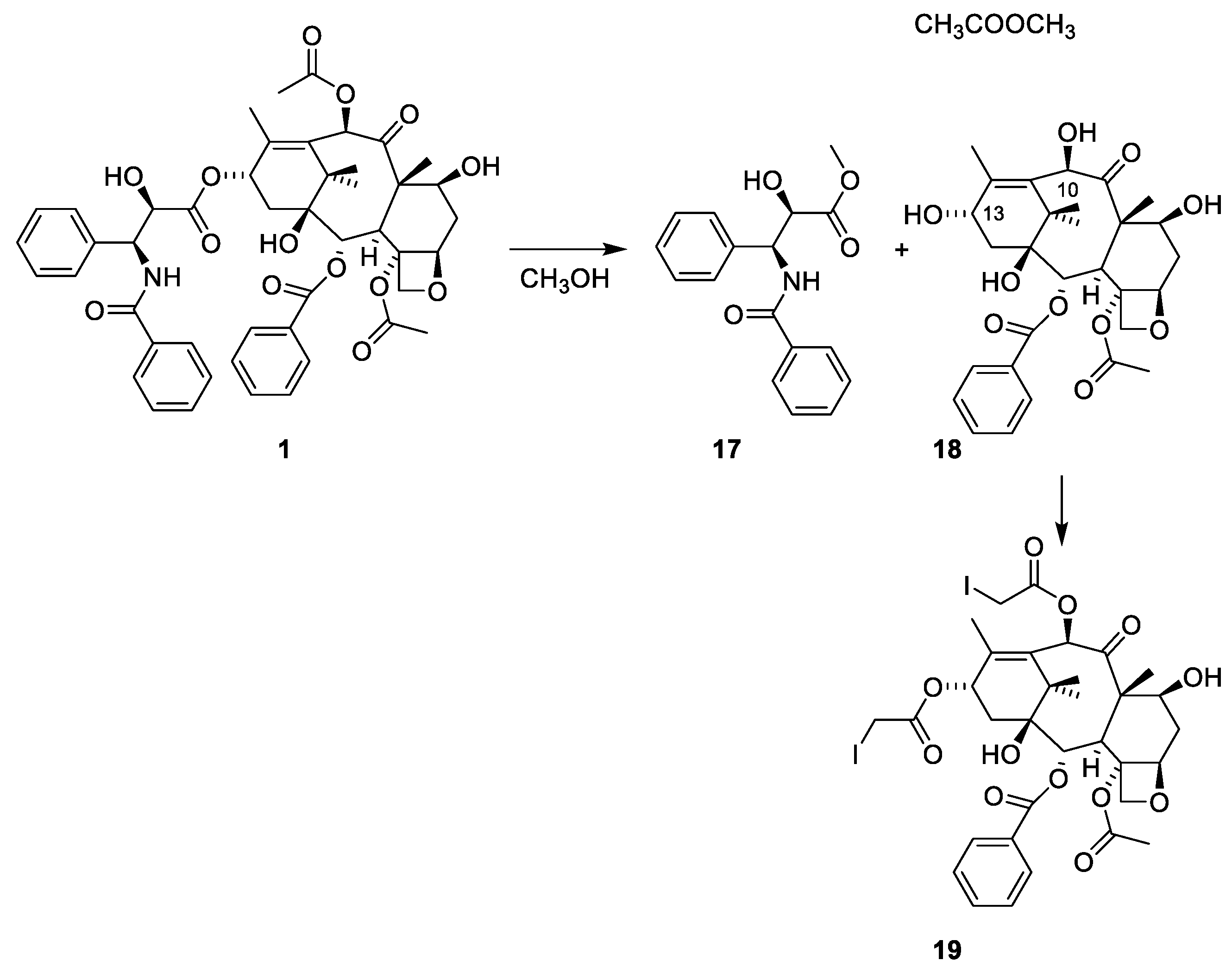
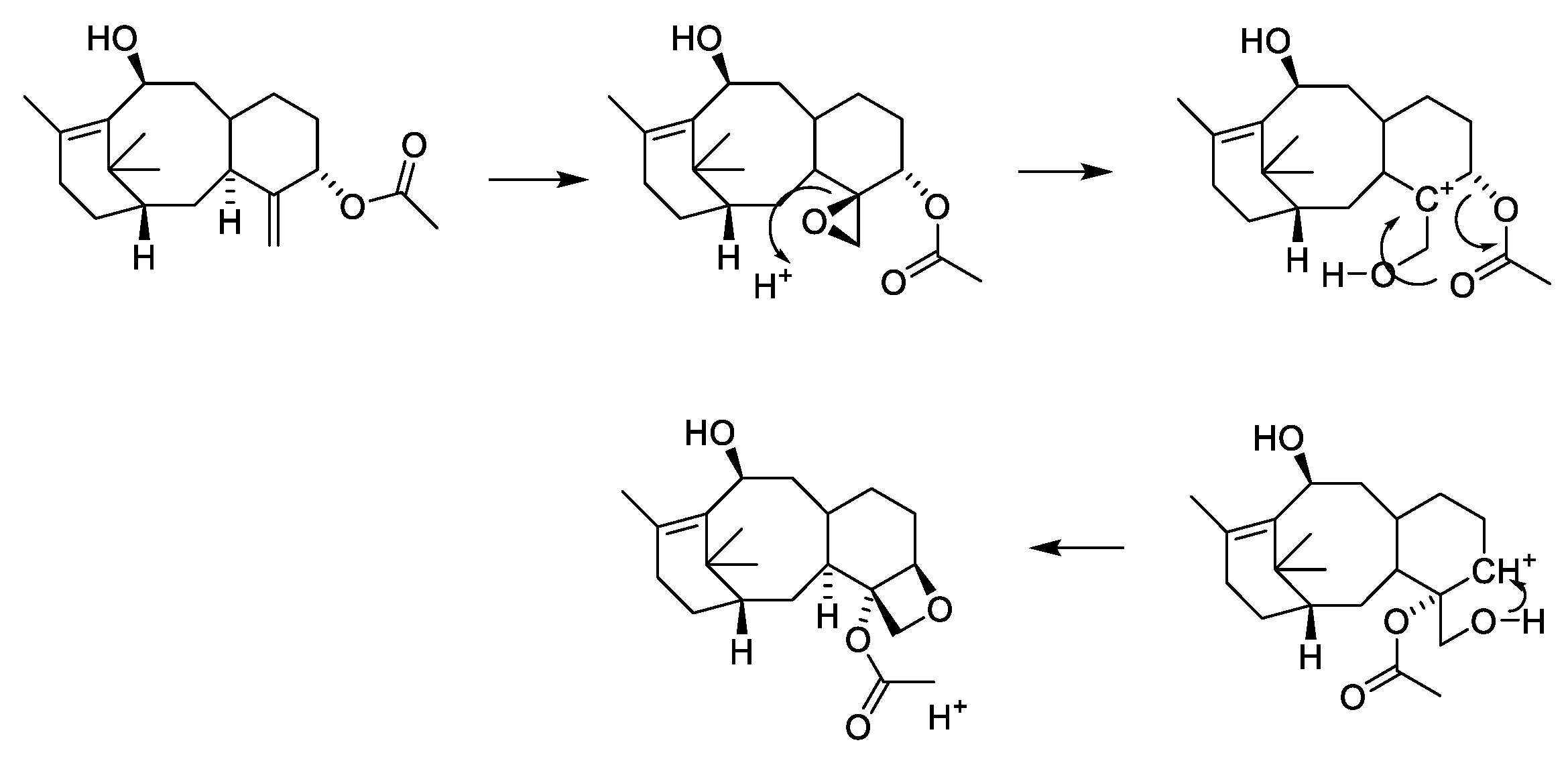
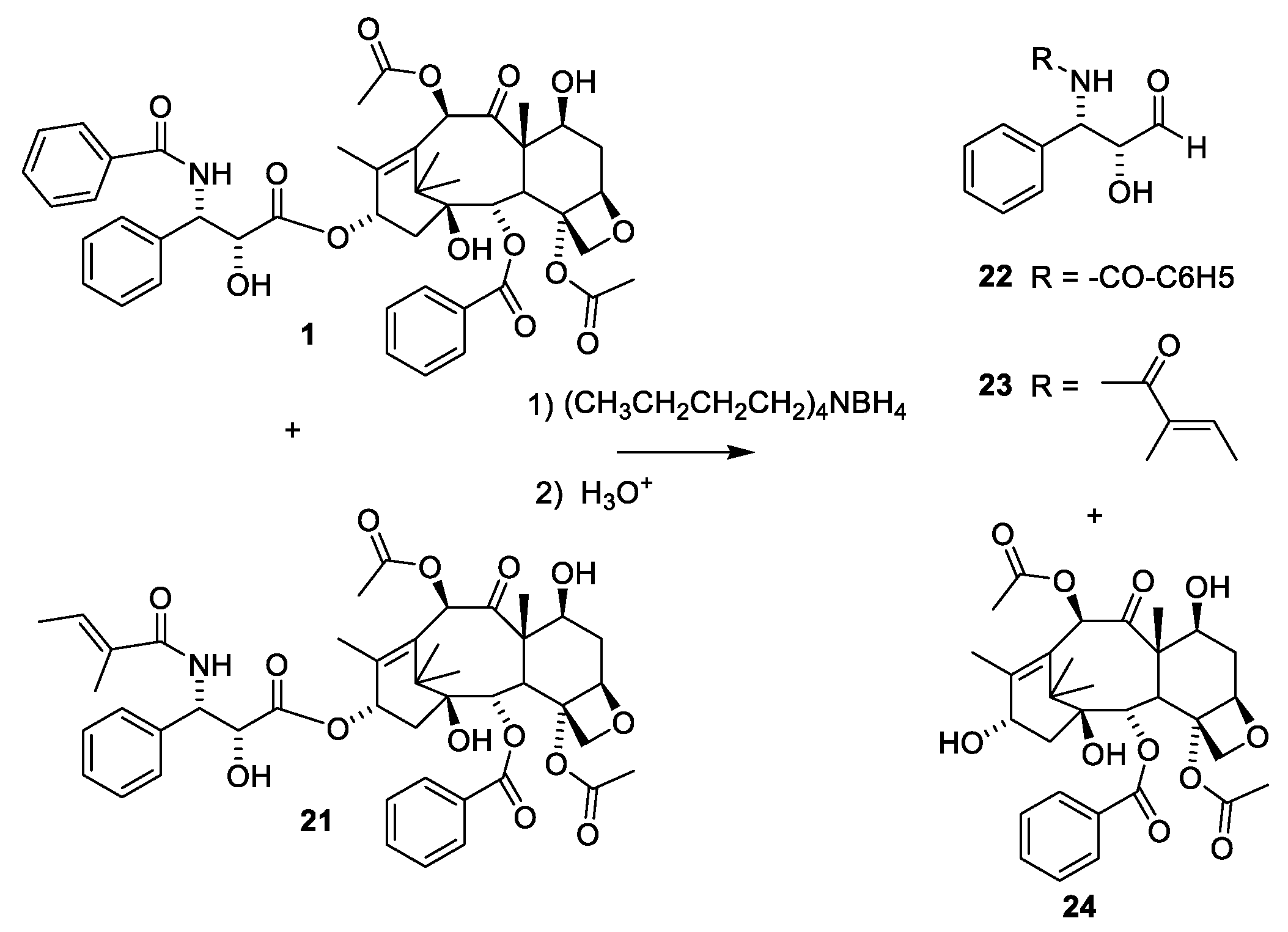
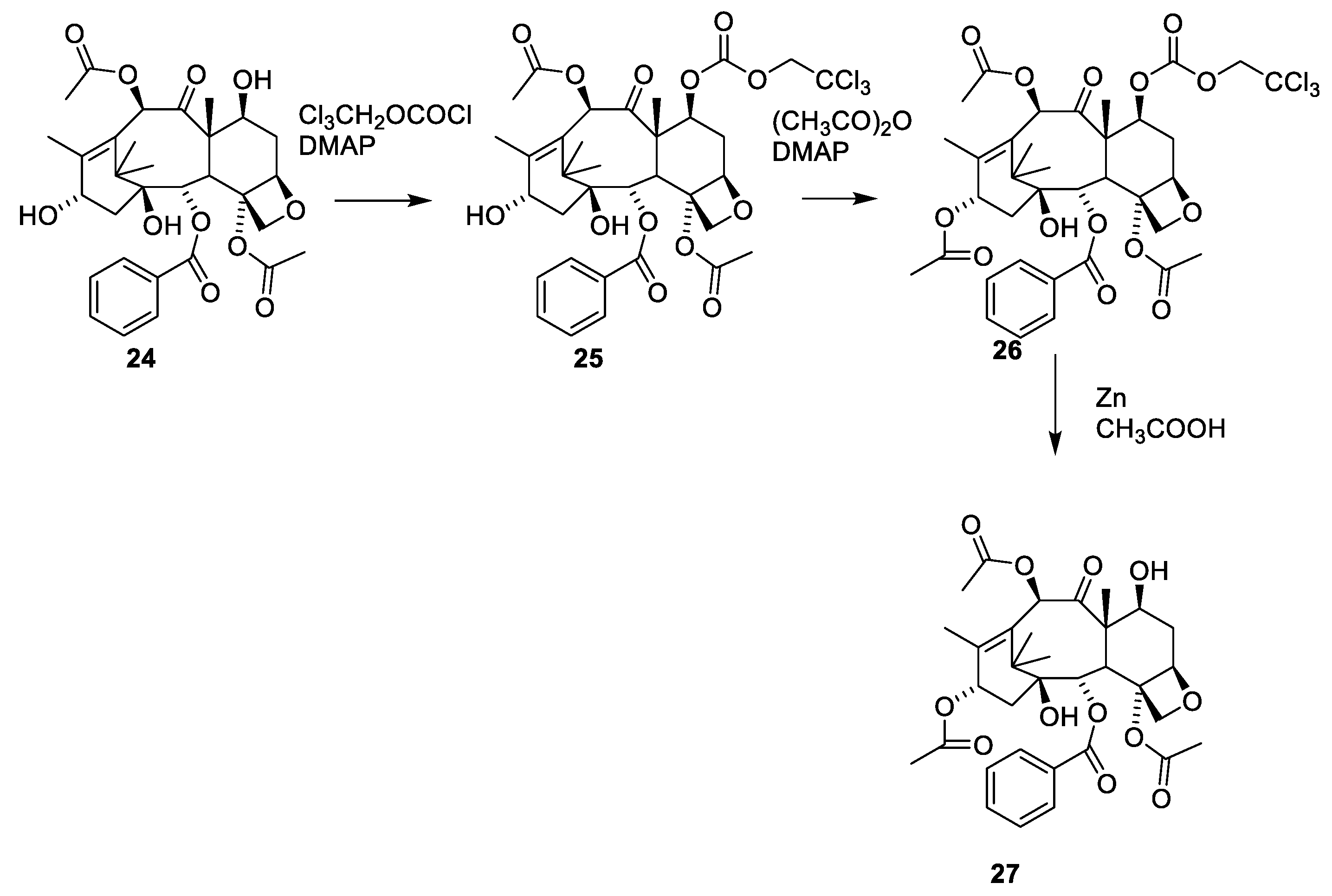
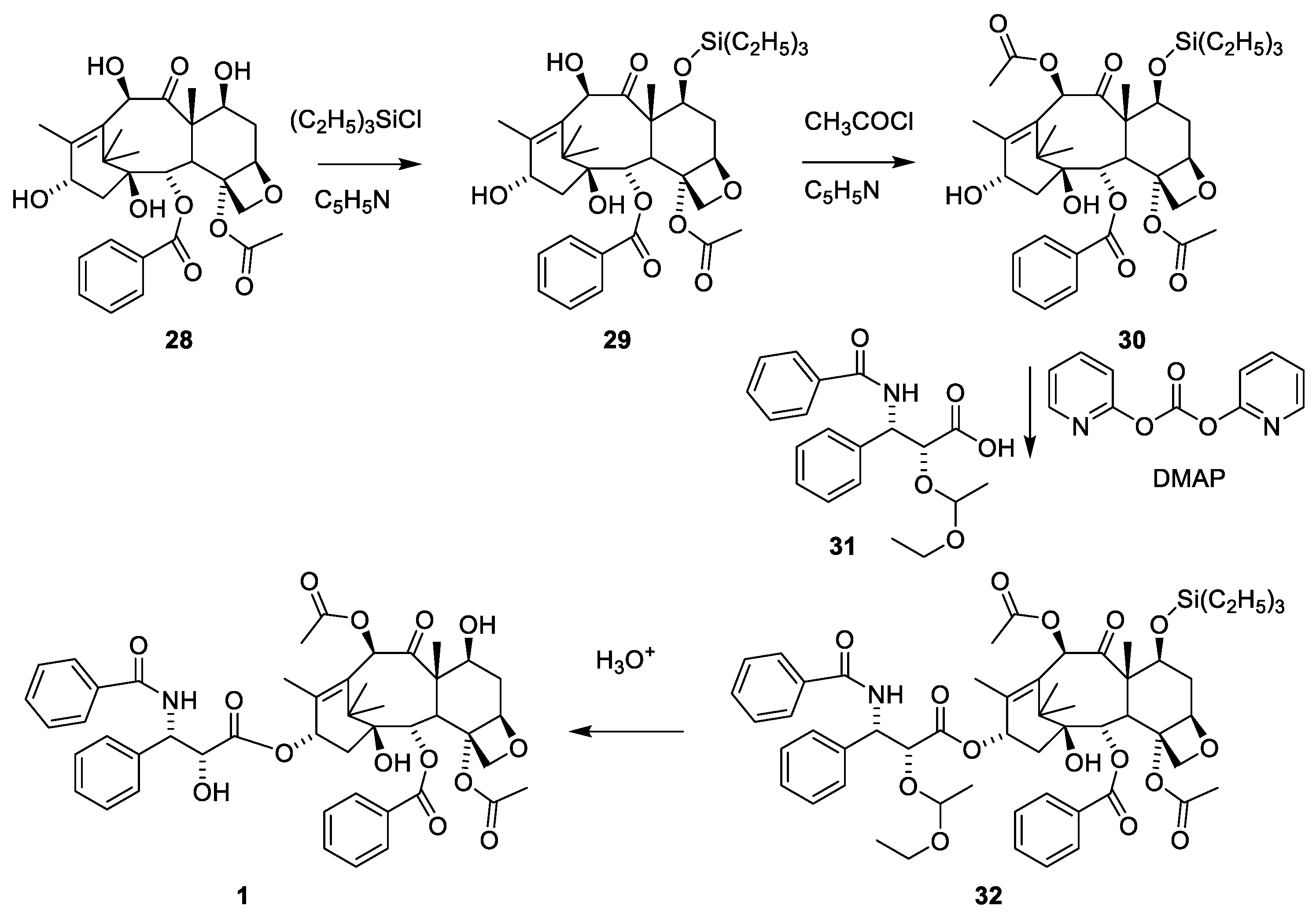
2.4.4. Chemistry of Taxanes
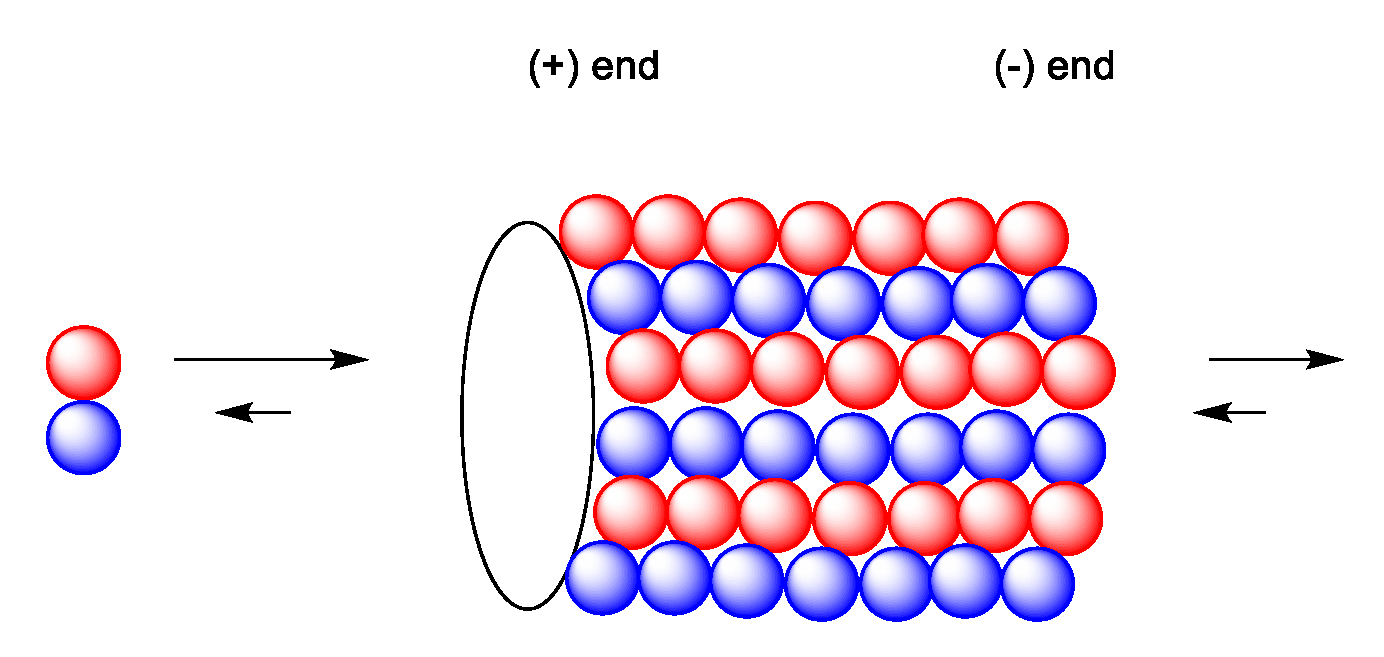
2.5. Mechanism of Action of Paclitaxel
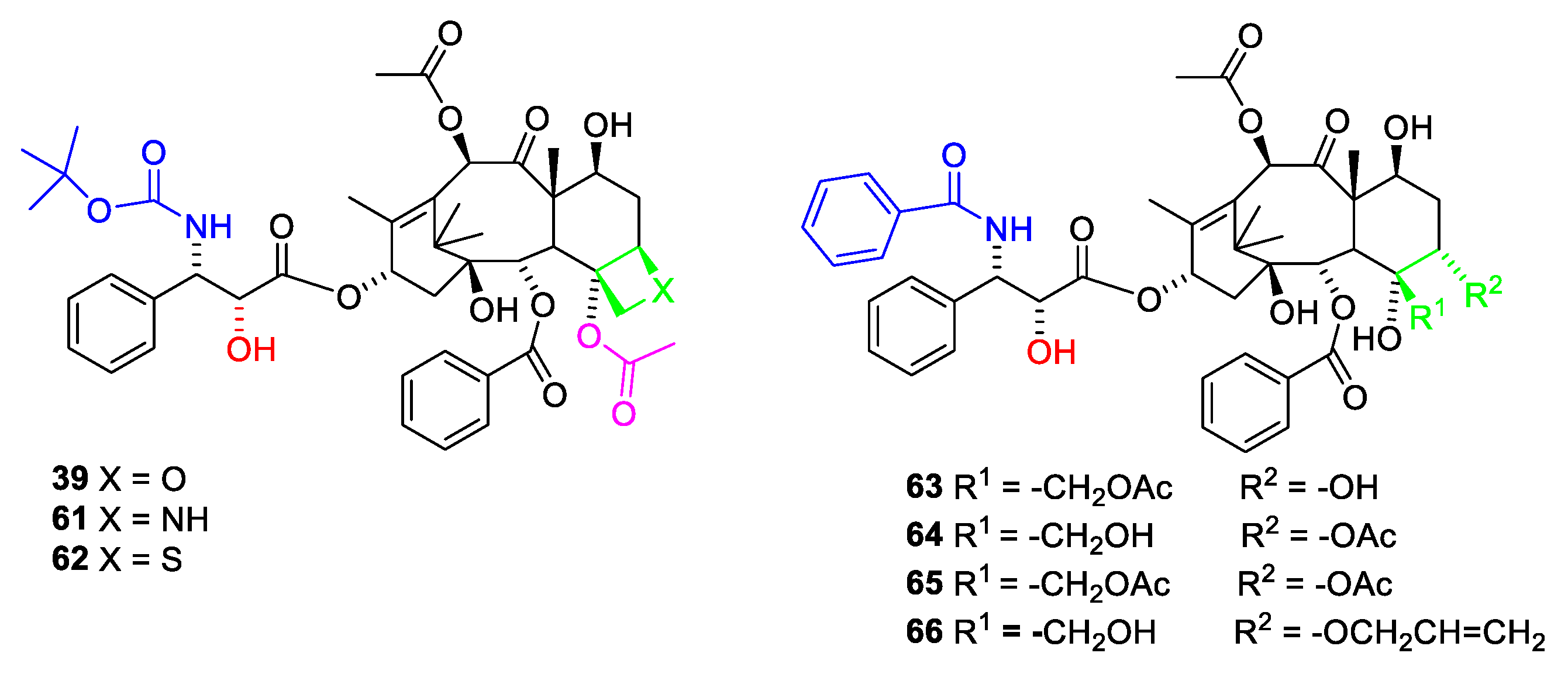
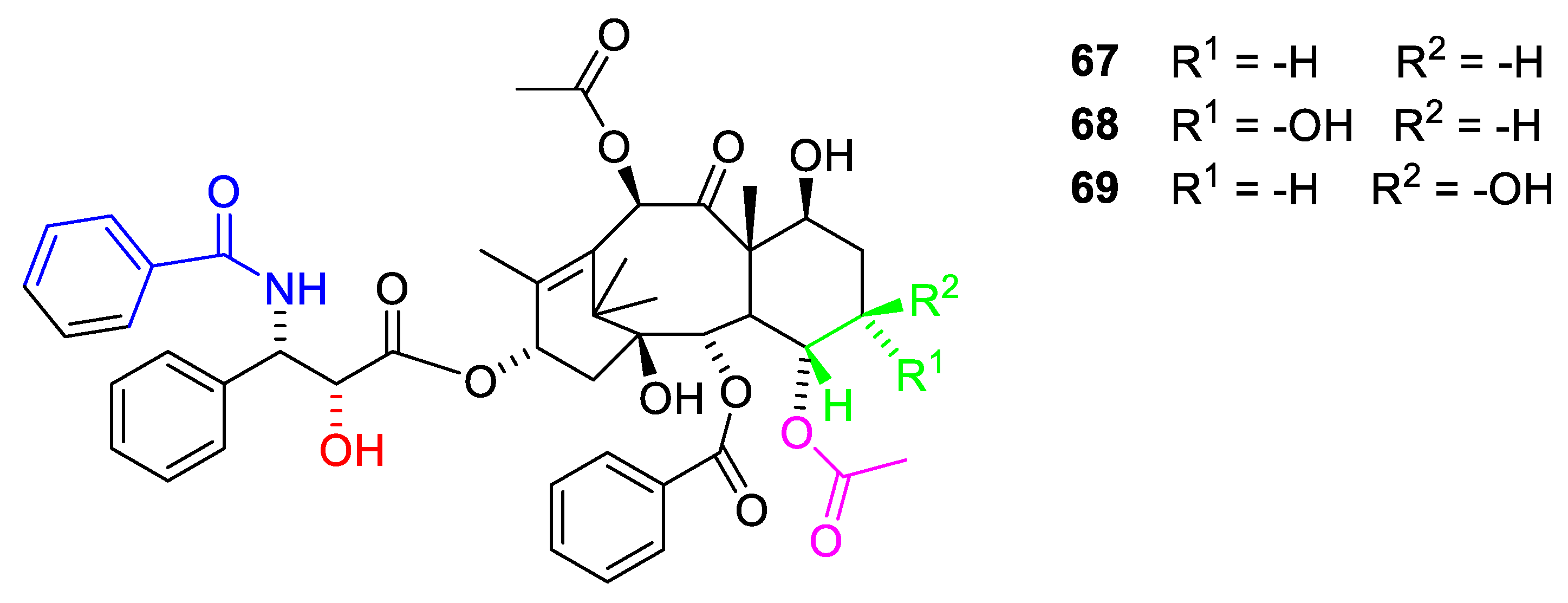
