


Mycobacterium Time-Series Genome Analysis Identifies AAC2′ as a Potential Drug Target with Naloxone Showing Potential Bait Drug Synergism
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
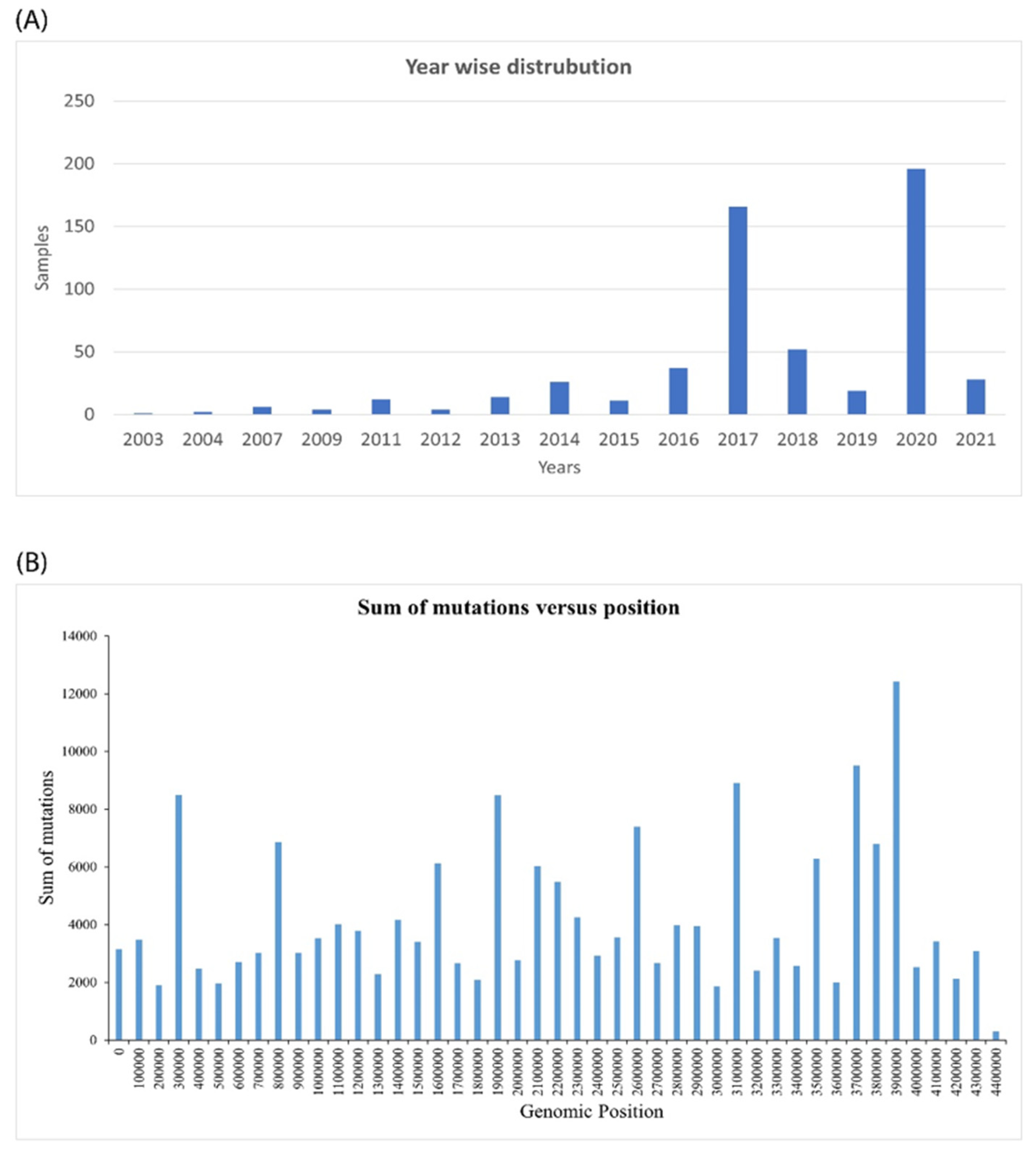
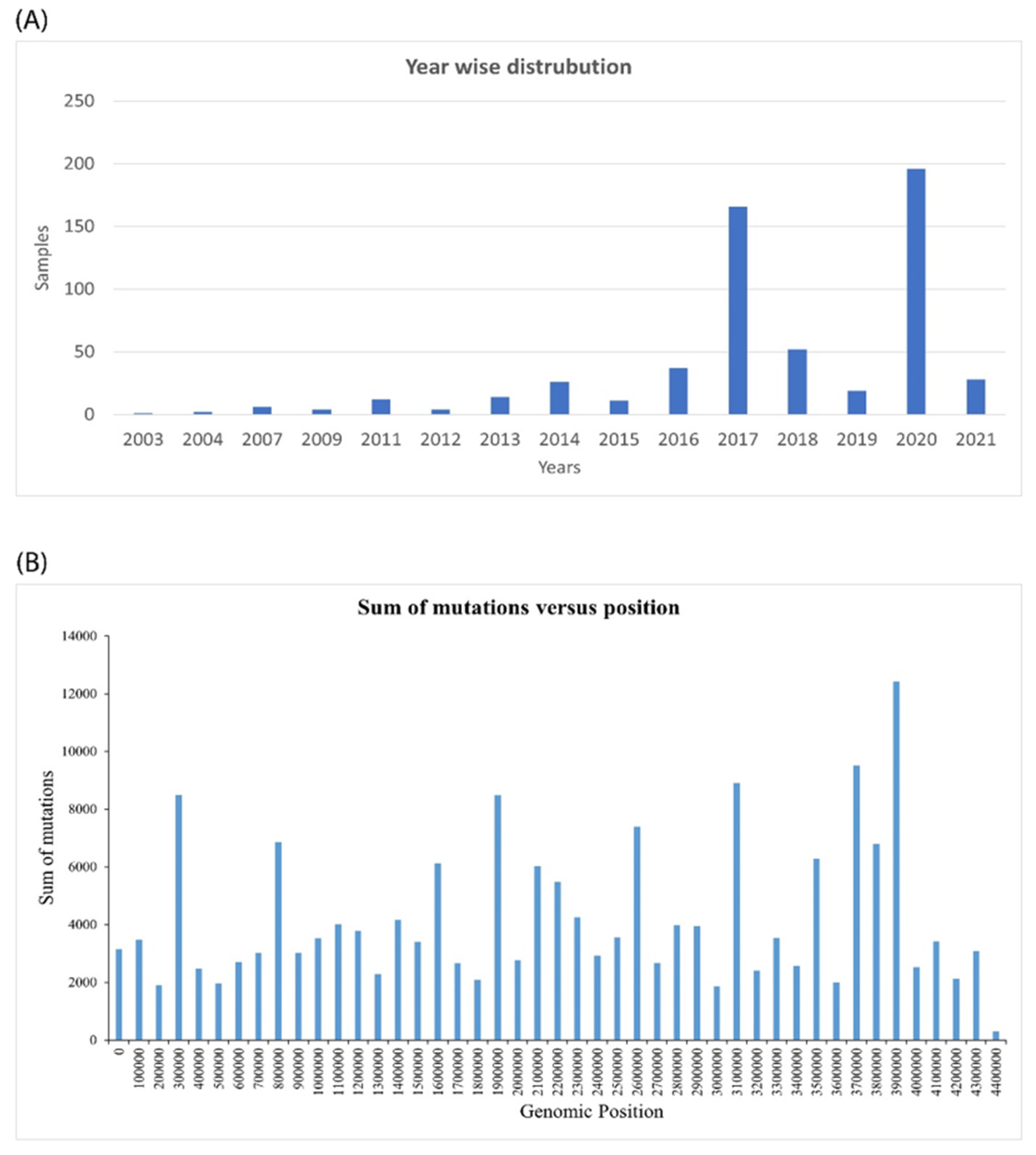
2.1. Genomic Analysis of Mycobacterium Tuberculosis Provides Insights into Mutation Patterns
2.2. 369 Genes with Unknown and Hypothetical Functions Were Annotated for Better Functional Insights and Target Selection
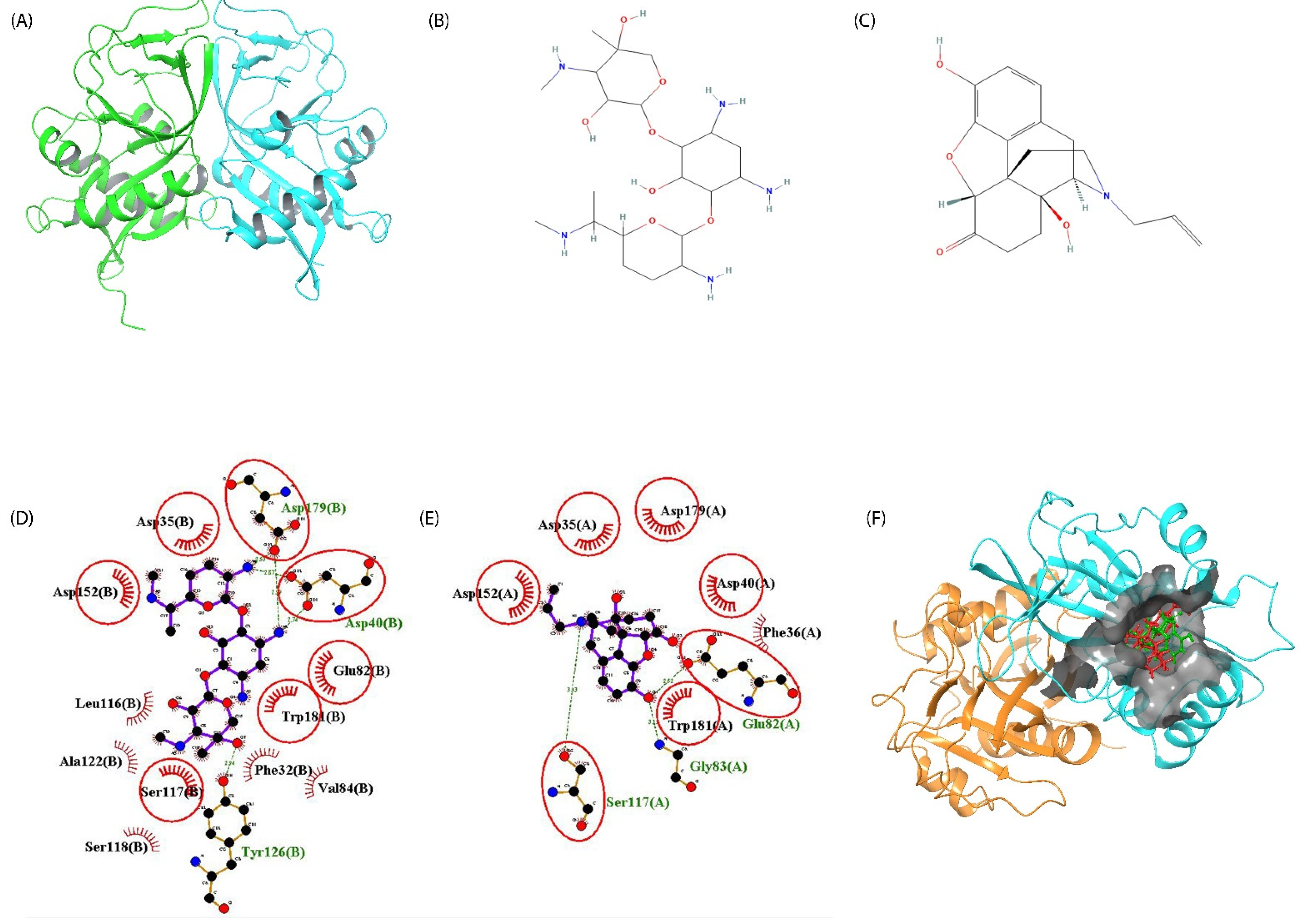
2.3. Aminoglycoside 2′-N-Acetyltransferase (AAC2′): Mode of Action toward the Resistance of Aminoglycosides
2.4. Screening of Phyto-Actives as Potential Drug Candidates with Potential Features as Aminoglycosides
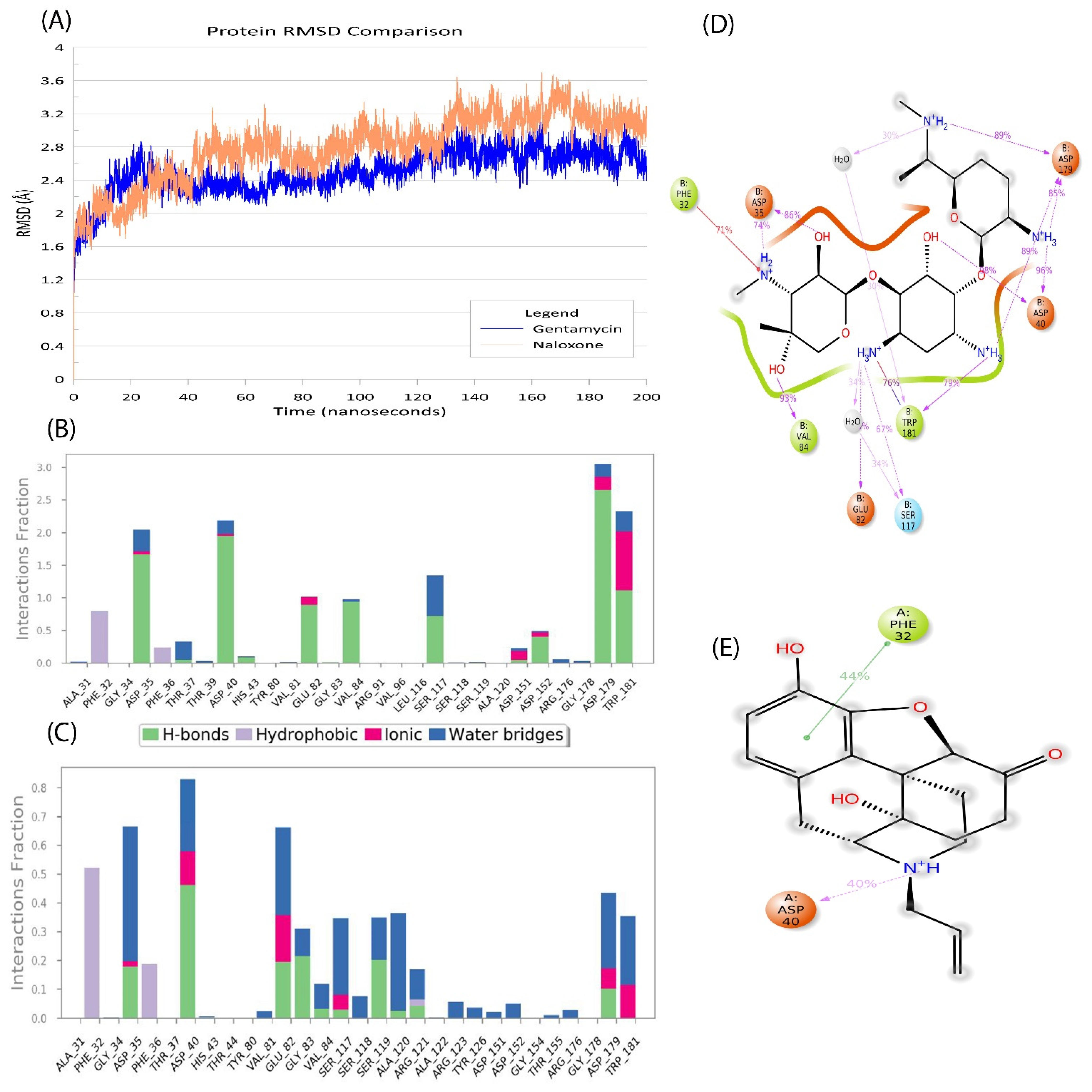
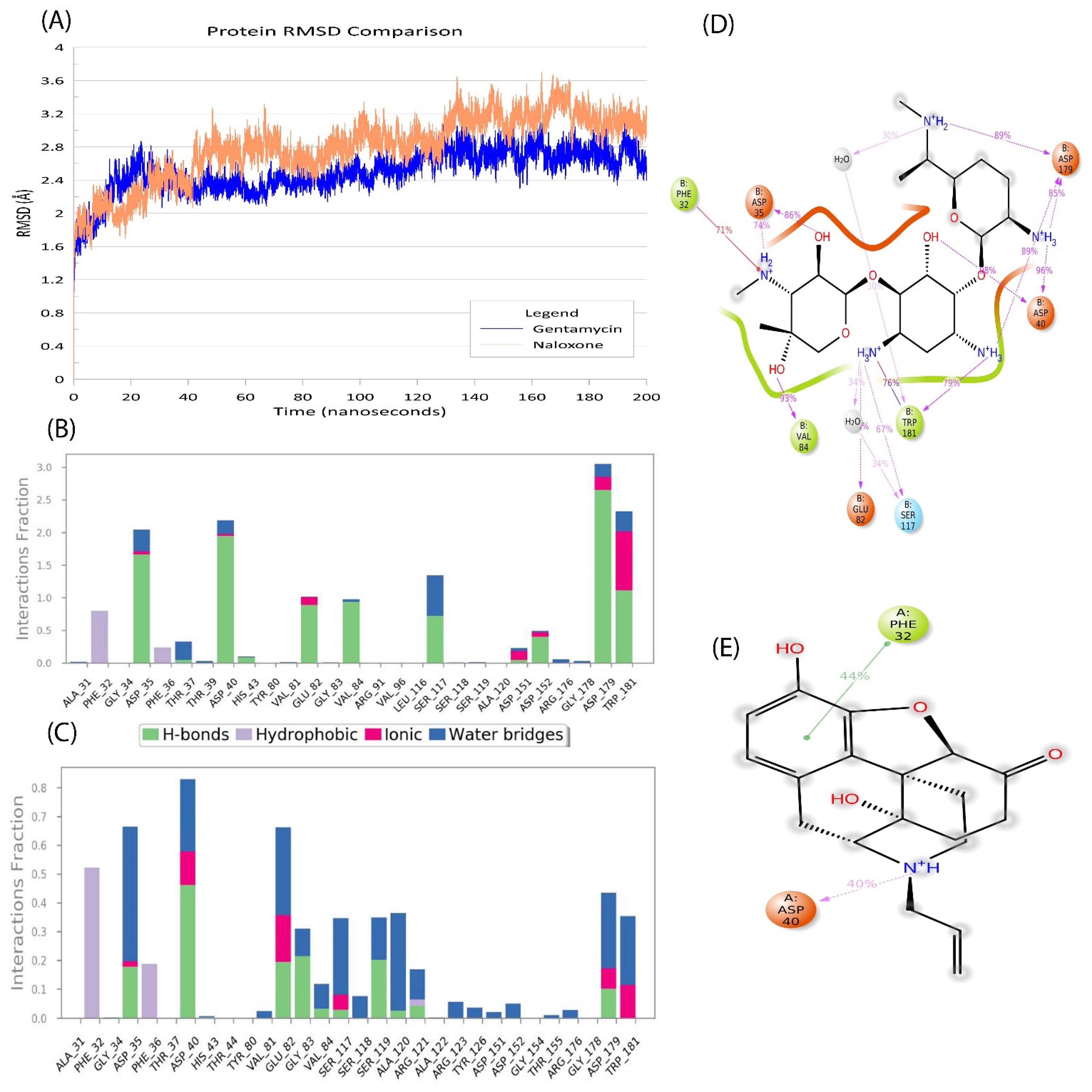
2.5. Naloxone–AAC2′ Interaction Profile as a Bait Drug Synergistic Effect with Aminoglycosides
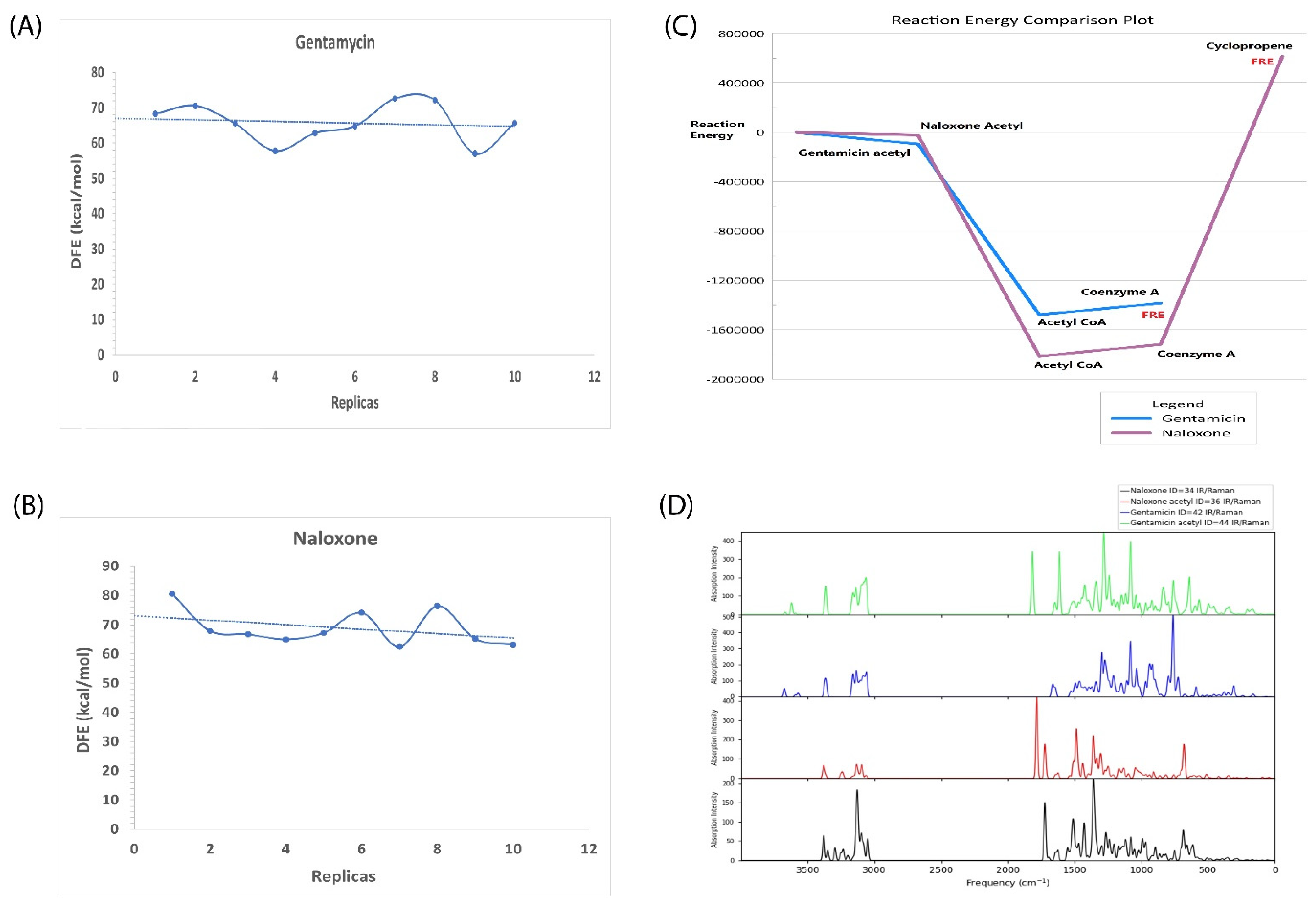
2.6. In Silico Validation of Naloxone Acetylation to Be Highly Unstable and Disordered
3. Discussion
4. Materials and Methods
4.1. Retrieving the Data from Public Datasets
4.2. Mapping of Samples with Reference Genomes and Variant Calling
Variants Calling
4.3. Annotation of Variants
4.4. Functional Annotation
- (a)
- Downloading the Genomic Sequences
- (b)
- InterPro Scan
- (c)
- NCBI BLAST Search
- (d)
- GO Mapping and Annotations
- (e)
- Merge Results from InterPro and GO Annotations
- (f)
- Functional Clustering of Genes
4.5. Molecular Docking
- (a)
- Protein Selection and Preparation
- (b)
- Ligand Preparation.
4.6. Feasibility of Reaction Studies
4.7. Molecular Dynamics Simulation Studies
4.8. Well-Tempered Metadynamics Simulation Studies
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keshavjee, S.; Farmer, P.E. Tuberculosis, Drug Resistance, and the History of Modern Medicine. N. Engl. J. Med. 2012, 367, 931–936. [Google Scholar] [CrossRef]
