
Oxidative Desulfurization Activity of NIT Nitroxide Radical Modified Metallophthalocyanine

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
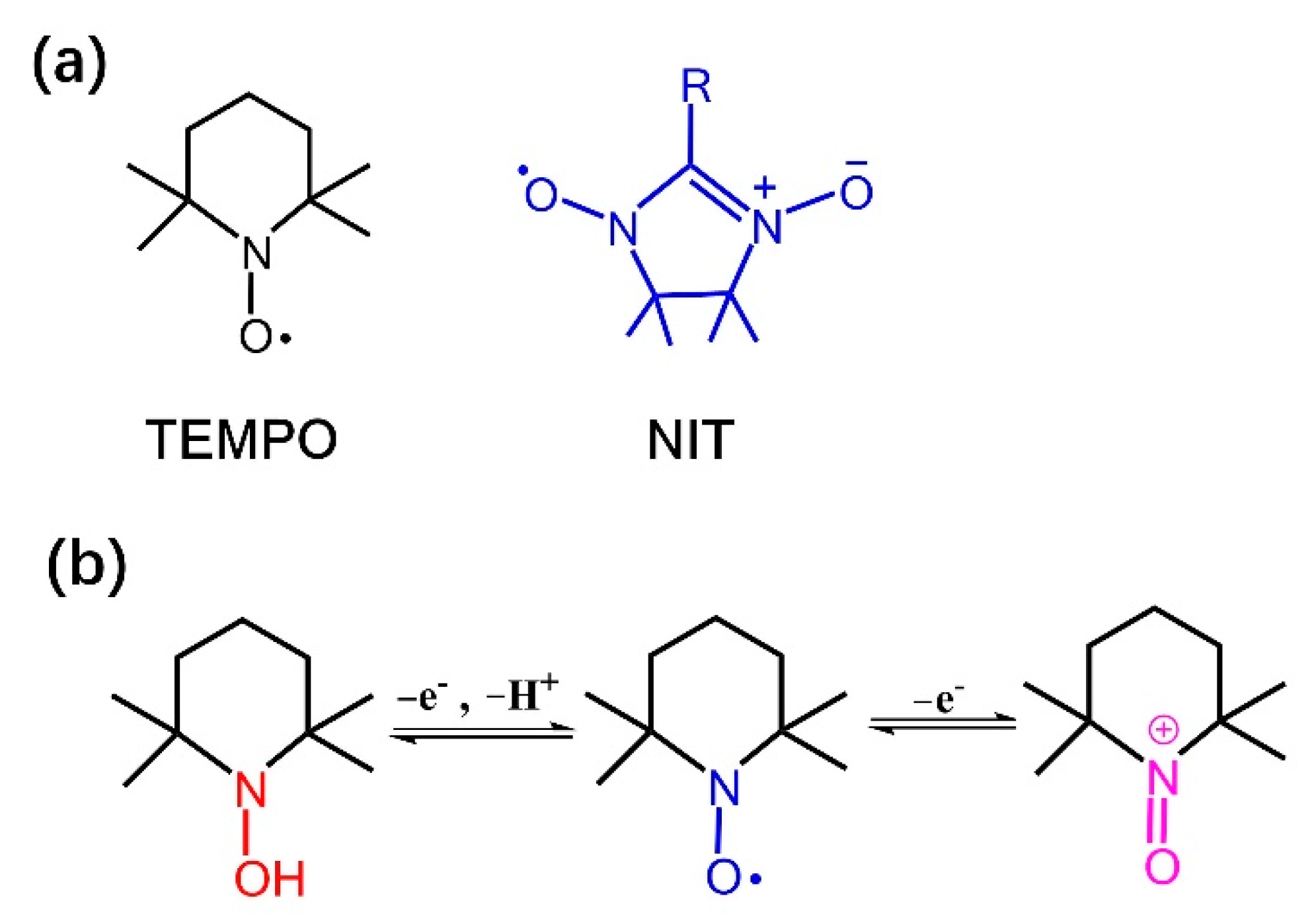
2.1. Synthesis of MPcTcCl8-NIT Catalysts
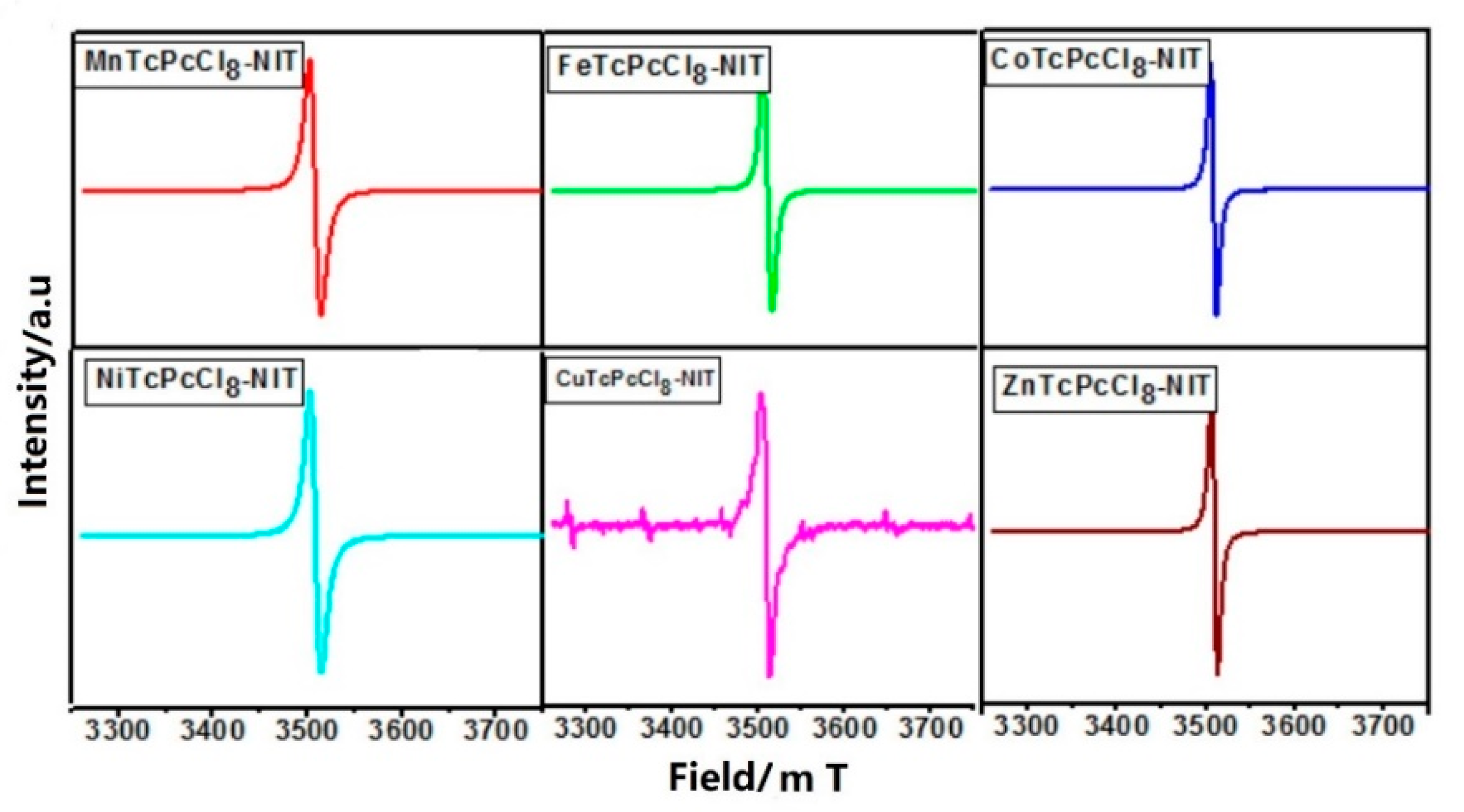
2.1.1. ESR Spectra
2.1.2. IR Spectra
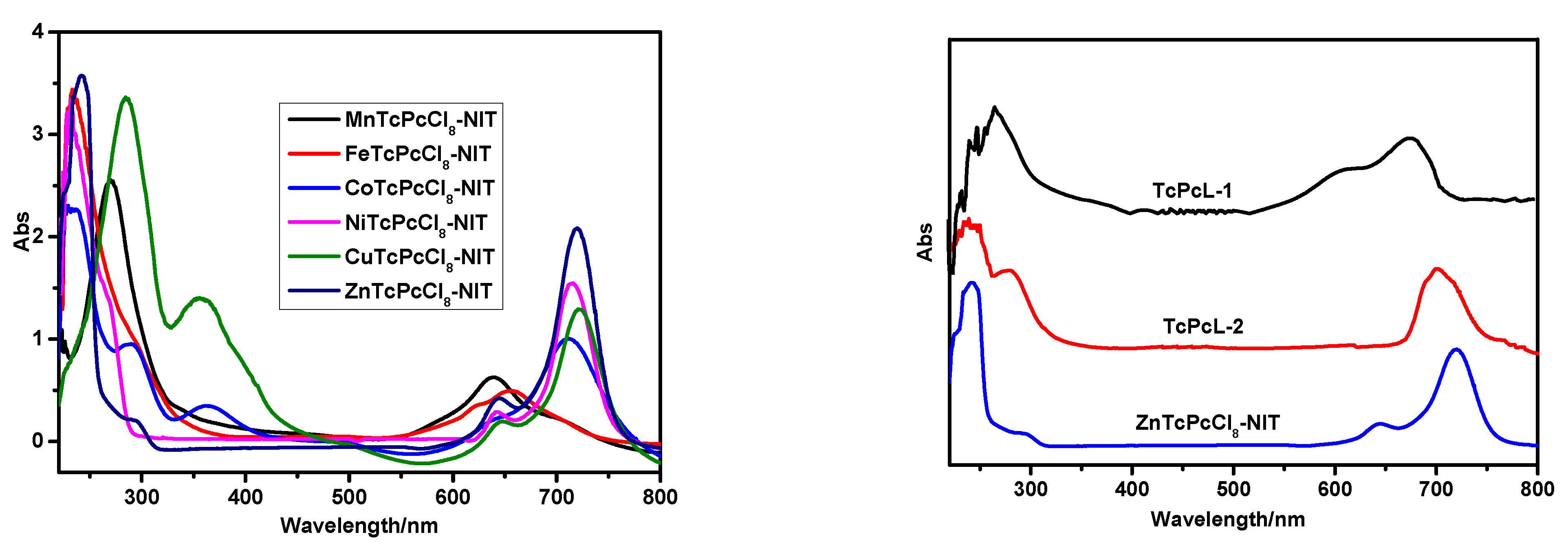
2.1.3. UV-Vis Spectra
2.1.4. XPS Spectra forMPcTcCl8-NIT
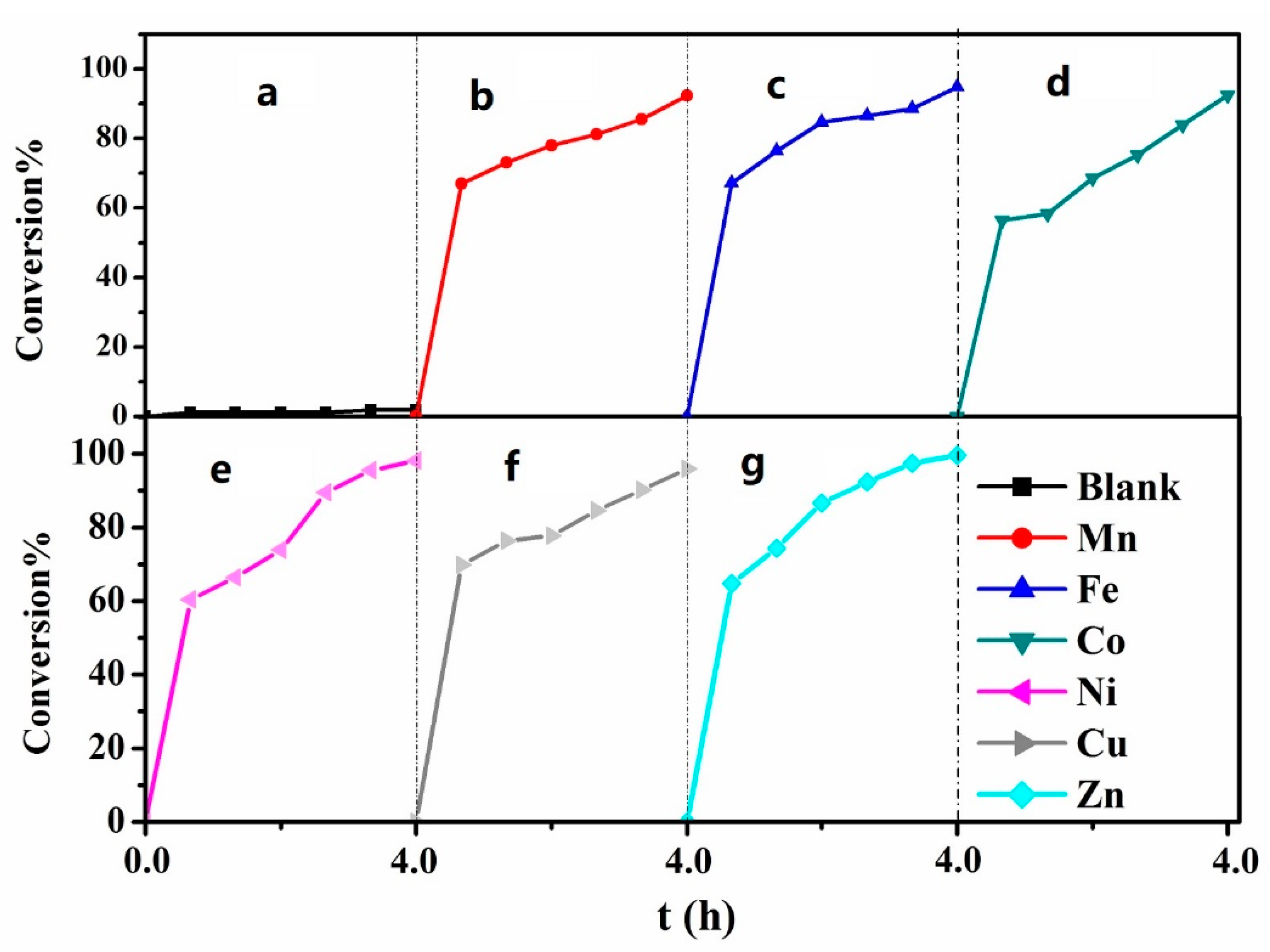
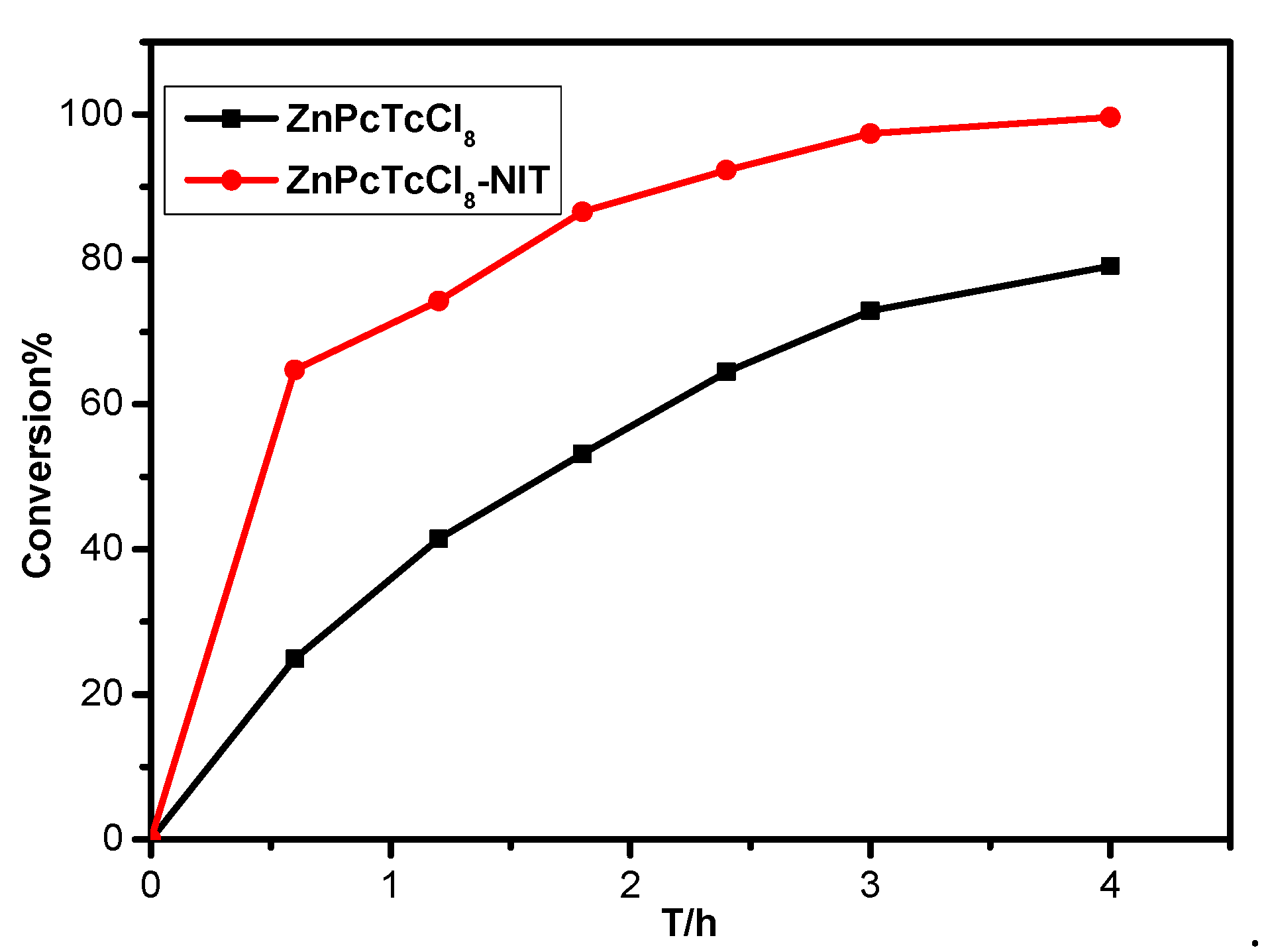
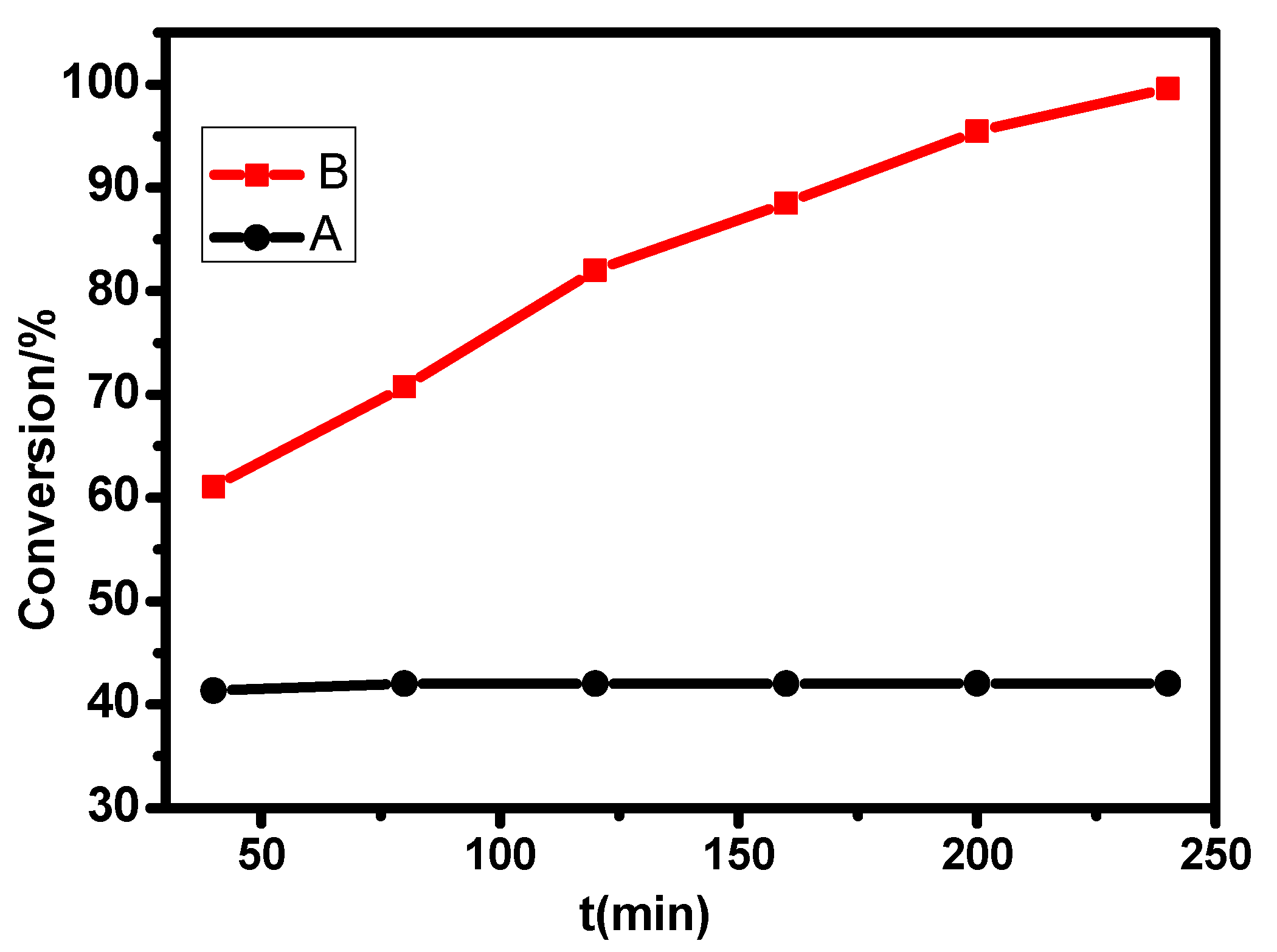
2.2. Desulfurization Effect of MPcTcCl8-NIT
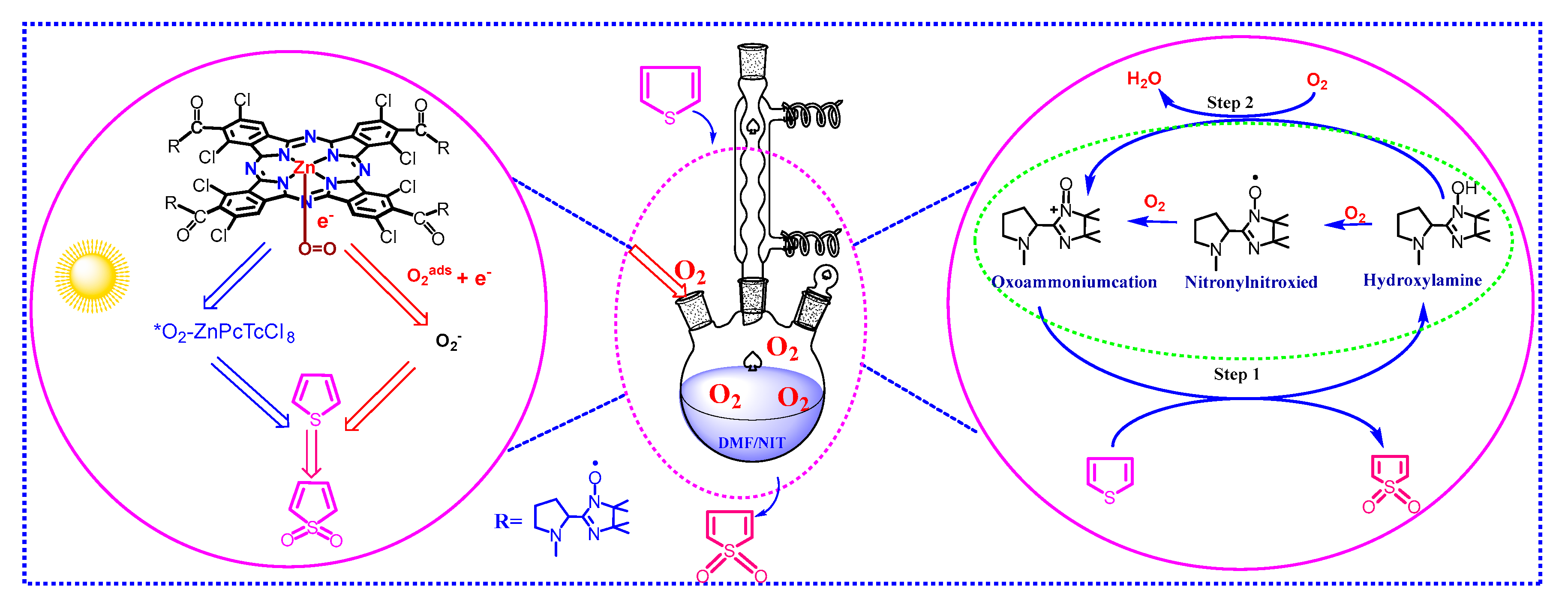
2.3. Photo-Catalytic Degradation Mechanism of the MPcTcCl8-NIT
3. Materials and Methods
3.1. Characterization
3.2. Synthesis Methods
3.2.1. Synthesis of ZnPcTcCl8
3.2.2. Synthesis of the TcPcL-1 Intermediate
3.2.3. Synthesis of the TcPcL-2 Intermediate
3.2.4. Synthesis of ZnPcTcCl8-NIT
3.3. Evaluation of the Photocatalytic Activity
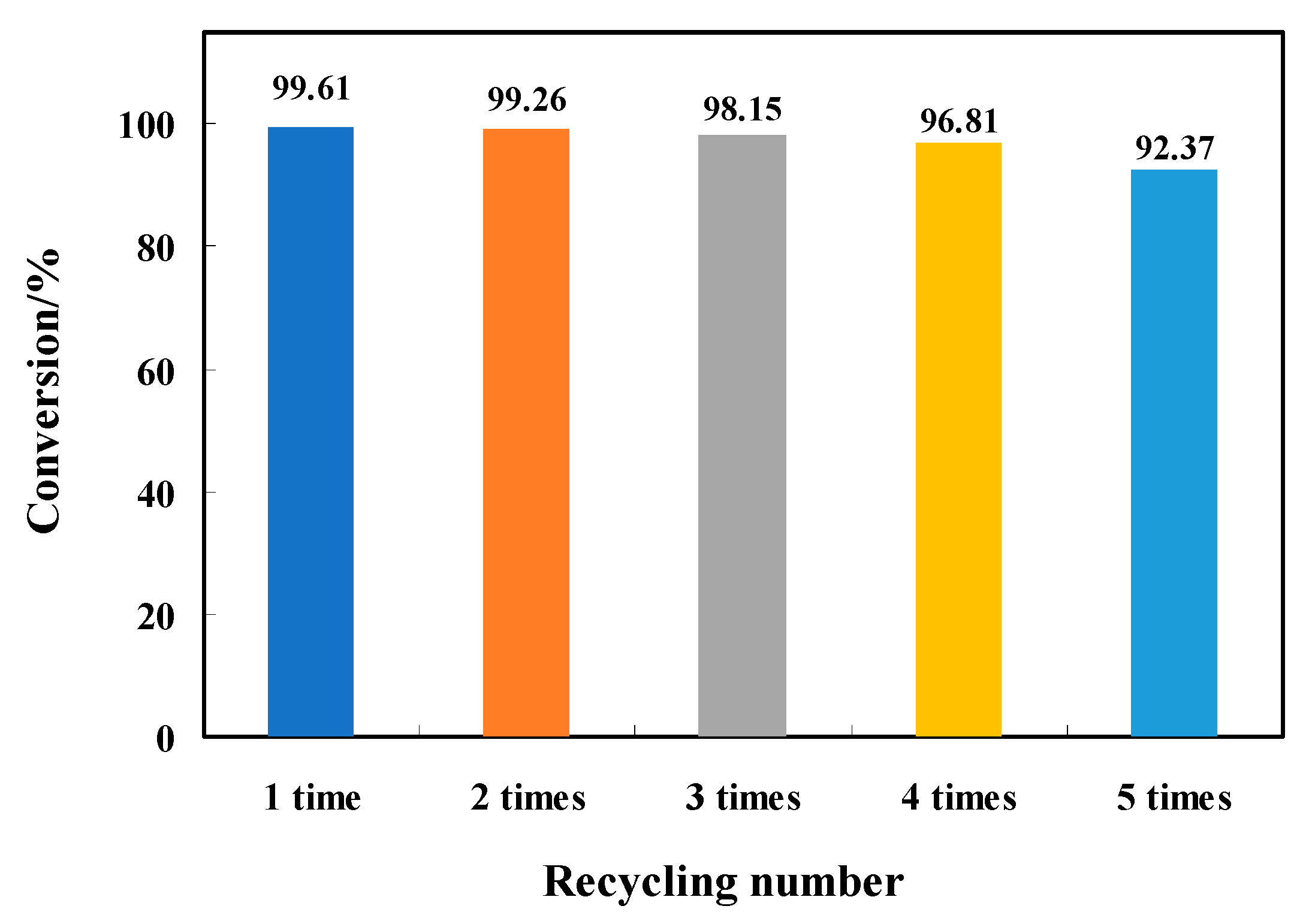
3.4. Evaluation of the Catalyst Recycling
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Stanislaus, A.; Marafi, A.; Rana, M.S. Recent advances in the science and technology of ultra low sulfur diesel (ULSD) production. Catal. Today 2010, 153, 1–68. [Google Scholar] [CrossRef]
- Kaufmann, T.G.; Kaldor, A.; Stuntz, G.F.; Kerby, M.C.; Ansell, L.L. Catalyswas science and technology for cleaner transportation fuels. Catal. Today 2000, 62, 77–90. [Google Scholar] [CrossRef]
