
Synthesis, Structural Determination, and Antioxidant Activities of Acyclic and Substituted Heterocyclic Phosphonates Linearly Linked 4-hydroxy-2(1H)-quinolinone
Abstract
:1. Introduction
2. Results and Discussion
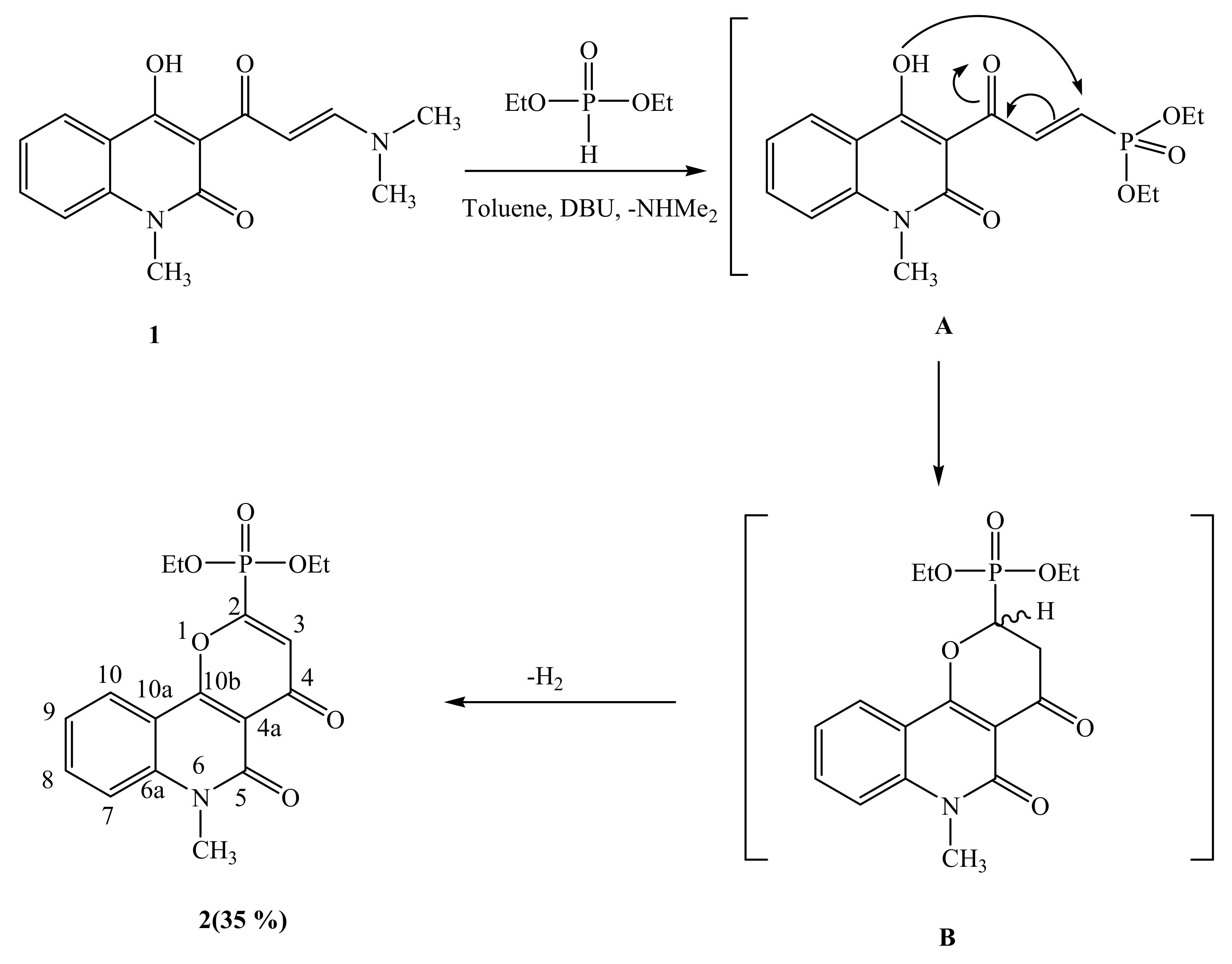
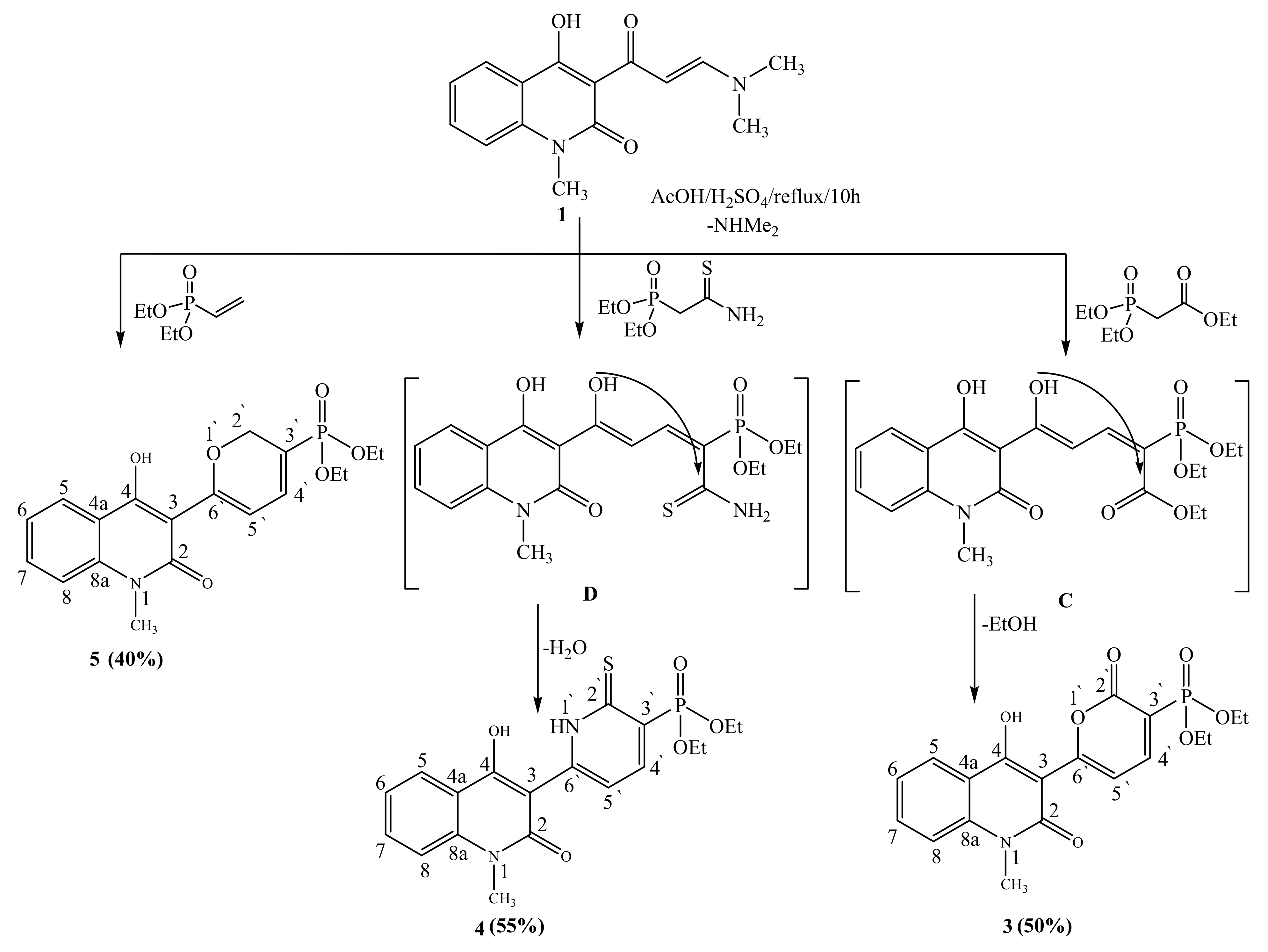
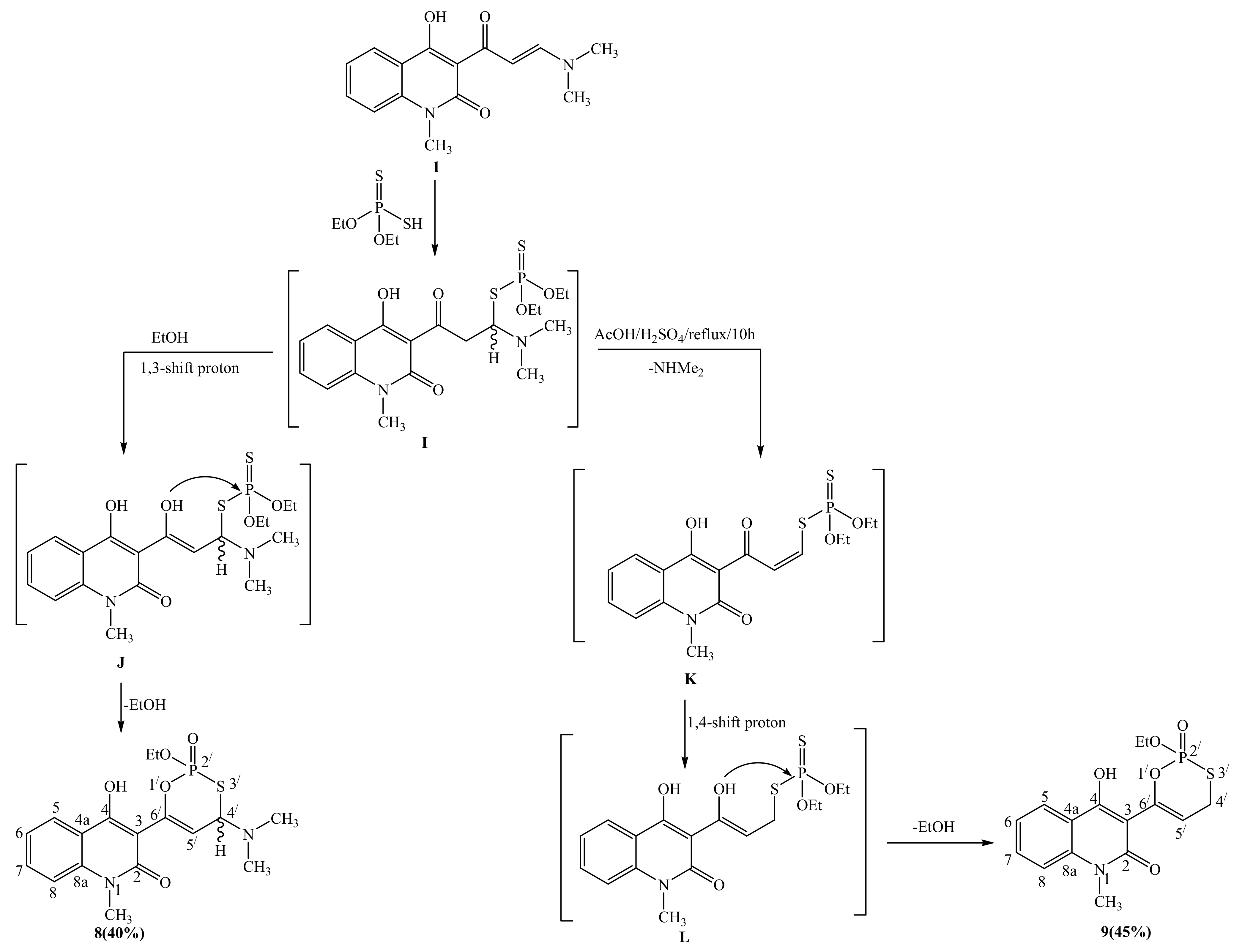
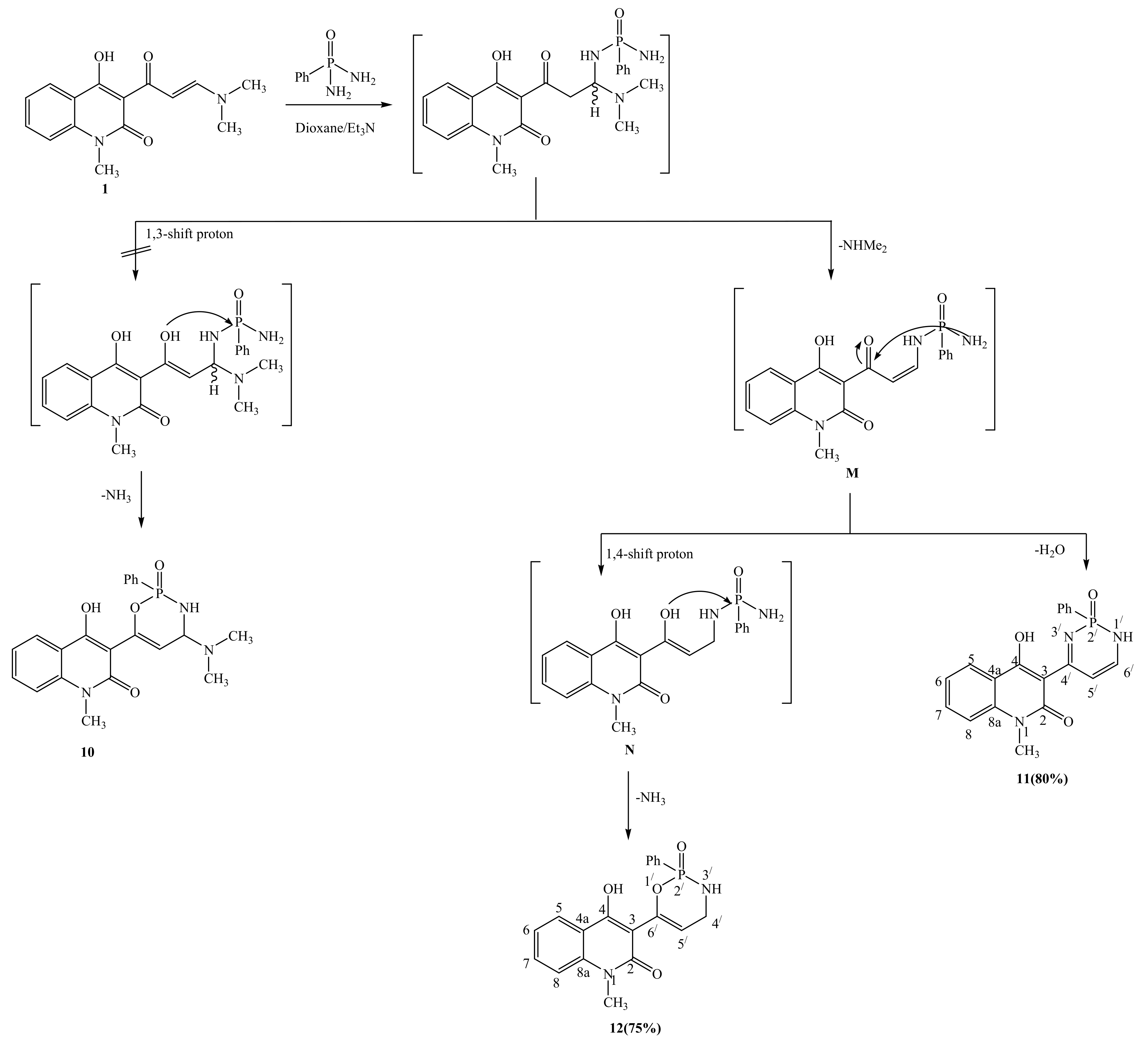
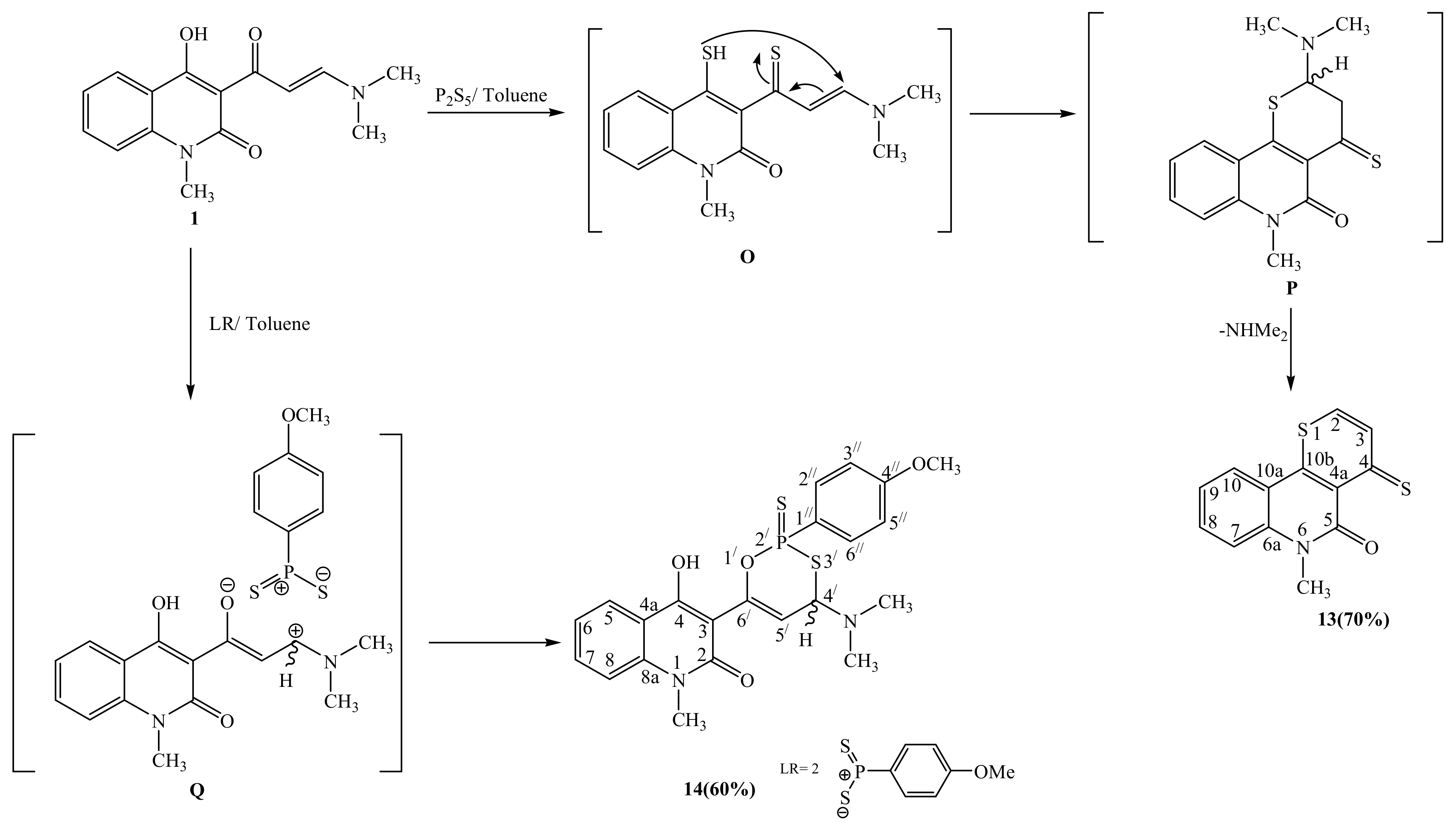
2.1. Synthetic Strategies
2.2. Pharmacology
Antioxidant Activity
3. Experimental
3.1. Instruments and Reagents
3.2. Synthesis
3.2.1. Synthesis of Diethyl 5,6-dihydro-6-methyl-4,5-dioxo-4H-pyrano[3,2-c]quinolin-2-yl-2-phosphonate (2)
3.2.2. General Procedure for the Synthesis of Compounds 3, 4, 5, 6, and 9
3.2.3. Diethyl [6-(4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinolin-3-yl)-2-oxo-2H-pyran-3-yl]phosphonate (3)
3.2.4. Diethyl[6-(4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinolin-3-yl)-2-thioxo-1,2-dihydropyridin-3-yl]phosphonate (4)
3.2.5. Diethyl[6-(4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinolin-3-yl)-2H-pyran-3-yl]phosphonate (5)
3.2.6. Diethyl [6-(4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinolin-3-yl)-2-oxo-1,2-dihydropyridin-3-yl]phosphonate (6)
3.2.7. General Procedure for Synthesis of Compounds 7 and 8
