
Computer-Aided Drug Design Boosts RAS Inhibitor Discovery


Abstract
:1. Introduction
2. Biochemical Features of RAS
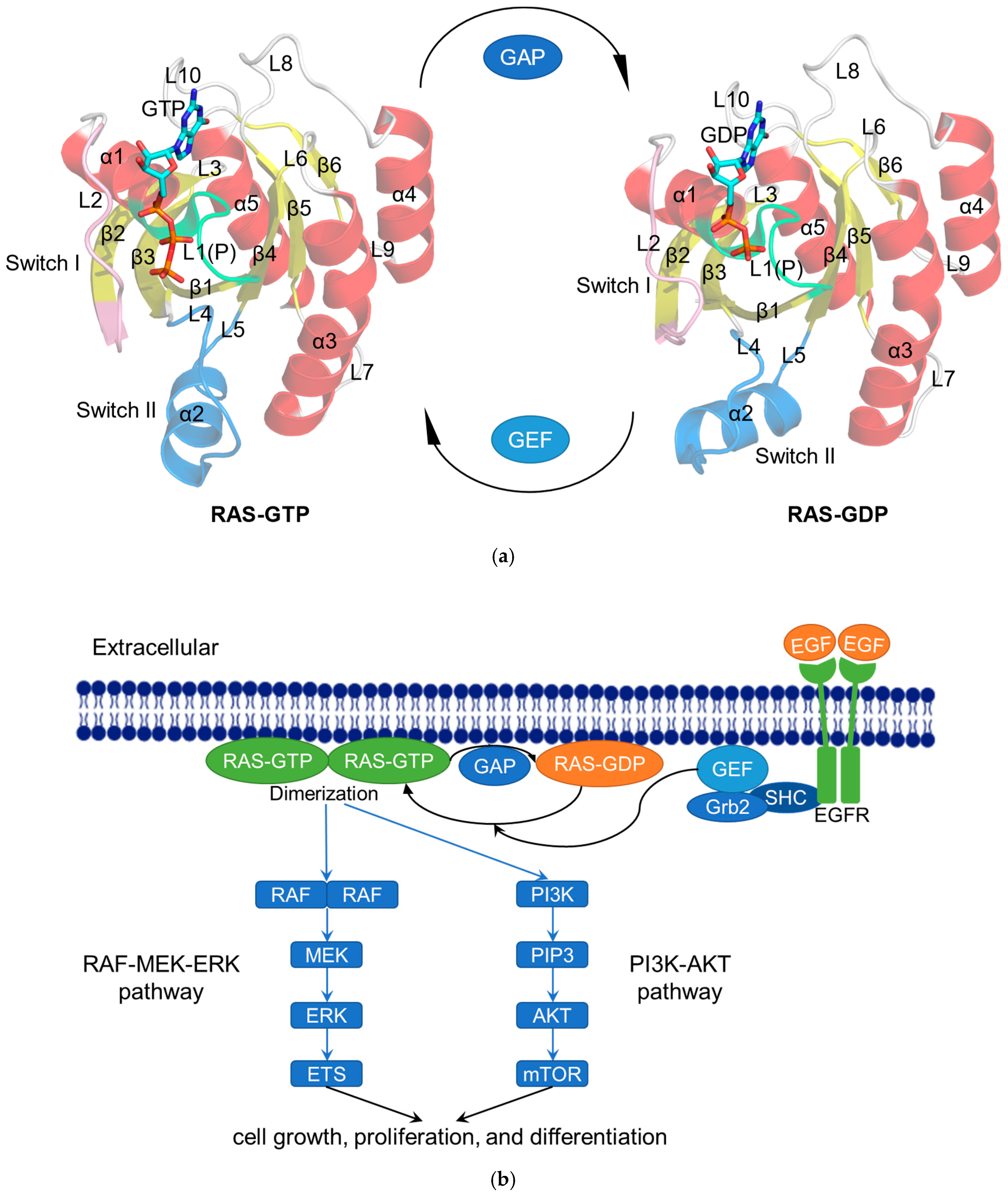
2.1. RAS in Normal Physiological Condition

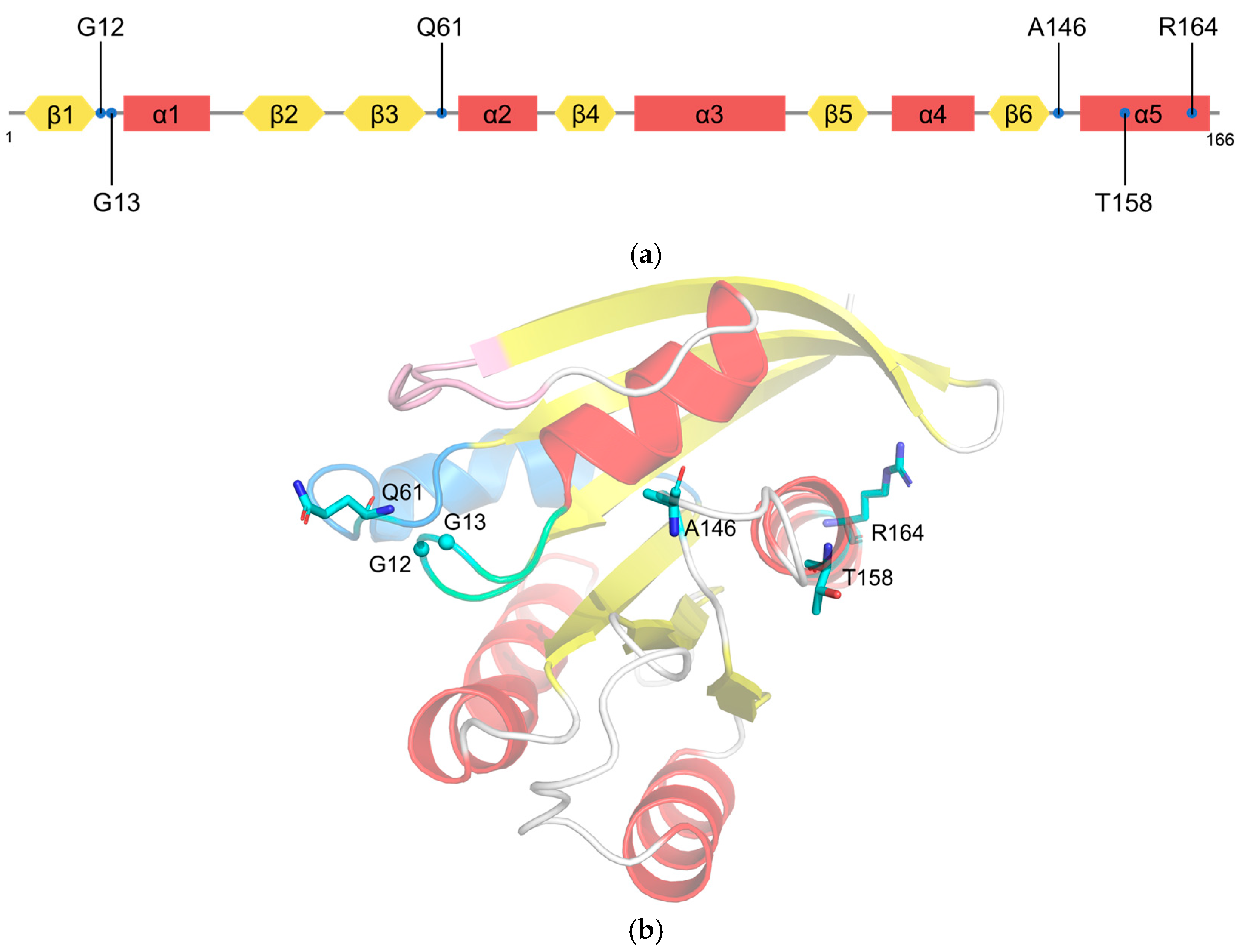
2.2. RAS Mutations Trigger Signaling Dysfunction
3. Application of CADD Methods in the Development of RAS Inhibitors
3.1. Determination of the Target Protein Structure
3.2. Identification of Binding Sites
3.3. Virtual Screening
3.4. Molecular Docking Studies
3.5. Molecular Dynamics (MD) Simulation
3.6. Quantitative Structure–Activity Relationship Study (QSAR)
3.7. Pharmacophore Modelling
3.8. Other CADD Applications
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Liu, S.; Alnammi, M.; Ericksen, S.S.; Voter, A.F.; Ananiev, G.E.; Keck, J.L.; Hoffmann, F.M.; Wildman, S.A.; Gitter, A. Practical Model Selection for Prospective Virtual Screening. J. Chem. Inf. Model. 2019, 59, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Yang, H.; Du Bois, D.R.; Ziller, J.W.; Nowick, J.S. X-ray crystallographic structure of a teixobactin analogue reveals key interactions of the teixobactin pharmacophore. Chem. Commun. 2017, 53, 2772–2775. [Google Scholar] [CrossRef]
- Polanski, J. Receptor dependent multidimensional QSAR for modeling drug—Receptor interactions. Curr. Med. Chem. 2009, 16, 3243–3257. [Google Scholar] [CrossRef] [PubMed]
- Nikonenko, A.; Zankov, D.; Baskin, I.; Madzhidov, T.; Polishchuk, P. Multiple Conformer Descriptors for QSAR Modeling. Mol. Inform. 2021, 40, e2060030. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- da Silva Rocha, S.F.L.; Olanda, C.G.; Fokoue, H.H.; Sant’Anna, C.M.R. Virtual Screening Techniques in Drug Discovery: Review and Recent Applications. Curr. Top. Med. Chem. 2019, 19, 1751–1767. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Walters, W.P.; Wang, R. New Trends in Virtual Screening. J. Chem. Inf. Model. 2020, 60, 4109–4111. [Google Scholar] [CrossRef] [PubMed]
- Cleves, A.E.; Jain, A.N. Structure- and Ligand-Based Virtual Screening on DUD-E(+): Performance Dependence on Approximations to the Binding Pocket. J. Chem. Inf. Model. 2020, 60, 4296–4310. [Google Scholar] [CrossRef]
- Palazzesi, F.; Pozzan, A. Deep Learning Applied to Ligand-Based De Novo Drug Design. Methods Mol. Biol. 2022, 2390, 273–299. [Google Scholar] [PubMed]
- Mo, S.P.; Coulson, J.M.; Prior, I.A. RAS variant signalling. Biochem. Soc. Trans. 2018, 46, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Pai, E.F.; Kabsch, W.; Krengel, U.; Holmes, K.C.; John, J.; Wittinghofer, A. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature 1989, 341, 209–214. [Google Scholar] [CrossRef]
- Ahmadian, M.R.; Hoffmann, U.; Goody, R.S.; Wittinghofer, A. Individual rate constants for the interaction of Ras proteins with GTPase-activating proteins determined by fluorescence spectroscopy. Biochemistry 1997, 36, 4535–4541. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef]
- Welman, A.; Burger, M.M.; Hagmann, J. Structure and function of the C-terminal hypervariable region of K-Ras4B in plasma membrane targetting and transformation. Oncogene 2000, 19, 4582–4591. [Google Scholar] [CrossRef] [Green Version]
- Spencer-Smith, R.; Koide, A.; Zhou, Y.; Eguchi, R.R.; Sha, F.; Gajwani, P.; Santana, D.; Gupta, A.; Jacobs, M.; Herrero-Garcia, E.; et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. [Google Scholar] [CrossRef]
- Casey, P.J.; Solski, P.A.; Der, C.J.; Buss, J.E. p21ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. USA 1989, 86, 8323–8327. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R.; Zhang, J. Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev. 2016, 116, 6607–6665. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmuller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Banerjee, A.; Jang, H.; Zhang, J.; Gaponenko, V.; Nussinov, R. GTP Binding and Oncogenic Mutations May Attenuate Hypervariable Region (HVR)-Catalytic Domain Interactions in Small GTPase K-Ras4B, Exposing the Effector Binding Site. J. Biol. Chem. 2015, 290, 28887–28900. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef]
