
Current Progress in the Chemoenzymatic Synthesis of Natural Products


Abstract
:1. Introduction
2. Selected Natural Product Syntheses Incorporating Chemoenzymatic Methods
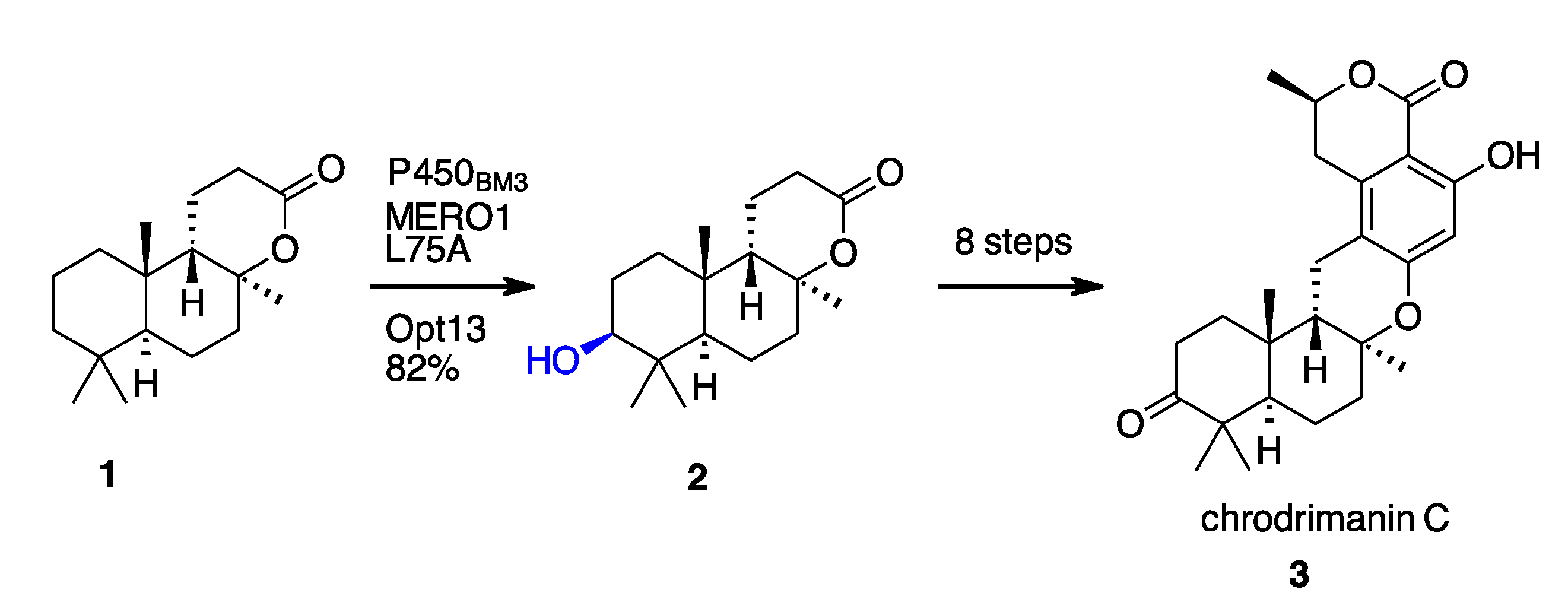
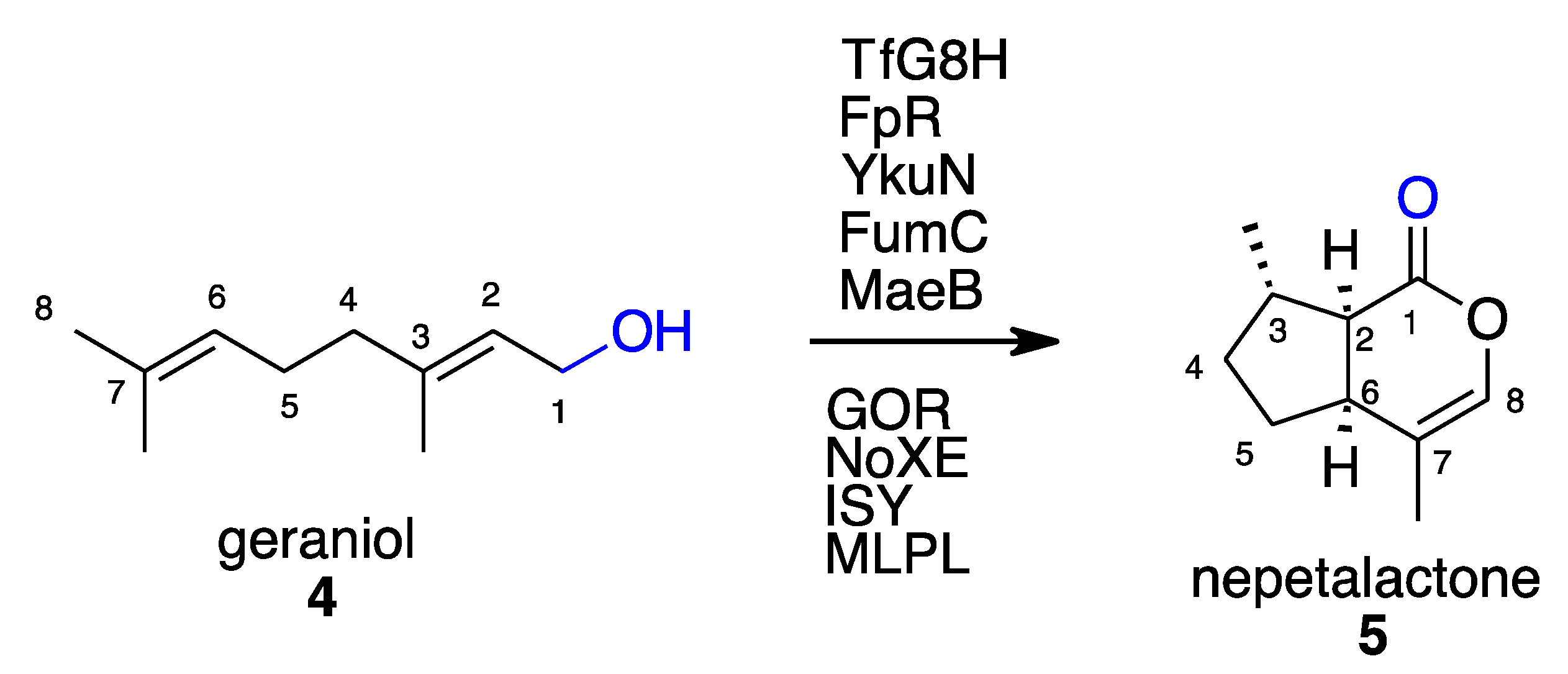
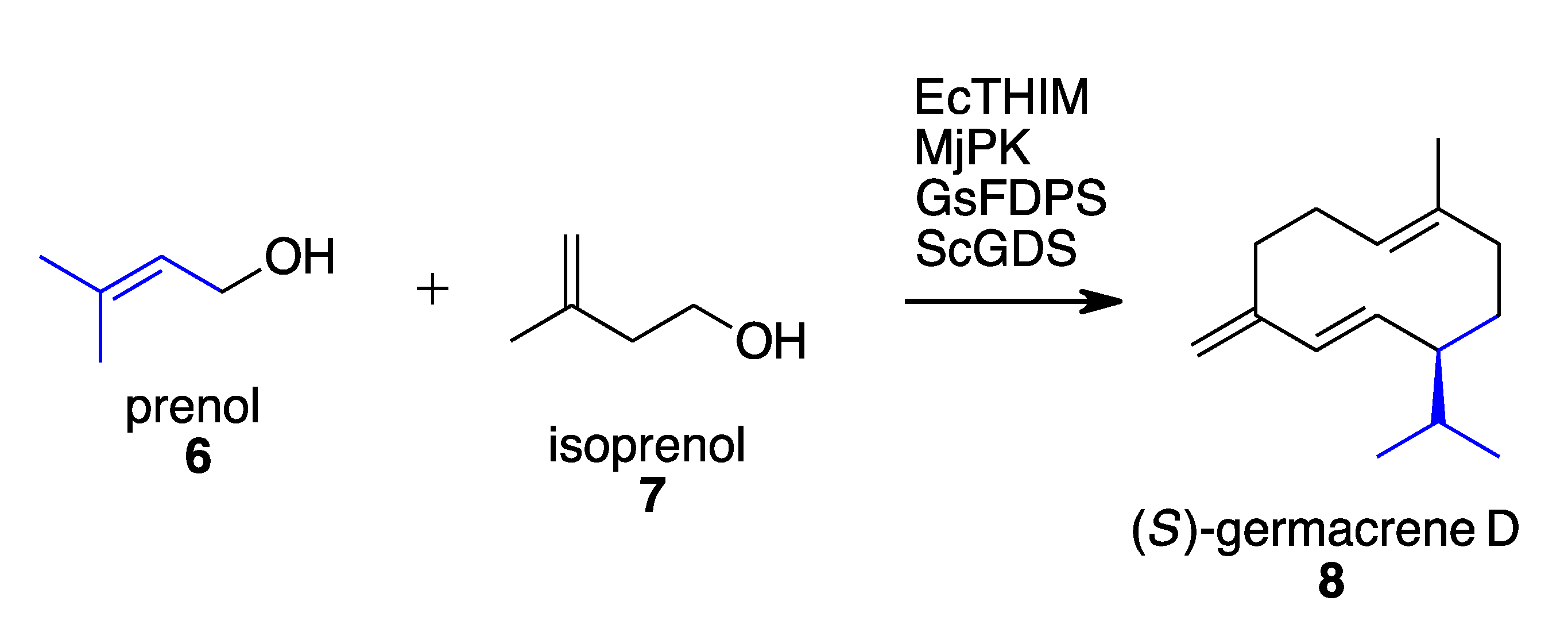
2.1. Terpenoids
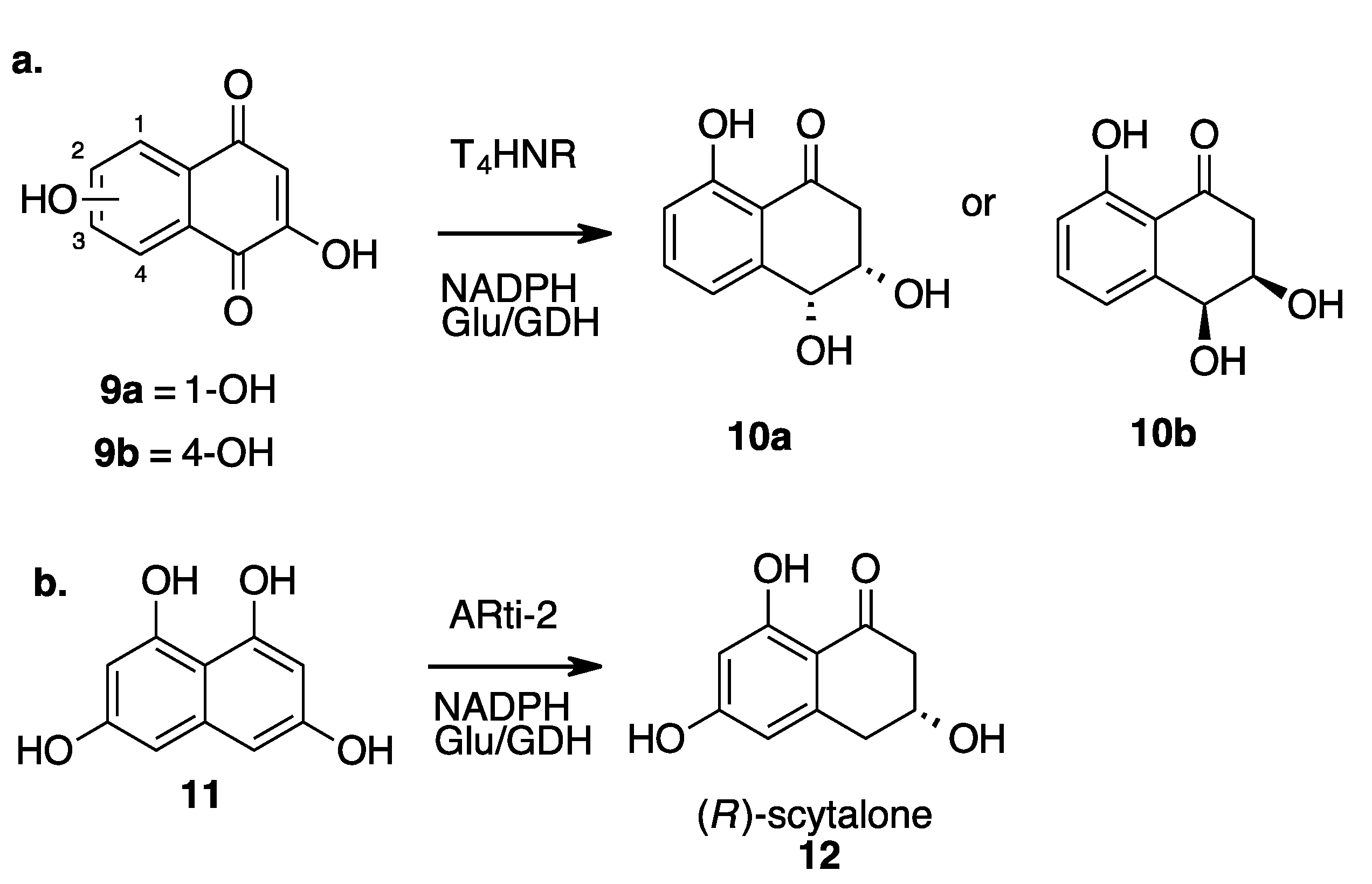
2.2. Polyketides
2.3. Glycans
2.4. Peptides and Amino Acids
2.5. Alkaloids
2.6. Miscellaneous
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Truax, N.J.; Romo, D. Bridging the Gap between Natural Product Synthesis and Drug Discovery. Nat. Prod. Rep. 2020, 37, 1436–1453. [Google Scholar] [CrossRef] [PubMed]
- Olaizola, M. Commercial Development of Microalgal Biotechnology: From the Test Tube to the Marketplace. Biomol. Eng. 2003, 20, 459–466. [Google Scholar] [CrossRef]
- Baran, P.S. Natural Product Total Synthesis: As Exciting as Ever and Here to Stay. J. Am. Chem. Soc. 2018, 140, 4751–4755. [Google Scholar] [CrossRef] [PubMed]
- Karimov, R.R.; Hartwig, J.F. Transition-Metal-Catalyzed Selective Functionalization of C(Sp3)−H Bonds in Natural Products. Angew. Chem. Int. Ed. 2018, 57, 4234–4241. [Google Scholar] [CrossRef]
- Harwood, S.J.; Palkowitz, M.D.; Gannett, C.N.; Perez, P.; Yao, Z.; Sun, L.; Abruña, H.D.; Anderson, S.L.; Baran, P.S. Modular Terpene Synthesis Enabled by Mild Electrochemical Couplings. Science 2022, 375, 745–752. [Google Scholar] [CrossRef]
- Pitre, S.P.; Overman, L.E. Strategic Use of Visible-Light Photoredox Catalysis in Natural Product Synthesis. Chem. Rev. 2022, 122, 1717–1751. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.W.; Hudlicky, T. The Quest for a Practical Synthesis of Morphine Alkaloids and Their Derivatives by Chemoenzymatic Methods. Acc. Chem. Res. 2015, 48, 674–687. [Google Scholar] [CrossRef]
- de Miranda, A.S.; Miranda, L.S.M.; de Souza, R.O.M.A. Lipases: Valuable Catalysts for Dynamic Kinetic Resolutions. Biotechnol. Adv. 2015, 33, 372–393. [Google Scholar] [CrossRef]
- Langvik, O.; Saloranta, T.; Murzin, D.Y.; Leino, R. Heterogeneous Chemoenzymatic Catalyst Combinations for One-Pot Dynamic Kinetic Resolution Applications. ChemCatChem 2015, 7, 4004–4015. [Google Scholar] [CrossRef]
- Muthana, S.; Cao, H.; Chen, X. Recent Progress in Chemical and Chemoenzymatic Synthesis of Carbohydrates. Curr. Opin. Chem. Biol. 2009, 13, 573–581. [Google Scholar] [CrossRef]