3.2.8. Diethyl-4-(dimethylamino)-1,2-dihydro-6-(1,2-dihydro-4-hydroxy-1-methyl-2-oxoquinolin-3-yl)-2-oxopyridin-3-yl-3-phosphonate (7)
3.2.9. 3-[4-(dimethylamino)-2-ethoxy-2-oxido-4H-1,3,2-oxathiaphosphinin-6-yl]-4-hydroxy-1-methylquinolin-2(1H)-one (8)
3.2.10. 3-(2-ethoxy-2-oxido-4H-1,3,2-oxathiaphosphinin-6-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (9)
3.2.11. General Procedure for Synthesis of Compounds 11 and 12
3.2.12. 4-hydroxy-1-methyl-3-(2-oxido-2-phenyl-1,2-dihydro-1,3,2-diazaphosphinin-4-yl)quinolin-2(1H)-one (11)
3.2.13. 4-hydroxy-1-methyl-3-(2-oxido-2-phenyl-3,4-dihydro-2H-1,3,2-oxazaphosphinin-6-yl)quinolin-2(1H)-one (12)
3.2.14. Synthesis of 6-methyl-4-thioxo-4H-thiopyrano[3,2-c]quinolin-5(6H)-one (13)
3.2.15. Synthesis of 3-(4-(dimethylamino)-2-(4-methoxyphenyl)-2-sulfanylene-4H-1,3,2-oxathiaphosphinin-6-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (14)
3.3. Antioxidant Activity
- Ao = the sample’s initial absorbance. (t = 0 min)
- At is the sample’s absorbance at time t. (t = 180 min)
- Aoo = the control’s initial absorbance. (t = 0 min)
- Ato is the absorption of control at time t. (t = 180 min)