- Edkins, S.; O’Meara, S.; Parker, A.; Stevens, C.; Reis, M.; Jones, S.; Greenman, C.; Davies, H.; Dalgliesh, G.; Forbes, S.; et al. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol. Ther. 2006, 5, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Tumurkhuu, M.; Saitoh, M.; Takita, J.; Mizuno, Y.; Mizuguchi, M. A novel SOS1 mutation in Costello/CFC syndrome affects signaling in both RAS and PI3K pathways. J. Recept. Signal Transduct. Res. 2013, 33, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Wang, Y.; Chai, Z.; Ni, D.; Li, X.; Pu, J.; Chen, J.; Zhang, J.; Lu, S.; Lv, C.; et al. Targeting RAS phosphorylation in cancer therapy: Mechanisms and modulators. Acta Pharm. Sin. B 2021, 11, 3433–3446. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, X.; Xia, X.; Zhang, W.; Tian, H. A benzoxazole compound as a novel MEK inhibitor for the treatment of RAS/RAF mutant cancer. Int. J. Cancer 2019, 145, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, B.S.; Durinck, S.; Stawiski, E.W.; Yin, J.; Wang, W.; Lin, E.; Moffat, J.; Martin, S.E.; Modrusan, Z.; Seshagiri, S. ERK Mutations and Amplification Confer Resistance to ERK-Inhibitor Therapy. Clin Cancer Res 2018, 24, 4044–4055. [Google Scholar] [CrossRef]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yu, X.; Martin, T.C.; Bansal, A.; Cheung, K.; Lubin, A.; Stratikopoulos, E.; Cahuzac, K.M.; Wang, L.; Xie, L.; et al. AKT Degradation Selectively Inhibits the Growth of PI3K/PTEN Pathway-Mutant Cancers with Wild-Type KRAS and BRAF by Destabilizing Aurora Kinase B. Cancer Discov. 2021, 11, 3064–3089. [Google Scholar] [CrossRef]
- Galoian, K.; Temple, H.T.; Galoyan, A. mTORC1 inhibition and ECM-cell adhesion-independent drug resistance via PI3K-AKT and PI3K-RAS-MAPK feedback loops. Tumour. Biol. 2012, 33, 885–890. [Google Scholar] [CrossRef]
- Ni, D.; Li, X.; He, X.; Zhang, H.; Zhang, J.; Lu, S. Drugging K-Ras(G12C) through covalent inhibitors: Mission possible? Pharmacol Ther. 2019, 202, 1–17. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Wang, Y.; Kaiser, C.E.; Frett, B.; Li, H.Y. Targeting mutant KRAS for anticancer therapeutics: A review of novel small molecule modulators. J. Med. Chem. 2013, 56, 5219–5230. [Google Scholar] [CrossRef]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.; et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Spencer-Smith, R.; O’Bryan, J.P. Targeting the alpha4-alpha5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene 2019, 38, 2984–2993. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef]
- Wang, C.X.; Wang, T.T.; Zhang, K.D.; Li, M.Y.; Shen, Q.C.; Lu, S.Y.; Zhang, J. Pan-KRAS inhibitors suppress proliferation through feedback regulation in pancreatic ductal adenocarcinoma. Acta Pharmacol. Sin. 2022. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2007, 50, 2.9.1–2.9.31. [Google Scholar] [CrossRef]
- Donninger, H.; Hesson, L.; Vos, M.; Beebe, K.; Gordon, L.; Sidransky, D.; Liu, J.W.; Schlegel, T.; Payne, S.; Hartmann, A.; et al. The Ras effector RASSF2 controls the PAR-4 tumor suppressor. Mol. Cell. Biol. 2010, 30, 2608–2620. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, S.; Jamil, F.; Ali, A.; Sehgal, S.A. Comparative Modeling, Molecular Docking, and Revealing of Potential Binding Pockets of RASSF2; a Candidate Cancer Gene. Interdiscip. Sci. 2017, 9, 214–223. [Google Scholar] [CrossRef]
- Collier, T.A.; Piggot, T.J.; Allison, J.R. Molecular Dynamics Simulation of Proteins. Methods Mol. Biol. 2020, 2073, 311–327. [Google Scholar]
- Prakash, P.; Sayyed-Ahmad, A.; Cho, K.J.; Dolino, D.M.; Chen, W.; Li, H.; Grant, B.J.; Hancock, J.F.; Gorfe, A.A. Computational and biochemical characterization of two partially overlapping interfaces and multiple weak-affinity K-Ras dimers. Sci. Rep. 2017, 7, 40109. [Google Scholar] [CrossRef]
- Tuncbag, N.; Gursoy, A.; Nussinov, R.; Keskin, O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat. Protoc. 2011, 6, 1341–1354. [Google Scholar] [CrossRef]
- Muratcioglu, S.; Chavan, T.S.; Freed, B.C.; Jang, H.; Khavrutskii, L.; Freed, R.N.; Dyba, M.A.; Stefanisko, K.; Tarasov, S.G.; Gursoy, A.; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Jisna, V.A.; Jayaraj, P.B. Protein Structure Prediction: Conventional and Deep Learning Perspectives. Protein J. 2021, 40, 522–544. [Google Scholar] [CrossRef]
