
Design, Synthesis, Electronic Properties, and X-ray Structural Characterization of Various Modified Electron-Rich Calixarene Derivatives and Their Conversion to Stable Cation Radical Salts

Abstract
:1. Introduction
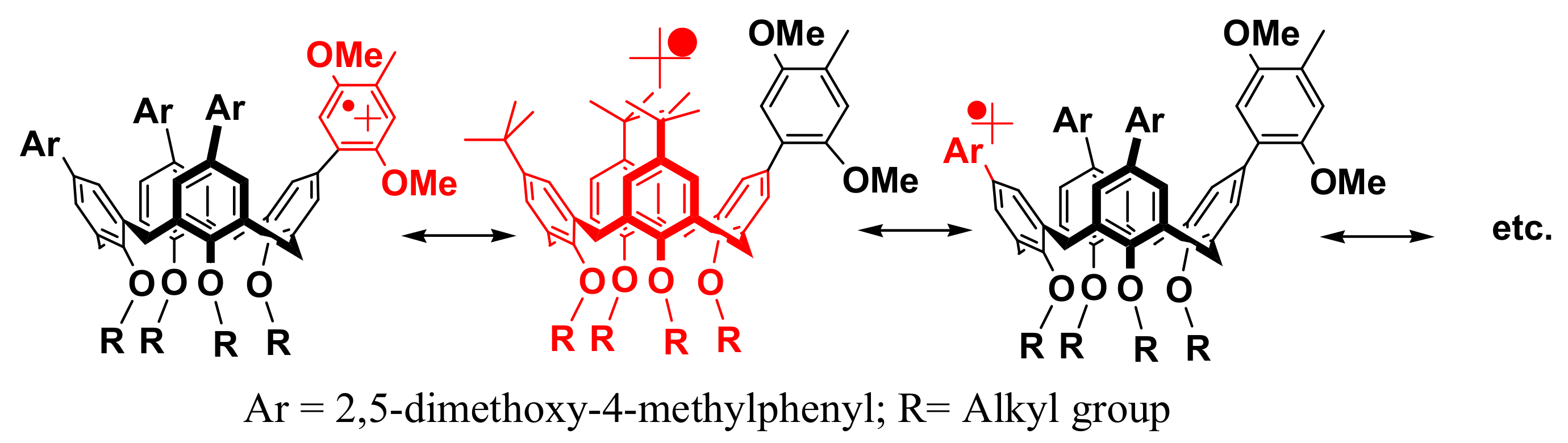
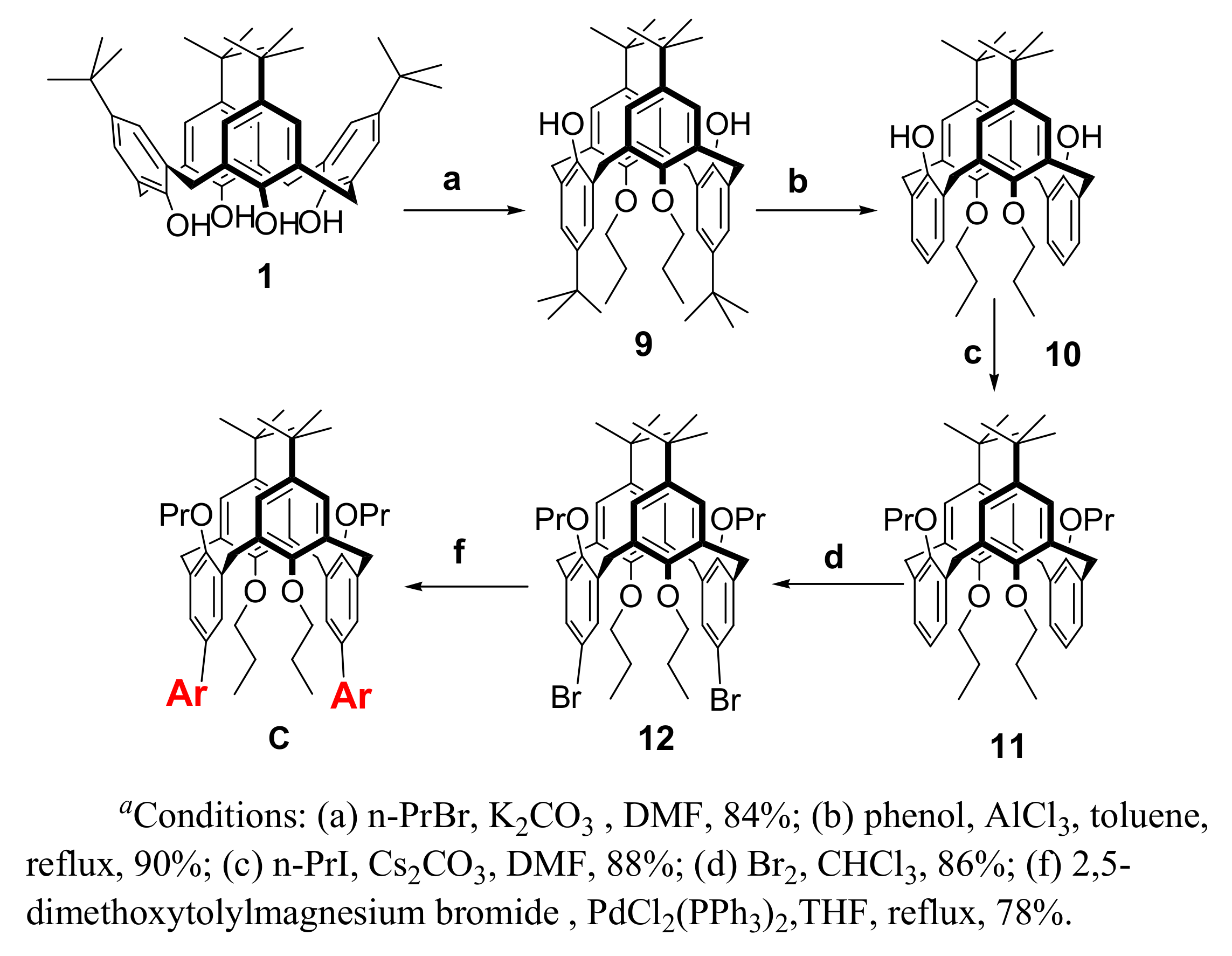
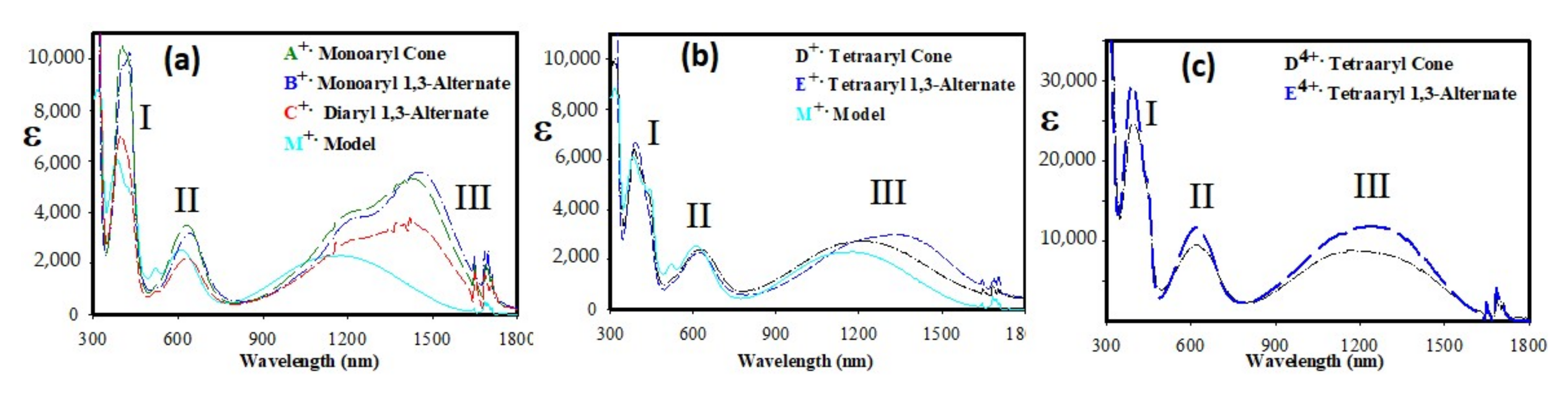
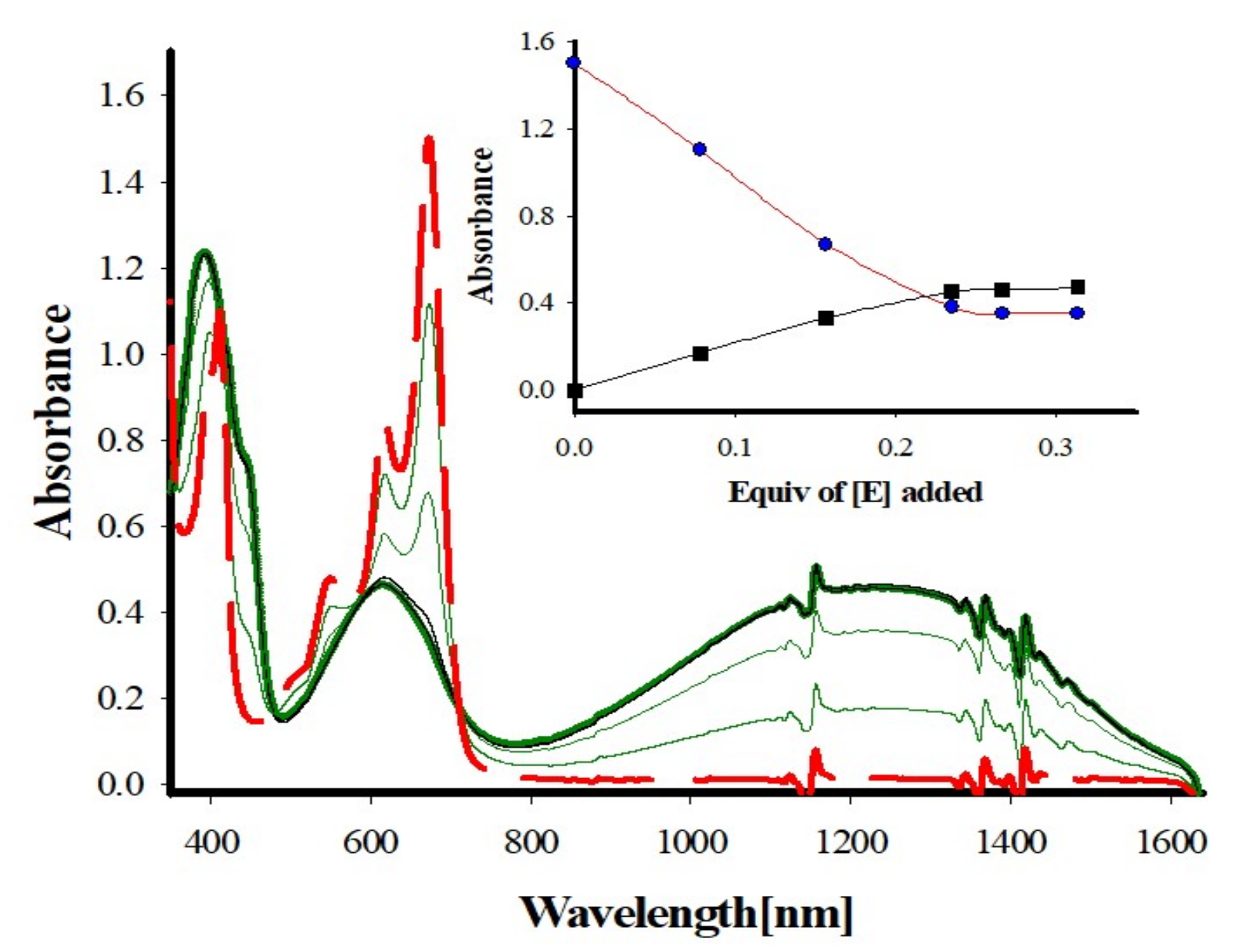
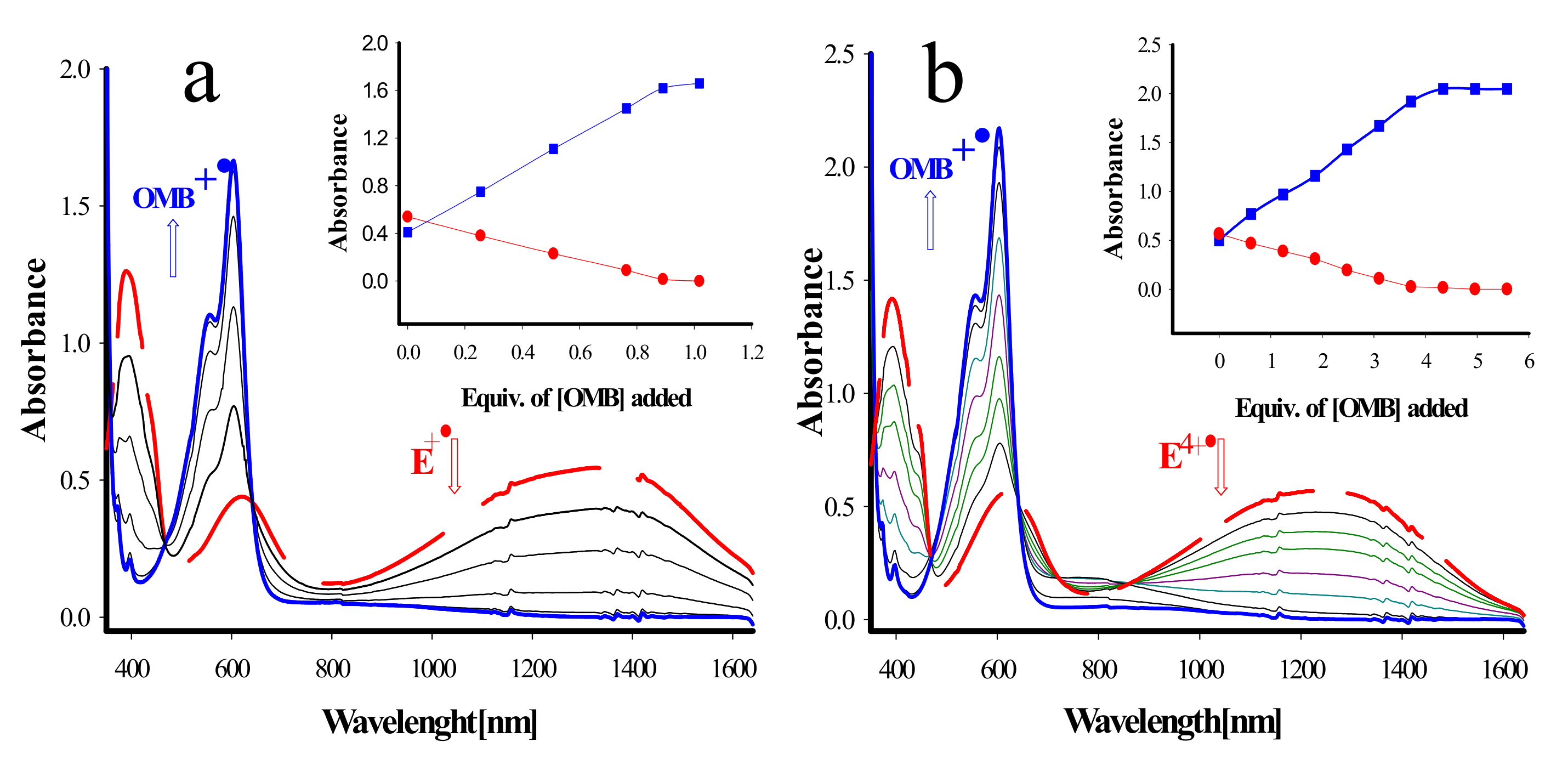
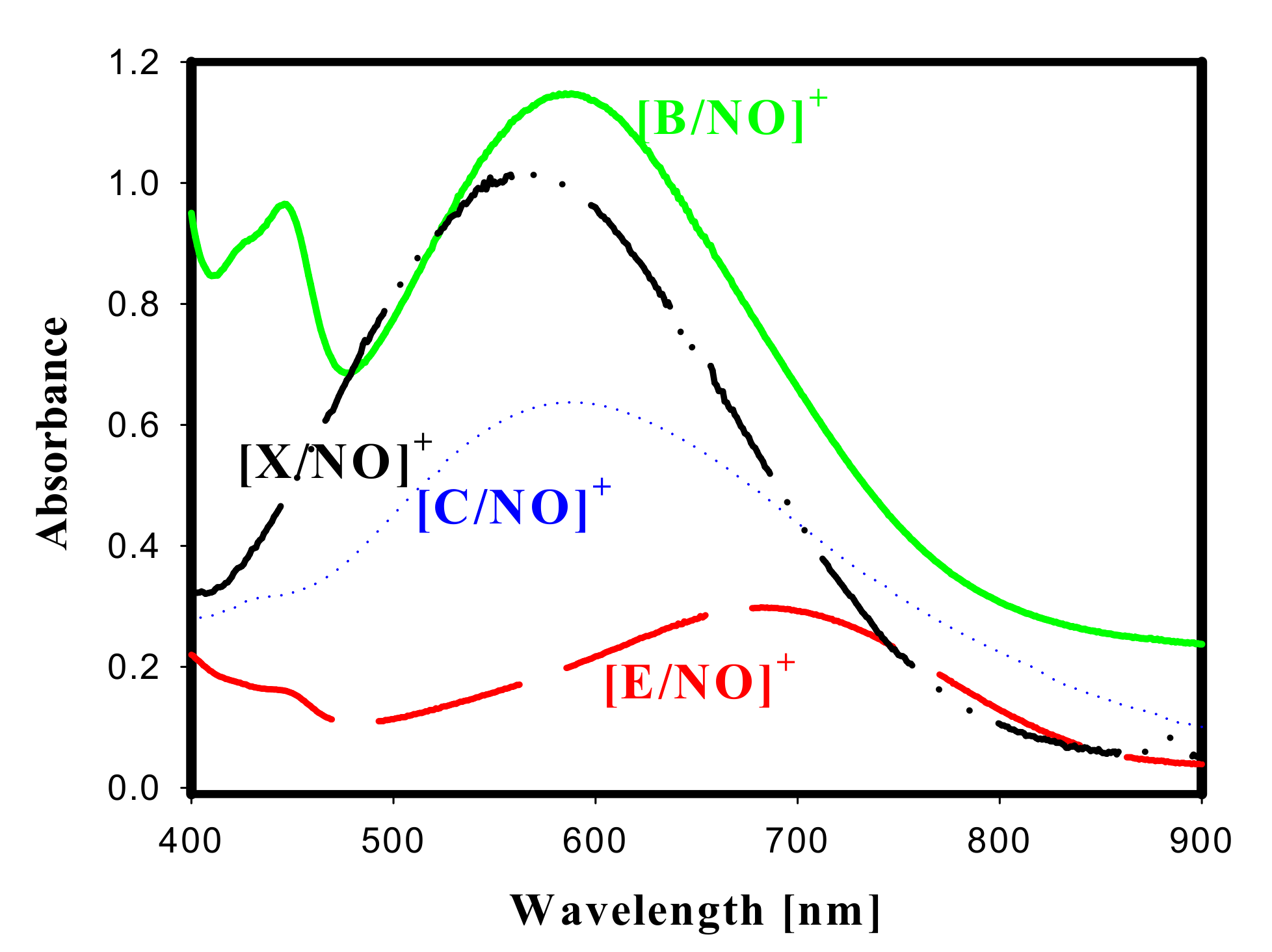
2. Results and Discussion
3. Materials and Methods
3.1. Cyclic Voltammetry
3.2. Oxidation of Various Calixarene Donors
3.2.1. Stoichiometric Oxidation of Tetraarylcalixarene Using MA+• as an Oxidant
3.2.2. Preparation of 1,2,3,4,7,8,9,10-octahydro-1,1,4,4,7,7,10,10-octamethylnaphthacene Cation Radical Salt (NAP+• SbCl6−)
3.2.3. Preparative Isolation of Stilbenoid Cation Radical Salts Using Et3O+ SbCl6−
3.3. Chemistry
3.4. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gutsche, C.D.; Stoddart, J.F. Monographs in Supramolecular Chemistry; Royal Society of Chemistry: Cambridge, UK, 1989; Volume 1. [Google Scholar]
- Makeiff, D.A.; Pope, D.J.; Sherman, J.C. Template Effects in the Formation of a Tetramethylene-Bridged Hemicarceplex. J. Am. Chem. Soc. 2000, 122, 1337–1342. [Google Scholar] [CrossRef]
- Ikeda, A.; Tsudera, T.; Shinkai, S. Molecular Design of a “Molecular Syringe” Mimic for Metal Cations Using a 1,3-Alternate Calix[4]arene Cavity. J. Org. Chem. 1997, 62, 3568–3574. [Google Scholar] [CrossRef]
