

Regioselective Synthesis, Structural Characterization, and Antiproliferative Activity of Novel Tetra-Substituted Phenylaminopyrazole Derivatives
 , ,
, ,  , and
, and
Abstract
:1. Introduction
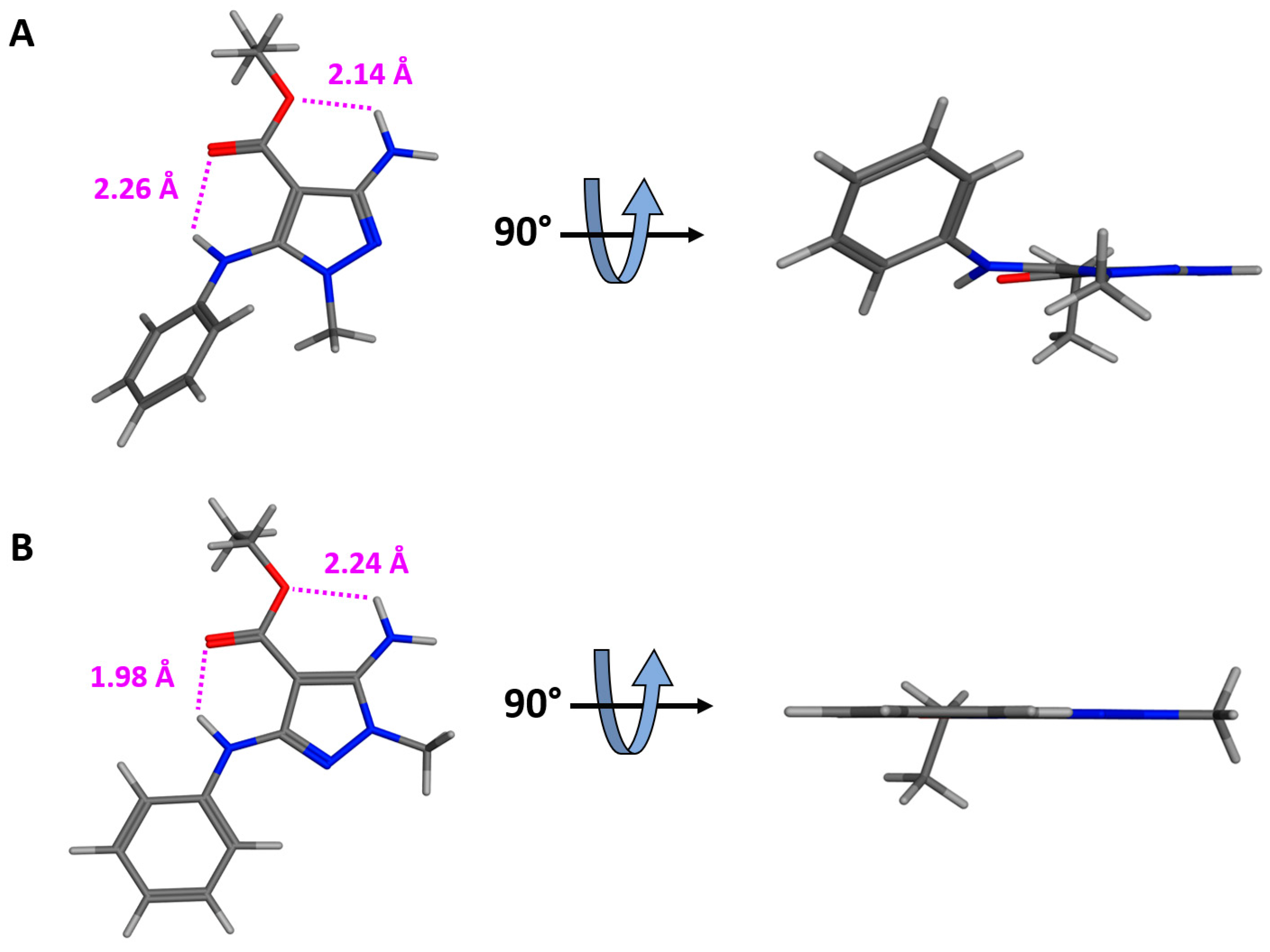
2. Results and Discussion
2.1. Chemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
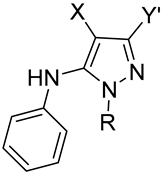 | ||||||
|---|---|---|---|---|---|---|
| AMR | X | Y | Cpd | Y′ | R | Yield (%) |
| I | CN | CN | 1 [43] | NH2 | Me | 56 |
| II | CN | COC(Me)3 | 2 | t-Bu | Me | 61 |
| III | CN | COPh | 3 | Ph | Me | 60 |
| III | CN | COPh | 4 | Ph | CH2Ph | 27 |
| IV | COPh | COPh | 5 | Ph | Me | 50 |
| V | COOMe | CN | 6 | NH2 | Me | 12 |
| VI | SO2Ph | CN | 7 | NH2 | Me | 61 |
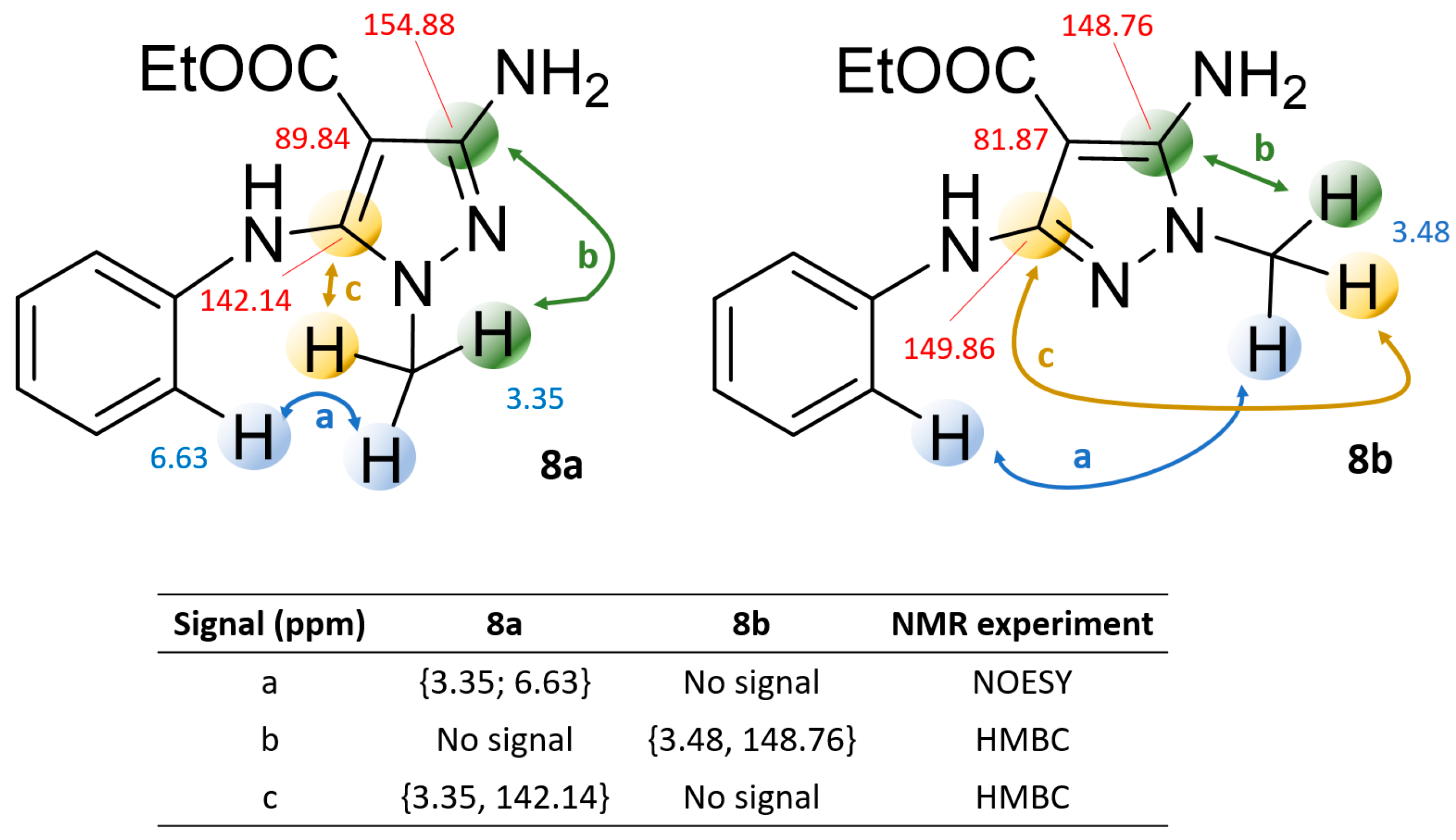
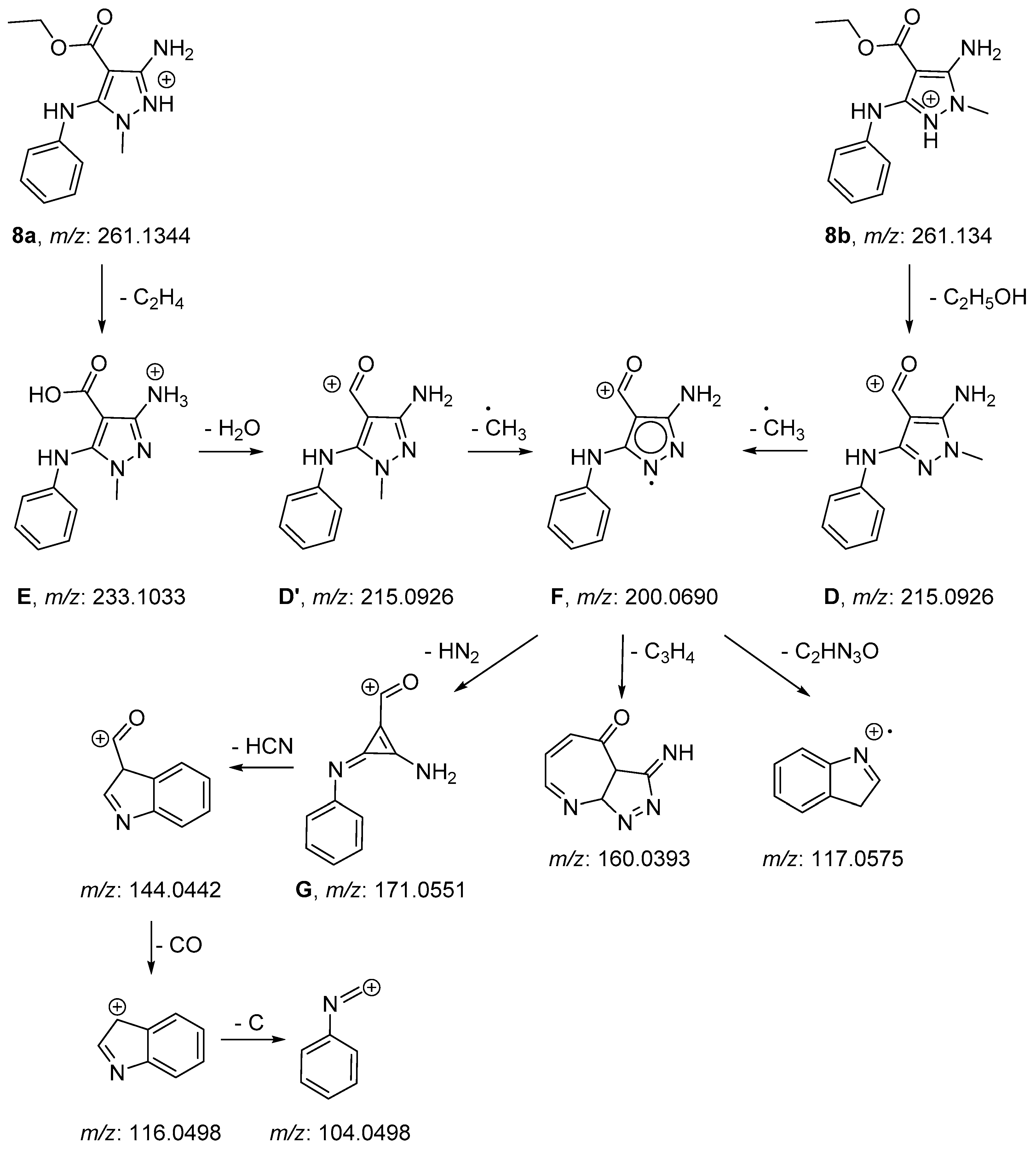
2.2. NMR and Mass Spectrometry Analyses of Derivatives 8a and 8b
2.3. Antiproliferative Activity
3. Materials and Methods
3.1. Chemistry
3.2. General Synthetic Procedure for the Preparation of Pyrazoles 1–7
3.3. Synthesis of Ethyl 2-Cyano-3-(methylthio)-3-(phenylamino)acrylate (BVI)
3.4. Synthesis of Compounds 8a and 8b
3.5. Synthesis of Compound 8b via Pyrazole Methylation
3.6. Mass Spectrometry Analysis
3.6.1. LC-HRMS
3.6.2. FIA MS/MS
3.6.3. Biology
3.7. Computational Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kuo, T.-H.; Lin, T.-H.; Yang, R.-S.; Kuo, S.-C.; Fu, W.-M.; Hung, H.-Y. Novel pyrazole derivatives effectively inhibit osteoclastogenesis, a potential target for treating osteoporosis. J. Med. Chem. 2015, 58, 4954–4963. [Google Scholar] [CrossRef] [PubMed]
- Tanitame, A.; Oyamada, Y.; Ofuji, K.; Fujimoto, M.; Iwai, N.; Hiyama, Y.; Suzuki, K.; Ito, H.; Terauchi, H.; Kawasaki, M.; et al. Synthesis and antibacterial activity of a novel series of potent DNA gyrase inhibitors. pyrazole derivatives. J. Med. Chem. 2004, 47, 3693–3696. [Google Scholar] [CrossRef] [PubMed]