- Li, L.; Meyer, C.; Zhou, Z.-W.; Elmezayen, A.; Westover, K. Therapeutic Targeting the Allosteric Cysteinome of RAS and Kinase Families. J. Mol. Biol. 2022, 434, 167626. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, B.V.; Rammohan, A.; Babu, T.M.; Zheng, G.Y.; Chen, W.; Rajendra, W.; Zyryanov, G.V.; Gu, W. Molecular insight into isoform specific inhibition of PI3K-alpha and PKC-eta with dietary agents through an ensemble pharmacophore and docking studies. Sci. Rep. 2021, 11, 12150. [Google Scholar] [CrossRef]
- Parca, L.; Mangone, I.; Gherardini, P.F.; Ausiello, G.; Helmer-Citterich, M. Phosfinder: A web server for the identification of phosphate-binding sites on protein structures. Nucleic Acids Res. 2011, 39, W278–W282. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Srivastava, H.K.; Raghava, G.P. A web server for analysis, comparison and prediction of protein ligand binding sites. Biol. Direct 2016, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, M.; Ghersi, D.; Sanchez, R. SITEHOUND-web: A server for ligand binding site identification in protein structures. Nucleic Acids Res. 2009, 37, W413–W416. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Skrlj, B.; Erzen, N.; Kunej, T.; Janezic, D. GenProBiS: Web server for mapping of sequence variants to protein binding sites. Nucleic Acids Res. 2017, 45, W253–W259. [Google Scholar] [CrossRef]
- Wang, Y.; Lupala, C.S.; Liu, H.; Lin, X. Identification of Drug Binding Sites and Action Mechanisms with Molecular Dynamics Simulations. Curr. Top. Med. Chem. 2018, 18, 2268–2277. [Google Scholar] [CrossRef] [PubMed]
- Prakash, P.; Hancock, J.F.; Gorfe, A.A. Binding hotspots on K-ras: Consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins 2015, 83, 898–909. [Google Scholar] [CrossRef]
- Brenke, R.; Kozakov, D.; Chuang, G.Y.; Beglov, D.; Hall, D.; Landon, M.R.; Mattos, C.; Vajda, S. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 25, 621–627. [Google Scholar] [CrossRef]
- Grant, B.J.; Lukman, S.; Hocker, H.J.; Sayyah, J.; Brown, J.H.; McCammon, J.A.; Gorfe, A.A. Novel allosteric sites on Ras for lead generation. PLoS ONE 2011, 6, e25711. [Google Scholar] [CrossRef]
- Mattos, C.; Bellamacina, C.R.; Peisach, E.; Pereira, A.; Vitkup, D.; Petsko, G.A.; Ringe, D. Multiple solvent crystal structures: Probing binding sites, plasticity and hydration. J. Mol. Biol. 2006, 357, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, G.; O’Connor, C.; Zerbe, B.; Kearney, B.M.; Napoleon, R.; Kovrigina, E.A.; Vajda, S.; Kozakov, D.; Kovrigin, E.L.; Mattos, C. Analysis of binding site hot spots on the surface of Ras GTPase. J. Mol. Biol. 2011, 413, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Broomhead, N.K.; Soliman, M.E. Can We Rely on Computational Predictions To Correctly Identify Ligand Binding Sites on Novel Protein Drug Targets? Assessment of Binding Site Prediction Methods and a Protocol for Validation of Predicted Binding Sites. Cell Biochem. Biophys. 2017, 75, 15–23. [Google Scholar] [CrossRef]
- Long, P.Q.; Quan, P.M. Virtual screening stategies in drug discovery–A brief overview. Vietnam. J. Sci. Technol. 2021, 59, 415–440. [Google Scholar]
- Vázquez, J.; López, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging ligand-based and structure-based methods in drug discovery: An overview of combined virtual screening approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Zhu, Z.; Wang, X.S.; Xia, J.; Wu, S. Computational representations of protein–ligand interfaces for structure-based virtual screening. Expert Opin. Drug Discov. 2021, 16, 1175–1192. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Pantsar, T.; Rissanen, S.; Dauch, D.; Laitinen, T.; Vattulainen, I.; Poso, A. Assessment of mutation probabilities of KRAS G12 missense mutants and their long-timescale dynamics by atomistic molecular simulations and Markov state modeling. PLoS Comput. Biol. 2018, 14, e1006458. [Google Scholar] [CrossRef]