- Stöckigt, J.; Chen, Z.; Ruppert, M. Enzymatic and Chemo-Enzymatic Approaches towards Natural and Non-Natural Alkaloids: Indoles, Isoquinolines, and Others. In BT—Natural Products via Enzymatic Reactions; Piel, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 67–103. ISBN 978-3-642-16427-9. [Google Scholar]
- Azerad, R.; Buisson, D. Dynamic Resolution and Stereoinversion of Secondary Alcohols by Chemo-Enzymatic Processes. Curr. Opin. Biotechnol. 2000, 11, 565–571. [Google Scholar] [CrossRef]
- Mortison, J.D.; Sherman, D.H. Frontiers and Opportunities in Chemoenzymatic Synthesis. J. Org. Chem. 2010, 75, 7041–7051. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Abdelraheem, E.M.M.; Busch, H.; Hanefeld, U.; Tonin, F. Biocatalysis Explained: From Pharmaceutical to Bulk Chemical Production. React. Chem. Eng. 2019, 4, 1878–1894. [Google Scholar] [CrossRef]
- Riehl, P.S.; Lim, J.; Finnigan, J.D.; Charnock, S.J.; Hyster, T.K. An Efficient Synthesis of the Bicyclic Darunavir Side Chain Using Chemoenzymatic Catalysis. Org. Process Res. Dev. 2022, 26, 2096–2101. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Romero, E.O.; Pyser, J.B.; Yazarians, J.A.; Narayan, A.R.H. Chemoenzymatic Total Synthesis of Natural Products. Acc. Chem. Res. 2021, 54, 1374–1384. [Google Scholar] [CrossRef]
- Roddan, R.; Carter, E.M.; Thair, B.; Hailes, H.C. Chemoenzymatic Approaches to Plant Natural Product Inspired Compounds. Nat. Prod. Rep. 2022, 39, 1375–1382. [Google Scholar] [CrossRef]
- Li, J.; Amatuni, A.; Renata, H. Recent Advances in the Chemoenzymatic Synthesis of Bioactive Natural Products. Curr. Opin. Chem. Biol. 2020, 55, 111–118. [Google Scholar] [CrossRef]
- Murray, L.A.M.; Mckinnie, S.M.K.; Moore, B.S.; George, J.H.; Cruz, S.; States, U.; Jolla, L.; States, U.; Jolla, L.; States, U. Meroterpenoid natural products from Streptomyces bacteria—The Evolution of Chemoenzymatic Syntheses. Nat. Prod. Rep. 2021, 37, 1334–1366. [Google Scholar] [CrossRef]
- Stout, C.N.; Renata, H. Reinvigorating the Chiral Pool: Chemoenzymatic Approaches to Complex Peptides and Terpenoids. Acc. Chem. Res. 2021, 54, 1143–1156. [Google Scholar] [CrossRef]
