
Bioactive Diarylpentanoids: Insights into the Biological Effects beyond Antitumor Activity and Structure–Activity Relationships


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
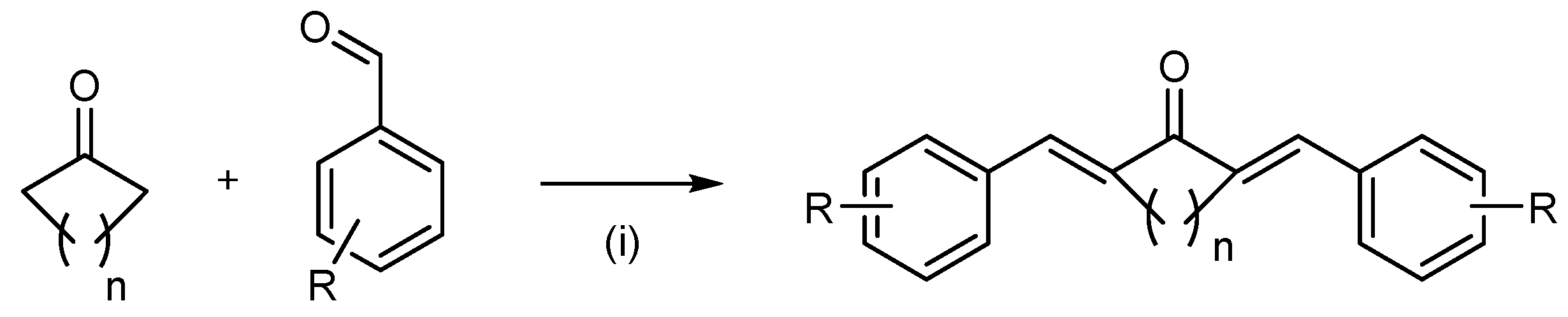
2. Synthesis of the Diarylpentanoids
3. Biological Activity of Diarylpentanoids
3.1. Anti-Infective Activity
3.1.1. Antibacterial Activity
3.1.2. Antiparasitic Activity
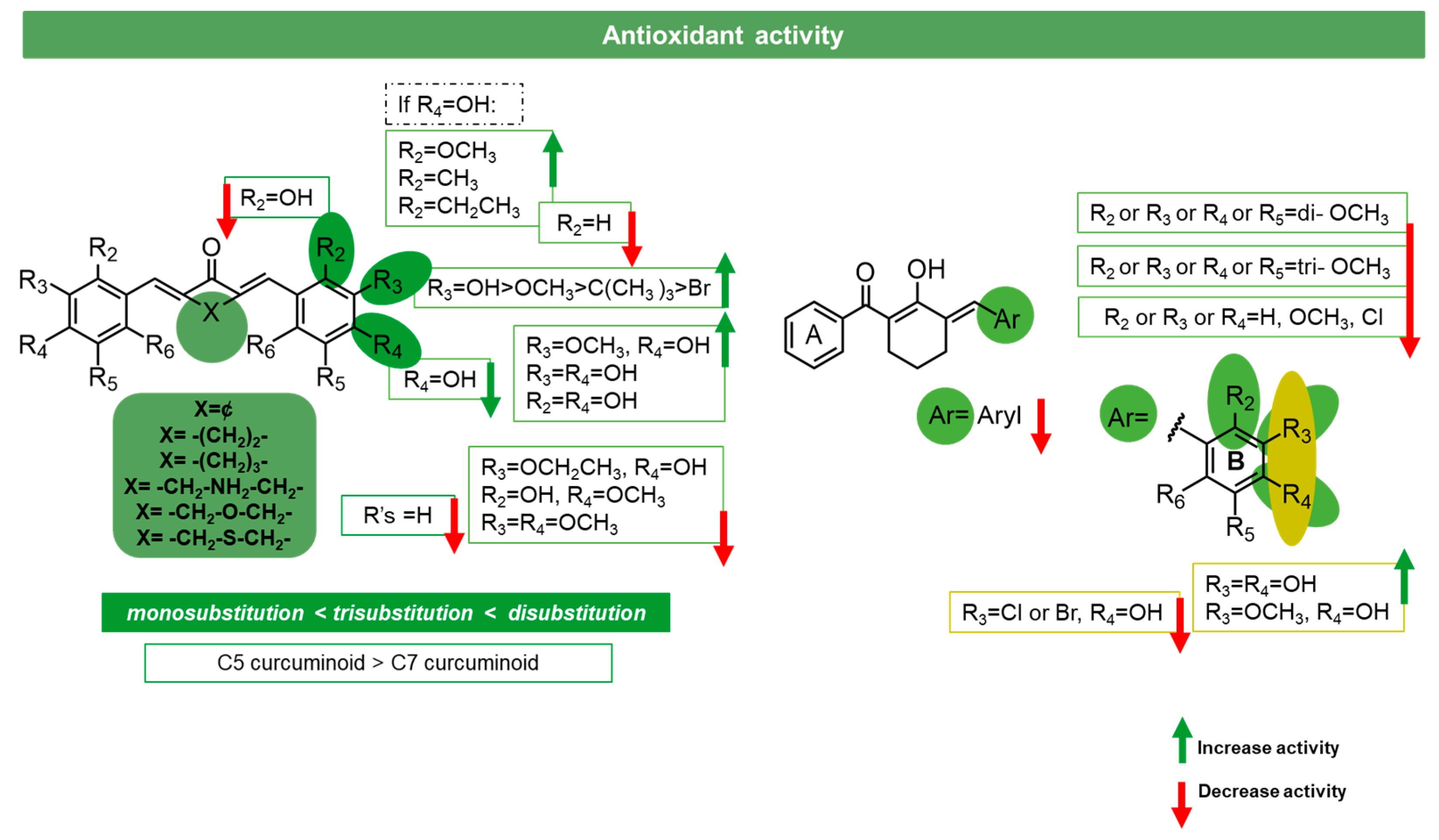
3.2. Antioxidant Activity
3.3. Anti-Inflammatory Activity
3.3.1. Effect of Diarylpentanoids on NO Production
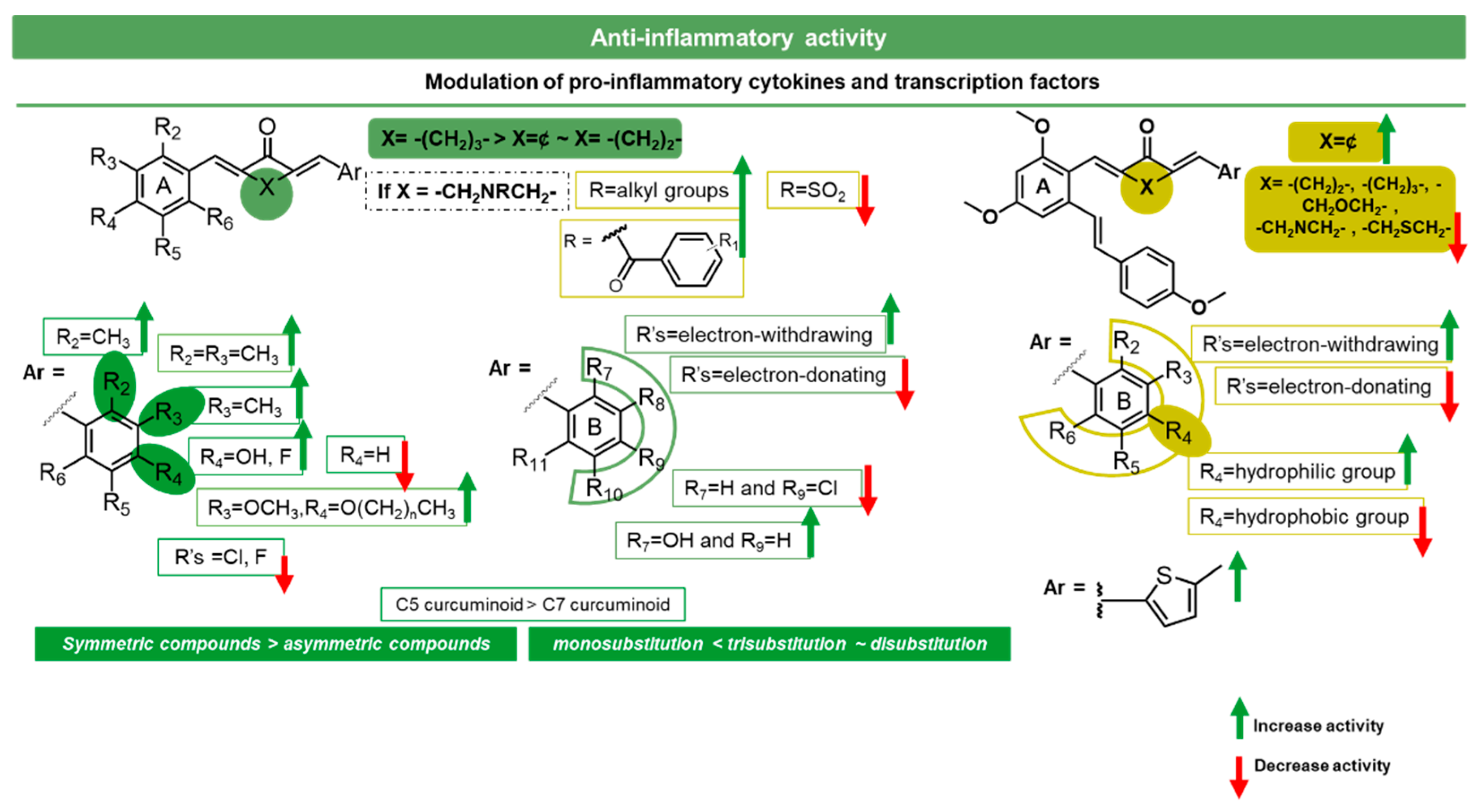
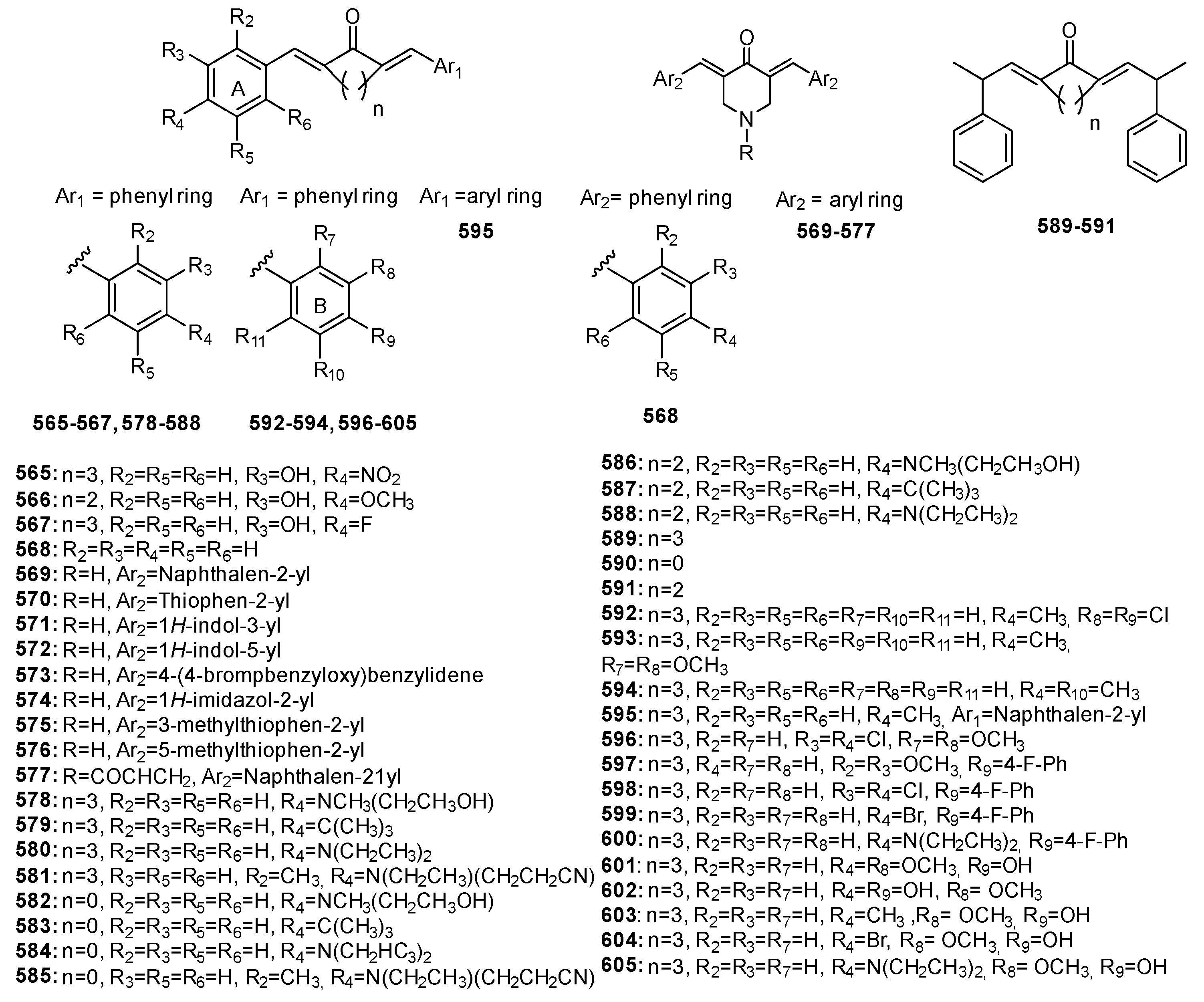
3.3.2. Modulation of Pro-Inflammatory Cytokines and Transcription Factors by Diarylpentanoids
3.3.3. Regulation of COX and LOX Pathways by Diarylpentanoids
3.4. Antidiabetic Activity
3.5. Anti-Hyperuricemic Activity
3.6. Neuroprotector Activity
3.7. Interference with Diverse Biochemical Targets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| ABTS+ | 2,2’-azinobis(3-ethylbenzothiazoline-6-sulfonicacid) radical cation assay |
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s disease |
| ADMET | Absorption, distribution, metabolism, excretion, toxicity |
| AP-1 | Activator protein 1 |
| BChE | Butyrylcholinesterase |
| CA-II | Carbonic anhydrase II |
| COX | Cyclooxygenase |
| DPPH | Diphenylpicrylhydrazyl assay |
| FRAP | Antioxidant power assay |
| IC50 | Half-maximal inhibitory concentration |
| IFN-γ | Interferon-gamma |
| iNOS | Nitric oxide synthase |
| IZ | Inhibition zone |
| LOX | Lipoxygenase |
| LPS | Lipopolysaccharide |
| LTs | Leukotrienes |
| NET | O2−• radical scavenging assay |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| pdb | Protein data bank |
| PMNs | Polymorphonuclear leukocytes |
| PGs | Prostaglandins |
| QSAR | Quantitative structure–activity relationship |
| SAR | Structure–activity relationship |
| STZHFD | Streptozocin and high-fat diet |
| TNF-α | Tumor necrosis factor-alpha |
| TRAP | ROO• radical scavenging assay |
| TRP | Transient receptor potential |
| TRPA1 | Transient receptor potential channels of ankyrin type-1 |
| TRPV1 | Transient receptor potential channels of vanilloid type-1 |
| Tx | Thromboxanes |
| URAT1 | Urate transporter 1 |
| XOD | Xanthine oxidase |
| 11β-HSD | 11β-hydroxysteroid dehydrogenase |
References
- Moreira, J.; Almeida, J.; Saraiva, L.; Cidade, H.; Pinto, M. Chalcones as Promising Antitumor Agents by Targeting the p53 Pathway: An Overview and New Insights in Drug-Likeness. Molecules 2021, 26, 3737. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone derivatives: Role in anticancer therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef] [PubMed]
