
Discovery of E3 Ligase Ligands for Target Protein Degradation
 ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CRBN Ligands
3. VHL Ligands
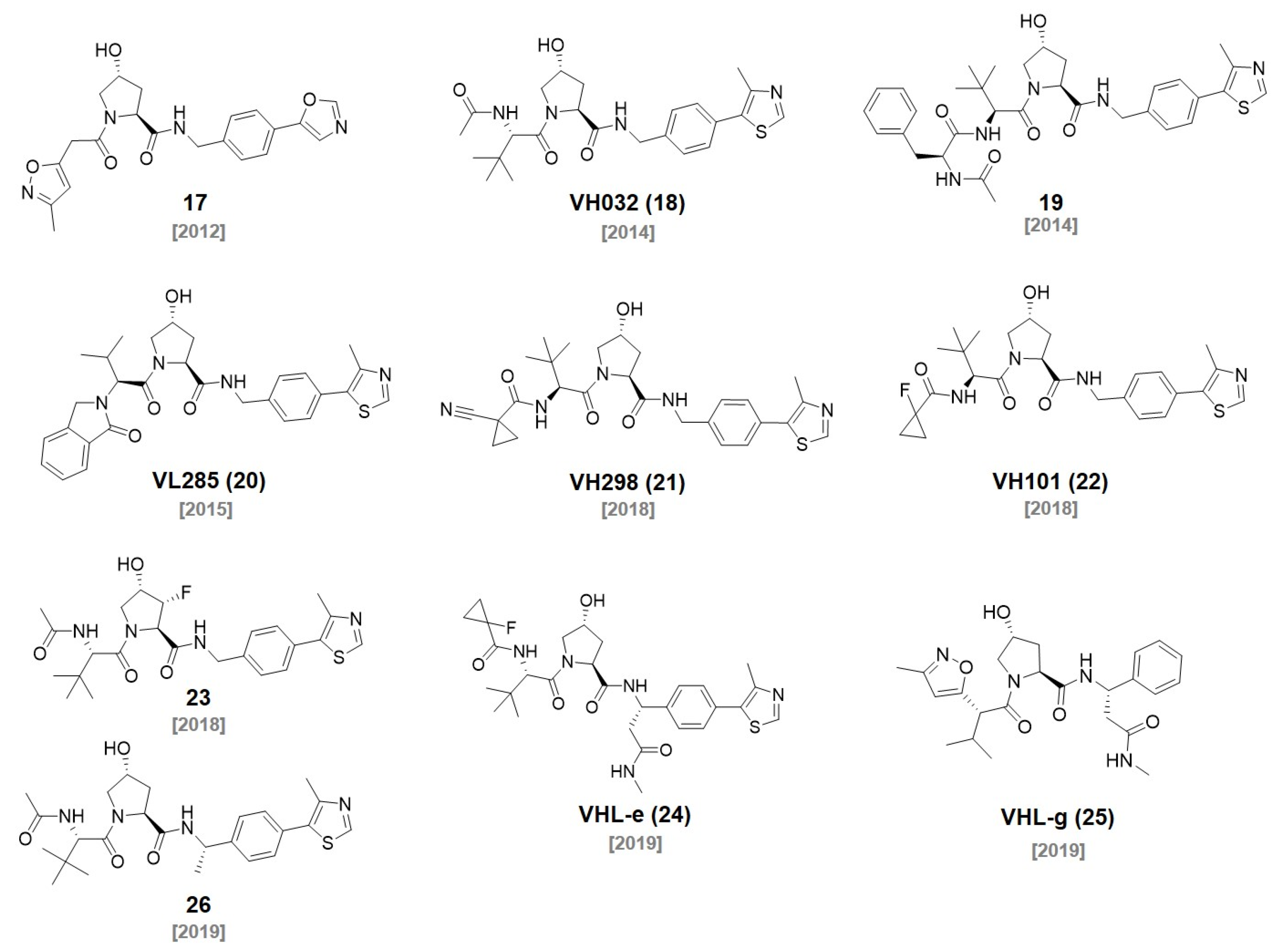
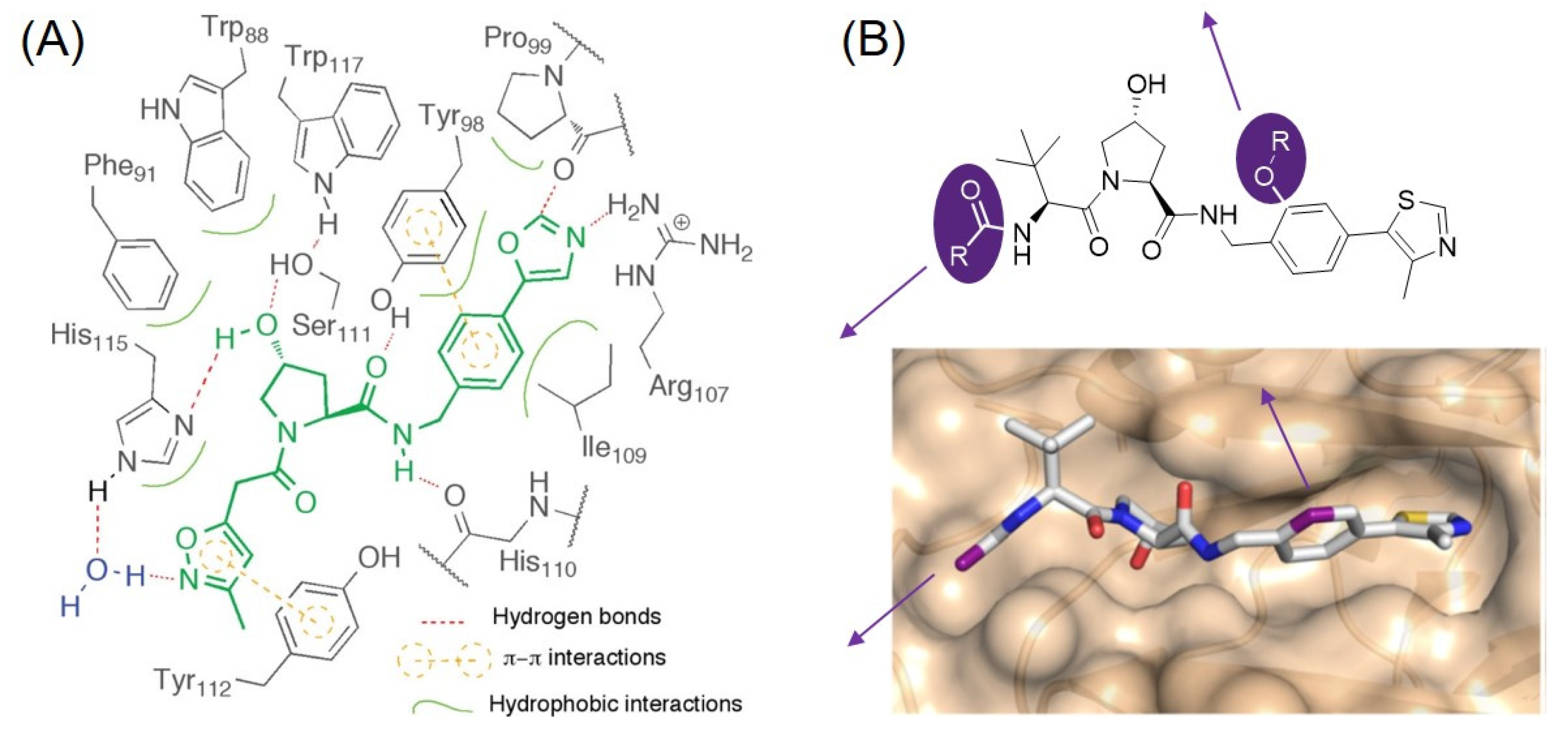
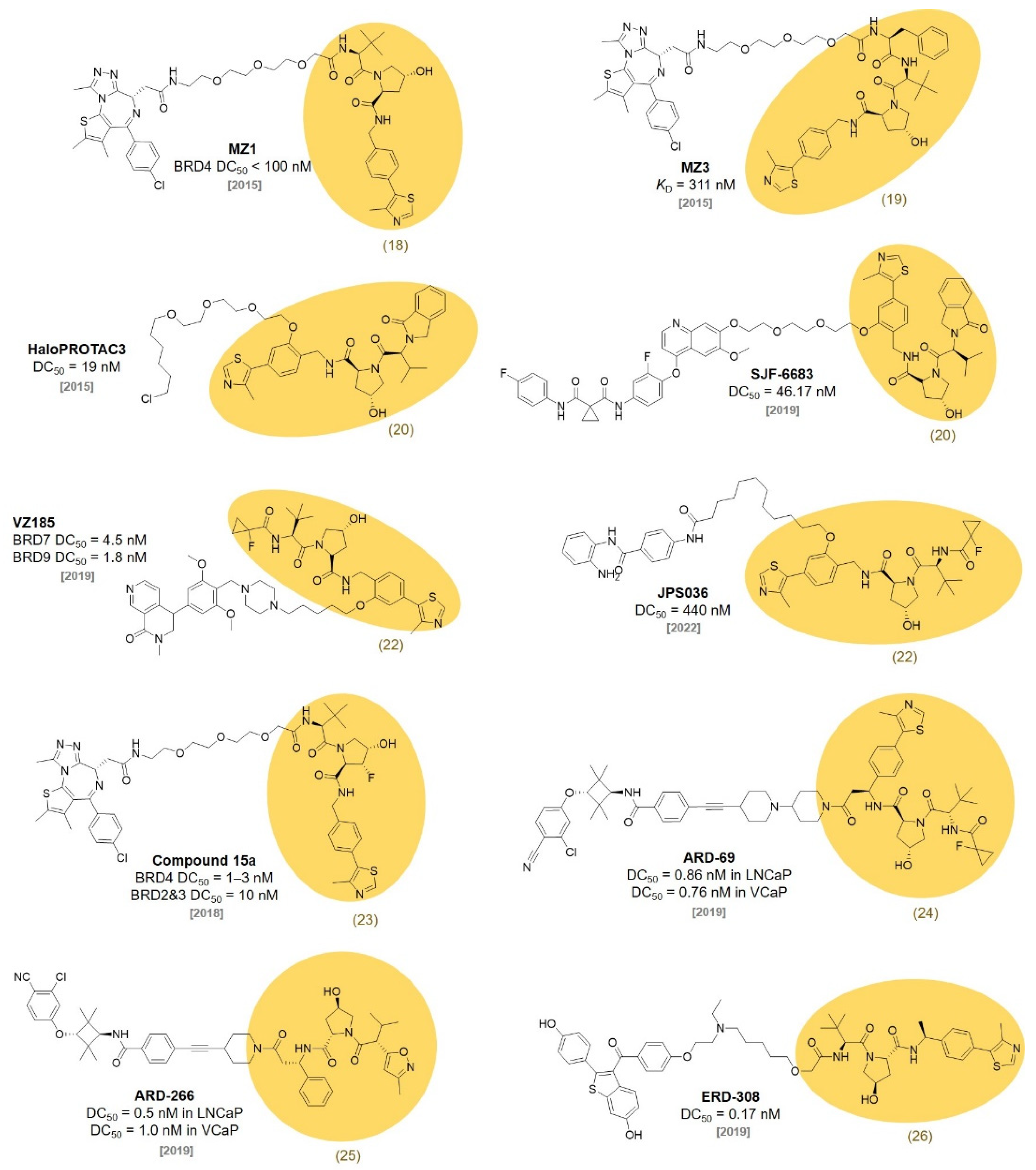
4. IAP Ligands
5. MDM2 Ligands
6. DCAF Ligands
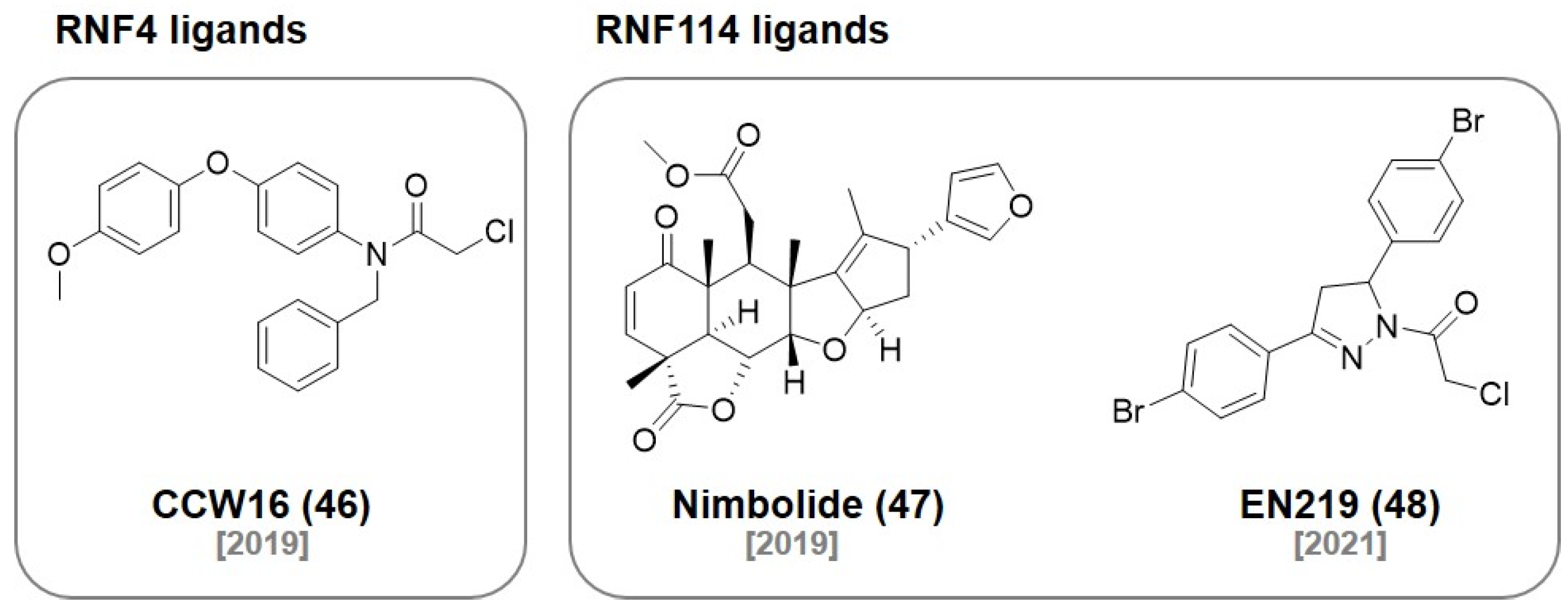
7. RNF Ligands
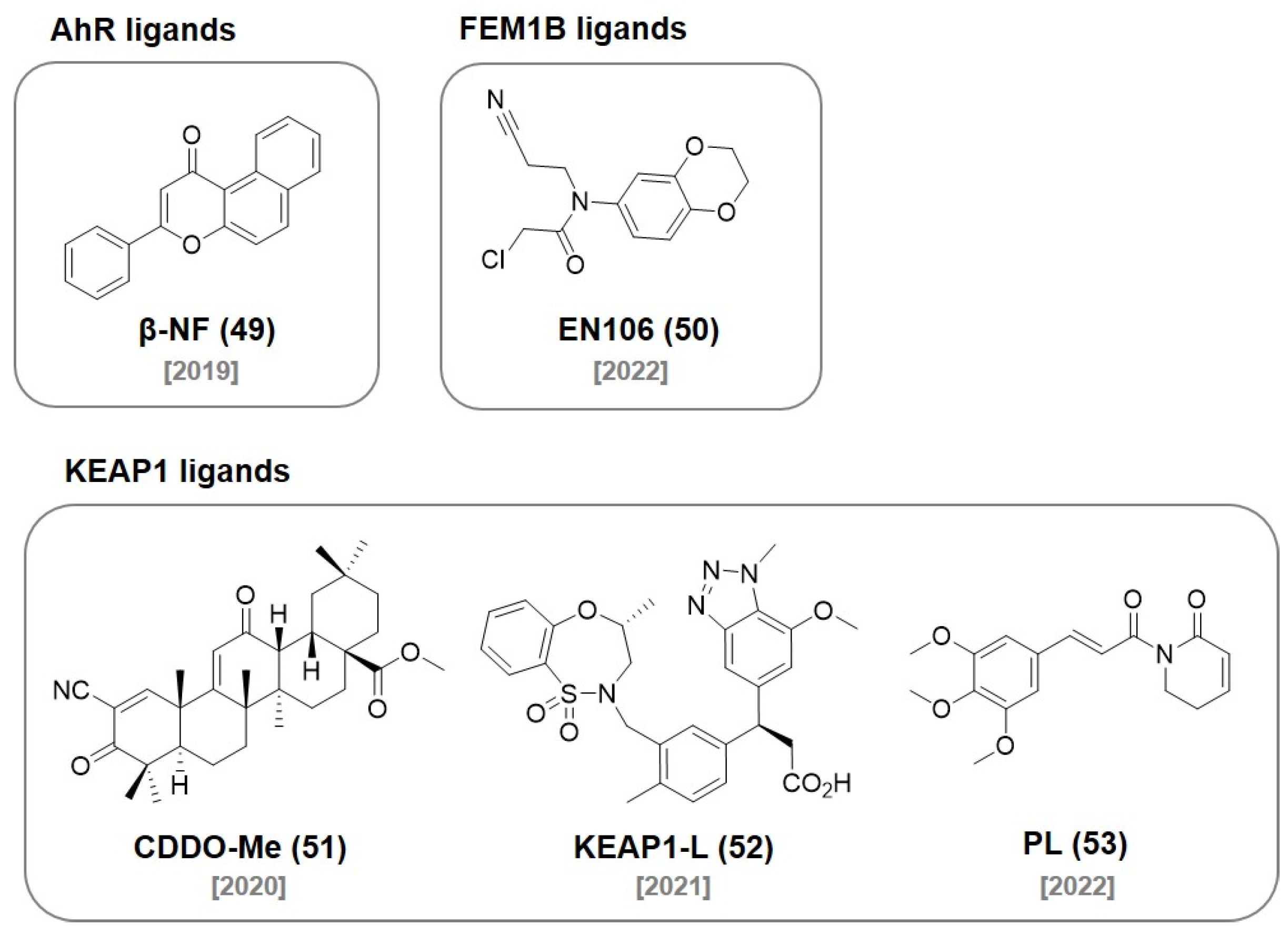
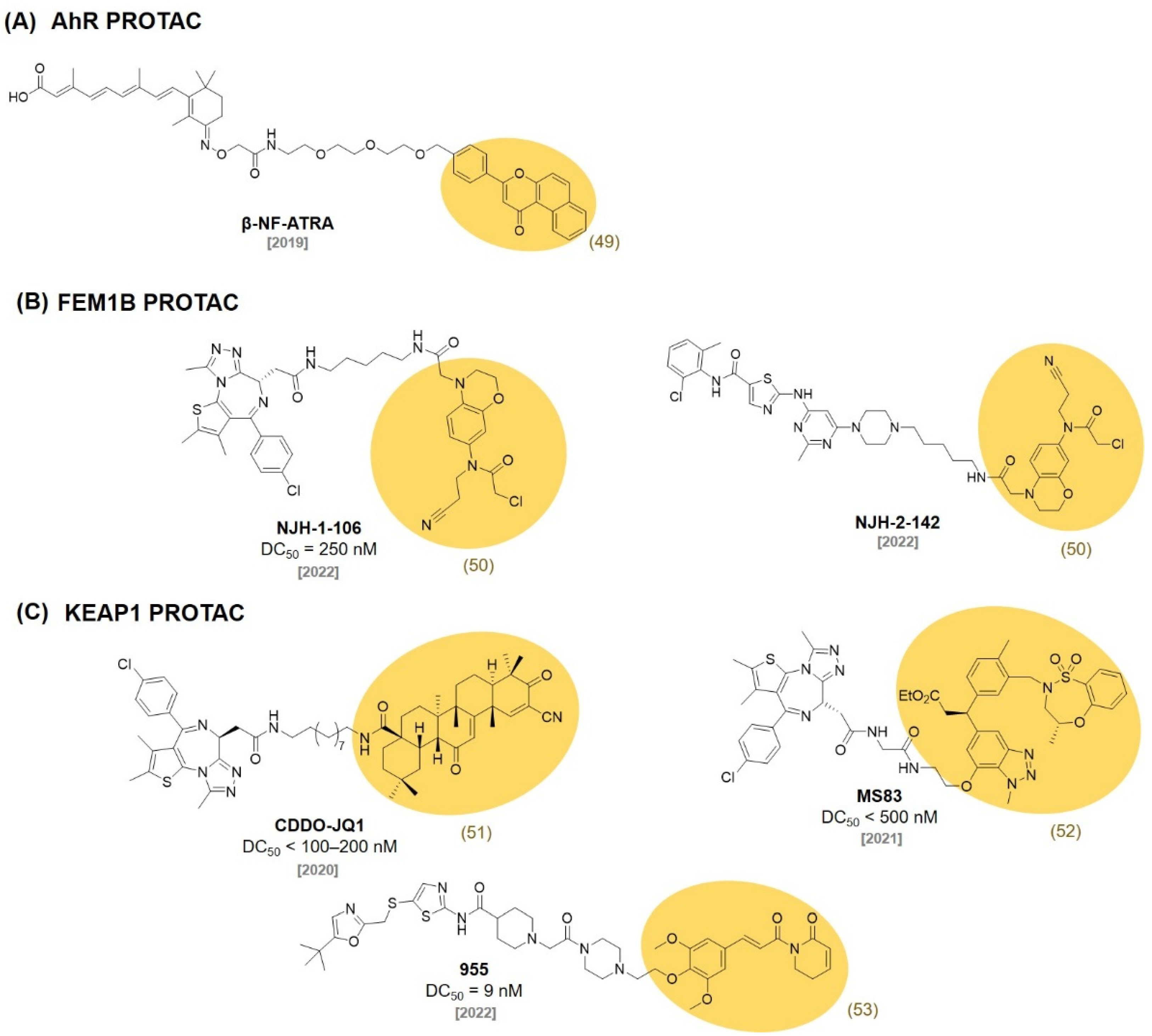
8. AhR Ligands
9. FEM1B Ligands
10. KEAP1 Ligands
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Röth, S.; Fulcher, L.J.; Sapkota, G.P. Advances in targeted degradation of endogenous proteins. Cell Mol. Life Sci. 2019, 76, 2761–2777. [Google Scholar] [CrossRef] [Green Version]
- Prozzillo, Y.; Fattorini, G.; Santopietro, M.V.; Suglia, L.; Ruggiero, A.; Ferreri, D.; Messina, G. Targeted Protein Degradation Tools: Overview and Future Perspectives. Biology 2020, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Ottis, P.; Palladino, C.; Thienger, P.; Britschgi, A.; Heichinger, C.; Berrera, M.; Julien-Laferriere, A.; Roudnicky, F.; Kam-Thong, T.; Bischoff, J.R.; et al. Cellular Resistance Mechanisms to Targeted Protein Degradation Converge Toward Impairment of the Engaged Ubiquitin Transfer Pathway. ACS Chem. Biol. 2019, 14, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Riley-Gillis, B.; Vijay, P.; Shen, Y. Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Mol. Cancer Ther. 2019, 18, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabi, S.B.; Crews, C.M. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Harjai, K.; Chhibber, S. Thalidomide treatment modulates macrophage pro-inflammatory function and cytokine levels in Klebsiella pneumoniae B5055 induced pneumonia in BALB/c mice. Int. Immunopharmacol. 2010, 10, 777–783. [Google Scholar] [CrossRef]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef] [Green Version]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Bruins, L.A.; Schmidt, J.E.; Van Wier, S.; Chang, X.-B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, G.; Ding, Y.; He, S.; Sheng, C. Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J. Med. Chem. 2021, 64, 10606–10620. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Burslem, G.M.; Ottis, P.; Jaime-Figueroa, S.; Morgan, A.; Cromm, P.M.; Toure, M.; Crews, C.M. Efficient Synthesis of Immunomodulatory Drug Analogues Enables Exploration of Structure-Degradation Relationships. ChemMedChem 2018, 13, 1508–1512. [Google Scholar] [CrossRef]