- King-Smith, E.; Zwick, C.R.; Renata, H. Applications of Oxygenases in the Chemoenzymatic Total Synthesis of Complex Natural Products. Biochemistry 2018, 57, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhou, Q.; Wu, Y.; Chen, X.; Zhong, F. Properties and Mechanisms of Flavin-Dependent Monooxygenases and Their Applications in Natural Product Synthesis. Int. J. Mol. Sci. 2022, 23, 2622. [Google Scholar] [CrossRef] [PubMed]
- Malico, A.A.; Calzini, M.A.; Gayen, A.K.; Williams, G.J. Synthetic Biology, Combinatorial Biosynthesis, and Chemo-enzymatic Synthesis of Isoprenoids. J. Ind. Microbiol. Biotechnol. 2020, 47, 675–702. [Google Scholar] [CrossRef]
- Zetzsche, L.E.; Chakrabarty, S.; Narayan, A.R.H. The Transformative Power of Biocatalysis in Convergent Synthesis. J. Am. Chem. Soc. 2022, 144, 5214–5225. [Google Scholar] [CrossRef]
- Cigan, E.; Eggbauer, B.; Schrittwieser, J.H.; Kroutil, W. The Role of Biocatalysis in the Asymmetric Synthesis of Alkaloids—An Update. RSC Adv. 2021, 11, 28223–28270. [Google Scholar] [CrossRef]
- Pyser, J.B.; Chakrabarty, S.; Romero, E.O.; Narayan, A.R.H. State-of-the-Art Biocatalysis. ACS Cent. Sci. 2021, 7, 1105–1116. [Google Scholar] [CrossRef]
- Zhao, F.; Masci, D.; Tomarelli, E.; Castagnolo, D. Biocatalytic and Chemo-Enzymatic Approaches for the Synthesis of Heterocycles. Synthesis 2020, 52, 2948–2961. [Google Scholar]
- Kaspar, F.; Schallmey, A. Chemo-Enzymatic Synthesis of Natural Products and Their Analogs. Curr. Opin. Biotechnol. 2022, 77, 102759. [Google Scholar] [CrossRef]
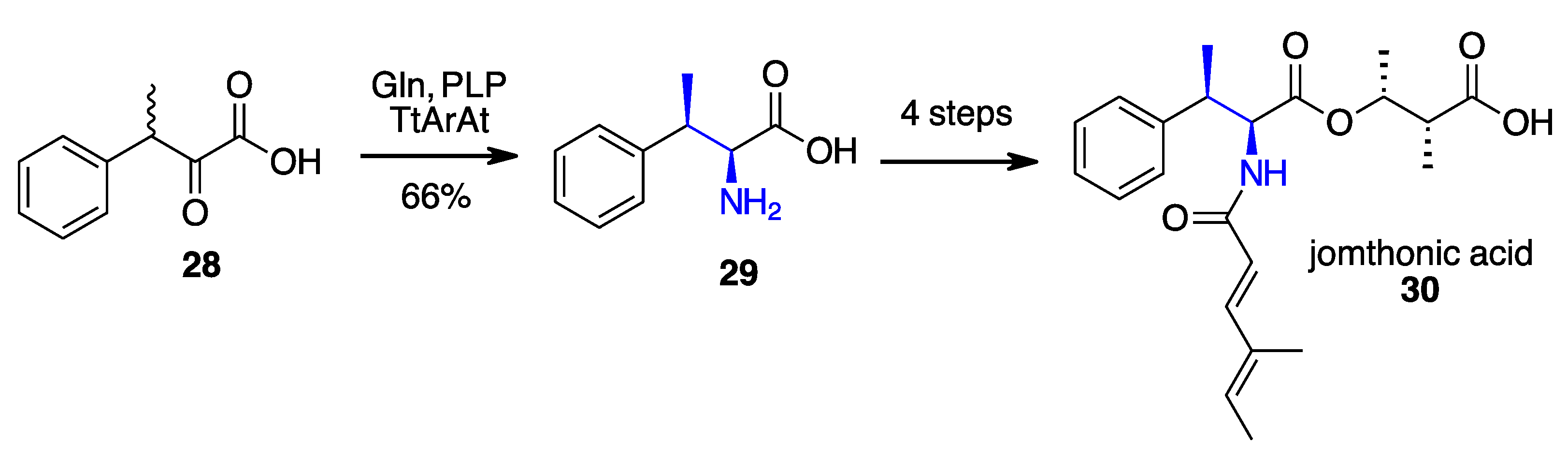
- Li, F.; Yang, L.C.; Zhang, J.; Chen, J.S.; Renata, H. Stereoselective Synthesis of β-Branched Aromatic α-Amino Acids by Biocatalytic Dynamic Kinetic Resolution**. Angew. Chem. Int. Ed. 2021, 60, 17680–17685. [Google Scholar] [CrossRef]
- Li, F.; Renata, H. A Chiral-Pool-Based Strategy to Access Trans-Syn-Fused Drimane Meroterpenoids: Chemoenzymatic Total Syntheses of Polysin, N-Acetyl-Polyveoline and the Chrodrimanins. J. Am. Chem. Soc. 2021, 143, 18280–18286. [Google Scholar] [CrossRef]
- Li, J.; Renata, H. Concise Chemoenzymatic Synthesis of Fasamycin A. J. Org. Chem. 2021, 86, 11206–11211. [Google Scholar] [CrossRef] [PubMed]