- Thapa, P.; Upadhyay, S.P.; Suo, W.Z.; Singh, V.; Gurung, P.; Lee, E.S.; Sharma, R.; Sharma, M. Chalcone and its analogs: Therapeutic and diagnostic applications in Alzheimer’s disease. Bioorg. Chem. 2021, 108, 104681. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, M.; Iqbal, M.; Docea, A.O. Pharmacological properties of chalcones: A review of preclinical including molecular mechanisms and clinical evidence. Front. Pharmacol. 2021, 11, 592654. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T.; Hoang, N.T.; Nguyen, T.P.D.; Chau, N.H.T.; Le, N.T.N.; Le, H.B.T.; Phung, H.T.; Nguyen, H.T.; Nguyen, H.M. Extraction of curcumin from turmeric residue (Curcuma longa L.) using deep eutectic solvents and surfactant solvents. Anal. Methods 2022, 14, 850–858. [Google Scholar] [CrossRef]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem. Pharmacol. 2008, 75, 787–809. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Thomas, S.G.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Sung, B.; Tharakan, S.T.; Misra, K.; Priyadarsini, I.K.; Rajasekharan, K.N. Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature. Biochem. Pharmacol. 2008, 76, 1590–1611. [Google Scholar] [CrossRef]
- Anand, P.; Sundaram, C.; Jhurani, S.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin and cancer: An “old-age” disease with an “age-old” solution. Cancer Lett. 2008, 267, 133–164. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Harikumar, K.B. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem. Cell Biol. 2009, 41, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, D.K.; Mishra, P.K. Curcumin and its analogues: Potential anticancer agents. Med. Res. Rev. 2010, 30, 818–860. [Google Scholar] [CrossRef]
- Oliveira, A.S.; Sousa, E.; Helena Vasconcelos, M.; Pinto, M. Curcumin: A natural lead for potential new drug candidates. Curr. Med. Chem. 2015, 22, 4196–4232. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.C.; Anilkumar, N.; Thakur, G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J. Med. Chem. 2019, 76–104. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, J.R.; Pandit, B.; Bhasin, D.; Etter, J.P.; Regan, N.; Abdelhamid, D.; Li, C.; Lin, J.; Li, P.-K. Structure–activity relationship studies of curcumin analogues. Biorg. Med. Chem. Lett. 2009, 19, 2065–2069. [Google Scholar] [CrossRef]
- Moreira, J.; Almeida, J.; Loureiro, J.B.; Ramos, H.; Palmeira, A.; Pinto, M.M.; Saraiva, L.; Cidade, H. A Diarylpentanoid with Potential Activation of the p53 Pathway: Combination of in silico Screening Studies, Synthesis, and Biological Activity Evaluation. ChemMedChem 2021, 16, 2969–2981. [Google Scholar] [CrossRef] [PubMed]