- De Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468. [Google Scholar] [CrossRef] [Green Version]
- Kargbo, R.B. PROTAC Molecules for the Treatment of Autoimmune Disorders. ACS Med. Chem. Lett. 2019, 10, 276–277. [Google Scholar] [CrossRef] [Green Version]
- Sabnis, R.W. Bifunctional Compounds as SMARCA2 Degraders for Treating Cancer. ACS Med. Chem. Lett. 2022, 13, 15–16. [Google Scholar] [CrossRef]
- Kim, S.A.; Go, A.; Jo, S.-H.; Park, S.J.; Jeon, Y.U.; Kim, J.E.; Lee, H.K.; Park, C.H.; Lee, C.-O.; Park, S.G.; et al. A novel cereblon modulator for targeted protein degradation. Eur. J. Med. Chem. 2019, 166, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Takwale, A.D.; Jo, S.-H.; Jeon, Y.U.; Kim, H.S.; Shin, C.H.; Lee, H.K.; Ahn, S.; Lee, C.O.; Ha, J.D.; Kim, J.-H.; et al. Design and characterization of cereblon-mediated androgen receptor proteolysis-targeting chimeras. Eur. J. Med. Chem. 2020, 208, 112769. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Burris III, H.A.; Vuky, J.; Dreicer, R.; Sartor, A.O.; Sternberg, C.N.; Percent, I.J.; Hussain, M.H.A.; Kalebasty, A.R.; Shen, J.; et al. Phase 1/2 study of ARV-110, an androgen receptor (AR) PROTAC degrader, in metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, 17. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Schott, A.F.; Nanda, R.; Lu, H.; Keung, C.F.; Gedrich, R.; Parameswaran, J.; Han, H.S.; Hurvitz, S.A. ARV-471, an estrogen receptor (ER) PROTACdegrader, combined with palbociclib in advanced ER+/human epidermal growth factor receptor 2–negative (HER2-) breast cancer: Phase 1b cohort (part C) of a phase 1/2 study. J. Clin. Oncol. 2022, 40, TPS1120. [Google Scholar] [CrossRef]
- Mainolfi, N.; JI, N.; Kluge, A.F. CRBN Ligands and Uses Thereof. International Patent WO 2019140387 A1, 18 July 2019. [Google Scholar]
- Kargbo, R.B. PROTAC Degradation of IRAK4 for the Treatment of Cancer. ACS Med. Chem. Lett. 2019, 10, 1370–1371. [Google Scholar] [CrossRef] [Green Version]