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aly, A.A.; Mohamed, A.H.; Ramadan, M. Synthesis and colon anticancer activity of some novel thiazole/-2-quinolone derivatives. J. Mol. Struct. 2020, 1207, 127798. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871. [Google Scholar] [CrossRef] [PubMed]
- Mandewale, M.C.; Patil, U.C.; Shedge, S.V.; Dappadwad, U.R.; Yamgar, R.S. A review on quinoline hydrazone derivatives as a new class of potent antitubercular and anticancer agents. Beni Suef Univ. J. Basic Appl. Sci. 2017, 6, 354–361. [Google Scholar] [CrossRef]
- Sankaran, M.; Kumarasamy, C.; Chokkalingam, U.; Mohan, P.S. Synthesis, antioxidant and toxicological study of novel pyrimido quinoline derivatives from 4-hydroxy-3-acyl quinolin-2-one. Bioorg. Med. Chem. Lett. 2010, 20, 7147. [Google Scholar] [CrossRef]
- Abdou, W.M.; Shaddy, A.A.; Kamel, A.A. Structure-based design and synthesis of acyclic and substituted heterocyclic phosphonates linearly linked to thiazolobenzimidazoles as potent hydrophilic antineoplastic agents. Chem. Pap. 2017, 71, 1961. [Google Scholar] [CrossRef]
- Savegnago, L.; Vieira, A.I.; Seus, N.; Goldani, B.S.; Castro, M.R.; Lenardão, E.J.; Alves, D. Synthesis and antioxidant properties of novel quinoline–chalcogenium compounds. Tetrahedron Lett. 2013, 54, 40. [Google Scholar] [CrossRef]
- Keri, R.S.; Patil, S.A. Quinoline: A promising antitubercular target. Biomed. Pharmacother. 2014, 68, 1161. [Google Scholar] [CrossRef]