- Rathore, R.; Lindeman, S.V.; Rao, K.S.S.P.; Sun, D.; Kochi, J.K. Guest Penetration Deep within the Cavity of Calix[4]arene Hosts: The Tight Binding of Nitric Oxide to Distal (Cofacial) Aromatic Groups. Angew. Chem. Int. Ed. 2000, 39, 2123–2127. [Google Scholar] [CrossRef]
- Cobben, P.L.H.M.; Egberink, R.J.M.; Bomer, J.G.; Bergveld, P.; Verboom, W.; Reinhoudt, D.N. Transduction of selective recognition of heavy metal ions by chemically modified field effect transistors (CHEMFETs). J. Am. Chem. Soc. 1992, 114, 10573–10582. [Google Scholar] [CrossRef]
- Mamardashvili, N.Z.; Koifman, O.I. Porphyrin-Calix[4]arenes. Russ. J. Org. Chem. 2005, 41, 787–806. [Google Scholar] [CrossRef]
- Verboom, W.; Vreekamp, R.H.; Rudkevich, D.M.; Reinhoudt, D.N. Functionalization and Application of Calixarenes. In Molecular Engineering for Advanced Materials; Becher, J., Schaumburg, K., Eds.; Springer: Dordrecht, The Netherlands, 1995; pp. 119–145. [Google Scholar]
- Beer, P.D.; Chen, Z.; Goulden, A.J.; Grieve, A.; Hesek, D.; Szemes, F.; Wear, T. Anion recognition by novel ruthenium(II) bipyridyl calix[4]arene receptor molecules. J. Chem. Soc. Chem. Commun. 1994, 10, 1269–1271. [Google Scholar] [CrossRef]
- Atwood, J.L.; Holman, K.T.; Steed, J.W. Laying traps for elusive prey: Recent advances in the non-covalent binding of anions. Chem. Commun. 1996, 12, 1401–1407. [Google Scholar] [CrossRef]
- Beer, P.D.; Drew, M.G.B.; Hesek, D.; Shade, M.; Szemes, F. Anion recognition properties of new-rim bis[rhenium(I) bipyridyl, ruthenium(II) bis(bipyridyl), cobaltocenium]calix[4]arene receptors dictated by lower-rim substituents. Chem. Commun. 1996, 18, 2161–2162. [Google Scholar] [CrossRef]