- Kargbo, R.B. PROTAC-Mediated Degradation of Bruton’s Tyrosine Kinase as a Therapeutic Strategy for Cancer. ACS Med. Chem. Lett. 2021, 12, 688–689. [Google Scholar] [CrossRef]
- Min, J.; Mayasundari, A.; Keramatnia, F.; Jonchere, B.; Yang, S.W.; Jarusiewicz, J.; Actis, M.; Das, S.; Young, B.; Slavish, J.; et al. Phenyl-Glutarimides: Alternative Cereblon Binders for the Design of PROTACs. Angew. Chem. Int. Ed. 2021, 60, 26663–26670. [Google Scholar] [CrossRef]
- Alcock, L.J.; Chang, Y.; Jarusiewicz, J.A.; Actis, M.; Nithianantham, S.; Mayasundari, A.; Min, J.; Maxwell, D.; Hunt, J.; Smart, B.; et al. Development of Potent and Selective Janus Kinase 2/3 Directing PG–PROTACs. ACS Med. Chem. Lett. 2022, 13, 475–482. [Google Scholar] [CrossRef]
- Wang, S.; Xu, T.; Wang, M.; Hu, J.; Han, X.; Xiang, W.; REJ, R. Cereblon E3 Ligase Inhibitors. International Patent WO 2021041664 A1, 4 March 2021. [Google Scholar]
- Kargbo, R.B. Discovery of Spirocyclic Androgen Receptor Protein Degraders for the Treatment of Prostate Cancer. ACS Med. Chem. Lett. 2021, 12, 1635–1636. [Google Scholar] [CrossRef]
- Sabnis, R.W. BRD9 Bifunctional Degraders for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 1879–1880. [Google Scholar] [CrossRef]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef]
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Soares, P.; Gadd, M.S.; Frost, J.; Galdeano, C.; Ellis, L.; Epemolu, O.; Rocha, S.; Read, K.D.; Ciulli, A. Group-Based Optimization of Potent and Cell-Active Inhibitors of the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase: Structure–Activity Relationships Leading to the Chemical Probe (2S,4R)-1-((S)-2-(1-Cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 2018, 61, 599–618. [Google Scholar]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef]
- Smalley, J.P.; Baker, I.M.; Pytel, W.A.; Lin, L.-Y.; Bowman, K.J.; Schwabe, J.W.R.; Cowley, S.M.; Hodgkinson, J.T. Optimization of Class I Histone Deacetylase PROTACs Reveals that HDAC1/2 Degradation is Critical to Induce Apoptosis and Cell Arrest in Cancer Cells. J. Med. Chem. 2022, 65, 5642–5659. [Google Scholar] [CrossRef]