- Khunt, R.C.; Khedkar, V.M.; Chawda, R.S.; Chauhan, N.A.; Parikh, A.R.; Coutinho, E.C. Synthesis, antitubercular evaluation and 3D-QSAR study of N-phenyl-3-(4-fluorophenyl)-4-substituted pyrazole derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Takate, S.J.; Shinde, A.D.; Karale, B.K.; Akolkar, H.; Nawale, L.; Sarkar, D.; Mhaske, P.C. Thiazolyl-pyrazole derivatives as potential antimycobacterial agents. Bioorg. Med. Chem. Lett. 2019, 29, 1199–1202. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kaur, K.; Gupta, G.K.; Sharma, A.K. Pyrazole containing natural products: Synthetic preview and biological significance. Eur. J. Med. Chem. 2013, 69, 735–753. [Google Scholar] [CrossRef]
- Ojwach, S.O.; Darkwa, J. Pyrazole and (pyrazol-1-yl)metal complexes as carbon–carbon coupling catalysts. Inorg. Chim. Acta 2010, 363, 1947–1964. [Google Scholar] [CrossRef]
- Singer, R.A.; Caron, S.; McDermott, R.E.; Arpin, P.; Do, N.M. Alternative biarylphosphines for use in the palladium-catalyzed amination of aryl halides. Synthesis 2003, 11, 1727–1731. [Google Scholar] [CrossRef]
- Singer, R.A.; Dore, M.; Sieser, J.E.; Berliner, M.A. Development of nonproprietary phosphine ligands for the Pd-catalyzed amination reaction. Tetrahedron Lett. 2006, 47, 3727–3731. [Google Scholar] [CrossRef]
- Kowalczyk, R.; Skarzewski, J. Regioselective synthesis of optically active (pyrazolyl)pyridines with adjacent quaternary carbon stereocenter: Chiral N,N-donating ligands. Tetrahedron 2005, 61, 623–628. [Google Scholar] [CrossRef]
- Ebenezer, O.; Shapi, M.; Tuszynski, J.A. A Review of the recent development in the synthesis and biological evaluations of pyrazole derivatives. Biomedicines 2022, 10, 1124. [Google Scholar] [CrossRef]
- Sui, Z.; Guan, J.; Ferro, M.P.; McCoy, K.; Wachter, M.P.; Murray, W.V.; Singer, M.; Steber, M.; Ritchie, D.M.; Argentieri, D.C. 1,3-Diarylcycloalkanopyrazoles and diphenyl hydrazides as selective inhibitors of cyclooxygenase-2. Bioorg. Med. Chem. Lett. 2000, 10, 601–604. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Abdel-Aziem, T. Design, synthesis and biological evaluation of some pyrazole derivatives as anti-inflammatory-antimicrobial agents. Bioorg. Med. Chem. 2004, 12, 1935–1945. [Google Scholar] [CrossRef]