- Bat-Erdene, U.; Billingsley, J.M.; Turner, W.C.; Lichman, B.R.; Ippoliti, F.M.; Garg, N.K.; O’Connor, S.E.; Tang, Y. Cell-Free Total Biosynthesis of Plant Terpene Natural Products Using an Orthogonal Cofactor Regeneration System. ACS Catal 2021, 11, 9898–9903. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Dunbabin, A.; Benton, J.C.R.; Mart, R.J.; Allemann, R.K. Modular Chemoenzymatic Synthesis of Terpenes and Their Analogues. Angew. Chem. Int. Ed. 2020, 59, 8486–8490. [Google Scholar] [CrossRef] [PubMed]
- Shinde, P.B.; Oh, H.-S.; Choi, H.; Rathwell, K.; Ban, Y.H.; Kim, E.J.; Yang, I.; Lee, D.G.; Sherman, D.H.; Kang, H.-W.; et al. Chemoenzymatic Synthesis of Glycosylated Macrolactam Analogues of the Macrolide Antibiotic YC-17. Adv. Synth. Catal. 2015, 357, 2697–2711. [Google Scholar] [CrossRef]
- Schmidt, J.J.; Khatri, Y.; Brody, S.I.; Zhu, C.; Pietraszkiewicz, H.; Valeriote, F.A.; Sherman, D.H. A Versatile Chemoenzymatic Synthesis for the Discovery of Potent Cryptophycin Analogs. ACS Chem. Biol. 2020, 15, 524–532. [Google Scholar] [CrossRef]
- Ding, Y.; Rath, C.M.; Bolduc, K.L.; Håkansson, K.; Sherman, D.H. Chemoenzymatic Synthesis of Cryptophycin Anticancer Agents by an Ester Bond-Forming Non-Ribosomal Peptide Synthetase Module. J. Am. Chem. Soc. 2011, 133, 14492–14495. [Google Scholar] [CrossRef]
- Lowell, A.N.; DeMars, M.D., 2nd; Slocum, S.T.; Yu, F.; Anand, K.; Chemler, J.A.; Korakavi, N.; Priessnitz, J.K.; Park, S.R.; Koch, A.A.; et al. Chemoenzymatic Total Synthesis and Structural Diversification of Tylactone-Based Macrolide Antibiotics through Late-Stage Polyketide Assembly, Tailoring, and C—H Functionalization. J. Am. Chem. Soc. 2017, 139, 7913–7920. [Google Scholar] [CrossRef]
- Rittner, A.; Joppe, M.; Schmidt, J.J.; Mayer, L.M.; Reiners, S.; Heid, E.; Herzberg, D.; Sherman, D.H.; Grininger, M. Chemoenzymatic Synthesis of Fluorinated Polyketides. Nat. Chem. 2022, 14, 1000–1006. [Google Scholar] [CrossRef]
- Saha, N.; Müller, M.; Husain, S.M. Asymmetric Synthesis of Natural Cis-Dihydroarenediols Using Tetrahydroxynaphthalene Reductase and Its Biosynthetic Implications. Org. Lett. 2019, 21, 2204–2208. [Google Scholar] [CrossRef]
- Singh, S.K.; Rajput, A.; De, A.; Chakraborti, T.; Husain, S.M. Promiscuity of an Unrelated Anthrol Reductase of Talaromyces Islandicus WF-38-12. Catal. Sci. Technol. 2021, 11, 474–478. [Google Scholar] [CrossRef]
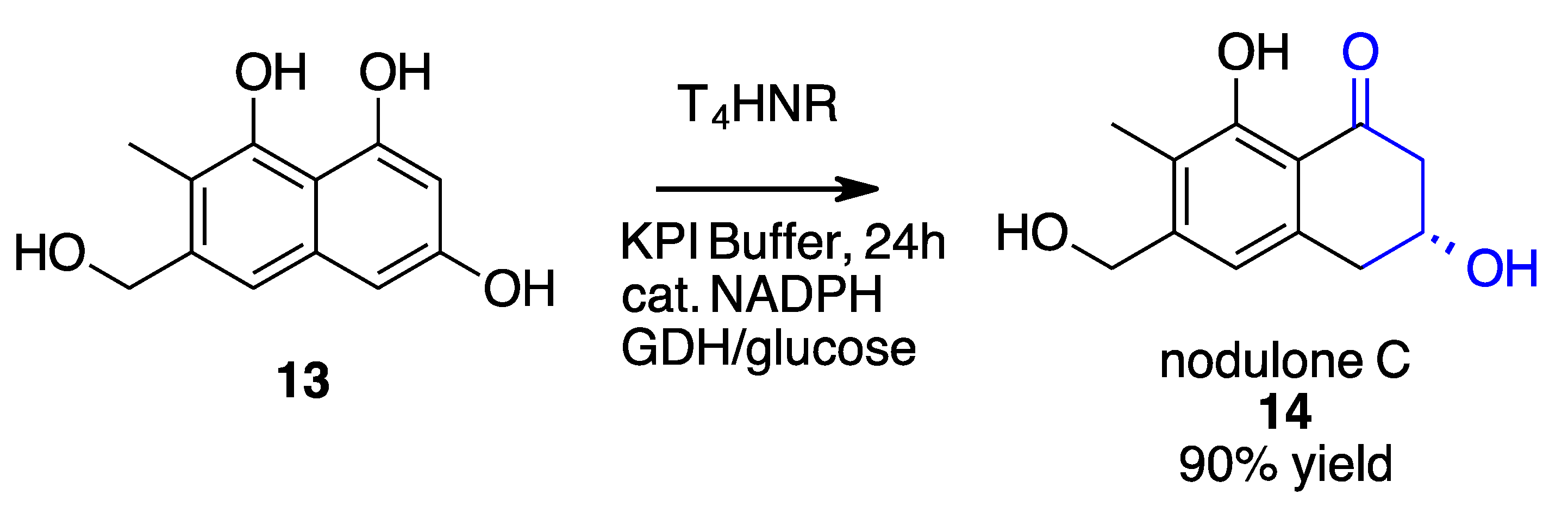