- Novais, P.; Silva, P.; Moreira, J.; Palmeira, A.; Amorim, I.; Pinto, M.; Cidade, H.; Bousbaa, H. BP-M345, a New Diarylpentanoid with Promising Antimitotic Activity. Molecules 2021, 26, 7139. [Google Scholar] [CrossRef]
- Pinto, P.; Machado, C.M.; Moreira, J.; Almeida, J.D.P.; Silva, P.M.; Henriques, A.C.; Soares, J.X.; Salvador, J.A.; Afonso, C.; Pinto, M. Chalcone derivatives targeting mitosis: Synthesis, evaluation of antitumor activity and lipophilicity. Eur. J. Med. Chem. 2019, 184, 111752. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.; Durães, F.; Freitas-Silva, J.; Szemerédi, N.; Resende, D.I.; Pinto, E.; da Costa, P.M.; Pinto, M.; Spengler, G.; Cidade, H.; et al. New diarylpentanoids and chalcones as potential antimicrobial adjuvants. Biorg. Med. Chem. Lett. 2022, 67, 128743. [Google Scholar] [CrossRef]
- Moreira, J.; Saraiva, L.; Pinto, M.M.; Cidade, H. Diarylpentanoids with antitumor activity: A critical review of structure-activity relationship studies. Eur. J. Med. Chem. 2020, 192, 112177. [Google Scholar] [CrossRef]
- Du, Z.-Y.; Liu, R.-R.; Shao, W.-Y.; Mao, X.-P.; Ma, L.; Gu, L.-Q.; Huang, Z.-S.; Chan, A.S. α-Glucosidase inhibition of natural curcuminoids and curcumin analogs. Eur. J. Med. Chem. 2006, 41, 213–218. [Google Scholar] [CrossRef]
- Lee, K.-H.; Aziz, F.H.A.; Syahida, A.; Abas, F.; Shaari, K.; Israf, D.A.; Lajis, N.H. Synthesis and biological evaluation of curcumin-like diarylpentanoid analogues for anti-inflammatory, antioxidant and anti-tyrosinase activities. Eur. J. Med. Chem. 2009, 44, 3195–3200. [Google Scholar] [CrossRef]
- Din, Z.U.; dos Santos, A.; Trapp, M.A.; Lazarin-Bidóia, D.; Garcia, F.P.; Peron, F.; Nakamura, C.V.; Rodrigues-Filho, E. Curcumin inspired synthesis of unsymmetrical diarylpentanoids with highly potent anti-parasitic activities: In silico studies and DFT-based stereochemical calculation. MedChemComm 2016, 7, 820–831. [Google Scholar] [CrossRef]
- Faudzi, S.M.; Leong, S.; Abas, F.; Aluwi, M.M.; Rullah, K.; Lam, K.W.; Ahmad, S.; Tham, C.; Shaari, K.; Lajis, N. Synthesis, biological evaluation and QSAR studies of diarylpentanoid analogues as potential nitric oxide inhibitors. MedChemComm 2015, 6, 1069–1080. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem 2009, 78, 119–146. [Google Scholar] [CrossRef]
- Liang, G.; Yang, S.; Jiang, L.; Zhao, Y.; Shao, L.; Xiao, J.; Ye, F.; Li, Y.; Li, X. Synthesis and anti-bacterial properties of mono-carbonyl analogues of curcumin. Chem. Pharm. Bull. 2008, 56, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, B.; Venkataraman, R. Synthesis and biological evaluation of some curcumin analogs and their derivatives. Rasāyan J. Chem. 2010, 3, 260–265. [Google Scholar]