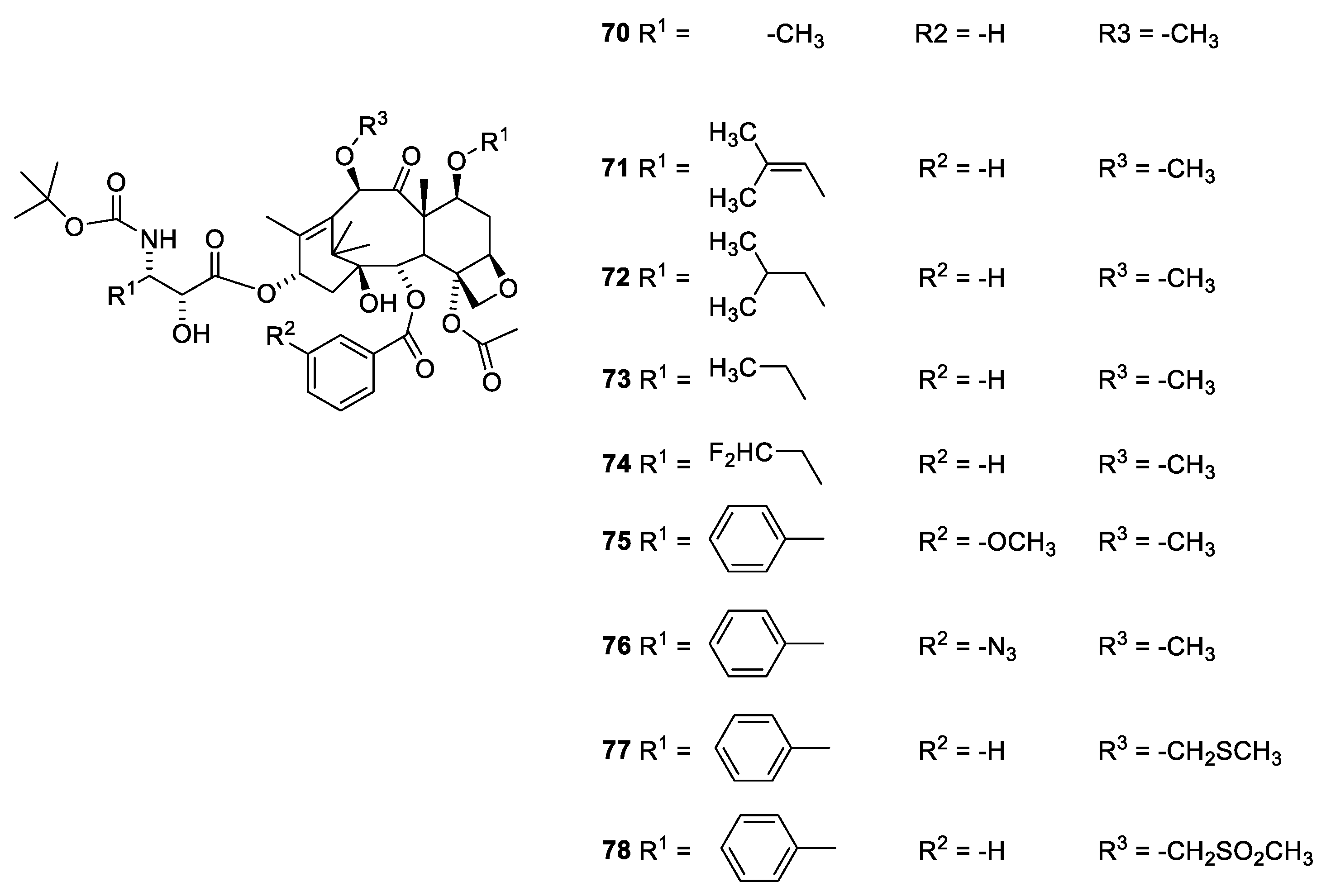
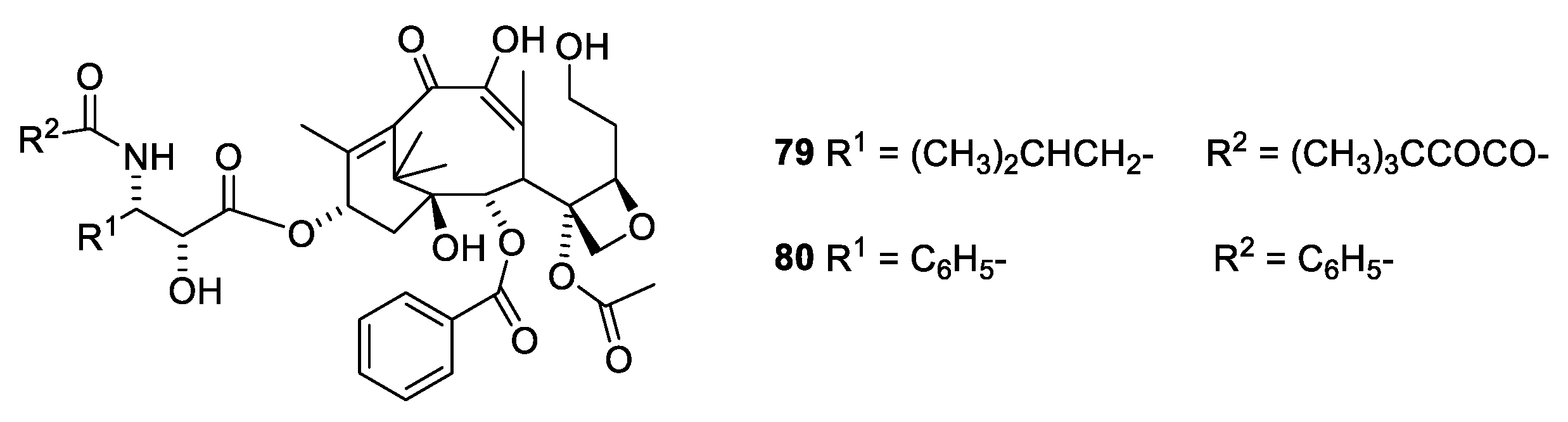
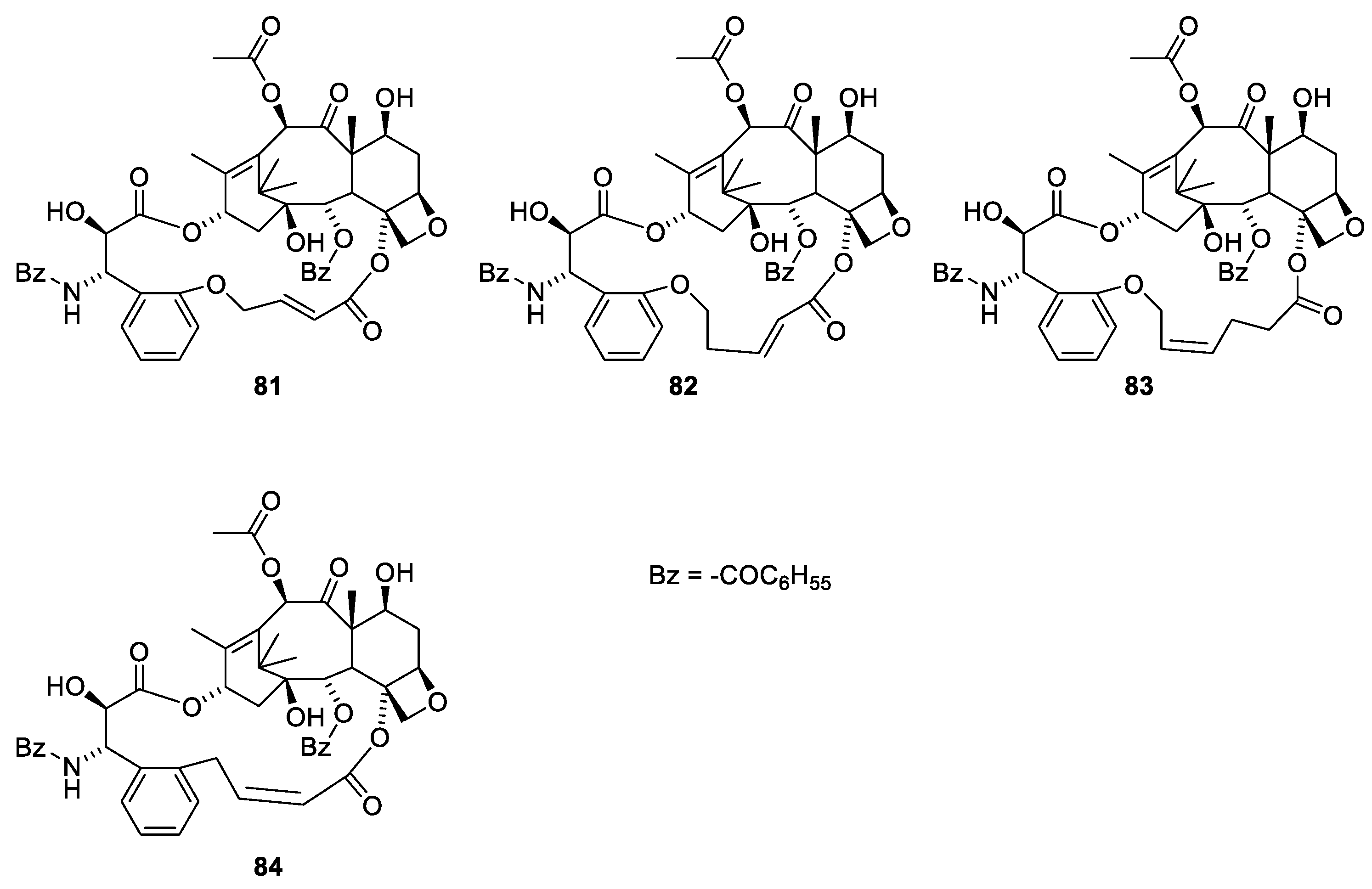
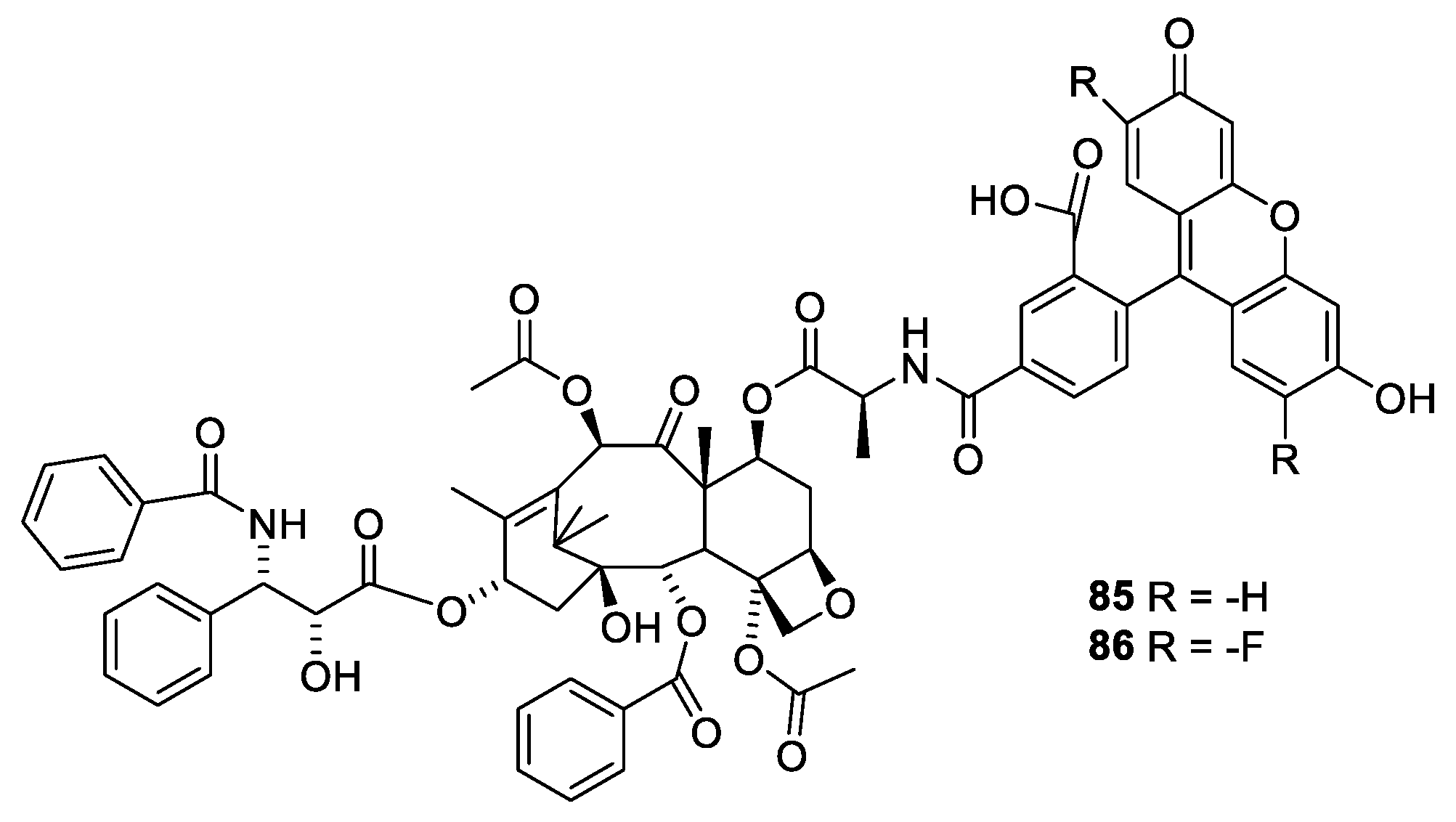
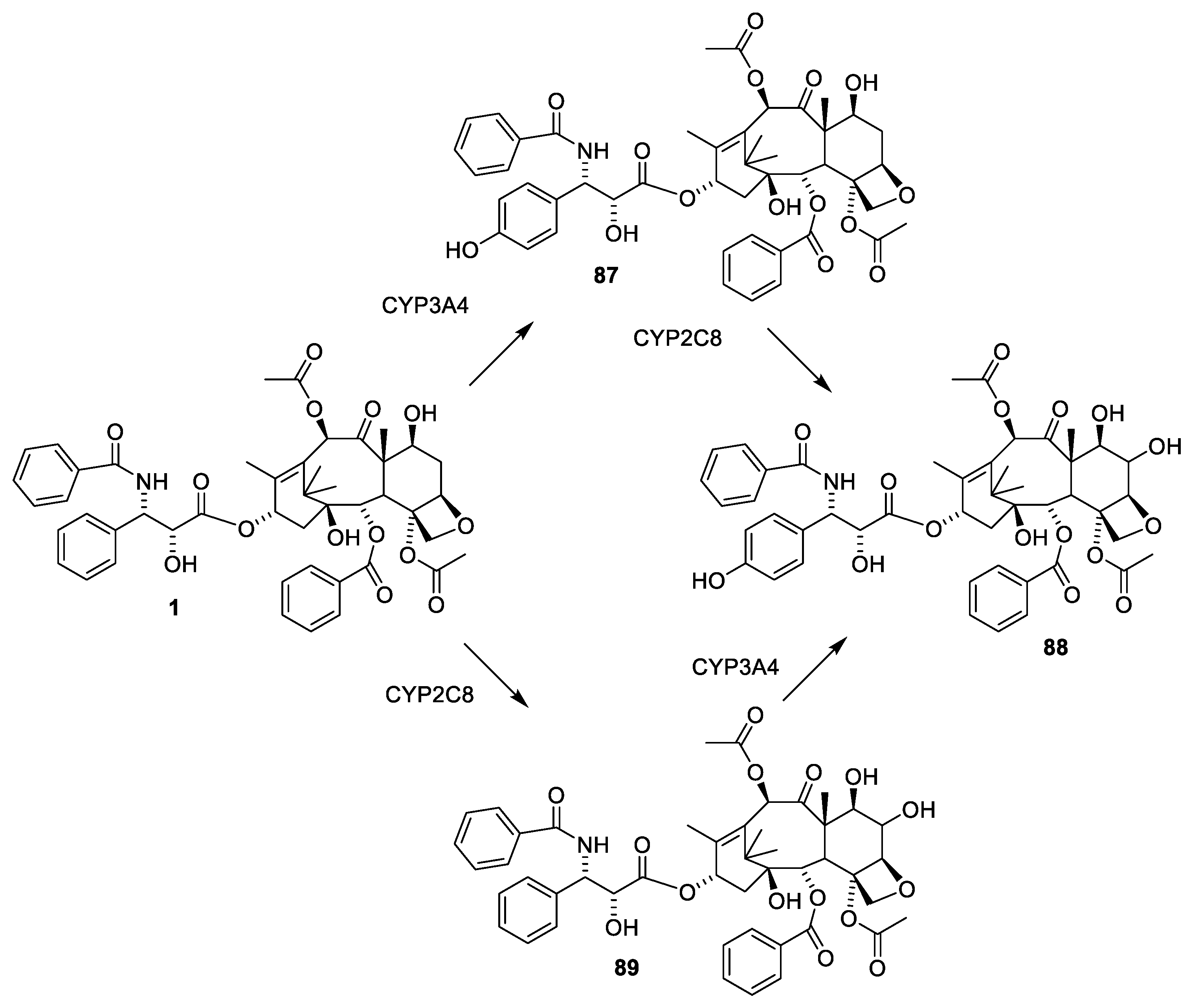
2.6. Structure Activity Relationships of Paclitaxel
2.7. Clinical Trials of Paclitaxel and Analogues
2.7.1. Clinical Use of Paclitaxel
2.7.2. Clinical Use of Docetazel
2.7.3. Clinical Use of Cabazitaxel