- Abd El-Karim, S.S.; Mohamed, H.S.; Abdelhameed, M.F.; Amr, A.E.; Almehizia, A.A.; Nossier, E.S. Design, synthesis and molecular docking of new pyrazole-thiazolidinones as potent anti-inflammatory and analgesic agents with TNF-alpha inhibitory activity. Bioorg. Chem. 2021, 111, 104827. [Google Scholar] [CrossRef]
- Shi, J.B.; Chen, L.Z.; Wang, B.S.; Huang, X.; Jiao, M.M.; Liu, M.M.; Tang, W.J.; Liu, X.H. Novel Pyrazolo [4,3-d] pyrimidine as Potent and Orally Active Inducible Nitric Oxide Synthase (iNOS) Dimerization Inhibitor with Efficacy in Rheumatoid Arthritis Mouse Model. J. Med. Chem. 2019, 62, 4013–4031. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; Abdelall, E.K.A.; Elshemy, H.A.H.; El-Nahass, E.; Abdel-Fattah, M.M.; Abdelgawad, Y.Y.M. New indomethacin analogs as selective COX-2 inhibitors: Synthesis, COX-1/2 inhibitory activity, anti-inflammatory, ulcerogenicity, histopathological, and docking studies. Arch. Pharm. 2021, 354, 2000328. [Google Scholar] [CrossRef]
- Harras, M.F.; Sabour, R.; Alkamali, O.M. Discovery of new non-acidic lonazolac analogues with COX-2 selectivity as potent anti-inflammatory agents. MedChemComm 2019, 10, 1775–1788. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; El-Saadi, M.T.; Elzayat, S.G.; Amin, N.H. New substituted pyrazole derivatives targeting COXs as potential safe anti-inflammatory agents. Future Med. Chem. 2019, 11, 1871–1887. [Google Scholar] [CrossRef]
- Taher, A.T.; Sarg, M.T.M.; Ali, N.R.E.; Elnagdi, N.H. Design, synthesis, modeling studies and biological screening of novel pyrazole derivatives as potential analgesic and anti-inflammatory agents. Bioorg. Chem. 2019, 89, 103023. [Google Scholar] [CrossRef]
- Zabiulla; Gulnaz, A.R.; Mohammed, Y.H.E.; Khanum, S.A. Design, synthesis and molecular docking of benzophenone conjugated with oxadiazole sulphur bridge pyrazole pharmacophores as anti inflammatory and analgesic agents. Bioorg. Chem. 2019, 92, 103220. [Google Scholar] [CrossRef]
- Katoch-Rouse, R.; Pavlova, O.A.; Caulder, T.; Hoffman, A.F.; Mukhin, A.G.; Horti, A.G. Synthesis, structure–activity relationship, and evaluation of SR141716 analogues: Development of central cannabinoid receptor ligands with lower lipophilicity. J. Med. Chem. 2003, 46, 642–645. [Google Scholar] [CrossRef]