- Arduini, A.; Domiano, L.; Pochini, A.; Secchi, A.; Ungaro, R.; Ugozzoli, F.; Struck, O.; Verboom, W.; Reinhoudt, D.N. Synthesis of 1,2-bridged calix[4]arene-biscrowns in the 1,2-alternate conformation. Tetrahedron 1997, 53, 3767–3776. [Google Scholar] [CrossRef]
- Arduin, A.; Pochini, A.; Secchi, A.; Ugozzoli, F. Recognition of Neutral Molecules. In Calixarenes 2001; Asfari, Z., Böhmer, V., Harrowfield, J., Vicens, J., Saadioui, M., Eds.; Springer: Dordrecht, The Netherlands, 2001; pp. 457–475. [Google Scholar]
- Hailu, S.T.; Butcher, R.J.; Hudrlik, P.F.; Hudrlik, A.M. Crystal structure of a mono-bridged calix[4]arene. Acta Crystallogr. E Crystallogr. Commun. 2015, 71 Pt 7, 772–775. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Johnston, D.E., Jr.; Stewart, D.R. Pathways for the Reversion of p-tert-Butylcalix[8]arene top-tert-Butylcalix[4]arene(1). J. Org. Chem. 1999, 64, 3747–3750. [Google Scholar] [PubMed]
- Collins, E.M.; McKervey, M.A.; Madigan, E.; Moran, M.B.; Owens, M.; Ferguson, G.; Harris, S.J. Chemically modified calix[4]arenes. Regioselective synthesis of 1,3-(distal) derivatives and related compounds. X-Ray crystal structure of a diphenol-dinitrile. J. Chem. Soc. Perkin Trans. 1 1991, 12, 3137–3142. [Google Scholar] [CrossRef]
- Nishimura, Y.; Takemura, T.; Arai, S. Effective fluorescent sensing of Na ion by calix[4]arene bearing pyrene and perylene based on energy transfer. ARKIVOC 2007, 2007, 259–268. [Google Scholar]
- Verboom, W.; Bodewes, P.J.; van Essen, G.; Timmerman, P.; van Hummel, G.J.; Harkema, S.; Reinhoudt, D.N. A novel approach to inherently chiral calix[4]arenes by direct introduction of a substituent at the meta position. Tetrahedron 1995, 51, 499–512. [Google Scholar]