- Manna, T.; Rajput, A.; Saha, N.; Mondal, A.; Debnath, S.C.; Husain, S.M. Chemoenzymatic Total Synthesis of Nodulones C and D Using a Naphthol Reductase of Magnaporthe Grisea. Org. Biomol. Chem. 2022, 20, 3737–3741. [Google Scholar] [CrossRef] [PubMed]
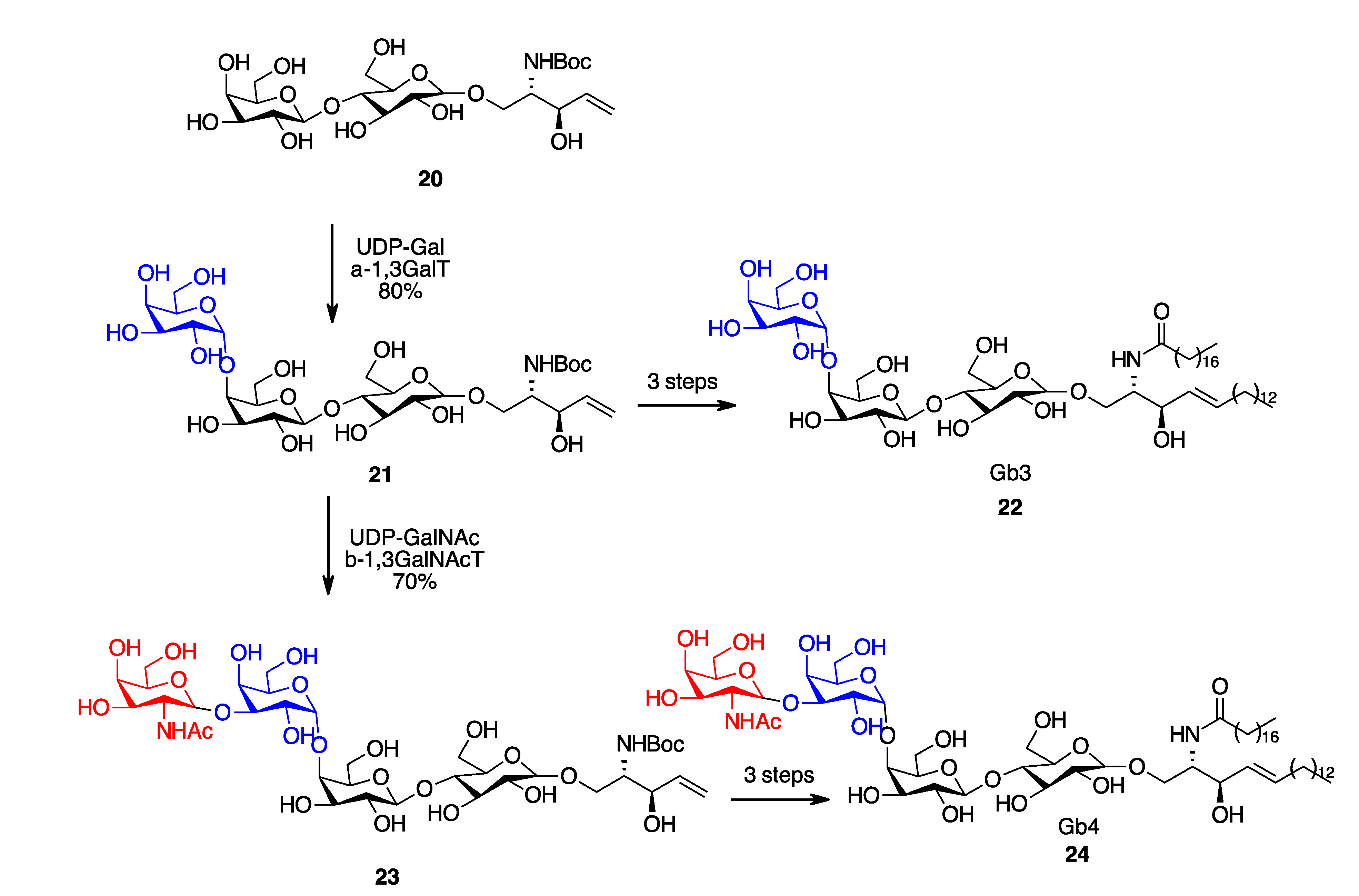
- Na, L.; Li, R.; Chen, X. Recent Progress in Synthesis of Carbohydrates with Sugar Nucleotide-Dependent Glycosyltransferases. Curr. Opin. Chem. Biol. 2021, 61, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Vacariu, C.M.; Tanner, M.E. Recent Advances in the Synthesis and Biological Applications of Peptidoglycan Fragments. Chem. A Eur. J. 2022, 28, e202200788. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xu, H.; Fang, J.; Zhang, X. Enzymatic and Chemoenzymatic Synthesis of Human Milk Oligosaccharides and Derivatives. Carbohydr. Polym. 2022, 291, 119564. [Google Scholar] [CrossRef]
- Li, B.-H.; Ye, X.-S. Recent Advances in Glycan Synthesis. Curr. Opin. Chem. Biol 2020, 58, 20–27. [Google Scholar] [CrossRef]
- Desmons, S.; Grayson-Steel, K.; Nuñez-Dallos, N.; Vendier, L.; Hurtado, J.; Clapés, P.; Fauré, R.; Dumon, C.; Bontemps, S. Enantioselective Reductive Oligomerization of Carbon Dioxide into L-Erythrulose via a Chemoenzymatic Catalysis. J. Am. Chem. Soc. 2021, 143, 16274–16283. [Google Scholar] [CrossRef]
- Srivastava, A.D.; Unione, L.; Bunyatov, M.; Gagarinov, I.A.; Delgado, S.; Abrescia, N.G.A.; Ardá, A.; Boons, G.J. Chemoenzymatic Synthesis of Complex N-Glycans of the Parasite S. Mansoni to Examine the Importance of Epitope Presentation on DC-SIGN Recognition. Angew. Chem. Int. Ed. 2021, 60, 19287–19296. [Google Scholar] [CrossRef]
- Hu, Z.; Benkoulouche, M.; Barel, L.A.; le Heiget, G.; ben Imeddourene, A.; le Guen, Y.; Monties, N.; Guerreiro, C.; Remaud-Siméon, M.; Moulis, C.; et al. Convergent Chemoenzymatic Strategy to Deliver a Diversity of Shigella Flexneri Serotype-Specific O-Antigen Segments from a Unique Lightly Protected Tetrasaccharide Core. J. Org. Chem. 2021, 86, 2058–2075. [Google Scholar] [CrossRef]
- Rath, P.; Rapp, J.; Brilisauer, K.; Braun, M.; Kolukisaoglu, Ü.; Forchhammer, K.; Grond, S. Hybrid Chemoenzymatic Synthesis of C 7 -Sugars for Molecular Evidence of in Vivo Shikimate Pathway Inhibition. ChemBioChem 2022, 23, 202200241. [Google Scholar] [CrossRef]