- Chen, L.; Liu, X.; Zou, Y. Recent Advances in the Construction of Phosphorus-Substituted Heterocycles. Adv. Synth. Catal. 2020, 362, 1724. [Google Scholar] [CrossRef]
- Ali, T.E.; Assiri, M.A. Synthesis of Novel 4-(1,3,2-Diazaphosphinin-5-yl)-1,2-Dihydrothieno[3,2-d][1,2,3] Diazaphosphinines: Cyclization of 5-[(2-Thienyl)Methyl]Hydrazono-2-Oxido-2-Phenyl-4-(Trifluoromethyl)-1H-1,3,2-Diazaphosphinine with Phosphorus Halides. Polycycl. Aromat. Compd. 2021, 1–9. [Google Scholar] [CrossRef]
- Fang, Y.-L.; Wu, Z.-L.; Xiao, M.-W.; Tang, Y.-T.; Li, K.-M.; Ye, J.; Hu, A.-X. One-Pot Three-Component Synthesis of Novel Diethyl((2-oxo-1,2-dihydroquinolin-3-yl)(arylamino)methyl)phosphonate as Potential Anticancer Agents. Int. J. Mol. Sci. 2016, 17, 653. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.M.; Abdel-Kariem, S.M.; Ali, T.E. Synthesis and antioxidant properties of some novel 1,3,4,2-oxadiazaphosphepino[6,7-c]quinolinones and pyrazolo[3,4:4′,3′]quinolino[5,1-c][1,4,2]oxazaphosphinine. Phosphorus Sulfur Silicon Relat. Elem. 2017, 92, 866. [Google Scholar] [CrossRef]
- Elassar, A.-Z.A.; El-Khair, A.A. Recent developments in the chemistry of enaminones. Tetrahedron 2003, 59, 8463. [Google Scholar] [CrossRef]