- Gao, S.R.; Yu, G.R.; Rashid, A.; Ahmed, A.A.; Salem, S.A.; Chen, X.C. Desulfurization of fuel oils: Mutual solubility of ionic liquids and fuel oil. Fuel 2016, 173, 164–171. [Google Scholar] [CrossRef]
- Tailleur, R.G.; Ravigli, J.; Quenza, S.; Valencia, N. Catalyst for ultra-low sulfur and aromatic diesel. Appl. Catal. A-Gen. 2005, 282, 227–235. [Google Scholar] [CrossRef]
- Madriz, L.; Carrero, H.; Domínguez, J.R.; Vargas, R.; Fernandez, L. Catalytic hydrotreatment in reverse microemulsions under microwave Irradiation. Fuel 2013, 112, 338–346. [Google Scholar] [CrossRef]
- Parviz, D.; Kazemeini, M.; Rashidi, A.M.; Jozani, K.J. Naphtha hydrodesulfurization over micro and nanostructure MoO3 catalysts. Sci. Iran. 2011, 18, 479–485. [Google Scholar] [CrossRef]
- Hai, M.; Mei, B.W.; Yen, T.F. A new method for obtaining ultra-low sulfur diesel fuel via ultrasound assisted oxidative desulfurization. Fuel 2003, 82, 405–414. [Google Scholar] [CrossRef]
- Wang, J.Y.; Li, J.L.; Zhang, S.W.; Zhao, R.L.; Gao, N. Synthesis and application of different phthalocyanine molecular sieve catalyst for oxidative desulfurization. J. Solid State Chem. 2015, 225, 347–353. [Google Scholar] [CrossRef]
- Yuan, Z.C.; Lan, Y.; Chen, S.Y.; Chen, D.J. Preparation of magnetically recyclable palygorskite Fe-octacarboxylic acid phthalocyanine nano-catalysts and their photocatalytic behavior for degradation of Rhodamine B. Appl. Clay Sci. 2017, 147, 153–159. [Google Scholar] [CrossRef]