- Testa, A.; Lucas, X.; Castro, G.V.; Chan, K.-H.; Wright, J.E.; Runcie, A.C.; Gadd, M.S.; Harrison, W.T.A.; Ko, E.-J.; Fletcher, D.; et al. 3-Fluoro-4-hydroxyprolines: Synthesis, Conformational Analysis, and Stereoselective Recognition by the VHL E3 Ubiquitin Ligase for Targeted Protein Degradation. J. Am. Chem. Soc. 2018, 140, 9299–9313. [Google Scholar] [CrossRef] [Green Version]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.-Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhao, L.; Xiang, W.; Qin, C.; Miao, B.; Xu, T.; Wang, M.; Yang, C.-Y.; Chinnaswamy, K.; Stuckey, J.; et al. Discovery of Highly Potent and Efficient PROTAC Degraders of Androgen Receptor (AR) by Employing Weak Binding Affinity VHL E3 Ligase Ligands. J. Med. Chem. 2019, 62, 11218–11231. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hu, B.; Wang, M.; Xu, F.; Miao, B.; Yang, C.-Y.; Wang, M.; Liu, Z.; Hayes, D.F.; Chinnaswamy, K.; et al. Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (ER). J. Med. Chem. 2019, 62, 1420–1442. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP Antagonists Induce Autoubiquitination of c-IAPs, NF-kB Activation, and TNFa-Dependent Apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, K.; Takubo, K.; Kikuchi, R.; Nishimoto, M.; Kitagawa, M.; Abe, F.; Nishikawa, K.; Tsuruo, T.; Naito, M. Small Molecules Destabilize cIAP1 by Activating Auto-ubiquitylation. J. Biol. Chem. 2008, 283, 8961–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Kitaguchi, R.; Okuhira, K.; Naito, M.; Hashimoto, Y. Double protein knockdown of cIAP1 and CRABP-II using a hybrid molecule consisting of ATRA and IAPs antagonist. Bioorg. Med. Chem. Lett. 2012, 22, 4453–4457. [Google Scholar] [CrossRef]
- Ohoka, N.; Okuhira, K.; Ito, M.; Nagai, K.; Shibata, N.; Hattori, T.; Ujikawa, O.; Shimokawa, K.; Sano, O.; Koyama, R.; et al. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs). J. Biol. Chem. 2017, 292, 4556–4570. [Google Scholar] [CrossRef] [Green Version]
- Ohoka, N.; Ujikawa, O.; Shimokawa, K.; Sameshima, T.; Shibata, N.; Hattori, T.; Nara, H.; Cho, N.; Naito, M. Different Degradation Mechanisms of Inhibitor of Apoptosis Proteins (IAPs) by the Specific and Nongenetic IAP-Dependent Protein Eraser (SNIPER). Chem. Pharm. Bull. 2019, 67, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Shimokawa, K.; Shibata, N.; Sameshima, T.; Miyamoto, N.; Ujikawa, O.; Nara, H.; Ohoka, N.; Hattori, T.; Cho, N.; Naito, M. Targeting the Allosteric Site of Oncoprotein BCR-ABL as an Alternative Strategy for Effective Target Protein Degradation. ACS Med. Chem. Lett. 2017, 8, 1042–1047. [Google Scholar] [CrossRef]