2.7.4. Clinical Use of Nanoparticle Formulations of Paclitaxel
3. Microtubule-Targeting Compounds
3.1. Microtubule-Destabilizing Compounds
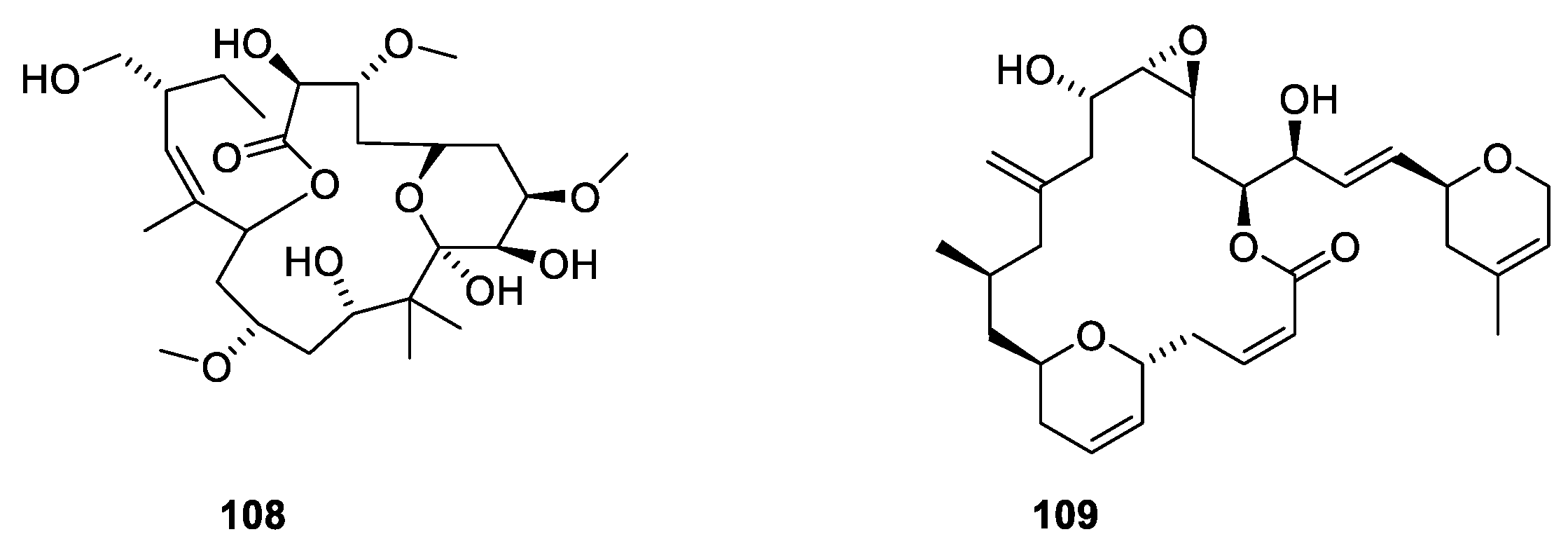
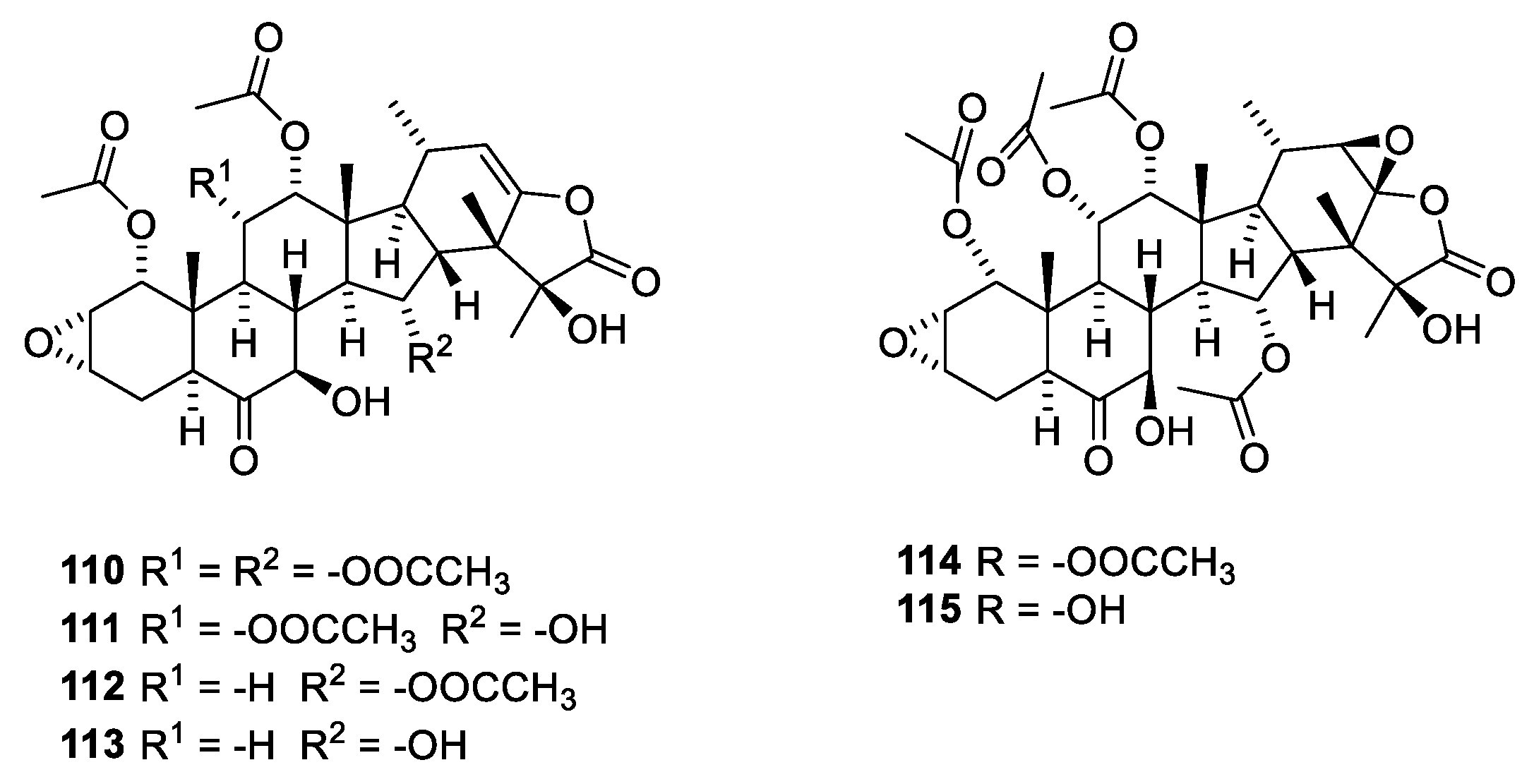
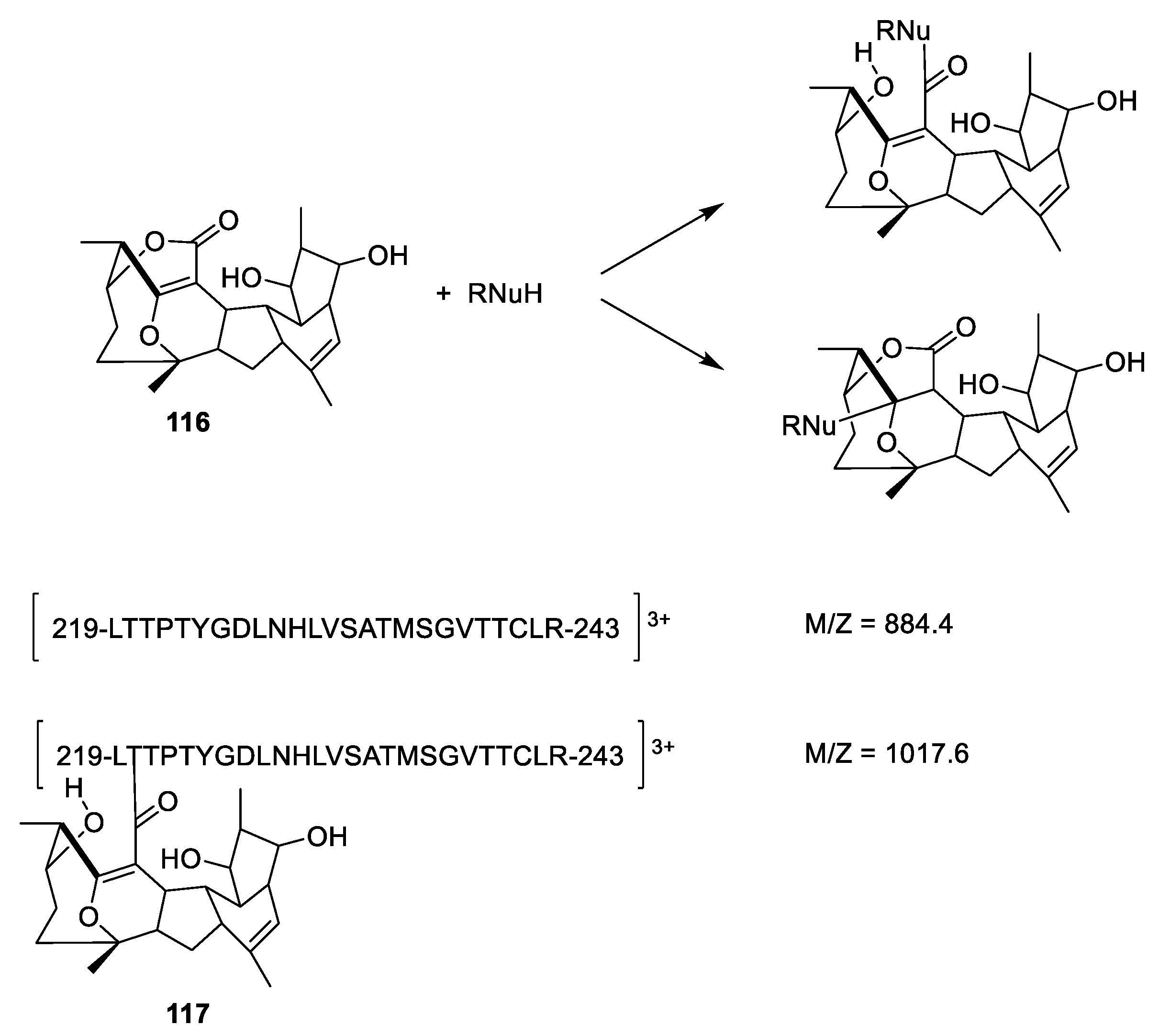
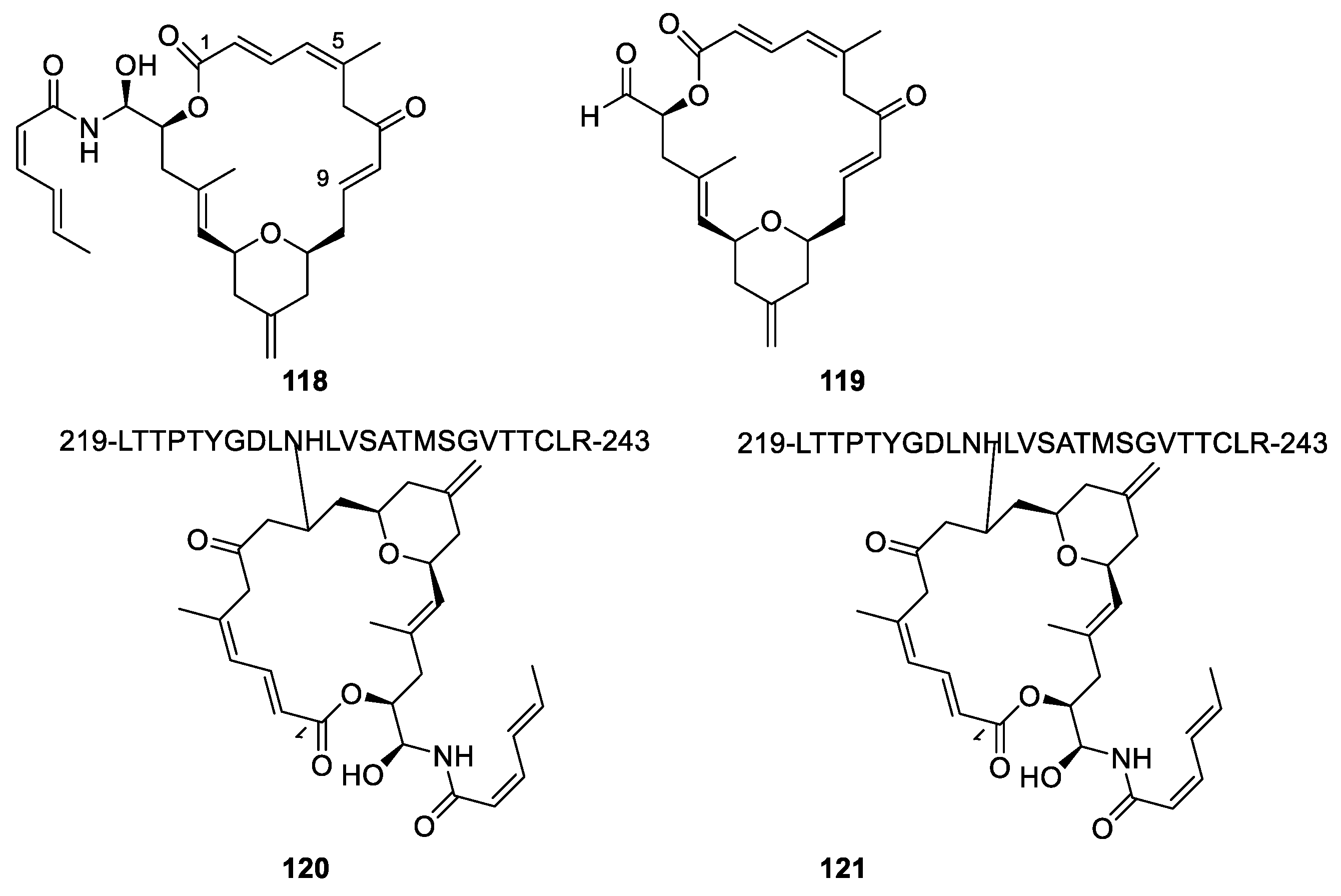
3.2. Microtubule-Stabilizing Compounds
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Conceição, P. Human Development Report 2019. In U.N.D. Programme; UN: San Francisco, CA, USA, 2019. Available online: https://hdr.undp.org/sites/default/files/hdr2019.pdf (accessed on 7 November 2021).
- Sung, H.; Siegel, R.L.; Jemal, A.; Ferlay, J.; Laversanne, M.; Soerjomataram, I.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.B. Drugs That Changed Society: History and Current Status of the Early Antibiotics: Salvarsan, Sulfonamides, and β-Lactams. Molecules 2021, 26, 6057. [Google Scholar] [CrossRef] [PubMed]
- Association of the Nordic Cancer Registries (Ed.) The Fact Sheets Provide Useful Information and Graphics on Specific Populations and Cancer Sites; Danish Cancer Society: Copenhagen, Denmark, 2021; Available online: https://nordcan.iarc.fr/en/factsheets (accessed on 7 November 2021).