- Tanitame, A.; Oyamada, Y.; Ofuji, K.; Fujimoto, M.; Suzuki, K.; Ueda, T.; Terauchi, H.; Kawasaki, M.; Nagai, K.; Wachi, M.; et al. Synthesis and antibacterial activity of novel and potent DNA gyrase inhibitors with azole ring. Bioorg. Med. Chem. 2004, 12, 5515–5524. [Google Scholar] [CrossRef]
- Liu, H.; Chu, Z.-W.; Xia, D.-G.; Cao, H.-Q.; Lv, X.-H. Discovery of novel multi-substituted benzo-indole pyrazole schiff base derivatives with antibacterial activity targeting DNA gyrase. Bioorg. Chem. 2020, 99, 103807. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Fayed, E.A.; Rizk, H.F.; Desouky, S.E.; Ragab, A. Hydrazonoyl bromide precursors as DHFR inhibitors for the synthesis of bis-thiazolyl pyrazole derivatives; antimicrobial activities, antibiofilm, and drug combination studies against MRSA. Bioorg. Chem. 2021, 116, 105339. [Google Scholar] [CrossRef]
- Stauffer, S.R.; Coletta, C.J.; Tedesco, R.; Nishiguchi, G.; Carlson, K.; Sun, J.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Pyrazole ligands: Structure–affinity/activity relationships and estrogen receptor-α-selective agonists. J. Med. Chem. 2000, 43, 4934–4947. [Google Scholar] [CrossRef]
- Stauffer, S.R.; Huang, Y.; Coletta, C.J.; Tedesco, R.; Katzenellenbogen, J.A. Estrogen pyrazoles: Defining the pyrazole core structure and the orientation of substituents in the ligand binding pocket of the estrogen receptor. Bioorg. Med. Chem. 2001, 9, 141–150. [Google Scholar] [CrossRef]
- Dawood, D.H.; Nossier, E.S.; Ali, M.M.; Mahmoud, A.E. Synthesis and molecular docking study of new pyrazole derivatives as potent anti-breast cancer agents targeting VEGFR-2 kinase. Bioorg. Chem. 2020, 101, 103916. [Google Scholar] [CrossRef]
- Cheng, C.; Yun, F.; Ullah, S.; Yuan, Q. Discovery of novel cyclin-dependent kinase (CDK) and histone deacetylase (HDAC) dual inhibitors with potent in vitro and in vivo anticancer activity. Eur. J. Med. Chem. 2020, 189, 112073. [Google Scholar] [CrossRef] [PubMed]