- Chao, Y.; Li, H.; Zhu, W.; Zhu, G.; Yan, Y. Deep oxidative desulfurization of dibenzothiophene in simulated diesel with tungstate and H2O2 in ionic liquids. Liquid Fuels Technol. 2010, 28, 1242–1249. [Google Scholar] [CrossRef]
- Yazu, K.; Makino, M.; Ukegawa, K. Oxidative desulfurization of diesel oil with hydrogen peroxide in the presence of acid catalyst in diesel oil/acetic acid biphasic system. Chem. Lett. 2004, 33, 1306–1307. [Google Scholar] [CrossRef]
- Campos-Martin, J.M.; Capel-Sanchez, M.C.; Fierro, J. Highly efficient deep desulfurization of fuels by chemical oxidation. Catal. Today 2004, 6, 557–562. [Google Scholar] [CrossRef]
- Lu, W.Y.; Xu, T.F.; Wang, Y.; Hu, H.G.; Li, N.; Jiang, X.M.; Chen, W.X. Synergistic photocatalytic properties and mechanism of g-C3N4 coupled with zinc phthalocyanine catalyst under visible light irradiation. Appl. Catal. B 2016, 180, 20–28. [Google Scholar] [CrossRef]
- Mensing, J.P.; Kerdcharoen, T.; Sriprachuabwong, C.; Wisitsoraat, A.; Phokharatkul, D.; Lomas, T.; Tuantranont, A. Facile preparationof graphene-metallophthalocyanine hybrid material by electrolytic exfoliation. J. Mater. Chem. 2012, 22, 17094–17099. [Google Scholar] [CrossRef]