- Alves, L.V.; Do Canto-Cavalheiro, M.M.; Cysne-Finkelstein, L.; Leon, L. In vitro antiproliferative effects of several diaryl derivatives on Leishmania spp. Biol. Pharm. Bull. 2003, 26, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martínez-Ruiz, A.; Hernansanz-Agustín, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef]
- McCord, J.M. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659. [Google Scholar] [CrossRef]
- Praticò, D. Alzheimer’s disease and oxygen radicals: New insights. Biochem. Pharmacol. 2002, 63, 563–567. [Google Scholar] [CrossRef]
- Youssef, K.M.; El-Sherbeny, M.A.; El-Shafie, F.S.; Farag, H.A.; Al-Deeb, O.A.; Awadalla, S.A.A. Synthesis of curcumin analogues as potential antioxidant, cancer chemopreventive agents. Arch. Pharm. Int. J. Pharm. Med. Chem. 2004, 337, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-Y.; Jiang, Y.-F.; Tang, Z.-K.; Mo, R.-Q.; Xue, G.-H.; Lu, Y.-J.; Zheng, X.; Dong, C.-Z.; Zhang, K. Antioxidation and tyrosinase inhibition of polyphenolic curcumin analogs. Biosci. Biotechnol. Biochem. 2011, 75, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
- Eryanti, Y.; Nurulita, Y.; Hendra, R.; Yuharmen, Y.; Syahri, J.; Zamri, A. Synthesizing derivatives from cyclopentanone analogue curcumin and their toxic, antioxidant and anti-inflammatory activities. Makara J. Sci. 2012, 117–123. [Google Scholar] [CrossRef]
- Bayomi, S.M.; El-Kashef, H.A.; El-Ashmawy, M.B.; Nasr, M.N.; El-Sherbeny, M.A.; Badria, F.A.; Abou-Zeid, L.A.; Ghaly, M.A.; Abdel-Aziz, N.I. Synthesis and biological evaluation of new curcumin derivatives as antioxidant and antitumor agents. Med. Chem. Res. 2013, 22, 1147–1162. [Google Scholar] [CrossRef]
- Chen, B.; Zhu, Z.; Chen, M.; Dong, W.; Li, Z. Three-dimensional quantitative structure–activity relationship study on antioxidant capacity of curcumin analogues. J. Mol. Struct. 2014, 1061, 134–139. [Google Scholar] [CrossRef]
- Li, Q.; Chen, J.; Luo, S.; Xu, J.; Huang, Q.; Liu, T. Synthesis and assessment of the antioxidant and antitumor properties of asymmetric curcumin analogues. Eur. J. Med. Chem. 2015, 93, 461–469. [Google Scholar] [CrossRef]
- Leong, S.W.; Faudzi, S.M.M.; Abas, F.; Aluwi, M.F.F.M.; Rullah, K.; Lam, K.W.; Bahari, M.N.A.; Ahmad, S.; Tham, C.L.; Shaari, K. Nitric oxide inhibitory activity and antioxidant evaluations of 2-benzoyl-6-benzylidenecyclohexanone analogs, a novel series of curcuminoid and diarylpentanoid derivatives. Biorg. Med. Chem. Lett. 2015, 25, 3330–3337. [Google Scholar] [CrossRef]
- Maskrey, B.H.; Megson, I.L.; Whitfield, P.D.; Rossi, A.G. Mechanisms of resolution of inflammation: A focus on cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1001–1006. [Google Scholar] [CrossRef]