- Hashemi, S.; Sharifi, A.; Zareei, S.; Mohamedi, G.; Biglar, M.; Amanlou, M. Discovery of direct inhibitor of KRAS oncogenic protein by natural products: A combination of pharmacophore search, molecular docking, and molecular dynamic studies. Res. Pharm. Sci. 2020, 15, 226–240. [Google Scholar]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef]
- Balajee, R.; Rajan, M.D. Molecular docking and simulation studies of farnesyl trasnferase with the potential inhibitor theflavin. J. Appl. Pharm. Sci. 2011, 1, 141–148. [Google Scholar]
- Chandra, A.; Grecco, H.E.; Pisupati, V.; Perera, D.; Cassidy, L.; Skoulidis, F.; Ismail, S.A.; Hedberg, C.; Hanzal-Bayer, M.; Venkitaraman, A.R.; et al. The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2011, 14, 148–158. [Google Scholar] [CrossRef]
- Leung, E.L.-H.; Luo, L.X.; Li, Y.; Liu, Z.-Q.; Li, L.L.; Shi, D.F.; Xie, Y.; Huang, M.; Lu, L.L.; Duan, F.G.; et al. Identification of a new inhibitor of KRAS-PDEδ interaction targeting KRAS mutant nonsmall cell lung cancer. Int. J. Cancer 2019, 145, 1334–1345. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Casique-Aguirre, D.; Briseño-Díaz, P.; García-Gutiérrez, P.; la Rosa, C.H.G.-D.; Quintero-Barceinas, R.S.; Rojo-Domínguez, A.; Vergara, I.; Medina, L.A.; Correa-Basurto, J.; Bello, M.; et al. KRas4B-PDE6δ complex stabilization by small molecules obtained by virtual screening affects Ras signaling in pancreatic cancer. BMC Cancer 2018, 18, 1299. [Google Scholar] [CrossRef]
- Zou, F.; Yang, Y.; Ma, T.; Xi, J.; Zhou, J.; Zha, X.M. Identification of novel MEK1 inhibitors by pharmacophore and docking based virtual screening. Med. Chem. Res. 2017, 26, 701–713. [Google Scholar] [CrossRef]
- Potluri, V.; Pradhan, D.; Umamaheswari, A. Ligand Based Virtual Screening to Design Novel Human MEK1 Protein Inhibitors for Potential Development of Drugs Against Melanoma. Nat. Preced. 2010. [Google Scholar] [CrossRef]
- Bhagat, R.T.; Butle, S.R.; Khobragade, D.S.; Wankhede, S.B.; Prasad, C.C.; Mahure, D.S.; Armarkar, A.V. Molecular Docking in Drug Discovery. J. Pharm. Res. 2021, 33, 46–58. [Google Scholar] [CrossRef]
- Mezei, M. A new method for mapping macromolecular topography. J. Mol. Graph. Model. 2003, 21, 463–472. [Google Scholar] [CrossRef]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28, 1071–1081.e3. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E., Jr.; Neet, K.E. The catalytic and regulatory properties of enzymes. Annu. Rev. Biochem. 1968, 37, 359–410. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Monticelli, L.; Tieleman, D.P. Force fields for classical molecular dynamics. Methods Mol. Biol. 2013, 924, 197–213. [Google Scholar] [PubMed]
- Hocker, H.J.; Cho, K.J.; Chen, C.Y.; Rambahal, N.; Sagineedu, S.R.; Shaari, K.; Stanslas, J.; Hancock, J.F.; Gorfe, A.A. Andrographolide derivatives inhibit guanine nucleotide exchange and abrogate oncogenic Ras function. Proc. Natl. Acad. Sci. USA 2013, 110, 10201–10206. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Ho, C.L.; Wang, J.L.; Lee, C.C.; Cheng, H.Y.; Wen, W.C.; Cheng, H.H.; Chen, M.C. Antroquinonol blocks Ras and Rho signaling via the inhibition of protein isoprenyltransferase activity in cancer cells. Biomed. Pharmacother. 2014, 68, 1007–1014. [Google Scholar] [CrossRef]
- Luo, C.; Xie, P.; Marmorstein, R. Identification of BRAF inhibitors through in silico screening. J. Med. Chem. 2008, 51, 6121–6127. [Google Scholar] [CrossRef] [Green Version]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef]
- Garrido Torres, J.A.; Jennings, P.C.; Hansen, M.H.; Boes, J.R.; Bligaard, T. Low-Scaling Algorithm for Nudged Elastic Band Calculations Using a Surrogate Machine Learning Model. Phys. Rev. Lett. 2019, 122, 156001. [Google Scholar] [CrossRef] [PubMed]
- Husic, B.E.; Pande, V.S. Markov State Models: From an Art to a Science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef]
- Bucher, D.; Pierce, L.C.; McCammon, J.A.; Markwick, P.R. On the Use of Accelerated Molecular Dynamics to Enhance Configurational Sampling in Ab Initio Simulations. J. Chem. Theory Comput. 2011, 7, 890–897. [Google Scholar] [CrossRef]
- Lu, S.; Ni, D.; Wang, C.; He, X.; Lin, H.; Wang, Z.; Zhang, J. Deactivation Pathway of Ras GTPase Underlies Conformational Substates as Targets for Drug Design. ACS Catal. 2019, 9, 7188–7196. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Zhang, J.; Nussinov, R. Inhibitors of Ras-SOS Interactions. ChemMedChem 2016, 11, 814–821. [Google Scholar] [CrossRef]