- Zhang, G.; Ren, J.J.; Liu, B.L.; Tian, M.; Zhou, H.W.; Zhao, J.S. In situ hydrothermal preparation and photocatalytic desulfurization performance of metallophthalocyanine sensitized SnO2. Inorg. Chim. Acta 2018, 471, 782–787. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, G.; Tian, M.; Liu, B.L.; Chen, W.X. Oxidative desulfurization of dibenzothiophene by central metal ions of chlorophthalocyanines-tetracarboxyl complexes. Inorg. Chim. Acta 2019, 485, 58–63. [Google Scholar] [CrossRef]
- Wang, F.; Wang, G.; Sun, W.; Wang, T.; Chen, X. Metallophthalocyanine functionalized magnetic mesoporous silica nanoparticles and its application in ultrasound-assisted oxidation of benzothiophene. Microporous Mesoporous Mater. 2015, 217, 203–209. [Google Scholar] [CrossRef]
- Rajagopal, G.; Kim, S.S.; Ju, M.K. Aluminum phthalocyanine: An active and simple catalyst for cyanosilylation of aldehydes. Appl. Organomet. Chem. 2007, 21, 198–202. [Google Scholar] [CrossRef]
- Ansari, I.A.; Gree, R. TEMPO-catalyzed aerobic oxidation of alcohols to aldehydes and ketones in ionic liquid [bmim][PF6]. Org. Lett. 2002, 4, 1507–1509. [Google Scholar] [CrossRef]
- Cotrim, A.P.; Hyodo, F.; Matsumoto, K.I.; Sowers, A.L.; Cook, J.A.; Baum, B.J.; Krishna, M.C.; Mitchell, J.B. Differential radiation protection of salivary glands versus tumor by Tempol with accompanying tissue assessment of Tempol by magnetic resonance imaging. Clin. Cancer Res. 2007, 13, 4928–4933. [Google Scholar] [CrossRef] [Green Version]