- Flygare, J.A.; Beresini, M.; Budha, N.; Chan, H.; Chan, I.T.; Cheeti, S.; Cohen, F.; Deshayes, K.; Doerner, K.; Eckhardt, S.G.; et al. Discovery of a Potent Small-Molecule Antagonist of Inhibitor of Apoptosis (IAP) Proteins and Clinical Candidate for the Treatment of Cancer (GDC-0152). J. Med. Chem. 2012, 55, 4101–4113. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.J.; Saeh, J.C.; Sha, L.; MacIntyre, T.; Wang, H.; Larsen, N.A.; Aquila, B.M.; Ferguson, A.D.; Laing, N.M.; Omer, C.A. Discovery of aminopiperidine-based Smac mimetics as IAP antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 1690–1694. [Google Scholar] [CrossRef] [PubMed]
- Vamos, M.; Welsh, K.; Finlay, D.; Lee, P.S.; Mace, P.D.; Snipas, S.J.; Gonzalez, M.L.; Ganji, S.R.; Ardecky, R.J.; Riedl, S.J.; et al. Expedient Synthesis of Highly Potent Antagonists of Inhibitor of Apoptosis Proteins (IAPs) with Unique Selectivity for ML-IAP. ACS Chem. Biol. 2013, 8, 725–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; He, Y.; Zhang, P.; Budamagunta, V.; Lv, D.; Thummuri, D.; Yang, Y.; Pei, J.; Yuan, Y.; Zhou, D.; et al. Discovery of IAP-recruiting BCL-XL PROTACs as potent degraders across multiple cancer cell lines. Eur. J. Med. Chem. 2020, 199, 112397. [Google Scholar] [CrossRef]
- Kim, K.S.; Zhang, L.; Williams, D.; Perez, H.L.; Stang, E.; Borzilleri, R.M.; Posy, S.; Lei, M.; Chaudhry, C.; Emanuel, S.; et al. Discovery of tetrahydroisoquinoline-based bivalent heterodimeric IAP antagonists. Bioorg. Med. Chem. Lett. 2014, 24, 5022–5029. [Google Scholar] [CrossRef]
- Schiemer, J.; Horst, R.; Meng, Y.; Montgomery, J.I.; Xu, Y.; Feng, X.; Borzilleri, K.; Uccello, D.P.; Leverett, C.; Brown, S.; et al. Snapshots and ensembles of BTK and cIAP1 protein degrader ternary complexes. Nat. Chem. Biol. 2021, 17, 152–160. [Google Scholar] [CrossRef]