- Fogel, N. Tuberculosis: A disease without boundaries. Tuberculosis 2015, 95, 527–531. [Google Scholar] [CrossRef]
- Comas, I.; Gagneux, S. The Past and Future of Tuberculosis Research. PLOS Pathog. 2009, 5, e1000600. [Google Scholar] [CrossRef]
- Murray, J.F.; Nadel, J.A.; Mason, R.J.; Broaddus, V.C.; Martin, T.R.; King, T.E. Murray and Nadel’s Textbook of Respiratory Medicine; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Berry, M.P.R.; Blankley, S.; Graham, C.; Bloom, C.; O’Garra, A. Systems approaches to studying the immune response in tuberculosis. Curr. Opin. Immunol. 2013, 25, 579–587. [Google Scholar] [CrossRef]
- Prasanna, A.; Niranjan, V. Classification of Mycobacterium tuberculosis DR, MDR, XDR Isolates and Identification of Signature MutationPattern of Drug Resistance. Bioinformation. 2019, 15, 261–268. [Google Scholar] [CrossRef]
- Elliott, A.; Beming, S.; Iseman, M.; Peloquin, C. Failure of drug penetration and acquisition of drug resistance in chronic tuberculous empyema. Tuber. Lung Dis. 1995, 76, 463–467. [Google Scholar] [CrossRef]
- Drake, J.W. The Distribution of Rates of Spontaneous Mutation over Viruses, Prokaryotes, and Eukaryotes. Ann. N. Y. Acad. Sci. 1999, 870, 100–107. [Google Scholar] [CrossRef]
- Anonymous. Five-Year Follow-Up of a Controlled Trial of Five 6-Month Regimens of Chemotherapy for Pulmonary Tuberculosis. Am. Rev. Respir. Dis. 1987, 136, 1339–1342. [Google Scholar] [CrossRef]
- Heep, M.; Rieger, U.; Beck, D.; Lehn, N. Mutations in the Beginning of the rpoB Gene Can Induce Resistance to Rifamycins in both Helicobacter pylori and Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2000, 44, 1075–1077. [Google Scholar] [CrossRef]