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martinez, R.A.; Canovas-Diaz, M.; Puente, T.D. A compressive review about Taxol: History and future challenges. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I.; Lichtenthal, B.; Lee, S.; Wang, C.; Wang, X. Taxane anticancer agents: A patent perspective. Expert Opin. Ther. Pat. 2016, 26, 1–20. [Google Scholar] [CrossRef]
- Kingston, D.G.I. My 60-Year Love Affair with Natural Products. J. Nat. Prod. 2021, 84, 932–948. [Google Scholar] [CrossRef]
- Kingston, D.G.I. Recent Advances in the Chemistry of Taxol. J. Nat. Prod. 2000, 63, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Sofias, A.M.; Dunne, M.; Storm, G.; Allen, C. The battle of "nano" paclitaxel. Adv. Drug Deliv. Rev. 2017, 122, 20–30. [Google Scholar] [CrossRef]
- Aldrich, L.N.; Burdette, J.E.; de Blanco, E.C.; Coss, C.C.; Eustaquio, A.S.; Fuchs, J.R.; Kinghorn, A.D.; MacFarlane, A.; Mize, B.K.; Oberlies, N.H.; et al. Discovery of Anticancer Agents of Diverse Natural Origin. J. Nat. Prod. 2022, 85, 702–719. [Google Scholar] [CrossRef]
- Hao, D.C.; Xiao, P.G.; Huang, B.; Ge, G.B.; Yang, L. Interspecific relationships and origins of Taxaceae and Cephalotaxaceae revealed by partitioned Bayesian analyses of chloroplast and nuclear DNA sequences. Plant Syst. Evol. 2008, 276, 89–104. [Google Scholar] [CrossRef]
- le Roux, M.; Guéritte, F. From the Pacific Yew (Taxus brevifolia) to the English Yew (Taxus baccata): Steps Towards the Discovery of Docetaxel (Taxotere®). In Navelbine and Taxotere Histories of Science; le Roux, M., Guéritte, F., Eds.; Elsevier: Oxford, UK, 2017; pp. 151–212. [Google Scholar]
- Wang, Y.-F.; Shi, Q.-W.; Dong, M.; Kiyota, H.; Gu, Y.-C.; Cong, B. Natural Taxanes: Developments Since 1828. Chem. Rev. 2011, 111, 7652–7709. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.J. The Efficacy of the English Longbow: A Reply to Kelly DeVries. War Hist. 1998, 5, 233–242. [Google Scholar] [CrossRef]
- Poudel, R.C.; Gao, L.-M.; Moller, M.; Baral, S.R.; Uprety, Y.; Liu, J.; Li, D.-Z. Yews (Taxus) along the Hindu Kush-Himalayan region: Exploring the ethnopharmacological relevance among communities of Mongol and Caucasian origins. J. Ethnopharmacol. 2013, 147, 190–203. [Google Scholar] [CrossRef]
- Ajaib, M.; Ishtiaq, M.; Bhatti, K.H.; Hussain, I.; Maqbool, M.; Hussain, T.; Mushtaq, W.; Ghani, A.; Azeem, M.; Khan, S.M.R.; et al. Inventorization of traditional ethnobotanical uses of wild plants of Dawarian and Ratti Gali areas of District Neelum, Azad Jammu and Kashmir Pakistan. PLoS ONE 2021, 16, e0255010. [Google Scholar] [CrossRef] [PubMed]
- Hilger, A.; Brande, F. Taxine, the alkaloid of the yew-tree (Taxus baccata). Ber. Dtsch. Chem. Ges. 1890, 23, 464–468. [Google Scholar] [CrossRef]
- Schaller, H. A. Beitrage Zur Konstitionsaufklärung Des Taxins B. Ûber Die Identitât Des ColieariDs Mil Demtrigonellin; ETH Zürich: Zürich, Switzerland, 1928. [Google Scholar]
- Guyer, A. Weitere Beiträge Zur Kenntnis Des Taxins; ETH Zürich: Zürich, Switzerland, 1922; pp. 1–86. [Google Scholar]
- Avendañol, C.; Menéndez, C. (Eds.) Anticancer Drugs Targeting Tubulin and Microtubule. In Medicinal Chemistry of Anticancer Drug; Elsevier: Amsterdam, The Netherland, 2015; pp. 359–390. [Google Scholar]
- Dewick, P.M. Medicinal Natural Products; John Wiley and Sons Ltd.: Chicester, UK, 2009. [Google Scholar]
- Lange, B.M.; Conner, C.F. Taxanes and Taxoids of the Genus Taxus—A Comprehensive Inventory of Chemical Diversity. Phytochemistry 2021, 190, 112829. [Google Scholar] [CrossRef]
- Sneader, W. Drug Discovery: A History; John Wiley and Sons Ltd.: West Sussex, UK, 2005. [Google Scholar]
- Hoffman, A.; Shahidi, F. Paclitaxel and other taxanes in hazelnut. J. Funct. Foods 2008, 1, 33–37. [Google Scholar] [CrossRef]
- Stierle, A.; Strobel, G.; Stierle, D. Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science 1993, 260, 214–217. [Google Scholar] [CrossRef]
- El-Sayed, A.S.A.; El-Sayed, M.T.; Rady, A.; Zein, N.; Enan, G.; Shindia, A.; El-Hefnawy, S.; Sitohy, M.; Sitohy, B. Exploiting the biosynthetic potency of taxol fromfungal endophytes of conifers plants; genomemining and metabolic manipulation. Molecules 2020, 25, 3000. [Google Scholar] [CrossRef]
- Stierle, A.; Stierle, D.; Stroble, G.; Bignami, G.; Grothaus, P. Bioactive metabolites of the endophytic fungi of Pacific yew, Taxus brevifolia. Paclitaxel, taxanes, and other bioactive compounds. ACS Symp. Ser. 1995, 583, 81–97. [Google Scholar]
- Yang, Y.; Zhao, H.; Barrero, R.A.; Zhang, B.; Sun, G.; Wilson, I.W.; Xie, F.; Walker, K.D.; Parks, J.W.; Bruce, R.; et al. Genome sequencing and analysis of the paclitaxel-producing endophytic fungus Penicillium aurantiogriseum NRRL 62431. BMC Genom. 2014, 15, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Stahlhut, R.; Park, G.; Petersen, R.; Ma, W.; Hylands, P. The occurrence of the anti-cancer diterpene taxol in Podocarpus gracilior Pilger (Podocarpaceae). Biochem. Syst. Ecol. 1999, 27, 613–622. [Google Scholar] [CrossRef]
- Qiao, F.; Cong, H.; Jiang, X.; Wang, R.; Yin, J.; Qian, D.; Wang, Z.; Nick, P. De novo characterization of a Cephalotaxus hainanensis transcriptome and genes related to paclitaxel biosynthesis. PLoS ONE 2014, 9, e106900. [Google Scholar] [CrossRef] [PubMed]
- Lucas, H. Ueber Ein in Den Blättern Von Taxus Baccata L. Enhaltenes Alkaloid (Das Taxin). In Archiv Der Pharmazie; Wiley-VCH: Weinheim, Germany, 1856; Volume 135, pp. 145–149. [Google Scholar]
- Graf, E. Taxin B, das Hauptalkaloid von Taxus baccata L. 4. Mitteilung: Taxus-Alkaloide. Arch. Pharm. Ber. Dtsch. Pharm. Ges. 1958, 291, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Graf, E.; Weinandy, S.; Koch, B.; Breitmaier, E. 13C NMR-Untersuchung von Taxin B aus Taxus baccata L. Liebigs Ann. Chem. 1986, 1986, 1147–1151. [Google Scholar] [CrossRef]
- Ettouati, L.; Ahond, A.; Poupat, C.; Potier, P. Revision Structurale de la Taxine B, alcoloide majoritaire des feuilles de Li´f d’Europe, Taxus baccata. J. Nat. Prod. 1991, 54, 1455–1458. [Google Scholar] [CrossRef]
- Lythgoe, B. Taxus alkaloids. Alkaloids 1968, 10, 597–626. [Google Scholar]
- Appendino, G.; Tagliapietra, S.; Ozen, H.C.; Gariboldi, P.; Gabetta, B.; Bombardelli, E. The chemistry and occurrence of taxane derivatives. IV. Taxanes from the seeds of Taxus baccata. J. Nat. Prod. 1993, 56, 514–520. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Kaspera, R.; Croteau, R. Cytochrome P450 oxygenases of Taxol biosynthesis. Phytochem. Rev. 2006, 5, 433–444. [Google Scholar] [CrossRef]
- Jennewein, S.; Croteau, R. Taxol: Biosynthesis, molecular genetics, and biotechnological applications. Appl. Microbiol. Biotechnol. 2001, 57, 13–19. [Google Scholar] [PubMed]