- Ohoka, N.; Morita, Y.; Nagai, K.; Shimokawa, K.; Ujikawa, O.; Fujimori, I.; Ito, M.; Hayase, Y.; Okuhira, K.; Shibata, N.; et al. Derivatization of inhibitor of apoptosis protein (IAP) ligands yields improved inducers of estrogen receptor alpha degradation. J. Biol. Chem. 2018, 293, 6776–6790. [Google Scholar] [CrossRef] [Green Version]
- Tamanini, E.; Buck, I.M.; Chessari, G.; Chiarparin, E.; Day, J.E.H.; Frederickson, M.; Griffiths-Jones, C.M.; Hearn, K.; Heightman, T.D.; Iqbal, A.; et al. Discovery of a Potent Nonpeptidomimetic, Small-Molecule Antagonist of Cellular Inhibitor of Apoptosis Protein 1 (cIAP1) and X-Linked Inhibitor of Apoptosis Protein (XIAP). J. Med. Chem. 2017, 60, 4611–4625. [Google Scholar] [CrossRef]
- Jiang, B.; Wang, E.S.; Donovan, K.A.; Liang, Y.; Fischer, E.S.; Zhang, T.; Gray, N.S. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. 2019, 58, 6321–6326. [Google Scholar] [CrossRef]
- Rana, S.; Bendjennat, M.; Kour, S.; King, H.M.; Kizhake, S.; Zahid, M.; Natarajan, A. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg. Med. Chem. Lett. 2019, 29, 1375–1379. [Google Scholar] [CrossRef]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.A.; Cryan, J.; Ahmed, A.; Dai, H.; McGonagle, G.A.; Rozier, C.; Benowitz, A.B. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg. Med. Chem. Lett. 2020, 30, 127106. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Liu, Y.-J.; Hao, L.; Mizoguchi, A.; Salzman, N.H.; Bevins, C.L.; Kobayashi, K.S. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc. Natl. Acad. Sci. USA 2010, 107, 14739–14744. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Williams, H.R.T.; Spagnolo, P.; Abdallah, A.; Ahmad, T.; Orchard, T.R.; Copley, S.J.; Desai, S.R.; Wells, A.U.; Du Bois, R.M.; et al. CARD15/NOD2 polymorphisms are associated with severe pulmonary sarcoidosis. Eur. Respir. J. 2010, 35, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Barr, M.J.; Lukens, J.R.; McGargill, M.A.; Chi, H.; Mak, T.W.; Kanneganti, T.-D. Signaling via the RIP2 Adaptor Protein in Central Nervous System-Infiltrating Dendritic Cells Promotes Inflammation and Autoimmunity. Immunity 2011, 34, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miah, A.H.; Smith, I.E.D.; Rackham, M.; Mares, A.; Thawani, A.R.; Nagilla, R.; Haile, P.A.; Votta, B.J.; Gordon, L.J.; Watt, G.; et al. Optimization of a Series of RIPK2 PROTACs. J. Med. Chem. 2021, 64, 12978–13003. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Ma, J.; Fang, Y.; Liu, Y.; Wu, S.; Dong, G.; Wang, W.; Sheng, C. Homo-PROTAC mediated suicide of MDM2 to treat non-small cell lung cancer. Acta Pharm. Sin. B. 2021, 11, 1617–1628. [Google Scholar] [CrossRef]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Zhang, Z.; Liu, J.-J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a Potent and Selective p53–MDM2 Inhibitor in Clinical Development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- De Dominici, M.; Porazzi, P.; Xiao, Y.; Chao, A.; Tang, H.-Y.; Kumar, G.; Fortina, P.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 2020, 135, 1560–1573. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef]
- Du, X.; Volkov, O.A.; Czerwinski, R.M.; Tan, H.; Huerta, C.; Morton, E.R.; Rizzi, J.P.; Wehn, P.M.; Xu, R.; Nijhawan, D.; et al. Structural Basis and Kinetic Pathway of RBM39 Recruitment to DCAF15 by a Sulfonamide Molecular Glue E7820. Structure 2019, 27, 1625–1633. [Google Scholar] [CrossRef]
- Uehara, T.; Minoshima, Y.; Sagane, K.; Sugi, N.H.; Mitsuhashi, K.O.; Yamamoto, N.; Kamiyama, H.; Takahashi, K.; Kotake, Y.; Uesugi, M.; et al. Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nat. Chem. Biol. 2017, 13, 675–680. [Google Scholar] [CrossRef]
- Li, L.; Mi, D.; Pei, H.; Duan, Q.; Wang, X.; Zhou, W.; Jin, J.; Li, D.; Liu, M.; Chen, Y. In vivo target protein degradation induced by PROTACs based on E3 ligase DCAF15. Signal Transduct. Target. Ther. 2020, 5, 129. [Google Scholar] [CrossRef]
- Zhang, X.; Crowley, V.M.; Wucherpfennig, T.G.; Dix, M.M.; Cravatt, B.F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019, 15, 737–746. [Google Scholar] [CrossRef]
- Zhang, X.; Luukkonen, L.M.; Eissler, C.L.; Crowley, V.M.; Yamashita, Y.; Schafroth, M.A.; Kikuchi, S.; Weinstein, D.S.; Symons, K.T.; Nordin, B.E.; et al. DCAF11 Supports Targeted Protein Degradation by Electrophilic Proteolysis-Targeting Chimeras. J. Am. Chem. Soc. 2021, 143, 5141–5149. [Google Scholar] [CrossRef]
- Ward, C.C.; Kleinman, J.I.; Brittain, S.M.; Lee, P.S.; Chung, C.Y.S.; Kim, K.; Petri, Y.; Thomas, J.R.; Tallarico, J.A.; McKenna, J.M.; et al. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol. 2019, 14, 2430–2440. [Google Scholar] [CrossRef]