- Morretta, E.; Sidibè, A.; Spallarossa, A.; Petrella, A.; Meta, E.; Bruno, O.; Monti, M.C.; Brullo, C. Synthesis, functional proteomics and biological evaluation of new 5-pyrazolyl ureas as potential anti-angiogenic compounds. Eur. J. Med. Chem. 2021, 226, 113872. [Google Scholar] [CrossRef]
- Long, J.K.; Gregory, V.; Gutteridge, S.; Taggi, A.E.; Bereznak, J.F. Fungicidal Pyrazoles and their Mixtures for Protection of Crops from Phytopathological Fungi. US Patent MY157327A, 31 May 2016. [Google Scholar]
- Taggi, A.E.; Bereznak, J.F.; Long, J.K. Preparation of Pyrazoles as Fungicides. WO Patent WO2014130241, 28 August 2014. [Google Scholar]
- Long, J.K.; Chittaboina, S.; McMahon, T.C. Preparation of Pyrazoles as Fungicides and Their Use in Fungicidal Mixtures. WO Patent WO2021183721, 16 September 2021. [Google Scholar]
- Cantin, L.D.; Ma, X.; Akuche, C.; Liang, S.X. Preparation of Anilino-Heteroaryl-Pyrazoles Useful for the Treatment of Diabetes. WO Patent WO2005112923, 1 December 2005. [Google Scholar]
- Rudolph, J.; Cantin, L.D.; Magnuson, S.; Bullock, W.; Bullion, A.M.; Chen, L.; Chuang, C.Y.; Liang, S.; Majumdar, D.; Ogutu, H.; et al. Preparation of Anilinopyrazoles for the Treatment of Diabetes. WO Patent WO2004050651A1, 17 June 2004. [Google Scholar]
- Lowe, D.; Shelekhin, T.; Wang, G.; Ma, X.; Iwuagwu, C.; Ying, S.; Magnuson, S.; Rudolph, J.; Koebberling, J.; Pernerstorfer, J.; et al. Preparation of Anilinopyrazoles for the Treatment of Diabetes. WO Patent WO2007027842-A1, 8 March 2007. [Google Scholar]
- Guisot, N. Preparation of Pyrazole Derivatives as Kinase Inhibitors. WO Patent WO2017103611A1, 22 June 2017. [Google Scholar]
- Garnier, J.M.D.; Brzozowski, M.; Feutrill, J.T.; Lessene, G.L.; Gardner, C.; Czabotar, P.E.; Cowan, A.; Sharma, P.; Schuster-Klein, C.A.; Poitevin, C. Preparation of Amido Compounds for Treating Necroptosis and/or Inhibiting RIP1 and/or MLKL. WO Patent WO2021253098 A1, 23 December 2021. [Google Scholar]
- Hassan, A.S.; Moustafa, G.O.; Awad, H.M.; Nossier, E.S.; Mady, M.F. Design, synthesis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole–indole hybrids. ACS Omega 2021, 6, 12361–12374. [Google Scholar] [CrossRef] [PubMed]