- Dussouy, C.; Téletchéa, S.; Lambert, A.; Charlier, C.; Botez, I.; de Ceuninck, F.; Grandjean, C. Access to Galectin-3 Inhibitors from Chemoenzymatic Synthons. J. Org. Chem. 2020, 85, 16099–16114. [Google Scholar] [CrossRef]
- Li, R.; Kooner, A.S.; Muthana, S.M.; Yuan, Y.; Yu, H.; Chen, X. A Chemoenzymatic Synthon Strategy for Synthesizing N-Acetyl Analogues of O-Acetylated N. Meningitidis W Capsular Polysaccharide Oligosaccharides. J. Org. Chem. 2020, 85, 16157–16165. [Google Scholar] [CrossRef]
- Yu, H.; Gadi, M.R.; Bai, Y.; Zhang, L.; Li, L.; Yin, J.; Wang, P.G.; Chen, X. Chemoenzymatic Total Synthesis of GM3 Gangliosides Containing Different Sialic Acid Forms and Various Fatty Acyl Chains. J. Org. Chem. 2021, 86, 8672–8682. [Google Scholar] [CrossRef]
- Li, Q.; Jaiswal, M.; Rohokale, R.S.; Guo, Z. A Diversity-Oriented Strategy for Chemoenzymatic Synthesis of Glycosphingolipids and Related Derivatives. Org. Lett. 2020, 22, 8245–8249. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Deng, Y.; Xu, Z.; Liu, X.; Chapla, D.G.; Moremen, K.W.; Wen, L.; Li, T. Integrated Chemoenzymatic Approach to Streamline the Assembly of Complex Glycopeptides in the Liquid Phase. J. Am. Chem. Soc. 2022, 144, 9057–9065. [Google Scholar] [CrossRef]
- Forneris, C.C.; Nguy, A.K.L.; Seyedsayamdost, M.R. Mapping and Exploiting the Promiscuity of OxyB toward the Biocatalytic Production of Vancomycin Aglycone Variants. ACS Catal. 2020, 10, 9287–9298. [Google Scholar] [CrossRef]
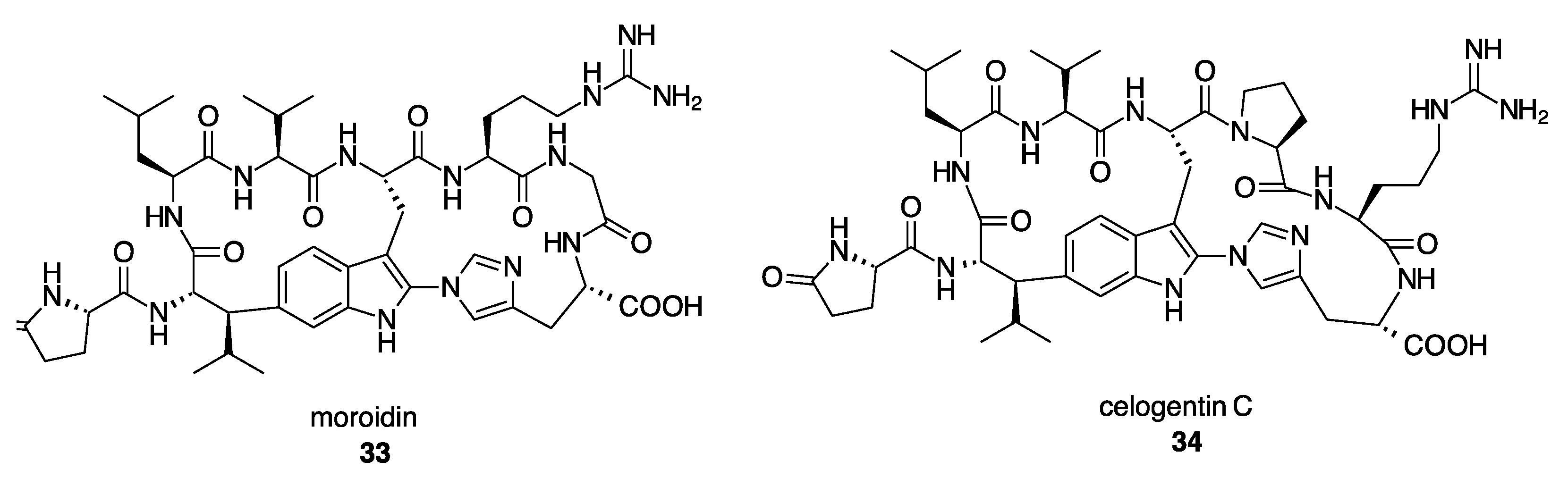
- Mohanty, I.; Nguyen, N.A.; Moore, S.G.; Biggs, J.S.; Gaul, D.A.; Garg, N.; Agarwal, V. Enzymatic Synthesis Assisted Discovery of Proline-Rich Macrocyclic Peptides in Marine Sponges. ChemBioChem 2021, 22, 2614–2618. [Google Scholar] [CrossRef]
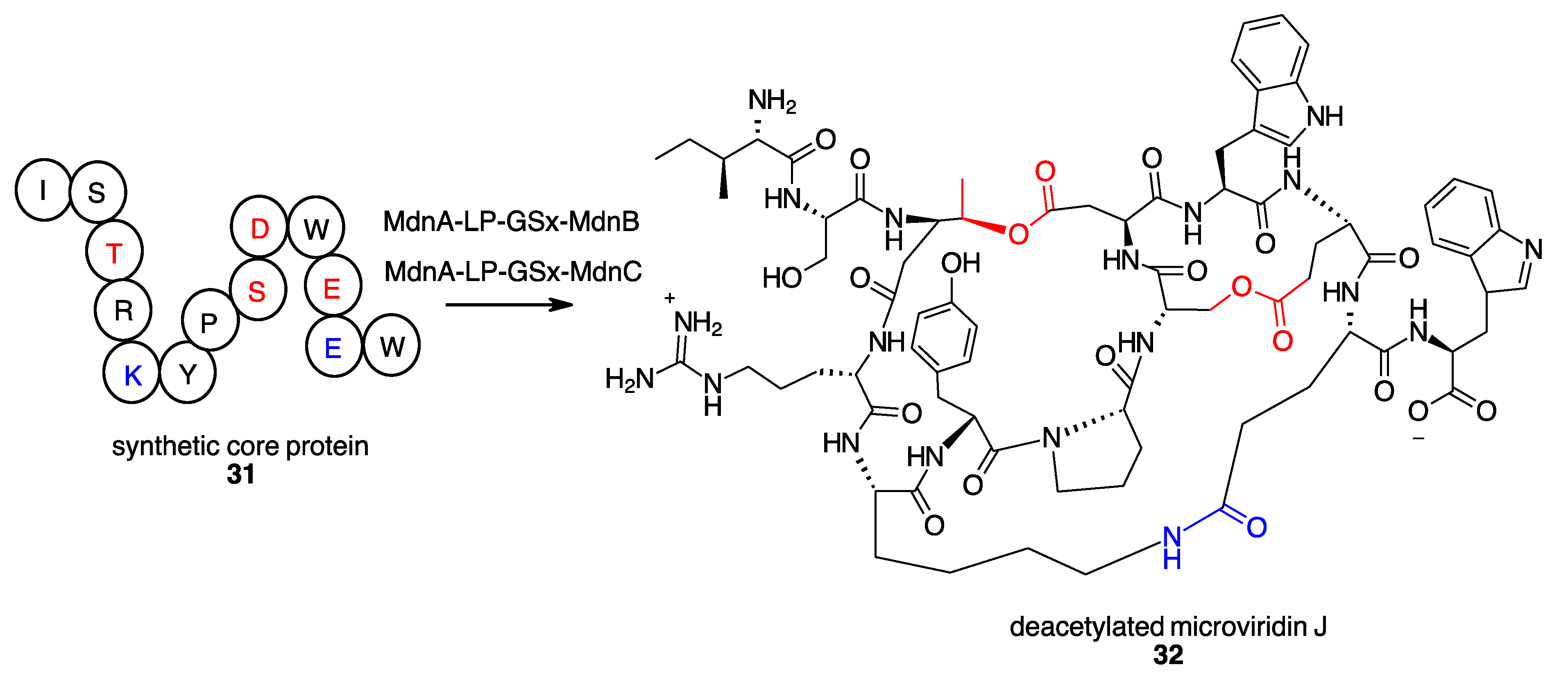
- Patel, K.P.; Silsby, L.M.; Li, G.; Bruner, S.D. Structure-Based Engineering of Peptide Macrocyclases for the Chemoenzymatic Synthesis of Microviridins. J. Org. Chem. 2021, 86, 11212–11219. [Google Scholar] [CrossRef] [PubMed]
- Kersten, R.D.; Mydy, L.S.; Fallon, T.R.; de Waal, F.; Shafiq, K.; Wotring, J.W.; Sexton, J.Z.; Weng, J.-K. Gene-Guided Discovery and Ribosomal Biosynthesis of Moroidin Peptides. J. Am. Chem. Soc. 2022, 144, 7686–7692. [Google Scholar] [CrossRef]
- Zheng, X.; Li, Y.; Guan, M.; Wang, L.; Wei, S.; Li, Y.-C.; Chang, C.-Y.; Xu, Z. Biomimetic Total Synthesis of the Spiroindimicin Family of Natural Products. Angew. Chem. Int. Ed. 2022, 61, e202208802. [Google Scholar] [CrossRef] [PubMed]