- Bull, J.A.; Croft, R.A.; Davis, O.A.; Doran, R.; Morgan, K.F. Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry. Chem. Rev. 2016, 116, 12150–12233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exposito, O.; Bonfill, M.; Moyano, E.; Onrubia, M.; Mirjalili, M.H.; Cusido, R.M.; Palazon, J. Biotechnological production of taxol and related taxoids: Current state and prospects. Anti-Cancer Agents Med. Chem. 2009, 9, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Singh, B.; Thakur, V.; Thakur, A.; Thakur, N.; Chand, D.; Kumar, P.; Pandey, D. Hyper-production of taxol from Aspergillus fumigatus, an endophytic fungus isolated from Taxus sp. of the Northern Himalayan region. Biotechnol. Rep. 2019, 24, e00395. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Drašar, P.; Rimpelová, S.; Christensen, S.B.; Khripach, V.A.; Jurášek, M. Large Scale Conversion of Trilobolide into the Payload of Mipsagargin: 8-O-(12-Aminododecanoyl)-8-O-Debutanoylthapsigargin. Biomolecules 2020, 10, 1640. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z. Artemisinin anti-malarial drugs in China. Acta Pharm. Sin. B 2016, 6, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Ajikumar, P.K.; Xiao, W.H.; Tyo, K.E.J.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T.H.; Pfeifer, B.; Stephanopoulos, G. Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science 2010, 330, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Magri, N.F.; Kingston, D.G.I.; Jitrangsri, C.; Piccariello, T. Modified taxols. 3. Preparation and acylation of baccatin III. J. Org. Chem. 1986, 51, 3239–3242. [Google Scholar] [CrossRef]
- Mathew, A.E.; Mejillano, M.R.; Nath, J.P.; Himes, R.H.; Stella, V.J. Synthesis and evaluation of some water-soluble prodrugs and derivatives of taxol with antitumor activity. J. Med. Chem. 1992, 35, 145–151. [Google Scholar] [CrossRef]
- Vellemae, E.; Stepanov, V.; Maeorg, U. Mild approach to the deprotection of troc from protected amines using mischmetal and TMSCl. Synth. Commun. 2010, 40, 3397–3404. [Google Scholar] [CrossRef]
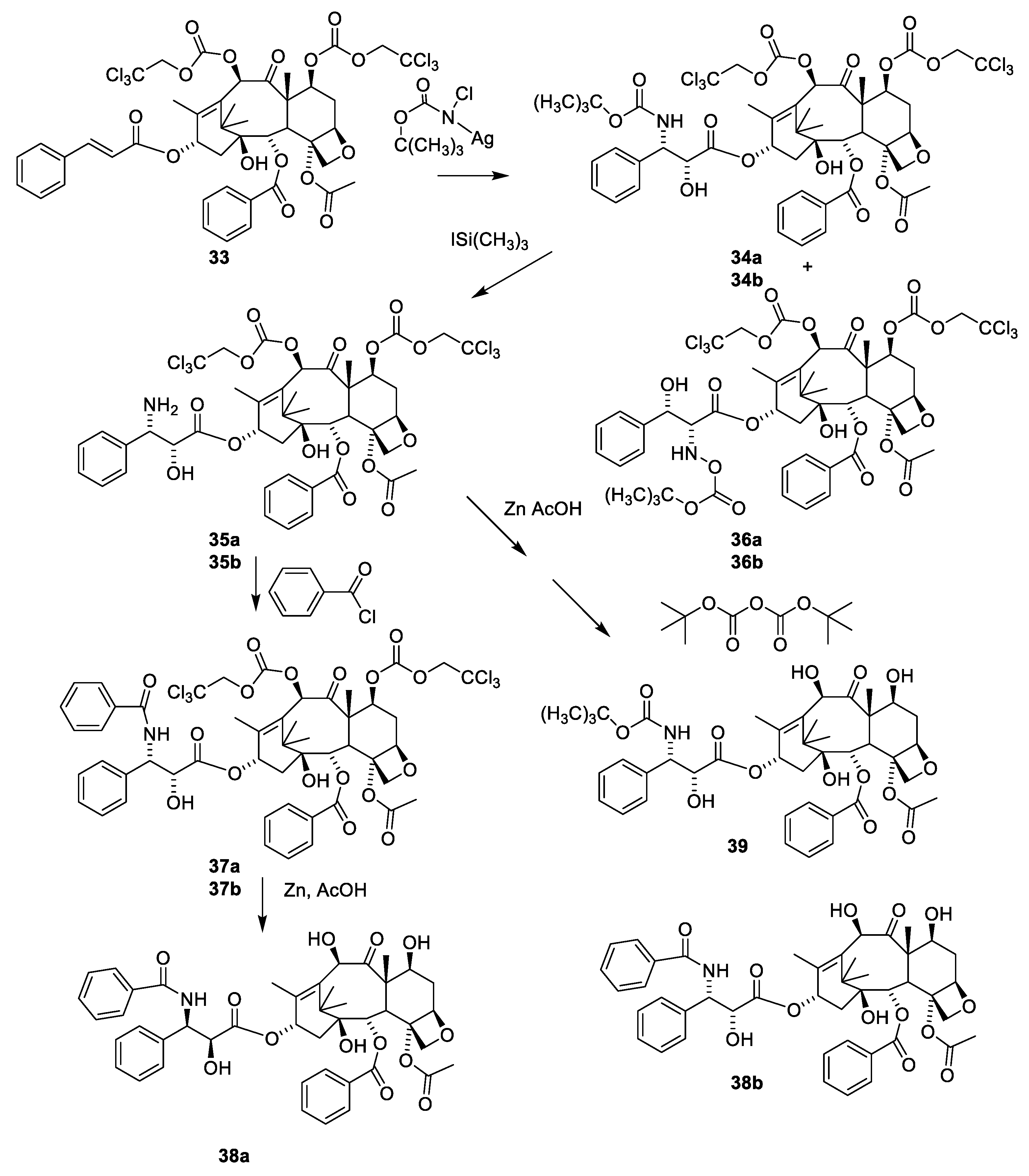
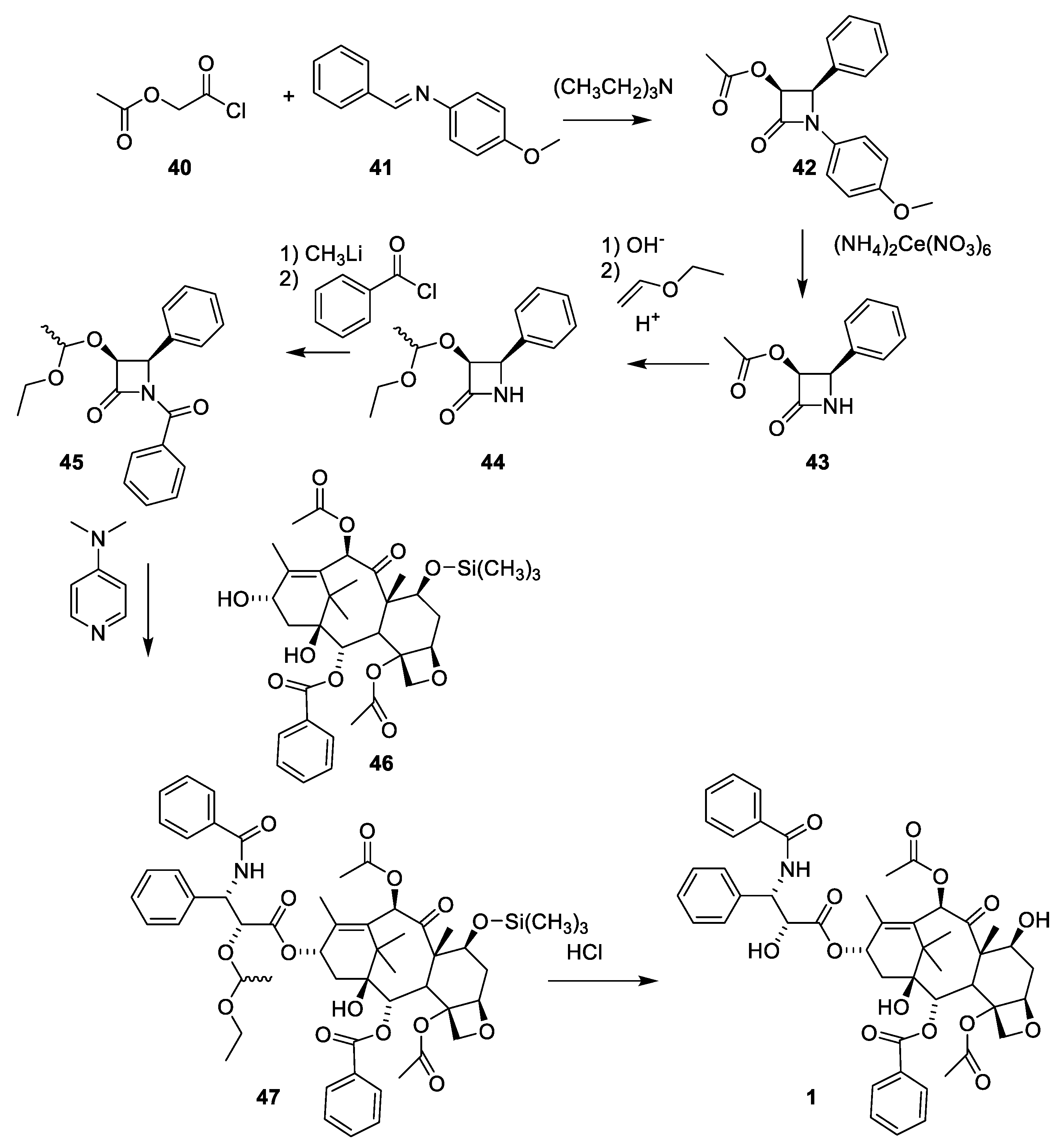
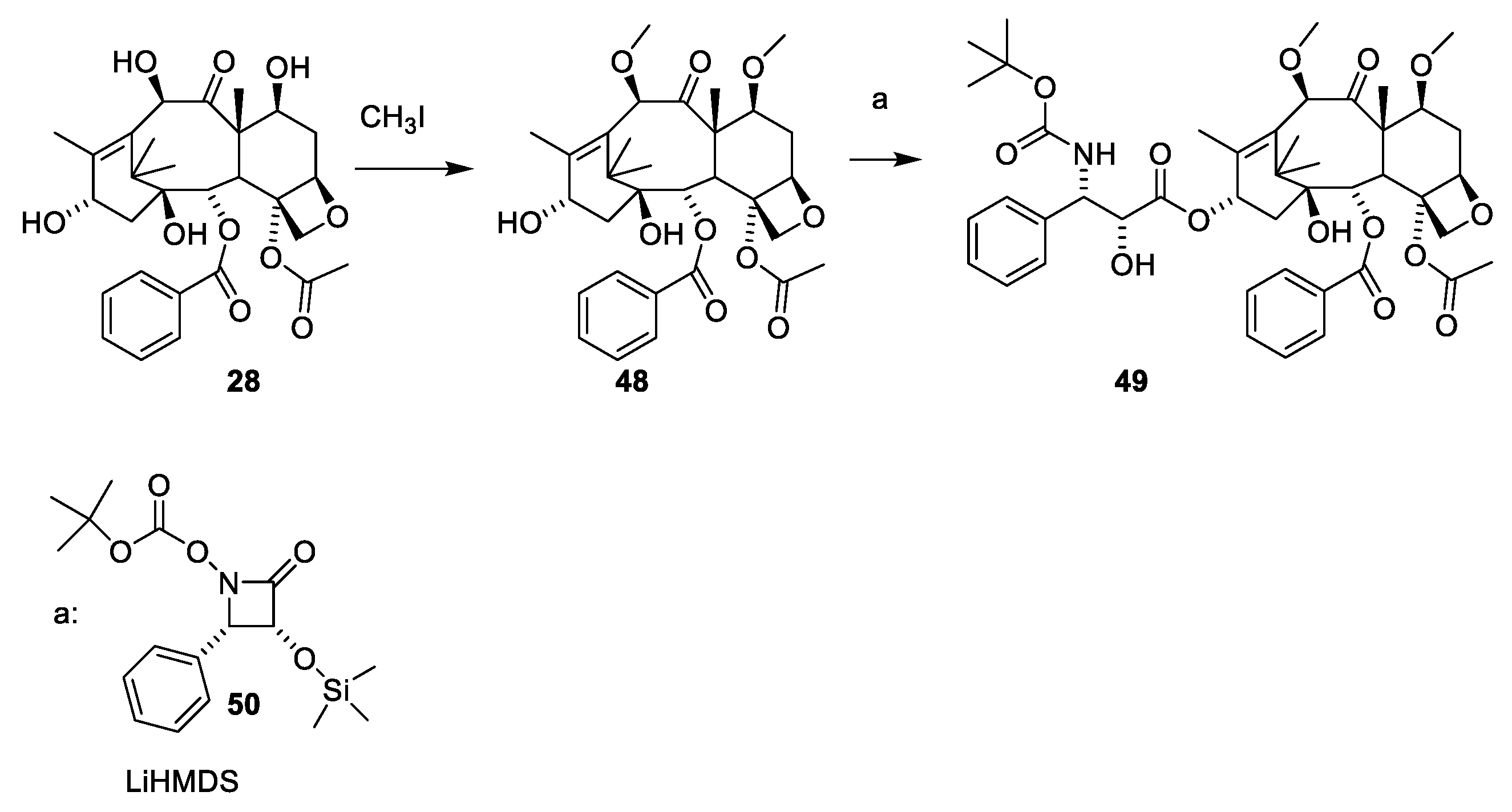
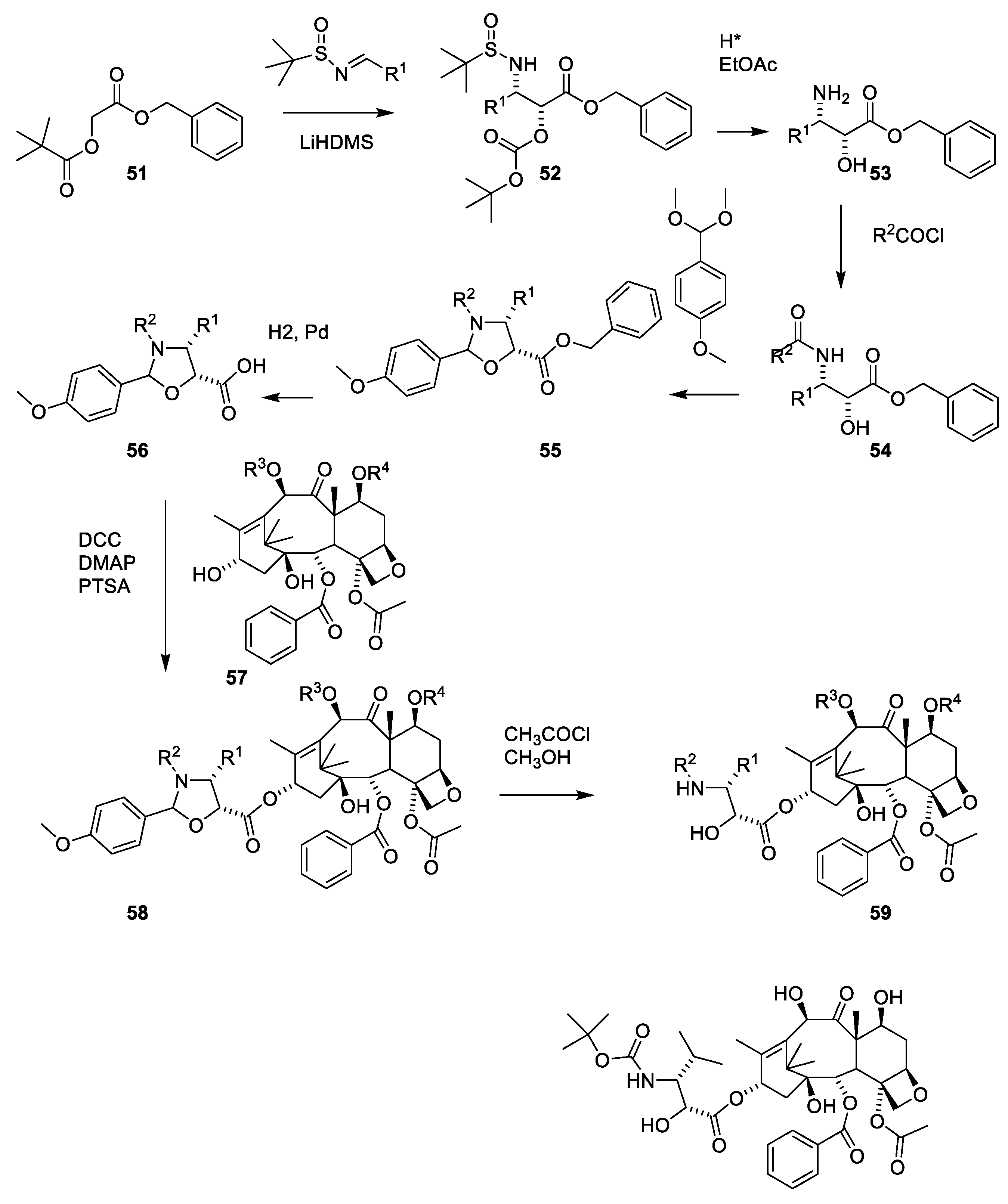
- Denis, J.N.; Greene, A.E.; Guenard, D.; Gueritte-Voegelein, F.; Mangatal, L.; Potier, P. Highly efficient, practical approach to natural taxol. J. Am. Chem. Soc. 1988, 110, 5917–5919. [Google Scholar] [CrossRef]
- Mangatal, L.; Adeline, M.T.; Guenard, D.; Gueritte-Voegelein, F.; Potier, P. Application of the vicinal hydroxyamination reaction with asymmetric induction to the hemisynthesis of taxol and analogs. Tetrahedron 1989, 45, 4177–4190. [Google Scholar] [CrossRef]
- Holton, R.A. Method for Preparation of Taxol; Florida State University: Tallahassee, FL, USA, 1990; p. 18. [Google Scholar]
- Didier, E.; Oddon, G.; Pauze, D.; Leon, P.; Riguet, D. Process for Preparing Drivatives of the Taxoid Famliy. U.S. Patent 5,962,705, 5 October 1999. [Google Scholar]
- Li, Y.; Liu, K.; Wang, J.; Ding, N.; Zhang, W. Method for Preparation of Taxol-Like Anti-Cancer Agent Cabazitaxel (XRP6258); Fudan University, Peop. Rep. China; Wuxi Target Drug Research Co., Ltd.: Wuxing, China, 2013; p. 14. [Google Scholar]
- Sun, S.; Zou, Q. Paclitaxel Derivative Cabazitaxel Synthesis Route Using Novel Catalyst; Tianjin University, Peop. Rep. China: Tianjin, China, 2020; p. 7. [Google Scholar]
- Holton, R.A. Preparation of C10 Ester Substituted Taxanes As Antitumor Agents; Florida State University Research Foundation, Inc.: Tallahassee, FL, USA, 2001; p. 87. [Google Scholar]
- Holton, R.A. Preparation and Formulation of Taxanes Having Improved Solubility for Pharmaceutical Use As Antitumor Agents; Florida State University Research Foundation, Inc.: Tallahassee, FL, USA, 2001; p. 319. [Google Scholar]
- Thottathil, J.K.; Trifunovich, I.D.; Kucera, D.J.; Li, W.-S. Beta-Lactams, Methods for the Preparation of Taxanes, and Sidechain-Bearing Taxanes; Bristol-Myers Squibb Co.: New York, NY, USA, 1994; p. 27. [Google Scholar]
- Jing, Y.-R.; Zhou, W.; Li, W.-l.; Zhao, L.-X.; Wang, Y.-F. The synthesis of novel taxoids for oral administration. Bioorg. Med. Chem. 2014, 22, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Kaul, R.; Risinger, A.L.; Mooberry, S.L. Microtubule-Targeting Drugs: More than Antimitotics. J. Nat. Prod. 2019, 82, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Kingston, D.G.I. Tubulin-Interactive Natural Products as Anticancer Agents. J. Nat. Prod. 2009, 72, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Meresse, P.; Dechaux, E.; Monneret, C.; Bertounesque, E. Etoposide: Discovery and medicinal chemistry. Curr. Med. Chem. 2004, 11, 2443–2466. [Google Scholar] [CrossRef]
- Yang, C.-P.H.; Horwitz, S.B. Taxol: The first microtubule stabilizing agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef]
- Wang, S.-R.; Yang, C.-G.; Sanchez-Murcia, P.A.; Snyder, J.P.; Yan, N.; Saez-Calvo, G.; Diaz, J.F.; Gago, F.; Fang, W.-S. Restoration of Microtubule Interaction and Cytotoxicity in D-seco Taxanes upon Incorporation of 20-Hydroxymethyl-4-allyloxy Groups. Org. Lett. 2015, 17, 6098–6101. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Hejab, N.M.A.; Howes, S.; Northcote, P.; Miller, J.H.; Diaz, J.F.; Downing, K.H.; Nogales, E. Insights into the Distinct Mechanisms of Action of Taxane and Non-Taxane Microtubule Stabilizers from Cryo-EM Structures. J. Mol. Biol. 2017, 429, 633–646. [Google Scholar] [CrossRef]
- Sharma, S.; Lagisetti, C.; Poliks, B.; Coates, R.M.; Kingston, D.G.I.; Bane, S. Dissecting Paclitaxel-Microtubule Association: Quantitative Assessment of the 2′-OH Group. Biochemistry 2013, 52, 2328–2336. [Google Scholar] [CrossRef] [PubMed]
- Suffness, M. Taxol: From Discovery to Clinical Use. In Annual Reports in Medicinal Chemistry; Bristol, J.A., Ed.; Academic Press: London, UK, 1993; pp. 305–314. [Google Scholar]
- Marder-Karsenti, R.; Dubois, J.; Bricard, L.; Guenard, D.; Gueritte-Voegelein, F. Synthesis and Biological Evaluation of D-Ring-Modified Taxanes: 5(20)-Azadocetaxel Analogs. J. Org. Chem. 1997, 62, 6631–6637. [Google Scholar] [CrossRef]
- Wang, M.; Cornett, B.; Nettles, J.; Liotta, D.C.; Snyder, J.P. The Oxetane Ring in Taxol. J. Org. Chem. 2000, 65, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Gunatilaka, A.A.L.; Ramdayal, F.D.; Sarragiotto, M.H.; Kingston, D.G.I.; Sackett, D.L.; Hamel, E. Synthesis and Biological Evaluation of Novel Paclitaxel (Taxol) D-Ring Modified Analogues. J. Org. Chem. 1999, 64, 2694–2703. [Google Scholar] [CrossRef]
- Pyo, S.-H.; Cho, J.-S.; Choi, H.-J.; Han, B.-H. Evaluation of paclitaxel rearrangement involving opening of the oxetane ring and migration of acetyl and benzoyl groups. J. Pharm. Biomed. Anal. 2007, 43, 1141–1145. [Google Scholar] [CrossRef]
- Ren, S.; Zhang, M.; Wang, Y.; Guo, J.; Wang, J.; Li, Y.; Ding, N. Synthesis and biological evaluation of novel cabazitaxel analogues. Bioorg. Med. Chem. 2021, 41, 116224. [Google Scholar]
- Tang, Y.; Rodriguez-Salarichs, J.; Zhao, Y.; Cai, P.; Estevez-Gallego, J.; Balaguer-Perez, F.; Horcajo, M.R.; Lucena-Agell, D.; Barasoain, I.; Diaz, J.F.; et al. Modification of C-seco taxoids through ring tethering and substituent replacement leading to effective agents against tumor drug resistance mediated by βIII-Tubulin and P-glycoprotein (P-gp) overexpressions. Eur. J. Med. Chem. 2017, 137, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, C.; Raspaglio, G.; Mozzetti, S.; Cicchillitti, L.; Filippetti, F.; Gallo, D.; Fattorusso, C.; Campiani, G.; Scambia, G. The seco-taxane IDN5390 is able to target class III β-tubulin and to overcome paclitaxel resistance. Cancer Res. 2005, 65, 2397–2405. [Google Scholar] [CrossRef]
- Yang, Y.; Alcaraz, A.A.; Snyder, J.P. The Tubulin-Bound Conformation of Paclitaxel: T-Taxol vs "PTX-NY". J. Nat. Prod. 2009, 72, 422–429. [Google Scholar] [CrossRef]
- Kingston, D.G.I.; Tamarkin, L.; Paciotti, G.F. Conformationally constrained and nanoparticle-targeted paclitaxels. Pure Appl. Chem. 2012, 84, 1455–1467. [Google Scholar] [CrossRef]
- Ganesh, T.; Guza, R.C.; Bane, S.; Ravindra, R.; Shanker, N.; Lakdawala, A.S.; Snyder, J.P.; Kingston, D.G.I. The bioactive taxol conformation on β-tubulin: Experimental evidence from highly active constrained analogs. Proc. Natl. Acad. Sci. USA 2004, 101, 10006–10011. [Google Scholar] [CrossRef] [PubMed]