- Tsudera, T.; Ikeda, A.; Shinkai, S. Light-switched metal-tunneling across a π-basic tube of 1,3-alternate-calix[4]arenes. Tetrahedron 1997, 53, 13609–13620. [Google Scholar]
- Iwamoto, K.; Araki, K.; Shinkai, S. Syntheses of all possible conformational isomers of O-alkyl-p-t-butylcalix[4]arenes. Tetrahedron 1991, 47, 4325–4342. [Google Scholar]
- Shinkai, S.; Fujimoto, K.; Otsuka, T.; Ammon, H.L. Syntheses and ion selectivity of conformational isomers derived from calix[4]arene. J. Org. Chem. 1992, 57, 1516–1523. [Google Scholar]
- Gutsche, C.D.; Reddy, P.A. Calixarenes. 25. Conformations and structures of the products of arylmethylation of calix[4]arenes. J. Org. Chem. 1991, 56, 4783–4791. [Google Scholar]
- Iqbal, M.; Mangiafico, T.; Gutsche, C.D. Calixarenes 21: The conformations and structures of the products of aroylation of the, calix[4]arenes. Tetrahedron 1987, 43, 4917–4930. [Google Scholar]
- Verboom, W.; Datta, S.; Asfari, Z.; Harkema, S.; Reinhoudt, D.N. Tetra-O-alkylated calix[4]arenes in the 1,3-alternate conformation. J. Org. Chem. 1992, 57, 5394–5398. [Google Scholar] [CrossRef]
- Dijkstra, P.J.; Brunink, J.A.J.; Bugge, K.E.; Reinhoudt, D.N.; Harkema, S.; Ungaro, R.; Ugozzoli, F.; Ghidini, E. Kinetically stable complexes of alkali cations with rigidified calix[4]arenes: Synthesis, x-ray structures, and complexation of calixcrowns and calixspherands. J. Am. Chem. Soc. 1989, 111, 7567–7575. [Google Scholar] [CrossRef]