- Honoré, N.; Cole, S.T. Streptomycin resistance in mycobacteria. Antimicrob. Agents Chemother. 1994, 38, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Heym, B.; Alzari, P.; Honore, N.; Cole, S.T. Missense mutations in the catalase-peroxidase gene, katG, are associated with isoniazid resistance in Mycobacterium tuberculosis. Mol. Microbiol. 1995, 15, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, N.; Sougakoff, W.; Truffot-Pernot, C.; Jarlier, V. Characterization of new mutations in pyrazinamide-resistant strains of Mycobacterium tuberculosis and identification of conserved regions important for the catalytic activity of the pyrazinamidase PncA. Antimicrob. Agents Chemother. 1999, 43, 1761–1763. [Google Scholar] [CrossRef]
- David, H.L. Probability distribution of drug-resistant mutants in unselected populations of Mycobacterium tuberculosis. Appl. Microbiol. 1970, 20, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Seung, K.J.; Gelmanova, I.E.; Peremitin, G.G.; Golubchikova, V.T.; Pavlova, V.E.; Sirotkina, O.B.; Yanova, G.; Strelis, A.K. The Effect of Initial Drug Resistance on Treatment Response and Acquired Drug Resistance during Standardized Short-Course Chemotherapy for Tuberculosis. Clin. Infect. Dis. 2004, 39, 1321–1328. [Google Scholar] [CrossRef]
- Gygli, S.M.; Borrell, S.; Trauner, A.; Gagneux, S. Antimicrobial resistance in Mycobacterium tuberculosis: Mechanistic and evolutionary perspectives. FEMS Microbiol. Rev. 2017, 41, 354–373. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.; Woodworth, J.S. Tuberculosis vaccines—Rethinking the current paradigm. Trends Immunol. 2014, 35, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Thillai, M.; Pollock, K.; Pareek, M.; Lalvani, A. Interferon-gamma release assays for tuberculosis: Current and future applications. Expert Rev. Respir. Med. 2013, 8, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, A.; Niranjan, V. MutVis: Automated framework for analysis and visualization of mutational signatures in pathogenic bacterial strains. Infect. Genet. Evol. 2021, 91, 104805. [Google Scholar] [CrossRef]