- Ban, H.S.; Suzuki, K.; Lim, S.S.; Jung, S.H.; Lee, S.; Ji, J.; Lee, H.S.; Lee, Y.S.; Shin, K.H.; Ohuchi, K. Inhibition of lipopolysaccharide-induced expression of inducible nitric oxide synthase and tumor necrosis factor-α by 2′-hydroxychalcone derivatives in RAW 264.7 cells. Biochem. Pharmacol. 2004, 67, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Morrison, D.C.; Parmely, T.J.; Russell, S.W.; Murphy, W.J. An interferon-γ-activated site (GAS) is necessary for full expression of the mouse iNOS gene in response to interferon-γ and lipopolysaccharide. J. Biol. Chem. 1997, 272, 1226–1230. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef]
- Guzik, T.; Korbut, R.; Adamek-Guzik, T. Nitric oxide and superoxide in inflammation. J. Physiol. Pharm. 2003, 54, 469–487. [Google Scholar]
- Napoli, C.; de Nigris, F.; Williams-Ignarro, S.; Pignalosa, O.; Sica, V.; Ignarro, L.J. Nitric oxide and atherosclerosis: An update. Nitric Oxide 2006, 15, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Sellebjerg, F.; Giovannoni, G.; Hand, A.; Madsen, H.; Jensen, C.; Garred, P. Cerebrospinal fluid levels of nitric oxide metabolites predict response to methylprednisolone treatment in multiple sclerosis and optic neuritis. J. Neuroimmunol. 2002, 125, 198–203. [Google Scholar] [CrossRef]
- Fitzpatrick, A.M.; Brown, L.A.S.; Holguin, F.; Teague, W.G.; Program, S.A.R.; Health, N.I.o. Levels of nitric oxide oxidation products are increased in the epithelial lining fluid of children with persistent asthma. J. Allergy Clin. Immunol. 2009, 124, 990–996.e9. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.W.; Faudzi, S.M.M.; Abas, F.; Aluwi, M.F.F.M.; Rullah, K.; Wai, L.K.; Bahari, M.N.A.; Ahmad, S.; Tham, C.L.; Shaari, K. Synthesis and sar study of diarylpentanoid analogues as new anti-inflammatory agents. Molecule 2014, 19, 16058–16081. [Google Scholar] [CrossRef] [PubMed]
- Weber, W.M.; Hunsaker, L.A.; Gonzales, A.M.; Heynekamp, J.J.; Orlando, R.A.; Deck, L.M.; Vander Jagt, D.L. TPA-induced up-regulation of activator protein-1 can be inhibited or enhanced by analogs of the natural product curcumin. Biochem. Pharmacol. 2006, 72, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Li, X.; Chen, L.; Yang, S.; Wu, X.; Studer, E.; Gurley, E.; Hylemon, P.B.; Ye, F.; Li, Y. Synthesis and anti-inflammatory activities of mono-carbonyl analogues of curcumin. Biorg. Med. Chem. Lett. 2008, 18, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Yang, S.; Zhou, H.; Shao, L.; Huang, K.; Xiao, J.; Huang, Z.; Li, X. Synthesis, crystal structure and anti-inflammatory properties of curcumin analogues. Eur. J. Med. Chem. 2009, 44, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yang, J.; Wang, Y.; Liang, D.; Yang, X.; Li, X.; Wu, J.; Wu, X.; Yang, S.; Li, X. Synthesis of mono-carbonyl analogues of curcumin and their effects on inhibition of cytokine release in LPS-stimulated RAW 264.7 macrophages. Bioorg. Med. Chem. 2010, 18, 2388–2393. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tang, L.; Zou, P.; Zhang, Y.; Wang, Z.; Fang, Q.; Jiang, L.; Chen, G.; Xu, Z.; Zhang, H. Synthesis and biological evaluation of allylated and prenylated mono-carbonyl analogs of curcumin as anti-inflammatory agents. Eur. J. Med. Chem. 2014, 74, 671–682. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, C.; He, W.; Wang, Z.; Fang, Q.; Xiao, B.; Liu, Z.; Liang, G.; Yang, S. Discovery and evaluation of asymmetrical monocarbonyl analogs of curcumin as anti-inflammatory agents. Drug Des. Devel. Ther. 2014, 8, 373. [Google Scholar]
- Zhang, Y.; Jiang, X.; Peng, K.; Chen, C.; Fu, L.; Wang, Z.; Feng, J.; Liu, Z.; Zhang, H.; Liang, G. Discovery and evaluation of novel anti-inflammatory derivatives of natural bioactive curcumin. Drug Des. Devel. Ther. 2014, 8, 2161. [Google Scholar]
- Zhao, C.; Zhang, Y.; Zou, P.; Wang, J.; He, W.; Shi, D.; Li, H.; Liang, G.; Yang, S. Synthesis and biological evaluation of a novel class of curcumin analogs as anti-inflammatory agents for prevention and treatment of sepsis in mouse model. Drug Des. Devel. Ther. 2015, 9, 1663. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Xu, T.; Qiu, C.; Wu, B.; Zhang, Y.; Chen, L.; Xia, Q.; Li, C.; Zhou, B.; Liu, Z. Synthesis and optimization of novel allylated mono-carbonyl analogs of curcumin (MACs) act as potent anti-inflammatory agents against LPS-induced acute lung injury (ALI) in rats. Eur. J. Med. Chem. 2016, 121, 181–193. [Google Scholar] [CrossRef]
- Pan, J.; Xu, T.; Xu, F.; Zhang, Y.; Liu, Z.; Chen, W.; Fu, W.; Dai, Y.; Zhao, Y.; Feng, J. Development of resveratrol-curcumin hybrids as potential therapeutic agents for inflammatory lung diseases. Eur. J. Med. Chem. 2017, 125, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Gafner, S.; Lee, S.-K.; Cuendet, M.; Barthélémy, S.; Vergnes, L.; Labidalle, S.; Mehta, R.G.; Boone, C.W.; Pezzuto, J.M. Biologic evaluation of curcumin and structural derivatives in cancer chemoprevention model systems. Phytochemistry. 2004, 65, 2849–2859. [Google Scholar] [CrossRef]
- Katsori, A.-M.; Chatzopoulou, M.; Dimas, K.; Kontogiorgis, C.; Patsilinakos, A.; Trangas, T.; Hadjipavlou-Litina, D. Curcumin analogues as possible anti-proliferative & anti-inflammatory agents. Eur. J. Med. Chem. 2011, 46, 2722–2735. [Google Scholar]