- Van Loon, J.D.; Groenen, L.C.; Wijmenga, S.S.; Verboom, W.; Reinhoudt, D.N. Upper rim calixcrowns: Elucidation of the mechanism of conformational interconversion of calix[4]arenes by quantitative 2-D EXSY NMR spectroscopy. J. Am. Chem. Soc. 1991, 113, 2378–2384. [Google Scholar]
- Alfieri, C.; Dradi, E.; Pochini, A.; Ungaro, R.; Andreetti, G.D. Synthesis, and X-ray crystal and molecular structure of a novel macro-bicyclic ligand: Crowned p-t-butyl-calix[4]arene. J. Chem. Soc. Chem. Commun. 1983, 19, 1075–1077. [Google Scholar] [CrossRef]
- Thuéry, P.; Nierlich, M.; Lamare, É.; Dozol, J.-F.; Asfari, Z.; Vicens, J. Bis(crown ether) and Azobenzocrown Derivatives of Calix[4]arene. A Review of Structural Information from Crystallographic and Modelling Studies. J. Incl. Phenom. Macrocycl. Chem. 2000, 36, 375–408. [Google Scholar]
- Ikeda, A.; Shinkai, S. On the Origin of High Ionophoricity of 1,3-Alternate Calix[4]arenes:.pi.-donor Participation in Complexation of Cations and Evidence for Metal-Tunneling through the Calix[4]arene Cavity. J. Am. Chem. Soc. 1994, 116, 3102–3110. [Google Scholar] [CrossRef]
- Xie, D.; Gutsche, C.D. Calixarene Anhydrides as Useful Synthetic Intermediates1. J. Org. Chem. 1997, 62, 2280–2284. [Google Scholar] [PubMed]
- Goldmann, H.; Vogt, W.; Paulus, E.; Boehmer, V. A series of calix[4]arenes, having two opposite para positions connected by an aliphatic chain. J. Am. Chem. Soc. 1988, 110, 6811–6817. [Google Scholar]
- Boehmer, V.; Vogt, W.; Goldmann, H.; McKervey, M.A.; Owens, M.; Cremin, S.; Collins, E.M. Control of alkali cation complexation in bridged calix[4]arene esters induced by small conformational changes. J. Org. Chem. 1990, 55, 2569–2570. [Google Scholar] [CrossRef]