- Wittinghofer, A.; Vetter, I.R. Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 2011, 80, 943–971. [Google Scholar] [CrossRef] [PubMed]
- Kano, Y.; Gebregiworgis, T.; Marshall, C.B.; Radulovich, N.; Poon, B.P.K.; St-Germain, J.; Cook, J.D.; Valencia-Sama, I.; Grant, B.M.M.; Herrera, S.G.; et al. Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat. Commun. 2019, 10, 224. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, D.; Lei, C.; Chen, Y.; Qiu, Y.; Li, X.; Li, M.; Ni, D.; Pu, J.; Zhang, J.; et al. Mechanistic insights into the effect of phosphorylation on Ras conformational dynamics and its interactions with cell signaling proteins. Comput. Struct. Biotechnol. J. 2021, 19, 1184–1199. [Google Scholar] [CrossRef]
- Pantsar, T. KRAS(G12C)-AMG 510 interaction dynamics revealed by all-atom molecular dynamics simulations. Sci. Rep. 2020, 10, 11992. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Kulakova, A.M.; Nemukhin, A.V. Proof of concept for poor inhibitor binding and efficient formation of covalent adducts of KRAS(G12C) and ARS compounds. Org. Biomol. Chem. 2020, 18, 3069–3081. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Kohler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef]
- Sarkar-Banerjee, S.; Sayyed-Ahmad, A.; Prakash, P.; Cho, K.J.; Waxham, M.N.; Hancock, J.F.; Gorfe, A.A. Spatiotemporal Analysis of K-Ras Plasma Membrane Interactions Reveals Multiple High Order Homo-oligomeric Complexes. J. Am. Chem. Soc. 2017, 139, 13466–13475. [Google Scholar] [CrossRef]
- Lu, S.; Shen, Q.; Zhang, J. Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms. Acc. Chem. Res. 2019, 52, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhu, L.; Cao, Y.; Wu, G.; Liu, X.; Chen, Y.; Wang, Q.; Shi, T.; Zhao, Y.; Wang, Y.; et al. ASD: A comprehensive database of allosteric proteins and modulators. Nucleic Acids Res. 2011, 39, D663–D669. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Ni, D.; Zhang, J. Allosteric Modulator Discovery: From Serendipity to Structure-Based Design. J. Med. Chem. 2019, 62, 6405–6421. [Google Scholar] [CrossRef]
- Feng, L.; Lu, S.; Zheng, Z.; Chen, Y.; Zhao, Y.; Song, K.; Xue, H.; Jin, L.; Li, Y.; Huang, C.; et al. Identification of an allosteric hotspot for additive activation of PPARγ in antidiabetic effects. Sci. Bull. 2021, 66, 1559–1570. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.; He, X.; Rehman, A.U.; Li, X.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 2020, 12, 464–476. [Google Scholar] [CrossRef]
- Lu, S.; Chen, Y.; Wei, J.; Zhao, M.; Ni, D.; He, X.; Zhang, J. Mechanism of allosteric activation of SIRT6 revealed by the action of rationally designed activators. Acta Pharm. Sin. B 2021, 11, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dai, J.; Ni, D.; He, X.; Zhang, H.; Zhang, J.; Fu, Q.; Liu, Y.; Lu, S. Insight into the mechanism of allosteric activation of PI3Kα by oncoprotein K-Ras4B. Int. J. Biol. Macromol. 2020, 144, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, C.; Peng, T.; Chai, Z.; Ni, D.; Liu, Y.; Zhang, J.; Chen, T.; Lu, S. Atomic-scale insights into allosteric inhibition and evolutional rescue mechanism of Streptococcus thermophilus Cas9 by the anti-CRISPR protein AcrIIA6. Comput. Struct. Biotechnol. J. 2021, 19, 6108–6124. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Jang, H. Does Ras Activate Raf and PI3K Allosterically? Front. Oncol. 2019, 9, 1231. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Yang, Z.; Chai, Z.; Zhou, S.; Wang, J.; Rehman, A.U.; Ni, D.; Pu, J.; Sun, J.; et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat. Commun. 2021, 12, 4721. [Google Scholar] [CrossRef]
- Gorfe, A.A. Mechanisms of allostery and membrane attachment in Ras GTPases: Implications for anti-cancer drug discovery. Curr. Med. Chem. 2010, 17, 1–9. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Y.; Ni, D.; Huang, Z.; Wei, J.; Feng, L.; Su, J.C.; Wei, Y.; Ning, S.; Yang, X.; et al. Targeting a cryptic allosteric site of SIRT6 with small-molecule inhibitors that inhibit the migration of pancreatic cancer cells. Acta Pharm. Sin. B 2022, 12, 876–889. [Google Scholar] [CrossRef]