- Poneti, G.; Bernot, K.; Bogani, L.; Caneschi, A.; Sessoli, R.; Wernsdorfer, W.; Gatteschi, D. A rational approach to the modulation of the dynamics of the magnetisation in a dysprosium–nitronyl-nitroxide radical complex. Chem. Commun. 2007, 18, 1807–1809. [Google Scholar] [CrossRef] [PubMed]
- Castagn, R.; Vita, F.; Daniele, E.; Criante, L.; Greci, L.; Ferraris, P.; Simoni, F. Nitroxide radicals reduce shrinkage in acrylate-based holographic gratings. Opt. Mater. 2007, 30, 539–544. [Google Scholar] [CrossRef]
- Nakatsuji, s.; Anzai, H. Recent progress in the development of organomagnetic materials based on neutral nitroxide radicals and charge transfer complexes derived from nitroxide radicals. J. Mater. Chem. 1997, 7, 2161–2174. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Russo, A.; Kuppusamy, P.; Krishna, M.C. Radiation, radicals, and images. Ann. N. Y. Acad. Sci. 2000, 899, 28–43. [Google Scholar] [CrossRef]
- Qin, X.Y.; Ding, G.R.; Wang, X.W.; Tan, J.; Guo, G.Z.; Sun, X.L. Synthesis, characterisation, cytotoxicity and radioprotective effect of novel chiral nitronyl nitroxyl radicals. J. Chem. Res. 2009, 8, 511–514. [Google Scholar] [CrossRef]
- Golubev, V.A.; Rozantsev, G.; Neiman, M.B. Some reactions of free iminoxyl radicals with the participation of the unpaired electron. Bull. Acad. Sci. USSR Div. Chem. Sci. 1965, 14, 1898–1904. [Google Scholar] [CrossRef]