- Borowiecki, P.; Zdun, B.; Dranka, M. Chemoenzymatic Enantioselective and Stereo-Convergent Syntheses of Lisofylline Enantiomers via Lipase-Catalyzed Kinetic Resolution and Optical Inversion Approach. Mol. Catal. 2021, 504, 111451. [Google Scholar] [CrossRef]
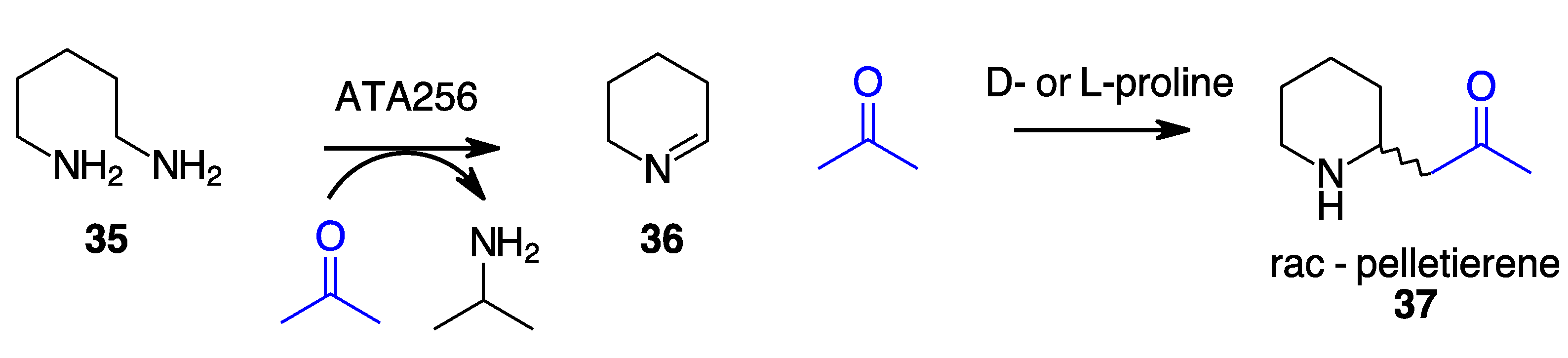
- Taday, F.; Cairns, R.; O’Connell, A.; O’Reilly, E. Combining Bio- and Organocatalysis for the Synthesis of Piperidine Alkaloids. Chem. Commun. 2022, 58, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Galman, J.L.; Slabu, I.; Parmeggiani, F.; Turner, N.J. Biomimetic Synthesis of 2-Substituted N-Heterocycle Alkaloids by One-Pot Hydrolysis, Transamination and Decarboxylative Mannich Reaction. Chem. Commun. 2018, 54, 11316–11319. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.C.; Grischek, B.; Zepeck, F.; Steinreiber, A.; Belaj, F.; Kroutil, W. Regio- and Stereoselective Monoamination of Diketones without Protecting Groups. Angew. Chem. Int. Ed. 2012, 51, 6713–6716. [Google Scholar] [CrossRef] [PubMed]
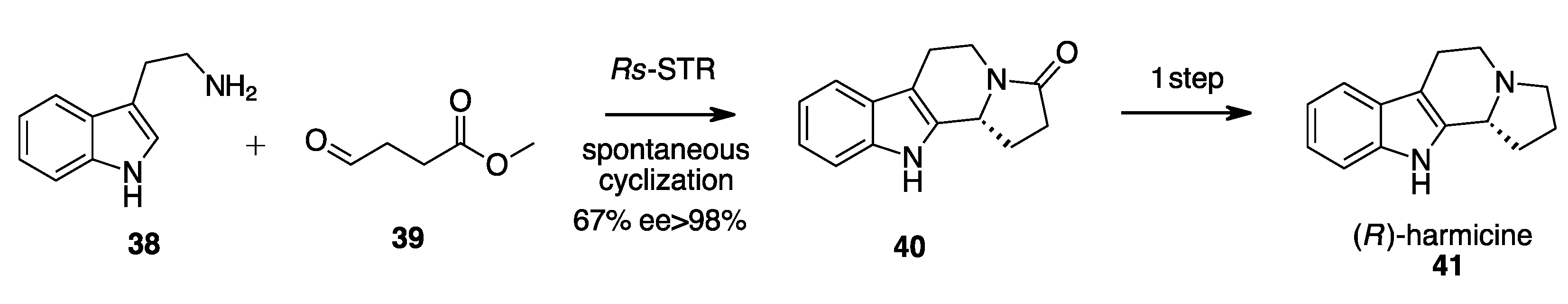
- Eger, E.; Schrittwieser, J.H.; Wetzl, D.; Iding, H.; Kuhn, B.; Kroutil, W. Asymmetric Biocatalytic Synthesis of 1-Aryltetrahydro-β-Carbolines Enabled by “Substrate Walking”. Chem. A Eur. J. 2020, 26, 16281–16285. [Google Scholar] [CrossRef]
- Walia, M.; Teijaro, C.N.; Gardner, A.; Tran, T.; Kang, J.; Zhao, S.; O’Connor, S.E.; Courdavault, V.; Andrade, R.B. Synthesis of (-)-Melodinine K: A Case Study of Efficiency in Natural Product Synthesis. J. Nat. Prod. 2020, 83, 2425–2433. [Google Scholar] [CrossRef]
- St-Pierre, B.; de Luca, V. A Cytochrome P-450 Monooxygenase Catalyzes the First Step in the Conversion of Tabersonine to Vindoline in Catharanthus Roseus. Plant Physiol. 1995, 109, 131–139. [Google Scholar] [CrossRef]
- Besseau, S.; Kellner, F.; Lanoue, A.; Thamm, A.M.K.; Salim, V.; Schneider, B.; Geu-Flores, F.; Höfer, R.; Guirimand, G.; Guihur, A.; et al. A Pair of Tabersonine 16-Hydroxylases Initiates the Synthesis of Vindoline in an Organ-Dependent Manner in Catharanthus Roseus. Plant Physiol. 2013, 163, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
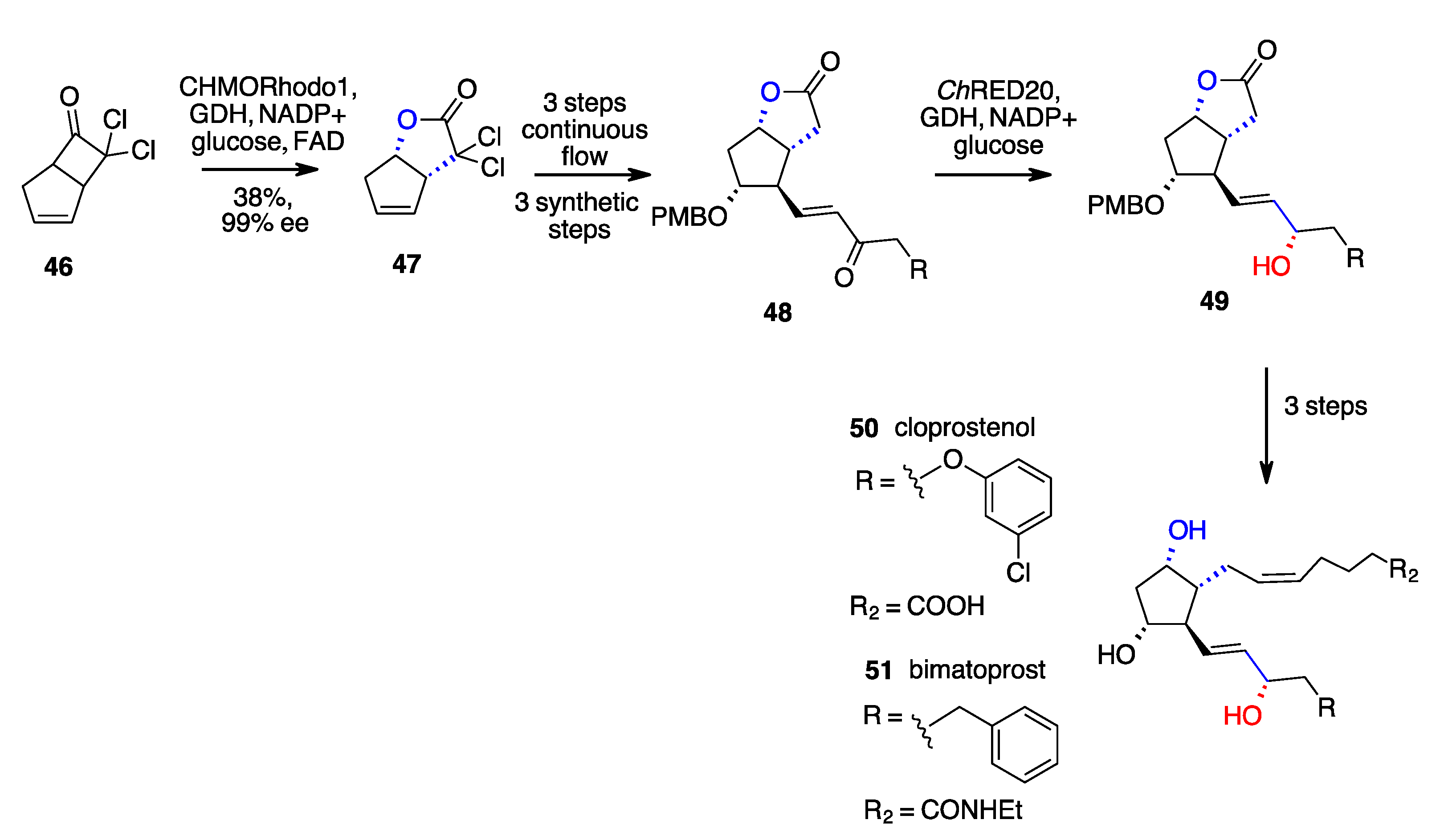