- Mullins, S. Calixarenes. J. Chem. Technol. Biotechnol. 1991, 50, 293–294. [Google Scholar]
- Iki, H.; Kikuchi, T.; Shinkai, S. Synthesis and spectral characterization of tricarbonylchromium complexes of calix[4]arene conformers. J. Chem. Soc. Perkin Trans. 1 1992, 6, 669–671. [Google Scholar] [CrossRef]
- Rathore, R.; Abdelwahed, S.H.; Guzei, I.A. Synthesis of a Calix[4]arene Derivative for Isolation of a Stable Cation Radical Salt for Use as a Colorimetric Sensor of Nitric Oxide. J. Am. Chem. Soc. 2004, 126, 13582–13583. [Google Scholar] [CrossRef] [PubMed]
- McHale, D.; Mamalis, P.; Green, J.; Marcinkiewicz, S. 319. Tocopherols. Part I. Synthesis of 7-methyltocol (η-tocopherol). J. Chem. Soc. Resum. 1958, 1600–1603. [Google Scholar] [CrossRef]
- Creutz, C.; Newton, M.D.; Sutin, N. Metal—lingad and metal—metal coupling elements. J. Photochem. Photobiol. A Chem. 1994, 82, 47–59. [Google Scholar] [CrossRef]
- Rathore, R.; Kumar, A.S.; Lindeman, S.V.; Kochi, J.K. Preparation and Structures of Crystalline Aromatic Cation-Radical Salts. Triethyloxonium Hexachloroantimonate as a Novel (One-Electron) Oxidant. J. Org. Chem. 1998, 63, 5847–5856. [Google Scholar] [CrossRef]
- Laverman, L.E.; Wanat, A.; Oszajca, J.; Stochel, G.; Ford, P.C.; van Eldik, R. Mechanistic Studies on the Reversible Binding of Nitric Oxide to Metmyoglobin. J. Am. Chem. Soc. 2001, 123, 285–293. [Google Scholar] [CrossRef]
- Culotta, E.; Koshland, D.E. NO News Is Good News. Science 1992, 258, 1862–1865. [Google Scholar] [CrossRef]