- Sun, T.T.; Liang, H.Z.; Liu, S.J.; Tang, E.J.; Fu, C.Y. Magnetic nanoparticles modified by polyamidoamine-immobilized TEMPO for catalytic oxidation of monomethoxy poly (ethylene glycol). J. Nanopart. Res. 2020, 22, 1–11. [Google Scholar] [CrossRef]
- Ye, W.; Yokota, S.; Fan, Y.; Kondo, T. A combination of aqueous counter collision and TEMPO-mediated oxidation for doubled carboxyl contents of α-chitin nanofibers. Cellulose 2021, 28, 2167–2181. [Google Scholar] [CrossRef]
- Tanaka, T.; Kiuchi, T.; Ooe, Y.; Iwamoto, H.; Takizawa, S.Y.; Murata, S.; Hasegawa, E. A Photocatalytic System Composed of Benzimidazolium Aryloxide and Tetramethylpiperidine 1-Oxyl to Promote Desulfonylative α-Oxyamination Reactions of α-Sulfonylketones. ACS Omega 2022, 7, 4655–4666. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, F.; Qian, L.Y.; Cao, X.H.; He, B.H.; Li, J.R. A fast-response electroactive actuator based on TEMPO-oxidized cellulose nanofibers. Smart Mater. Struct. 2022, 31, 025005. [Google Scholar] [CrossRef]
- Kenly, A.C.; Ruth, R.; Karla, R.A.; Belkis, S.R.; Orlando, R.; Marianelly, E.A. Cellulose Nanofibers as Functional Biomaterial from Pineapple Stubbles via TEMPO Oxidation and Mechanical Process. Waste Biomass Valoriz. 2022, 13, 1749–1758. [Google Scholar]
- Schatzschneider, U.; Rentschler, E. Electronic and conformational influences on the spin density distribution in substituted tert-butyl phenyl nitroxides. J. Mol. Struct. Theochem 2003, 638, 163–168. [Google Scholar] [CrossRef]