- Ahmad, W.; Kumolosasi, E.; Jantan, I.; Bukhari, S.N.; Jasamai, M. Effects of novel diarylpentanoid analogues of curcumin on secretory phospholipase A2, cyclooxygenases, lipo-oxygenase, and microsomal prostaglandin E synthase-1. Chem. Biol. Drug Des. 2014, 83, 670–681. [Google Scholar] [CrossRef]
- Aluwi, M.F.F.M.; Rullah, K.; Yamin, B.M.; Leong, S.W.; Bahari, M.N.A.; Lim, S.J.; Faudzi, S.M.M.; Jalil, J.; Abas, F.; Fauzi, N.M. Synthesis of unsymmetrical monocarbonyl curcumin analogues with potent inhibition on prostaglandin E 2 production in LPS-induced murine and human macrophages cell lines. Biorg. Med. Chem. Lett. 2016, 26, 2531–2538. [Google Scholar] [CrossRef]
- Raj, C.G.D.; Sarojini, B.K.; Khan, M.T.H.; Raghavendra, R. In vivo antidiabetic activity and in silico studies on adenosine monophosphate-activated protein kinase (AMPK) of (2E, 5E)-2, 5-bis (4-hydroxy-3-methoxybenzylidene) cyclopentanone. Med. Chem. Res. 2013, 22, 2430–2436. [Google Scholar] [CrossRef]
- Yuan, X.; Li, H.; Bai, H.; Su, Z.; Xiang, Q.; Wang, C.; Zhao, B.; Zhang, Y.; Zhang, Q.; Chu, Y. Synthesis of novel curcumin analogues for inhibition of 11β-hydroxysteroid dehydrogenase type 1 with anti-diabetic properties. Eur. J. Med. Chem. 2014, 77, 223–230. [Google Scholar] [CrossRef]
- Chen, H.; Yang, X.; Lu, K.; Lu, C.; Zhao, Y.; Zheng, S.; Li, J.; Huang, Z.; Huang, Y.; Zhang, Y. Inhibition of high glucose-induced inflammation and fibrosis by a novel curcumin derivative prevents renal and heart injury in diabetic mice. Toxicol. Lett. 2017, 278, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.W.; Abas, F.; Lam, K.W.; Yusoff, K. In vitro and in silico evaluations of diarylpentanoid series as α-glucosidase inhibitor. Biorg. Med. Chem. Lett. 2018, 28, 302–309. [Google Scholar] [CrossRef]
- Ao, G.-Z.; Zhou, M.-Z.; Li, Y.-Y.; Li, S.-N.; Wang, H.-N.; Wan, Q.-W.; Li, H.-Q.; Hu, Q.-H. Discovery of novel curcumin derivatives targeting xanthine oxidase and urate transporter 1 as anti-hyperuricemic agents. Bioorg. Med. Chem. 2017, 25, 166–174. [Google Scholar] [CrossRef]
- Association, A.s. 2016 Alzheimer’s disease facts and figures. Alzheimers. Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Guzior, N.; Wieckowska, A.; Panek, D.; Malawska, B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef]
- Cruz, M.I.; Cidade, H.; Pinto, M. Dual/multitargeted xanthone derivatives for Alzheimer’s disease: Where do we stand? Future Med. Chem. 2017, 9, 1611–1630. [Google Scholar] [CrossRef]
- Leong, S.W.; Abas, F.; Lam, K.W.; Shaari, K.; Lajis, N.H. 2-Benzoyl-6-benzylidenecyclohexanone analogs as potent dual inhibitors of acetylcholinesterase and butyrylcholinesterase. Bioorg. Med. Chem. 2016, 24, 3742–3751. [Google Scholar] [CrossRef]