- Ni, D.; Song, K.; Zhang, J.; Lu, S. Molecular Dynamics Simulations and Dynamic Network Analysis Reveal the Allosteric Unbinding of Monobody to H-Ras Triggered by R135K Mutation. Int. J. Mol. Sci. 2017, 18, 2249. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Fan, J.; Zhuang, H.; Liu, Y.; Ji, D.; Lu, S. Mechanistic Insights into the Long-range Allosteric Regulation of KRAS Via Neurofibromatosis Type 1 (NF1) Scaffold Upon SPRED1 Loading. J. Mol. Biol. 2022, 434, 167730. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Du, K.; Wang, Y.; Fan, J.; Li, M.; Ni, D.; Lu, S.; Bian, X.; Liu, Y. Autopromotion of K-Ras4B Feedback Activation Through an SOS-Mediated Long-Range Allosteric Effect. Front. Mol. Biosci. 2022, 9, 860962. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Jilek, R.J.; Cramer, R.D. Topomers: A validated protocol for their self-consistent generation. J. Chem. Inf. Comput. Sci. 2004, 44, 1221–1227. [Google Scholar] [CrossRef]
- Ul-Haq, Z.; Mahmood, U.; Reza, S. A combined 3D-QSAR and molecular docking strategy to understand the binding mechanism of (V600E)B-RAF inhibitors. Mol. Divers. 2012, 16, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Seidel, T.; Schuetz, D.A.; Garon, A.; Langer, T. The Pharmacophore Concept and Its Applications in Computer-Aided Drug Design. Prog. Chem. Org. Nat. Prod. 2019, 110, 99–141. [Google Scholar]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Wei, D.; Jiang, X.; Zhou, L.; Chen, J.; Chen, Z.; He, C.; Yang, K.; Liu, Y.; Pei, J.; Lai, L. Discovery of multitarget inhibitors by combining molecular docking with common pharmacophore matching. J. Med. Chem. 2008, 51, 7882–7888. [Google Scholar] [CrossRef]
- Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Investigation of marine-derived natural products as Raf kinase inhibitory protein (RKIP)-binding ligands. Mar. Drugs 2021, 19, 581. [Google Scholar] [CrossRef]
- Xie, H.; Chen, L.; Zhang, J.; Xie, X.; Qiu, K.; Fu, J. A combined pharmacophore modeling, 3D QSAR and virtual screening studies on imidazopyridines as B-Raf inhibitors. Int. J. Mol. Sci. 2015, 16, 12307–12323. [Google Scholar] [CrossRef]
- Jin, Y.; Li, L.; Yang, Z.; Liu, M.; Guo, H.; Shen, W. The discovery of a novel compound with potent antitumor activity: Virtual screening, synthesis, biological evaluation and preliminary mechanism study. Oncotarget 2017, 8, 24635–24643. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.L.; Yang, G.H.; Xiang, S.; Gao, D.D.; Zeng, C. In silico analysis of the effect of mutation on epidermal growth factor receptor in non-small-cell lung carcinoma: From mutational analysis to drug designing. J. Biomol. Struct. Dyn. 2017, 35, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Wang, M.H.; Wang, F.; Chen, P.Y.; Ke, X.G.; Yu, B.; Yang, Y.F.; You, P.T.; Wu, H.Z. Network pharmacology and molecular docking reveal the mechanism of Scopoletin against non-small cell lung cancer. Life Sci. 2021, 270, 119105. [Google Scholar] [CrossRef]
- Rehan, M.; Bajouh, O.S. Virtual screening of naphthoquinone analogs for potent inhibitors against the cancer-signaling PI3K/AKT/mTOR pathway. J. Cell. Biochem. 2019, 120, 1328–1339. [Google Scholar] [CrossRef]
- Wang, L.; Chen, L.; Yu, M.; Xu, L.-H.; Cheng, B.; Lin, Y.-S.; Gu, Q.; He, X.-H.; Xu, J. Discovering new mTOR inhibitors for cancer treatment through virtual screening methods and in vitro assays. Sci. Rep. 2016, 6, 18987. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.; Vorobieva, A.A.; Sheffler, W.; Doyle, L.A.; Park, H.; Bick, M.J.; Mao, B.; Foight, G.W.; Lee, M.Y.; Gagnon, L.A.; et al. De novo design of a fluorescence-activating beta-barrel. Nature 2018, 561, 485–491. [Google Scholar] [CrossRef]
- Liu, R.; Wang, J.; Xiong, P.; Chen, Q.; Liu, H. De novo sequence redesign of a functional Ras-binding domain globally inverted the surface charge distribution and led to extreme thermostability. Biotechnol. Bioeng. 2021, 118, 2031–2042. [Google Scholar] [CrossRef]