- Klueken, M.; Geist, J.; Montagne, C.; Millet, A.; Nicolas, L.; Tsuchiya, T. Active Compound Combinations. WO Patent WO2021255070 A1, 23 December 2021. [Google Scholar]
- Venkatesha, H.M.; Saragur, R.S.; Garg, R.; Pabba, J.; Autkar, S.S. A Pesticidally Active Mixture Comprising Pyrazolopyridine Anthranilamide Compound, Oxides or Salts thereof, with Inseciticides or Fungicides. WO Patent WO2022023931 A1, 3 February 2022. [Google Scholar]
- Lim, F.P.L.; Gan, R.X.Y.; Dolzhenko, A.V. Highly selective and efficient synthesis of 3-arylamino-substituted 5-aminopyrazole-4-carboxylates under microwave irradiation. Tetrahedron Lett. 2017, 58, 775–778. [Google Scholar] [CrossRef]
- Elgemeiea, G.; Fathyb, N.; Zagharyc, W.; Farag, A. S-Glycosides in medicinal chemistry: Novel synthesis of cyanoethylene thioglycosides and their pyrazole derivatives. Nucleosides Nucleotides Nucleic Acids 2017, 36, 198–212. [Google Scholar] [CrossRef]
- Lusardi, M.; Rotolo, C.; Ponassi, M.; Iervasi, E.; Rosano, C.; Spallarossa, A. One-pot synthesis and antiproliferative activity of highly functionalized pyrazole derivatives. ChemMedChem 2022, 17, e202100670. [Google Scholar] [CrossRef]
- Hassan, A.; Mohamed, N.; Ibrahim, Y.; Mourad, A.-F.; Aboul-Fetouh, S. Charge-transfer complexes of heterocyclics of biological interest—II. Aminopyrazoles. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1991, 47, 1635–1636. [Google Scholar] [CrossRef]
- Fischer, G.; Rudorf, W.-D.; Kleinpeter, E. Study of the distribution of the π-electrons in push-pull alkenes by 1H and 13C NMR spectroscopy. Magn. Reson. Chem. 1991, 29, 212–222. [Google Scholar] [CrossRef]
- Fischer, G.; Kleinpeter, E. Application of 2D EXSY NMR spectroscopy to the study of the dynamic behaviour of aroylcyanoketene-S,S-dimethylacetals. Magn. Reson. Chem. 1991, 29, 204–206. [Google Scholar] [CrossRef]
- Shizheng, Z.; Chaoyue, Q.; Guoling, X.; Qianli, C. convenient synthesis of a new push-pull alkenes: β-alkoxyl vinyl trifluoromethyl sulfones. Tetrahedron Lett. 1998, 39, 5265–5268. [Google Scholar] [CrossRef]
- Kleinpeter, E.; Klod, S.; Rudorf, W.-D. Electronic state of push−pull alkenes: An experimental dynamic NMR and theoretical ab initio MO study. J. Org. Chem. 2004, 69, 4317–4329. [Google Scholar] [CrossRef]