- Jiang, Y.; Du, Z.; Xue, G.; Chen, Q.; Lu, Y.; Zheng, X.; Conney, A.H.; Zhang, K. Synthesis and biological evaluation of unsymmetrical curcumin analogues as tyrosinase inhibitors. Molecules 2013, 18, 3948–3961. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Pal, D.; Prasad, R. Alkaline phosphatase: An overview. Indian J. Clin. Biochem. 2014, 29, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Srivastava, P.K. A molecular description of acid phosphatase. Appl. Biochem. Biotechnol. 2012, 167, 2174–2197. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Raghav, N. Synthesis, docking, and in vitro studies of some substituted bischalcones on acid and alkaline phosphatases. Med. Chem. Res. 2014, 23, 1781–1788. [Google Scholar] [CrossRef]
- Lomelino, C.; McKenna, R. Carbonic anhydrase inhibitors: A review on the progress of patent literature (2011–2016). Expert Opin. Ther. Pat. 2016, 26, 947–956. [Google Scholar] [CrossRef]
- Aditama, R.; Eryanti, Y.; Mujahidin, D.; Syah, Y.M.; Hertadi, R. Determination of activities of human carbonic anhydrase II inhibitors from curcumin analogs. Trop. J. Pharm. Res. 2017, 16, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Nalli, M.; Ortar, G.; Moriello, A.S.; Di Marzo, V.; De Petrocellis, L. Effects of curcumin and curcumin analogues on TRP channels. Fitoterapia 2017, 122, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhou, H.; Wang, Y.; Gurley, E.C.; Feng, B.; Chen, L.; Xiao, J.; Yang, S.; Li, X. Inhibition of LPS-induced production of inflammatory factors in the macrophages by mono-carbonyl analogues of curcumin. J. Cell. Mol. Med. 2009, 13, 3370–3379. [Google Scholar] [CrossRef] [Green Version]















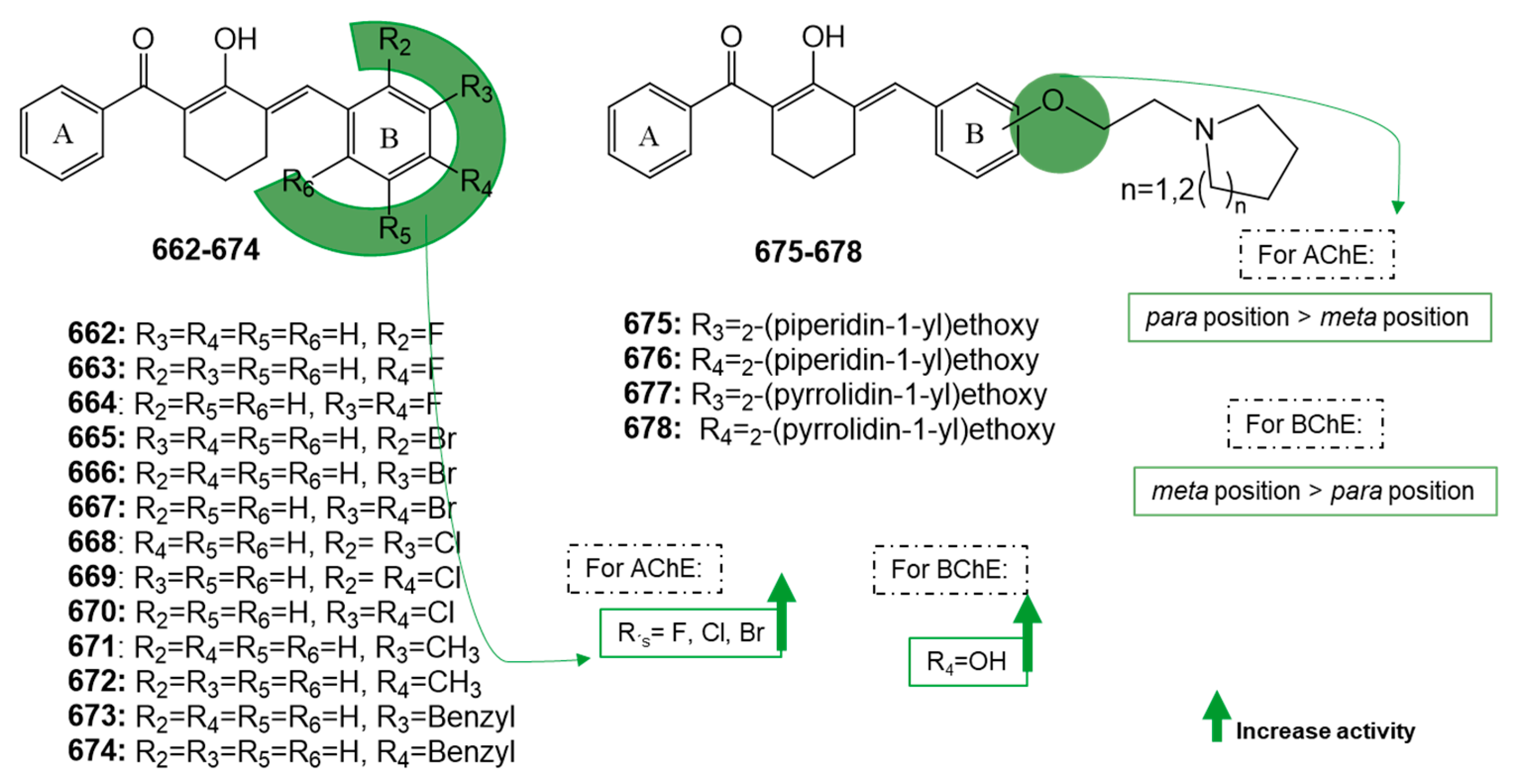
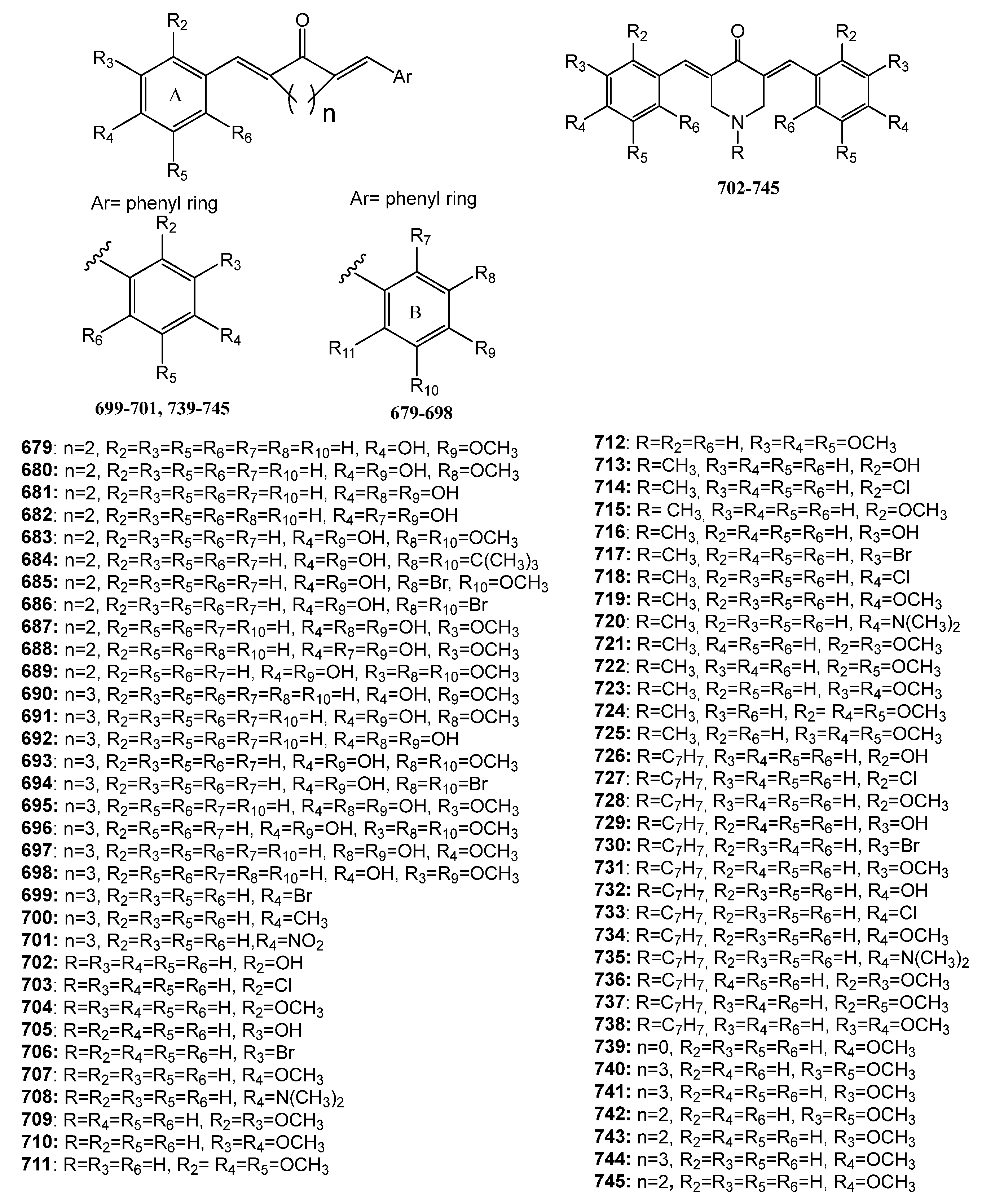


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, J.; Saraiva, L.; Pinto, M.M.; Cidade, H. Bioactive Diarylpentanoids: Insights into the Biological Effects beyond Antitumor Activity and Structure–Activity Relationships. Molecules 2022, 27, 6340. https://doi.org/10.3390/molecules27196340
Moreira J, Saraiva L, Pinto MM, Cidade H. Bioactive Diarylpentanoids: Insights into the Biological Effects beyond Antitumor Activity and Structure–Activity Relationships. Molecules. 2022; 27(19):6340. https://doi.org/10.3390/molecules27196340
Chicago/Turabian StyleMoreira, Joana, Lucilia Saraiva, Madalena M. Pinto, and Honorina Cidade. 2022. "Bioactive Diarylpentanoids: Insights into the Biological Effects beyond Antitumor Activity and Structure–Activity Relationships" Molecules 27, no. 19: 6340. https://doi.org/10.3390/molecules27196340