- Lew, J.M.; Kapopoulou, A.; Jones, L.M.; Cole, S.T. TubercuList-10 years after. Tuberculosis 2011, 91, 1–7. [Google Scholar] [CrossRef]
- Alampalli, S.V.; Uttarkar, A.; Ventakesh, S.; Chettinar, S.T.; Nageshan, R.K.; Niranjan, V.; Tatu, U.S. Genome Resequencing Reveals Single Nucleotide Polymorphism and Repeat Regions in Giardia lamblia Indian Isolate. J. Next Gener. Seq. Appl. 2017, 4, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Aínsa, J.A.; Pérez, E.; Pelicic, V.; Berthet, F.X.; Gicquel, B.; Martín, C. Aminoglycoside 2′-N -acetyltransferase genes are universally present in mycobacteria: Characterization of the aac-Ic gene from Mycobacterium tuberculosis and the aac(2)-Id gene from Mycobacterium smegmatis. Molecular Microbiol. 1997, 24, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Rather, P.N.; Orosz, E.; Shaw, K.J.; Hare, R.; Miller, G. Characterization and transcriptional regulation of the 2’-N-acetyltransferase gene from Providencia stuartii. J. Bacteriol. 1993, 175, 6492–6498. [Google Scholar] [CrossRef] [PubMed]
- Payie, K.G.; Clarke, A.J. Characterization of gentamicin 2’-N-acetyltransferase from Providencia stuartii: Its use of peptidoglycan metabolites for acetylation of both aminoglycosides and peptidoglycan. J. Bacteriol. 1997, 179, 4106–4114. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist Updat. 2010, 13, 151–171. [Google Scholar] [CrossRef]
- Franklin, K.; Clarke, A.J. Overexpression and Characterization of the Chromosomal Aminoglycoside 2′- N -Acetyltransferase of Providencia stuartii. Antimicrob. Agents Chemother. 2001, 45, 2238–2244. [Google Scholar] [CrossRef] [PubMed]