- Stanovnik, B. Enaminone, Enaminoesters, and Related Compounds in the Metal-Free Synthesis of Pyridines and Fused Pyridines. Eur. J. Org. Chem. 2019, 2019, 5120. [Google Scholar] [CrossRef]
- Šinkovec, R.; Grošelj, U.; Prek, B.; Počkaj, M.; Ričko, S.; Svete, J.; Stanovnik, B. A simple synthesis of dimethyl 2-[(Z)-3-amino-1-oxo-1-(substituted)but-2-en-2-yl]fumarates: Potential intermediates in the synthesis of polysubstituted five- and six-membered heterocycles. Zeitschrift Für Naturforschung B 2016, 71, 677. [Google Scholar] [CrossRef]
- Chimichi, S.; Boccalini, M.; Hassan, M.M.M.; Viola, G.; Dall’Acqua, F.; Curini, M. Synthesis, structural determination and photo-antiproliferative activity of new 3-pyrazolyl or -isoxazolyl substituted 4-hydroxy-2(1H)-quinolinones. Tetrahedron 2006, 62, 90. [Google Scholar] [CrossRef]
- Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Preparation and characterization of α-fluorinated-γ-aminophosphonates. J. Fluor. Chem. 2014, 167, 128. [Google Scholar] [CrossRef]
- Shawali, A.S. Bis-Enaminones as versatile precursors for terheterocycles: Synthesis and reactions. Arkivoc 2012, 2012, 383–431. [Google Scholar] [CrossRef]
- Ali, T.E. Synthesis and antibacterial activity of some new thiadiaza/triazaphospholes, thiadiaza/triaza/tetrazaphosphinines and thiadiaza/tetrazaphosphepines containing 1,2,4-triazinone moiety. Eur. J. Med. Chem. 2009, 44, 4539. [Google Scholar] [CrossRef]