- Hubig, S.M.; Rathore, R.; Kochi, J.K. Steric Control of Electron Transfer. Changeover from Outer-Sphere to Inner-Sphere Mechanisms in Arene/Quinone Redox Pairs. J. Am. Chem. Soc. 1999, 121, 617–626. [Google Scholar] [CrossRef]
- Wang, D.; Ivanova, L.V.; Ivanov, M.V.; Mirzaei, S.; Timerghazin, Q.K.; Reid, S.A.; Rathore, R. An Electron-Rich Calix[4]arene-Based Receptor with Unprecedented Binding Affinity for Nitric Oxide. Chemistry 2018, 24, 17439–17443. [Google Scholar] [CrossRef]
- Kochi, J.K.; Rathore, R.; Maguères, P.L. Stable Dimeric Aromatic Cation−Radicals. Structural and Spectral Characterization of Through-Space Charge Delocalization. J. Org. Chem. 2000, 65, 6826–6836. [Google Scholar] [CrossRef]
- Bruson, H.A.; Kroeger, J.W. Cycli-Alkylation of Aromatic Compounds by the Friedel and Crafts Reaction. J. Am. Chem. Soc. 1940, 62, 36–44. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calixarene | E1oxa | E2ox | E3ox | Calixarene | E1oxa | E2ox | E3ox | ||
|---|---|---|---|---|---|---|---|---|---|
| A |  | 1.08 | -- | -- | E |  | 1.07 | 1.25 | -- |
| B |  | 1.08 | -- | -- | W |  | 1.39 | -- | -- |
| C | 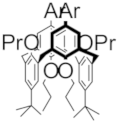 | 1.08 | 1.18 | -- | X | 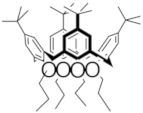 | 1.39 | -- | -- |
| D | 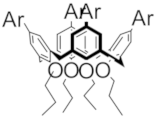 | 1.00 | 1.17 | 1.21 | M |  Model Model | 1.13 | -- | -- |
| Cation Radical (Calix+•) | Cation Radical (Calix+•) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Calixarene (Calix) | I, λmax (ε) | II, λmax (ε) | III, λmax (ε) | Calixarene (Calix) | I, λmax (ε) | II, λmax (ε) | III, λmax (ε) | ||