- Song, L.F.; Merz, K.M., Jr. Evolution of Alchemical Free Energy Methods in Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5308–5318. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Mortier, J.; Friberg, A.; Badock, V.; Moosmayer, D.; Schroeder, J.; Steigemann, P.; Siegel, F.; Gradl, S.; Bauser, M.; Hillig, R.C.; et al. Computationally Empowered Workflow Identifies Novel Covalent Allosteric Binders for KRAS(G12C). ChemMedChem 2020, 15, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Melville, J.L.; Burke, E.K.; Hirst, J.D. Machine learning in virtual screening. Comb. Chem. High Throughput Screen. 2009, 12, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Eckert, H.; Bajorath, J. Molecular similarity analysis in virtual screening: Foundations, limitations and novel approaches. Drug Discov. Today 2007, 12, 225–233. [Google Scholar] [CrossRef]
- Drosten, M.; Dhawahir, A.; Sum, E.Y.; Urosevic, J.; Lechuga, C.G.; Esteban, L.M.; Castellano, E.; Guerra, C.; Santos, E.; Barbacid, M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010, 29, 1091–1104. [Google Scholar] [CrossRef]
- Chen, S.; Li, F.; Xu, D.; Hou, K.; Fang, W.; Li, Y. The Function of RAS Mutation in Cancer and Advances in its Drug Research. Curr. Pharm. Des. 2019, 25, 1105–1114. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Ou, S.-H.I.; Johnson, M.L.; Christensen, J.; Velastegui, K.; Potvin, D.; Faltaos, D.; Chao, R.C. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. J. Clin. Oncol. 2019, 37, 3161. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Constituents | Location | |
|---|---|---|
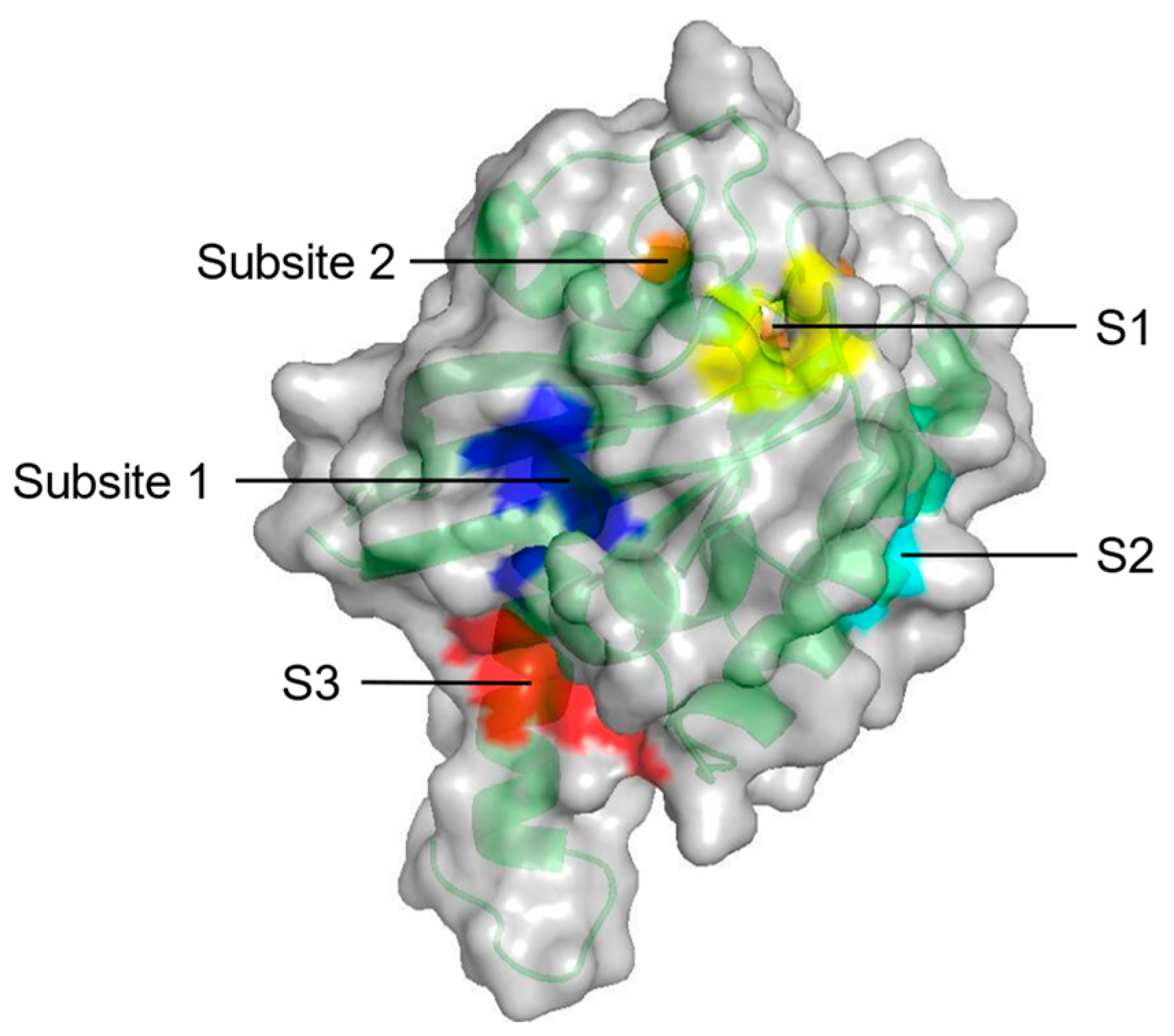
| S1 + Subsite 1 | V7, L56, M67, K5, D54, T74, Y71, E37, D38 | In the core β-strand region behind Switch II |
| S2 | V7, V9, G60, F78, M72, Q99, I100 | Near Switch II and α3 |
| S3 | D105, S106, D107, D108, M111, E162, Q165, H166 | Between L7 and α5 |
| Subsite 2 | D30, D33, D38, S39, Y40, I21, I36 | At the back of Switch I |
| Site | Constituents | Location |
|---|---|---|
| P1 | K5, L6, V7, S39, D54, I55, L56, M67, Q70, Y71, M72, R73, T74, G75 | Between β1–3 and α2 |
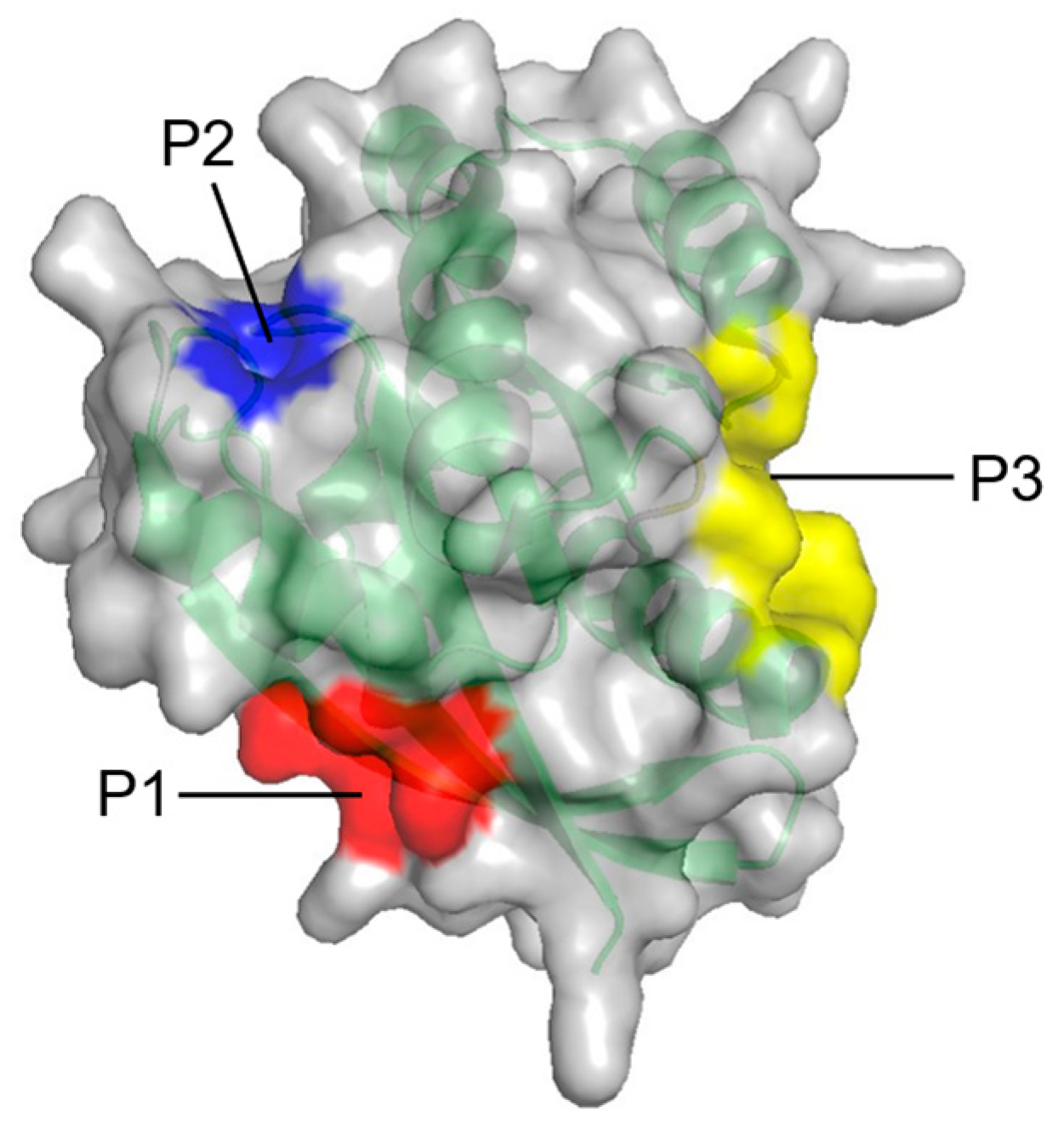
| P2 | Q61, E62, E63, Y64, S65, F90, E91, D92, I93, H94, H95, Y96, R97, E98, Q99 | Between L2, α2, and α3 |
| P3 | R97, K101, E107, D108, V109, P110, M111, S136, Y137, G138, I139, P140, R161, E162, I163, R164, K165, H166 | Between L7, L9, and α5 |
| Site | Consist | Location |
|---|---|---|
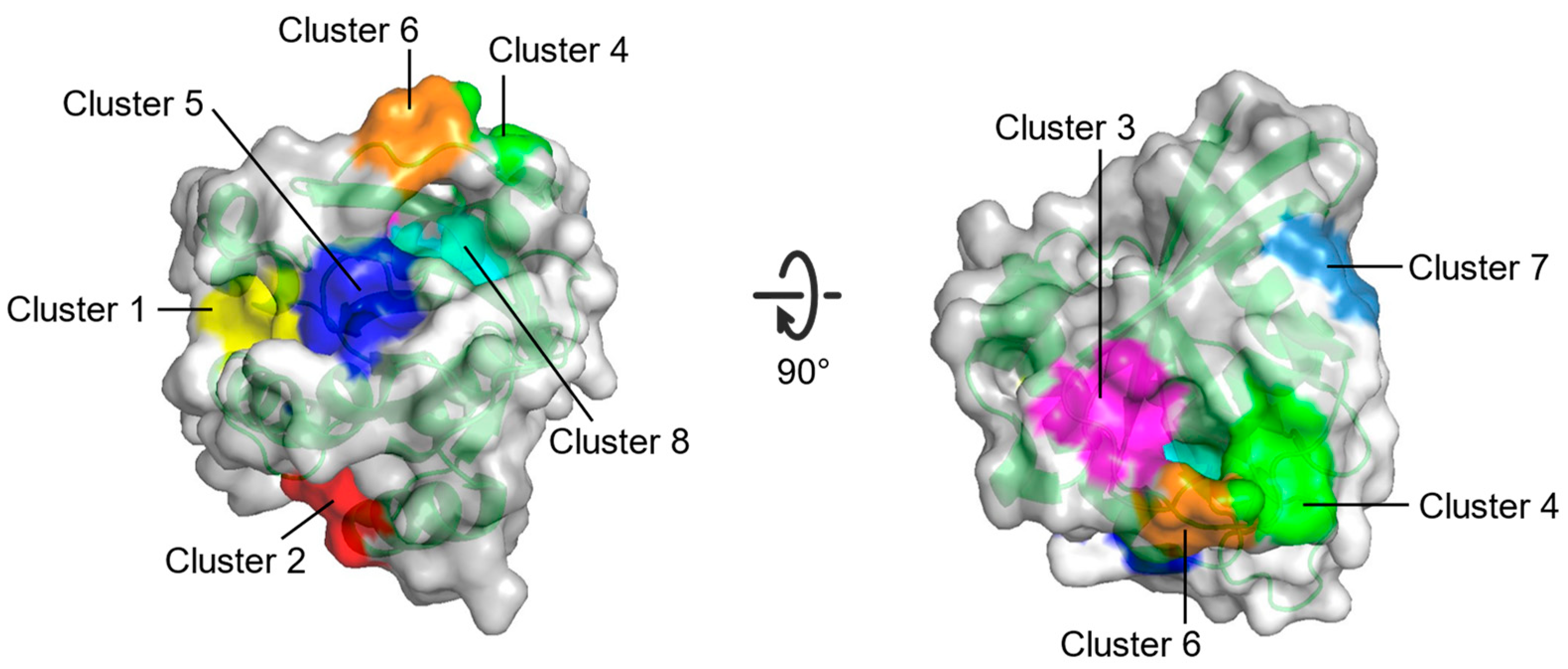
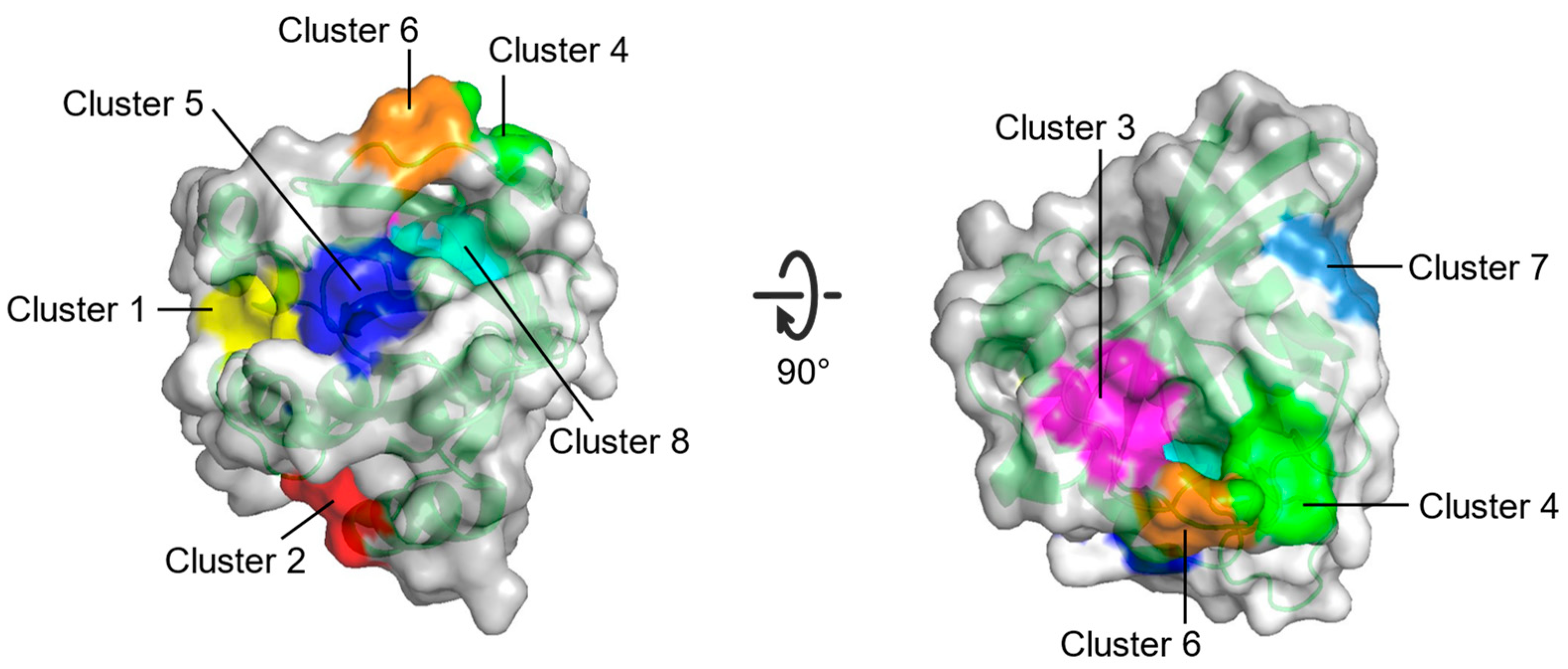
| Cluster 1 | R68, Q95, Y96, Q99, D92, E62, R68, D92, Q95, Y96, Q99, R102 | Between switch II and α3 |
| Cluster 2 | H94, L133, S136, Y137, F90, E91, I93, H94, L133, Y137 | Between α3 and α4 |
| Cluster 3 | S17, I21, Q25, H27, V29, D33, T35, D38, Y40 | Opposite to Switch I relative to gppnhp |
| Cluster 4 | F28, D30, K147 | Near L8 |
| Cluster 5 | A11, G12, N86, K88, S89, D92 | Between P-loop and N-terminus of α3 |
| Cluster 6 | D30, E31, Tyr32, GppNHp | Near N-terminus of switch I |
| Cluster 7 | L23, N26, K42, V44, V45, R149, E153, Y157 | Near C-terminus of α1 |
| Cluster 8 | G13, Y32, N86, K117, GppNHp | Between P-loop and switch I |
| CADD Methods | Results | References |
|---|---|---|
| Homology modeling | The 3D structure of RASSF2 | [51] |
| Molecular dynamics simulation | The stability of the prediction model | [53] |
| Template-based protein–protein complex structure prediction algorithm (PRISM) | The structure of KRAS4B-GTP homodimer | [55] |
| AlphaFold | Models of 145 RAS superfamily members | [57] |
| Web server (Sitehound-Web) | Top 10 binding pockets on RASSF2 | [51] |
| Probe-based molecular dynamics (PMD) simulation | Five potential druggable sites (S1–S3, Subsite 1 and Subsite 2) on KRAS | [53] |
| Fragment-based approach (FTMAP) | Three potential allosteric sites (P1–P3) on RAS | [67] |
| Multiple solvent crystal structures (MSCS) | Eight potential binding sites (Cluster 1–Cluster 8) on HRAS | [69] |
| Targeting Strategy | Drug | Targeting Information | CADD Methods | Reference | ||
|---|---|---|---|---|---|---|
| Virtual Screening | Ligand-Based | Receptor-Based | ||||
| Direct targeting KRAS | Andrographolide (AGP) and its benzylidene derivatives | Binding to a transient pocket on KRAS, blocking GDP–GTP exchange | Molecular docking; Molecular dynamics | [91] | ||