- Ozturk, T.; Ertas, E.; Mert, O. Use of Lawesson’s Reagent in Organic Syntheses. Chem. Rev. 2007, 107, 5210. [Google Scholar] [CrossRef]
- Ozturk, T.; Ertas, E.; Mert, O. A Berzelius Reagent, Phosphorus Decasulfide(P4S10), in Organic Syntheses. Chem. Rev. 2010, 110, 3419. [Google Scholar] [CrossRef]
- Kato, K.; Terao, S.; Shimamoto, N.; Hirata, M. Synthesis and biological activity of 2-O-alkylascorbic acids. J. Med. Chem. 1988, 31, 793. [Google Scholar] [CrossRef]
- Siddhuraju, P.; Becker, K. The antioxidant and free radical scavenging activities of processed cowpea (Vigna unguiculata (L.) Walp.) seed extracts. Food Chem. 2007, 101, 10. [Google Scholar] [CrossRef]
- Guo, Z.; Xing, R.; Liu, S.; Yu, H.; Wang, P.; Li, C.; Li, P. The synthesis and antioxidant activity of the Schiff bases of chitosan and carboxymethyl chitosan. Bioorg. Med. Chem. Lett. 2005, 15, 4600. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | The Percentage (%) Antioxidant Activity ± SD | ||
|---|---|---|---|
| 50 (µg/mL) | 75 (µg/mL) | 100 (µg/mL) | |
| 2 | 42.12 ± 0.60 | 46.70 ± 0.54 | 48.71 ± 0.50 |
| 3 | 44.11 ± 0.61 | 46.77 ± 0.51 | 50.71 ± 0.64 |
| 4 | 47.10 ± 055 | 49.77 ± 0.59 | 51.70 ± 0.44 |