- Kolanji, K.; Baumgarten, M. Dithienopyrrole Derivatives with Nitronyl Nitroxide Radicals and Their Oxidation to Cationic High-Spin Molecules. Chem. Eur. J. 2020, 26, 3626–3632. [Google Scholar] [CrossRef]
- Soule, B.P.; Hyodo, F.; Matsumoto, K.; Simone, N.L.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. The chemistry and biology of nitroxide compounds. Free Radical Biology & Medicine 2007, 42, 1632–1650. [Google Scholar] [CrossRef]
- Li, L.; Matsuda, R.; Tanaka, I.; Sato, H.; Kanoo, P.; Jeon, H.J.; Foo, M.L.; Wakamiya, A.; Murata, Y.; Kitagawa, S.A. Crystalline porous coordination polymer decorated with nitroxyl radicals catalyzes aerobic oxidation of alcohols. J. Am. Chem. Soc. 2014, 21, 7543–7546. [Google Scholar] [CrossRef] [PubMed]
- Augusto, O.; Trindade, D.F.; Linares, E.; Vaz, S.M. Cyclic nitroxides inhibit the toxicity of nitric oxide-derived oxidants: Mechanisms and implications. An. Acad. Brasil. Ciênci. 2008, 80, 179–189. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Jiang, Y.; Shi, M.; Bawa, M.; Wang, X.; Zhu, S. Assembly of metallophthalocyanine-polyoxometalate hybrid for highly efficient desulfurization of organic and inorganic sulfur under aerobic conditions. Fuel 2019, 241, 861–869. [Google Scholar] [CrossRef]
- Fang, Z.; Zhao, Z.; Li, N.; Zhu, Z.; Chen, W. Low-temperature catalytic oxidative desulfurization by two-phase system with O-bridged diiron perfluorophthalocyanine. Fuel 2021, 306, 121649. [Google Scholar] [CrossRef]