- Pappalardo, R.R.; Marcos, E.S.; Ruiz-Lopez, M.F.; Rinaldi, D.; Rivail, J.L. Solvent effects on molecular geometries and isomerization processes: A study of push-pull ethylenes in solution. J. Am. Chem. Soc. 1993, 115, 3722–3730. [Google Scholar] [CrossRef]
- Kleinpeter, E.; Schulenburg, A. Quantification of the push–pull effect in substituted alkenes. Tetrahedron Lett. 2005, 46, 5995–5997. [Google Scholar] [CrossRef]
- Savych, V.I.; Mykhalchuk, V.L.; Melnychuk, P.V.; Isakov, A.O.; Savchuk, T.; Timoshenko, V.M.; Siry, S.A.; Pavlenko, S.O.; Kovalenko, D.V.; Hryshchuk, O.V.; et al. Bicyclic pyrrolidines for medicinal chemistry via [3+2]-cycloaddition. J. Org. Chem. 2021, 86, 13289–13309. [Google Scholar] [CrossRef]
- Sofan, M.A.; El-Mekabaty, A.; Hasel, A.M.; Said, S.B. Synthesis, cytotoxicity assessment and antioxidant activity of some new thiazol-2-yl carboxamides. J. Heterocycl. Chem. 2021, 58, 1645–1655. [Google Scholar] [CrossRef]
- Frizzo, C.P.; Hennemann, B.L.; Kuhn, B.L.; Wust, K.M.; Paz, A.V.; Martins, M.A.P.; Zanatta, N.; Bonacorso, H.G. Trends for pyrazole fragmentation determined by gas chromatography coupled with mass spectrometry. In Gas Chromatography—Derivatization, Sample Preparation, Application; Kusch, P., Ed.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Elgemeie, G.H.; Elghandour, A.H.; Elaziz, G.W.A. Novel cyanoketene N,S-acetals and pyrazole derivatives using potassium 2-cyanoethylene-1-thiolates. Synth. Commun. 2007, 37, 2827–2834. [Google Scholar] [CrossRef]


| Cpd | Mean Growth Percentage ± SD | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| MCF7 | MDA-MB231 | Sk-Br3 | SKMEL28 | SKOV3 | Hep-G2 | HeLa | A549 | GM-6114 | |
| 1 | 108.43 ± 7.38 | 113.77 ± 4.39 | 92.72 ± 2.94 | 91.62 ± 3.38 | 107.78 ± 3.35 | 105.55 ± 7.40 | 97.39 ± 10.47 | 111.66 ± 3.77 | 89.08 ± 11.38 |
| 2 | 107.37 ± 10.99 | 112.16 ± 6.85 | 85.66 ± 3.28 | 92.87 ± 4.06 | 107.12 ± 5.64 | 94.15 ± 3.81 | 96.42 ± 7.05 | 113.10 ± 5.64 | 90.65 ± 3.32 |