- Kinsgton, D.G.I.; Ganesh, T.; Snyder, J.P.; Lakdawala, A.S.; Bane, S. Preparation of Conformationally Constrained Paclitaxel Analogs As Anticancer and Anti-Alzheimer’s Agents; Virginia Tech Intellectual Properties, Inc.: Blacksburg, VA, USA, 2005; p. 78. [Google Scholar]
- Giannakakou, P.; Gussio, R.; Nogales, E.; Downing, K.H.; Zaharevitz, D.; Bollbuck, B.; Poy, G.; Sackett, D.; Nicolaou, K.C.; Fojo, T. A common pharmacophore for epothilone and taxanes: Molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2904–2909. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.F.; Strobe, R.; Engelborghs, Y.; Souto, A.A.; Andreu, J.M. Molecular recognition of taxol by microtubules. Kinetics and thermodynamics of binding of fluorescent taxol derivatives to an exposed site. J. Biol. Chem. 2000, 275, 26265–26276. [Google Scholar] [PubMed] [Green Version]
- Gradishar, W.J. Albumin-bound paclitaxel: A next-generation taxane. Expert Opin. Pharmacother. 2006, 7, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-M. To Market, To Market-1993. In Annual Reports in Medicinal Chemistry; Bristol, J.A., Ed.; Academic Press: London, UK, 1994; p. 342. [Google Scholar]
- de Weger, V.A.; Beijnen, J.H.; Schellens, J.H.M. Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel-a review. Anti-Cancer Drugs 2014, 25, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-M. To Market, To Market-1995. In Annual Reports in Medicinal Chemistry; Academic Press: London, UK, 1996; p. 341. [Google Scholar]
- Manning, R.; Selck, F. Oncology Product Sales and Patent Expiration: A Policy Brief, Life Sciences, Bates White Economic Consultaning. 2017. Available online: https://www.bateswhite.com/media/publication/135_oncology%20policy%20brief%20white%20paper.pdf (accessed on 14 May 2022).
- Kuznetsova, L.; Sun, L.; Chen, J.; Zhao, X.; Seitz, J.; Das, M.; Li, Y.; Veith, J.M.; Pera, P.; Bernacki, R.J.; et al. Synthesis and biological evaluation of novel 3′-difluorovinyl taxoids. J. Fluor. Chem. 2012, 143, 177–188. [Google Scholar] [CrossRef]
- Bronson, J.; Dhar, M.; Ewing, W.; Lonberg, N. To Market, To Market-2010. In Annual Reports in Medicinal Chemistry; Macor, J.E., Ed.; Elsevier: London, UK, 2011; p. 451. [Google Scholar]
- Starpharma, D.E.P. Cabazitaxel. 2021. Available online: https://starpharma.com/drug_delivery/dep_cabazitaxel (accessed on 14 May 2022).
- Yuan, H.; Guo, H.; Luan, X.; He, M.; Li, F.; Burnett, J.; Truchan, N.; Sun, D. Albumin Nanoparticle of Paclitaxel (Abraxane) Decreases while Taxol Increases Breast Cancer Stem Cells in Treatment of Triple Negative Breast Cancer. Mol. Pharm. 2020, 17, 2275–2286. [Google Scholar] [CrossRef]
- Brennan, Z. Some Cancer Patients Now Have to Find Other Options as BRISTOL Myers’ Abraxane Falls Into Shortage from Manufacturing Woes, Endpoints News. 2021. Available online: https://endpts.com/some-cancer-patients-now-have-to-find-other-options-as-bristol-myers-abraxane-falls-into-shortage-from-manufacturing-woes/ (accessed on 13 May 2022).
- Gigant, B.; Cormier, A.; Dorleans, A.; Raveili, R.B.G.; Knossow, M. Microtubule-destabilizing agents: Structural and mechanistic insights from the interaction of colchicine and vinblastine with tubulin. Top. Curr. Chem. 2009, 286, 259–278. [Google Scholar] [PubMed]
- Denny, W.A. Deoxyribonucleic acid topoisomerase inhibitors. Compr. Med. Chem. II 2006, 7, 111–128. [Google Scholar]
- Dholwani, K.K.; Saluja, A.K.; Gupta, A.R.; Shah, D.R. A review on plant-derived natural products and their analogs with anti-tumor activity. Indian J. Pharmacol. 2008, 40, 49–58. [Google Scholar] [CrossRef]
- Christensen, S.B. Natural Products that Changed Society. Biomedicines 2021, 9, 472. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Kurkjian, C.D.; Yamada, H.Y. Mitosis-targeting natural products for cancer prevention and therapy. Curr. Drug. Targets 2012, 13, 1820–1830. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dong, G.; Sheng, C. Structural Simplification of Natural Products. Chem. Rev. 2019, 119, 4180–4220. [Google Scholar] [CrossRef] [PubMed]
- Bronson, J.; Black, A.; Dhar, M.; Ellsworth, B.A.; Merritt, J.R. To Market, to Market-2013. In Medicinal Chemistry Reviews 2014; Desai, M.C., Ed.; American Chemical Society: Washington, DC, USA, 2014; pp. 437–508. [Google Scholar]
- Kanakkanthara, A.; Northcote, P.T.; Miller, J.H. Peloruside A: A lead non-taxoid-site microtubule-stabilizing agent with potential activity against cancer, neurodegeneration, and autoimmune disease. Nat. Prod. Rep. 2016, 33, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Risinger, A.L.; Li, J.; Du, L.; Benavides, R.; Robles, A.J.; Cichewicz, R.H.; Kuhn, J.G.; Mooberry, S.L. Pharmacokinetic Analysis and in Vivo Antitumor Efficacy of Taccalonolides AF and AJ. J. Nat. Prod. 2017, 80, 409–414. [Google Scholar] [CrossRef]
- He, L.; Orr, G.A.; Horwitz, S.B. Novel molecules that interact with microtubules and have functional activity similar to Taxol. Drug Discov. Today 2001, 6, 1153–1164. [Google Scholar] [CrossRef]
- Li, J.; Risinger, A.L.; Mooberry, S.L. Taccalonolide microtubule stabilizers. Bioorg. Med. Chem. 2014, 22, 5091–5096. [Google Scholar] [CrossRef]
- Li, J.; Risinger, A.L.; Peng, J.; Chen, Z.; Hu, L.; Mooberry, S.L. Potent Taccalonolides, AF and AJ, Inform Significant Structure-Activity Relationships and Tubulin as the Binding Site of These Microtubule Stabilizers. J. Am. Chem. Soc. 2011, 133, 19064–19067. [Google Scholar] [CrossRef]
- Risinger, A.L.; Hastings, S.D.; Du, L. Taccalonolide C-6 Analogues, Including Paclitaxel Hybrids, Demonstrate Improved Microtubule Polymerizing Activities. J. Nat. Prod. 2021, 84, 1799–1805. [Google Scholar] [CrossRef]
- Buey, R.M.; Calvo, E.; Barasoain, I.; Pineda, O.; Edler, M.C.; Matesanz, R.; Cerezo, G.; Vanderwal, C.D.; Day, B.W.; Sorensen, E.J.; et al. Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nat. Chem. Biol. 2006, 3, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-H.; Kingston, D.G.I. Zampanolide and dactylolide: Cytotoxic tubulin-assembly agents and promising anticancer leads. Nat. Prod. Rep. 2014, 31, 1202–1226. [Google Scholar] [CrossRef] [PubMed]
- Field, J.J.; Pera, B.; Calvo, E.; Canales, A.; Zurwerra, D.; Trigili, C.; Rodriguez-Salarichs, J.; Matesanz, R.; Kanakkanthara, A.; Wakefield, S.J.; et al. Zampanolide, a Potent New Microtubule-Stabilizing Agent, Covalently Reacts with the Taxane Luminal Site in Tubulin α,β-Heterodimers and Microtubules. Chem. Biol. 2012, 19, 686–698. [Google Scholar] [CrossRef] [PubMed]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christensen, S.B. Drugs That Changed Society: Microtubule-Targeting Agents Belonging to Taxanoids, Macrolides and Non-Ribosomal Peptides. Molecules 2022, 27, 5648. https://doi.org/10.3390/molecules27175648
Christensen SB. Drugs That Changed Society: Microtubule-Targeting Agents Belonging to Taxanoids, Macrolides and Non-Ribosomal Peptides. Molecules. 2022; 27(17):5648. https://doi.org/10.3390/molecules27175648
Chicago/Turabian StyleChristensen, Søren Brøgger. 2022. "Drugs That Changed Society: Microtubule-Targeting Agents Belonging to Taxanoids, Macrolides and Non-Ribosomal Peptides" Molecules 27, no. 17: 5648. https://doi.org/10.3390/molecules27175648
APA StyleChristensen, S. B. (2022). Drugs That Changed Society: Microtubule-Targeting Agents Belonging to Taxanoids, Macrolides and Non-Ribosomal Peptides. Molecules, 27(17), 5648. https://doi.org/10.3390/molecules27175648