- Fang, Z.; Li, N.; Zhao, Z.; Zhu, Z.; Lu, W.; Chen, F.; Wang, J.; Chen, W. Bio-inspired strategy to enhance catalytic oxidative desulfurization by O-bridged diiron perfluorophthalocyanine axially coordinated with 4-mercaptopyridine. Chem. Eng. J. 2022, 433, 133569. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.F.; Tan, A.M.; Yang, Y.; Tian, M. Effects of MN4-Type Coordination Structure in Metallophthalocyanine for Bio-Inspired Oxidative Desulfurization Performance. Molecules 2022, 27, 904. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, M.; He, Y.; Zhang, G.; Wang, H. Oxidative Desulfurization Activity of NIT Nitroxide Radical Modified Metallophthalocyanine. Molecules 2022, 27, 5964. https://doi.org/10.3390/molecules27185964
Tian M, He Y, Zhang G, Wang H. Oxidative Desulfurization Activity of NIT Nitroxide Radical Modified Metallophthalocyanine. Molecules. 2022; 27(18):5964. https://doi.org/10.3390/molecules27185964
Chicago/Turabian StyleTian, Min, Yang He, Gai Zhang, and Haibo Wang. 2022. "Oxidative Desulfurization Activity of NIT Nitroxide Radical Modified Metallophthalocyanine" Molecules 27, no. 18: 5964. https://doi.org/10.3390/molecules27185964
APA StyleTian, M., He, Y., Zhang, G., & Wang, H. (2022). Oxidative Desulfurization Activity of NIT Nitroxide Radical Modified Metallophthalocyanine. Molecules, 27(18), 5964. https://doi.org/10.3390/molecules27185964