| 3 | 118.46 ± 6.19 | 115.98 ± 2.45 | 83.05 ± 2.41 | 92.97 ± 3.68 | 115.25 ± 3.37 | 112.20 ± 7.47 | 110.40 ± 2.49 | 115.25 ± 5.96 | 109.73 ± 8.12 |
| 4 | 92.21 ± 5.41 | 89.69 ± 2.90 | 84.72 ± 4.38 | 68.36 ± 3.13 | 80.45 ± 3.38 | 105.51 ± 8.80 | 73.77 ± 6.86 | 71.00 ± 3.40 | 83.00 ± 6.51 |
| 5 | 111.88 ± 5.19 | 111.89 ± 2.95 | 101.95 ± 3.18 | 119.57 ± 1.57 | 105.35 ± 6.08 | 110.03 ± 2.11 | 115.87 ± 9.17 | 112.22 ± 4.10 | 102.18 ± 8.31 |
| 6 | 105.46 ± 5.68 | 109.32 ± 6.43 | 81.74 ± 4.92 | 105.04 ± 2.76 | 109.12 ± 5.64 | 102.04 ± 4.42 | 92.32 ± 2.62 | 121.90 ± 5.92 | 84.57 ± 4.03 |
| 7 | 104.20 ± 3.10 | 110.14 ± 4.88 | 89.26 ± 3.54 | 109.99 ± 3.30 | 106.54 ± 4.47 | 107.16 ± 12.07 | 85.42 ± 6.98 | 115.77 ± 3.02 | 87.45 ± 7.73 |
| 8a | 98.06 ± 3.31 | 87.02 ± 5.76 | 94.76 ± 4.00 | 82.21 ± 4.00 | 106.46 ± 6.18 | 101.20 ± 9.16 | 103.26 ± 9.57 | 99.70 ± 9.20 | 107.65 ± 3.92 |
| 8b | 100.13 ± 6.20 | 89.99 ± 5.16 | 100.28 ± 4.10 | 81.21 ± 4.31 | 102.97 ± 4.37 | 98.37 ± 8.17 | 110.61 ± 7.72 | 86.65 ± 9.83 | 118.99 ± 9.70 |
| cisplatin | 72.74 ± 5.48 | 86.07 ± 7.04 | 70.59 ± 3.83 | 44.40 ± 2.53 | 36.83 ± 4.35 | 38.07 ± 2.22 | 29.33 ± 2.23 | 59.09 ± 6.03 | 39.52 ± 2.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lusardi, M.; Profumo, A.; Rotolo, C.; Iervasi, E.; Rosano, C.; Spallarossa, A.; Ponassi, M. Regioselective Synthesis, Structural Characterization, and Antiproliferative Activity of Novel Tetra-Substituted Phenylaminopyrazole Derivatives. Molecules 2022, 27, 5814. https://doi.org/10.3390/molecules27185814
Lusardi M, Profumo A, Rotolo C, Iervasi E, Rosano C, Spallarossa A, Ponassi M. Regioselective Synthesis, Structural Characterization, and Antiproliferative Activity of Novel Tetra-Substituted Phenylaminopyrazole Derivatives. Molecules. 2022; 27(18):5814. https://doi.org/10.3390/molecules27185814
Chicago/Turabian StyleLusardi, Matteo, Aldo Profumo, Chiara Rotolo, Erika Iervasi, Camillo Rosano, Andrea Spallarossa, and Marco Ponassi. 2022. "Regioselective Synthesis, Structural Characterization, and Antiproliferative Activity of Novel Tetra-Substituted Phenylaminopyrazole Derivatives" Molecules 27, no. 18: 5814. https://doi.org/10.3390/molecules27185814