- Zárate, S.G.; De la Cruz Claure, M.L.; Benito-Arenas, R.; Revuelta, J.; Santana, A.G.; Bastida, A. Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors. Molecules 2018, 23, 284. [Google Scholar] [CrossRef]
- D’Costa, V.; Wright, G.D. Biochemical Logic of Antibiotic Inactivation and Modification. In Antimicrobial Drug Resistance. Infectious Disease; Humana Press: Totowa, NJ, USA, 2009; pp. 81–95. [Google Scholar] [CrossRef]
- Srivastava, P.; Khandokar, Y.B.; Swarbrick, C.M.D.; Roman, N.; Himiari, Z.; Sarker, S.; Raidal, S.; Forwood, J.K. Structural Characterization of a Gcn5-Related N-Acetyltransferase from Staphylococcus aureus. PLoS ONE 2014, 9, e102348. [Google Scholar] [CrossRef]
- Smith, C.A.; Bhattacharya, M.; Toth, M.; Stewart, N.K.; Vakulenko, S.B. Aminoglycoside resistance profile and structural architecture of the aminoglycoside acetyltransferase AAC(6′)-Im. Microb. Cell 2017, 4, 402–410. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Skariyachan, S.; Muddebihalkar, A.G.; Badrinath, V.; Umashankar, B.; Eram, D.; Uttarkar, A.; Niranjan, V. Natural epiestriol-16 act as potential lead molecule against prospective molecular targets of multidrug resistant Acinetobacter baumannii-Insight from in silico modelling and in vitro investigations. Infect. Genet. Evol. 2020, 82, 104314. [Google Scholar] [CrossRef]
- Skariyachan, S.; Ravishankar, R.; Gopal, D.; Muddebihalkar, A.G.; Uttarkar, A.; Praveen, P.K.U.; Niranjan, V. Response regulator GacA and transcriptional activator RhlR proteins involved in biofilm formation of Pseudomonas aeruginosa are prospective targets for natural lead molecules: Computational modelling, molecular docking and dynamic simulation studies. Infect. Genet. Evol. 2020, 85, 104448. [Google Scholar] [CrossRef]