| A+• | 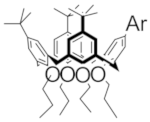 | 1420 (5600) | 620 (3600) | 370 (10,090) | E+• | 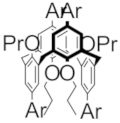 | 1300 (2950) | 620 (2100) | 375 (6500) |
| B+• |  | 1460 (5600) | 630 (3000) | 380 (10,000) | E4+• |  | 1240 (11,400) | 600 (11,370) | 400 (29,600) |
| C+• |  | 1340 (3500) | 632 (2300) | 388 (7000) | X |  | 440 | >1100 | -- |
| D+• | 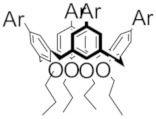 | 1200 (2600) | 626 (2210) | 376 (6300) | M |  Model Model | 1190 (2000) | 610 (2450) | 370 (6061) |
| D4+• | 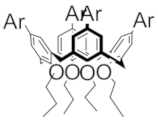 | 1180 (9800) | 600 (9810) | 400 (25,000) | Ar = 2,5-dimethoxy-4-methylphenyl | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rathore, R.; Lindeman, S.V.; Abdelwahed, S.H. Design, Synthesis, Electronic Properties, and X-ray Structural Characterization of Various Modified Electron-Rich Calixarene Derivatives and Their Conversion to Stable Cation Radical Salts. Molecules 2022, 27, 5994. https://doi.org/10.3390/molecules27185994
Rathore R, Lindeman SV, Abdelwahed SH. Design, Synthesis, Electronic Properties, and X-ray Structural Characterization of Various Modified Electron-Rich Calixarene Derivatives and Their Conversion to Stable Cation Radical Salts. Molecules. 2022; 27(18):5994. https://doi.org/10.3390/molecules27185994
Chicago/Turabian StyleRathore, Rajendra, Sergey V. Lindeman, and Sameh H. Abdelwahed. 2022. "Design, Synthesis, Electronic Properties, and X-ray Structural Characterization of Various Modified Electron-Rich Calixarene Derivatives and Their Conversion to Stable Cation Radical Salts" Molecules 27, no. 18: 5994. https://doi.org/10.3390/molecules27185994