- Zhu, K.; Jiang, M.; Ye, B.; Zhang, G.T.; Li, W.; Tang, P.; Huang, Z.; Chen, F. A Unified Strategy to Prostaglandins: Chemoenzymatic Total Synthesis of Cloprostenol, Bimatoprost, PGF2α, Fluprostenol, and Travoprost Guided by Biocatalytic Retrosynthesis. Chem. Sci. 2021, 12, 10362–10370. [Google Scholar] [CrossRef]
- Milzarek, T.M.; Schuler, S.; Matura, A.; Gulder, T.A.M. Evaluation of the Substrate Promiscuity of SorbC for the Chemo-Enzymatic Total Synthesis of Structurally Diverse Sorbicillinoids. ACS Catal. 2022, 12, 1898–1904. [Google Scholar] [CrossRef]
- Oelschlägel, M.; Stuhr, A.; Pollender, A.; Ganz, D.; Schlömann, M. Process for the Biotechnological Production of 2-Phenylethanols from Plant Sources. PCT/EP2021/084999, 16 June 2022. [Google Scholar]
- Guillamón Navarro, J.M.; Muñiz Calvo, S.; Bisquert Alcaraz, R. Recombinant Saccharomyces Cerevisiae for the Production of Hydroxytyrosol. PCT/ES2021/070769, 2 June 2022. [Google Scholar]
- A Kind of Efficient Synthesis of Hydroxytyrosol by Bacillus Licheniformis, Construction Method and Application. CN114891820A, 14 January 2020.
- Catinella, G.; Donzella, S.; Borgonovo, G.; Dallavalle, S.; Contente, M.L.; Pinto, A. Efficient 2-Step Enzymatic Cascade for the Bioconversion of Oleuropein into Hydroxytyrosol. Antioxidants 2022, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, F.; Contente, M.L.; Pinna, C.; Tamborini, L.; Pinto, A. Biocatalyzed Flow Oxidation of Tyrosol to Hydroxytyrosol and Efficient Production of Their Acetate Esters. Antioxidants 2021, 10, 1142. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanable, E.P.; Habgood, L.G.; Patrone, J.D. Current Progress in the Chemoenzymatic Synthesis of Natural Products. Molecules 2022, 27, 6373. https://doi.org/10.3390/molecules27196373
Vanable EP, Habgood LG, Patrone JD. Current Progress in the Chemoenzymatic Synthesis of Natural Products. Molecules. 2022; 27(19):6373. https://doi.org/10.3390/molecules27196373
Chicago/Turabian StyleVanable, Evan P., Laurel G. Habgood, and James D. Patrone. 2022. "Current Progress in the Chemoenzymatic Synthesis of Natural Products" Molecules 27, no. 19: 6373. https://doi.org/10.3390/molecules27196373
APA StyleVanable, E. P., Habgood, L. G., & Patrone, J. D. (2022). Current Progress in the Chemoenzymatic Synthesis of Natural Products. Molecules, 27(19), 6373. https://doi.org/10.3390/molecules27196373