- Patagar, D.; Uttarkar, A.; Patra, S.M.; Patil, J.H.; Kusanur, R.; Niranjan, V.; Kumar, H.G.A. Spiro Benzodiazepine Substituted Fluorocoumarins as Potent Anti-Anxiety Agents. Russ. J. Bioorganic Chem. 2021, 47, 390–398. [Google Scholar] [CrossRef]
- Niranjan, V.; Rao, A.; Janaki, B.; Uttarkar, A.; Setlur, A.S.; Chandrashekar, K.; Udayakumar, M. Molecular Docking and Interaction Studies of Identified Abscisic Acid Receptors in Oryza sativa: An In-Silico Perspective on Comprehending Stress Tolerance Mechanisms. Curr. Genom. 2021, 22, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Laio, A. Using Meta-dynamics to explore complex free-energy landscapes. Nature Reviews Physics. 2020, 2, 200–212. [Google Scholar] [CrossRef]
- Raniolo, S.; Limongelli, V. Ligand binding free-energy calculations with funnel Meta-dynamics. Nature Protocols. 2020, 15, 2837–2866. [Google Scholar] [CrossRef]
- Labby, K.J.; Tsodikova, G.S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med. Chem. 2013, 5, 1–40. [Google Scholar] [CrossRef]
- Sanz-García, F.; Anoz-Carbonell, E.; Pérez-Herrán, E.; Martín, C.; Lucía, A.; Rodrigues, L.; Aínsa, J.A. Mycobacterial Aminoglycoside Acetyltransferases: A Little of Drug Resistance, and a Lot of Other Roles. Front. Microbiol. 2019, 10, 46. [Google Scholar] [CrossRef]
- Smith, T.; Wolff, K.A.; Nguyen, L. Molecular Biology of Drug Resistance in Mycobacterium tuberculosis. In Pathogenesis of Mycobacterium tuberculosis and its Interaction with the Host Organism; Springer: Berlin/Heidelberg, Germany, 2012; Volume 374, pp. 53–80. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol Spectr. 2016, 4, 1–37. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Xu, Y. BitMapperBS: A fast and accurate read aligner for whole-genome bisulfite sequencing. Cold Spring Harbor Laboratory. 2018. Available online: https://github.com/BioInfoTools/BBMap (accessed on 24 February 2022).