| 5 | 45.14 ± 0.54 | 47.77 ± 0.52 | 49.73 ± 0.38 |
| 6 | 50.22 ± 0.56 | 51.23 ± 0.61 | 55.52 ± 0.60 |
| 7 | 62.60 ± 0.29 | 63.41 ± 0.30 | 66.75 ± 0.25 |
| 8 | 60.20 ± 0.33 | 61.80 ± 0.33 | 63.41 ± 0.22 |
| 9 | 52.41 ± 0.51 | 54.27 ± 0.52 | 56.68 ± 0.44 |
| 11 | 77.30 ± 0.25 | 80.31 ± 0.16 | 83.34 ± 0.14 |
| 12 | 75.56 ± 0.22 | 75.85 ± 0.12 | 76.35 ± 0.16 |
| 13 | 33.41 ± 0.50 | 34.25 ± 0.51 | 36.64 ± 0.41 |
| 14 | 80.70 ± 0.12 | 84.88 ± 0.11 | 86.30 ± 0.13 |
| Ascorbic acid | 68.21 ± 0.20 | 71.62 ± 0.15 | 73.50 ± 0.14 |
| Compound No. | The Percentage (%) Antioxidant Activity ± SD | ||
|---|---|---|---|
| 50 (µg/mL) | 75 (µg/mL) | 100 (µg/mL) | |
| 2 | 44.11 ± 0.59 | 45.66 ± 0.54 | 48.73 ± 0.51 |
| 3 | 45.14 ± 0.55 | 46.53 ± 0.52 | 50.70 ± 0.58 |
| 4 | 48.12 ± 054 | 50.72 ± 0.55 | 52.66 ± 0.33 |
| 5 | 46.12 ± 0.55 | 47.70 ± 0.53 | 50.72 ± 034 |
| 6 | 52.23 ± 0.54 | 53.22 ± 0.56 | 55.50 ± 0.60 |
| 7 | 64.61 ± 0.27 | 66.40 ± 0.30 | 66.60 ± 0.23 |
| 8 | 62.52 ± 0.30 | 63.66 ± 0.31 | 65.40 ± 0.21 |
| 9 | 55.33 ± 0.51 | 56.26 ± 0.51 | 58.55 ± 0.55 |
| 11 | 77.33 ± 0.22 | 81.28 ± 0.14 | 82.32 ± 0.12 |
| 12 | 76.54 ± 0.21 | 77.80 ± 0.15 | 78.33 ± 0.12 |
| 13 | 32.35 ± 0.52 | 34.20 ± 0.50 | 37.68 ± 0.33 |
| 14 | 81.73 ± 0.14 | 83.82 ± 0.14 | 85.33 ± 0.11 |