| Auriculasin | Blocking iKRASG12D–SOS1 interaction, inhibiting the guanylate cycle | Similarity searching; Pharmacophore modelling (via ligand–receptor complex fingerprint) | Molecular docking; Molecular dynamics | [76] | ||
| ARS-853, ARS-1620 | Targeting the SII-P of RAS proteins in the GDP-bound state formation, interfering with the region of Switch 1 and Switch 2, blocking SOS-mediated GTP binding and effector proteins involvement, | √ | Molecular docking | [44] | ||
| Compound D14 and C22 | stabilizing the KRAS4B–PDE6δ molecular complex, and blocking the release of abnormal KRAS with mutations | √ | Molecular docking; Molecular dynamics | [82] | ||
| Indirect targeting KRAS | IMB-1406 | Inducing apoptosis in HepG2 cells by arresting the cell cycle at the S phase and altering anti- and pro-apoptotic proteins leading to mitochondrial dysfunction and activation of caspase-3, one of the possible targets being protein farnesyltransferase | Molecular docking | [132] | ||
| NHTD | disrupting KRAS–PDEδ interaction, redistributing the localization of KRAS to endomembranes by targeting the prenyl-binding pocket of PDEδ | √ | [80] | |||
| Antroquinonol | Inhibiting prenyltransferase activity, blocking RAS and RAS-related GTP-binding protein activation | √ | Molecular docking | [93] | ||
| Theaflavin | Targeting farnesyltransferase, inhibiting PTM process | Molecular docking; Molecular dynamics | [78] | |||
| Upstream signaling pathway | Daidzein | Interacting with the kinase domain of the EGFR protein | √ | Molecular docking; Molecular dynamics | [133] | |
| Scopoletin | Iargeting EGFR, BRAF, and AKT1 in NSCLC | Molecular docking | [134] | |||
| Downstream signaling pathway | Purine-2,6-dione analogues | Inhibiting BRAF protein kinase (a molecule in the RAS–RAF–MEK–ERK signaling pathway) | Molecular docking | [94] | ||
| phosphoaminophosphonic acid adenylate ester (ANP), phosphoaminophosphonic acid guanylate ester (GNP) | Stabilizing RASSF2 (a KRAS-specific effector protein, promoting apoptosis and cell cycle arrest) | √ | Molecular docking | [51] | ||
| newly designed 2,6-disubstituted pyrazine derivatives | Inhibiting V600E BRAF | QSAR | Molecular docking (for the consideration of the similarity and alignment) | [125] | ||
| Dehydrocoelenterazine | Interacting with the RAF kinase inhibitor protein (RKIP) ligand-binding pocket, thus inhibiting RKIP | √ | Pharmacophore Modelling | Molecular docking; Molecular dynamics | [130] | |
| NCI 94680NCI 527880NCI 183519 | BRAF inhibitor | √ | QSAR; Pharmacophore modeling (used in the structural alignment step of QSAR modelling) | Molecular docking | [131] | |
| Pictilisib | PI3K-α inhibitor | √ | Pharmacophore Modelling | Molecular docking | [59] | |
| Staurosporine | PKC-η inhibitor | √ | Pharmacophore Modelling | Molecular docking | [59] | |
| Compound M4 | MEK1 inhibitor | √ | Pharmacophore Modelling | Molecular docking | [83] | |
| Catechin | MEK1 inhibitor | √ | Similarity searching | Molecular docking (using the drug library obtained from similarity searching); Molecular dynamics | [84] | |
| CID-20759629 | PI3Kγ/AKT/mTOR pan-inhibitor | √ | Similarity searching | Molecular docking; Molecular dynamics | [135] | |
| Compound 17 | mTOR inhibitor | √ | Similarity searching | Molecular docking; Molecular dynamics | [136] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Bai, Y.; Cui, J.; Zong, Z.; Gao, Y.; Zheng, Z. Computer-Aided Drug Design Boosts RAS Inhibitor Discovery. Molecules 2022, 27, 5710. https://doi.org/10.3390/molecules27175710
Wang G, Bai Y, Cui J, Zong Z, Gao Y, Zheng Z. Computer-Aided Drug Design Boosts RAS Inhibitor Discovery. Molecules. 2022; 27(17):5710. https://doi.org/10.3390/molecules27175710
Chicago/Turabian StyleWang, Ge, Yuhao Bai, Jiarui Cui, Zirui Zong, Yuan Gao, and Zhen Zheng. 2022. "Computer-Aided Drug Design Boosts RAS Inhibitor Discovery" Molecules 27, no. 17: 5710. https://doi.org/10.3390/molecules27175710
APA StyleWang, G., Bai, Y., Cui, J., Zong, Z., Gao, Y., & Zheng, Z. (2022). Computer-Aided Drug Design Boosts RAS Inhibitor Discovery. Molecules, 27(17), 5710. https://doi.org/10.3390/molecules27175710