- Križanović, K.; Echchiki, A.; Roux, J.; Šikić, M. Evaluation of tools for long read RNA-seq splice-aware alignment. Bioinformatics 2017, 34, 748–754. [Google Scholar] [CrossRef]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 1–19. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- OmicsBox—Bioinformatics Made Easy. Available online: https://www.biobam.com/omicsbox (accessed on 1 December 2021).
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. -Aided Mol. Design. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Pa, V.; Vijayaraghavareddy, P.; Uttarkar, A.; Dawane, A.; Sujitha, D.; Ashwin, V.; Babitha, K.C.; Niranjan, V.; Ms, S.; Cv, A.; et al. Novel small molecules targeting bZIP23 TF improve stomatal conductance and photosynthesis under mild drought stress by regulating ABA. FEBS J. 2022. [Google Scholar] [CrossRef]
- Uttarkar, A.; Niranjan, V. Re-profiling of natural inhibitor via combinatorial drug screening: Brefeldin A variant design as an effective antagonist leading to EPAC2 structure modification and antibody design for identification. bioRxiv 2021. [Google Scholar] [CrossRef]
- Uttarkar, A.; Niranjan, V. Brefeldin A variant via combinatorial screening acts as an effective antagonist inducing structural modification in EPAC2. Mol. Simul. 2022, 1–12. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- PubChem Compound Summary for CID 37768, Amikacin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Amikacin (accessed on 21 May 2022).
- PubChem Compound Summary for CID 3467, Gentamicin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Gentamicin (accessed on 21 May 2022).
- PubChem Compound Summary for CID 6032, Kanamycin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Kanamycin (accessed on 21 May 2022).
- PubChem Compound Summary for CID 8378, Neomycin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Neomycin (accessed on 21 May 2022).
- PubChem Compound Summary for CID 165580, Paromomycin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Paromomycin (accessed on 21 May 2022).
- PubChem Compound Summary for CID 42613186, Plazomicin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Plazomicin (accessed on 21 May 2022).
- PubChem Compound Summary for CID 19649, Streptomycin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Streptomycin (accessed on 21 May 2022).
- PubChem Compound Summary for CID 36294, Tobramycin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Tobramycin (accessed on 21 May 2022).
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-ype Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sac-erdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; Association for Computing Machinery, New York, NY, USA, 11 November 2006. [Google Scholar]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Uttarkar, A.; Kishore, A.P.; Srinivas, S.M.; Rangappa, S.; Kusanur, R.; Niranjan, V. Coumarin derivative as a potent drug candidate against triple negative breast cancer targeting the frizzled receptor of wingless-related integration site signaling pathway. J. Biomol. Struct. Dyn. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gopal, D.; Muddebihalkar, A.G.; Skariyachan, S.; Akshay Uttarkar, C.; Kaveramma, P.; Praveen, U.; Shankar, R.R.; Venkatesan, T.; Niranjan, V. Mitogen activated protein kinase-1 and cell division control protein-42 are putative targets for the binding of novel natural lead molecules: A therapeutic intervention against Candida albicans. J. Biomol. Struct. Dyn. 2019, 38, 4584–4599. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef]
- Niranjan, V.; Uttarkar, A. Well-Tempered Meta-Dynamics Protocol v2; ZappyLab, Inc.: Berkeley, CA, USA, 2022; 3p. [Google Scholar]
- Wang, J.; Ishchenko, A.; Zhang, W.; Razavi, A.; Langley, D. A highly accurate Meta-dynamics-based Dissociation Free Energy method to calculate protein-protein and protein-ligand binding potencies. Sci. Rep. 2022, 12, 2024. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl No. | Type of Variation | Percentage | Count |
|---|---|---|---|
| 1 | Conservative inframe insertion | 0% | 19 |
| 2 | Disruptive inframe deletion | 0% | 1 |
| 3 | Disruptive inframe insertion | 0% | 3 |
| 4 | Downstream gene variant | 44.91% | 2,103,270 |
| 5 | Frameshift variant | 0.00% | 125 |
| 6 | Initiator codon variant | 0.00% | 183 |
| 7 | Intergenic region | 1.00% | 46,659 |
| 8 | Intragenic variant | 0.09% | 4113 |