| Ascorbic acid | 66.80 ± 0.23 | 68.12 ± 0.20 | 72.32 ± 0.16 |
| Compound No. | IC50 (µmol/mL) |
|---|---|
| 2 | 0.1203 ± 0.0010 |
| 3 | 0.1111 ± 0.0012 |
| 4 | 0.1240 ± 0.0014 |
| 5 | 0.1266 ± 0.0014 |
| 6 | 0.1187 ± 0.0011 |
| 7 | 0.0978 ± 0.0005 |
| 8 | 0.0966 ± 0.0013 |
| 9 | 0.1132 ± 0.0012 |
| 11 | 0.0741 ± 0.0018 |
| 12 | 0.0763 ± 0.0018 |
| 13 | 0.1616 ± 0.0010 |
| 14 | 0.0601 ± 0.0014 |
| Ascorbic acid | 0.2255 ± 0.0070 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, M.M.; Alhalafi, M.H. Synthesis, Structural Determination, and Antioxidant Activities of Acyclic and Substituted Heterocyclic Phosphonates Linearly Linked 4-hydroxy-2(1H)-quinolinone. Molecules 2022, 27, 5960. https://doi.org/10.3390/molecules27185960
Hassan MM, Alhalafi MH. Synthesis, Structural Determination, and Antioxidant Activities of Acyclic and Substituted Heterocyclic Phosphonates Linearly Linked 4-hydroxy-2(1H)-quinolinone. Molecules. 2022; 27(18):5960. https://doi.org/10.3390/molecules27185960
Chicago/Turabian StyleHassan, Mohamed M., and Mona H. Alhalafi. 2022. "Synthesis, Structural Determination, and Antioxidant Activities of Acyclic and Substituted Heterocyclic Phosphonates Linearly Linked 4-hydroxy-2(1H)-quinolinone" Molecules 27, no. 18: 5960. https://doi.org/10.3390/molecules27185960