| 9 | Missense variant | 6.99% | 327,309 |
| 10 | Splice region variant | 0.03% | 1560 |
| 11 | Start lost | 0.02% | 818 |
| 12 | Start retained variant | 0% | 13 |
| 13 | Stop gained | 0.15% | 7177 |
| 14 | Stop lost | 0.03% | 1444 |
| 15 | Stop retained variant | 0.00% | 116 |
| 16 | Synonymous variant | 1.93% | 90,276 |
| 17 | Upstream gene variant | 44.84% | 2,099,823 |
| Protein Name | Gene ID | High Impact Mutations | Protein Function | Pathway | |
|---|---|---|---|---|---|
| 1 | pks2 | Mb3855c | 46 | Function unknown; supposedly involved in lipid metabolism. | Lipid metabolism |
| 2 | pks1 | Mb2971c | 45 | Polyketide synthase possibly involved in lipid synthesis. | Lipid metabolism |
| 3 | Mb3933c | Mb3933c | 40 | Hypothetical alanine and proline-rich protein. | Conserved hypotheticals |
| 4 | mmpL8 | Mb3853c | 40 | Thought to be involved in the transport of lipids; it is required in the production of a sulfated glycolipid, sulfolipid-1 (SL-1). | Cell wall and cell processes |
| 5 | pks13 | Mb3830c | 38 | Involved in the final steps of mycolic acid biosynthesis. Catalyzes the condensation of two fatty acyl chains. | Lipid metabolism |
| 6 | gltB | Mb3889c | 37 | Probably involved in glutamate biosynthesis [catalytic activity: 2 L-glutamate + NADP(+) = L-glutamine + 2-oxoglutarate + NADPH]. | Intermediary metabolism and respiration |
| 7 | pks12 | Mb2074c | 33 | Involved in the biosynthesis of mannosyl-beta-1-phosphomycoketide (MPM). | Lipid metabolism |
| 8 | Mb3018 | Mb3018 | 31 | Unknown; COULB is involved in the efflux system (possibly drug). | Cell wall and cell processes |
| 9 | embA | Mb3823 | 30 | Involved in the biosynthesis of the mycobacterial cell wall arabinan and resistance to ethambutol (EMB; Dextro-2,2′-(ethylenediimino)-DI-1-butanol). Polymerizes arabinose into the arabinan of arabinogalactan [catalytic activity: UDP-L-arabinose + indol-3-ylacetyl-Myo-inositol = UDP + indol-3-ylacetyl-myo-inositol L-arabinoside]. | Cell wall and cell processes |
| 10 | glnE | Mb2245c | 29 | Regulatory protein is involved in the regulation of glutamine synthetase activity. Adenylation and deadenylation of glutamine synthetase. Possibly regulates GLNB|Rv2919c [catalytic activity: ATP + [L-glutamate:ammonia ligase (ADP-forming)] = pyrophosphate + adenylyl-[L-glutamate:ammonia ligase (ADP-forming)]]. | Intermediary metabolism and respiration |
| Sl No. | Protein Function Category | Number of Proteins |
|---|---|---|
| 1 | Cell wall and cell processes | 305 |
| 2 | Conserved hypotheticals | 588 |
| 3 | Information pathways | 35 |
| 4 | Insertion seqs and phages | 39 |
| 5 | Intermediary metabolism and respiration | 371 |
| 6 | Lipid metabolism | 105 |
| 7 | PE/PPE | 40 |
| 8 | Regulatory proteins | 87 |
| 9 | Stable RNAs | 34 |
| 10 | Unknown | 2 |
| 11 | Virulence, detoxification, adaptation | 37 |
| Gentamycin | Asp40 | Tyr126 | Asp179 | |||
|---|---|---|---|---|---|---|
| Atom | Atom | Distance (Å) | Atoms | Distance (Å) | Atom | Distance (Å) |
| N2 | OD1 | 2.74 | OD2 | 2.99 | ||
| N4 | OD2 | 2.87 | OD2 | 2.53 | ||
| O7 | OH | 2.94 | ||||
| Naloxone | Glu82 | Gly83 | Ser117 | |||
| O1 | OE1 | 2.62 | N | 3.12 | ||
| N1 | OG | 3.03 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niranjan, V.; Uttarkar, A.; Murali, K.; Niranjan, S.; Gopal, J.; Kumar, J. Mycobacterium Time-Series Genome Analysis Identifies AAC2′ as a Potential Drug Target with Naloxone Showing Potential Bait Drug Synergism. Molecules 2022, 27, 6150. https://doi.org/10.3390/molecules27196150
Niranjan V, Uttarkar A, Murali K, Niranjan S, Gopal J, Kumar J. Mycobacterium Time-Series Genome Analysis Identifies AAC2′ as a Potential Drug Target with Naloxone Showing Potential Bait Drug Synergism. Molecules. 2022; 27(19):6150. https://doi.org/10.3390/molecules27196150
Chicago/Turabian StyleNiranjan, Vidya, Akshay Uttarkar, Keerthana Murali, Swarna Niranjan, Jayalatha Gopal, and Jitendra Kumar. 2022. "Mycobacterium Time-Series Genome Analysis Identifies AAC2′ as a Potential Drug Target with Naloxone Showing Potential Bait Drug Synergism" Molecules 27, no. 19: 6150. https://doi.org/10.3390/molecules27196150
APA StyleNiranjan, V., Uttarkar, A., Murali, K., Niranjan, S., Gopal, J., & Kumar, J. (2022). Mycobacterium Time-Series Genome Analysis Identifies AAC2′ as a Potential Drug Target with Naloxone Showing Potential Bait Drug Synergism. Molecules, 27(19), 6150. https://doi.org/10.3390/molecules27196150