- Spradlin, J.N.; Hu, X.; Ward, C.C.; Brittain, S.M.; Jones, M.D.; Ou, L.; To, M.; Proudfoot, A.; Ornelas, E.; Woldegiorgis, M.; et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019, 15, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.; Spradlin, J.N.; Novaes, L.F.T.; Zhang, E.; Hu, X.; Moeller, M.; Brittain, S.M.; McGregor, L.M.; McKenna, J.M.; Tallarico, J.A.; et al. A Nimbolide-Based Kinase Degrader Preferentially Degrades Oncogenic BCR-ABL. ACS Chem. Biol. 2020, 15, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Spradlin, J.N.; Boike, L.; Tong, B.; Brittain, S.M.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Maimone, T.J.; Nomura, D.K. Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function. Cell Chem. Biol. 2021, 28, 559–566. [Google Scholar] [CrossRef]
- Ohoka, N.; Tsuji, G.; Shoda, T.; Fujisato, T.; Kurihara, M.; Demizu, Y.; Naito, M. Development of Small Molecule Chimeras That Recruit AhR E3 Ligase to Target Proteins. ACS Chem. Biol. 2019, 14, 2822–2832. [Google Scholar] [CrossRef] [PubMed]
- Henning, N.J.; Manford, A.G.; Spradlin, J.N.; Brittain, S.M.; Zhang, E.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Rape, M.; Nomura, D.K. Discovery of a Covalent FEM1B Recruiter for Targeted Protein Degradation Applications. J. Am. Chem. Soc. 2022, 144, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hu, L. Nrf2 activation through the inhibition of Keap1–Nrf2 protein–protein interaction. Med. Chem. Res. 2020, 29, 846–867. [Google Scholar] [CrossRef]
- Tong, B.; Luo, M.; Xie, Y.; Spradlin, J.N.; Tallarico, J.A.; McKenna, J.M.; Schirle, M.; Maimone, T.J.; Nomura, D.K. Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci. Rep. 2020, 10, 15543. [Google Scholar] [CrossRef]
- Davies, T.G.; Wixted, W.E.; Coyle, J.E.; Griffiths-Jones, C.; Hearn, K.; McMenamin, R.; Norton, D.; Rich, S.J.; Richardson, C.; Saxty, G.; et al. Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein–Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J. Med. Chem. 2016, 59, 3991–4006. [Google Scholar] [CrossRef]
- Wei, J.; Meng, F.; Park, K.-S.; Yim, H.; Velez, J.; Kumar, P.; Wang, L.; Xie, L.; Chen, H.; Shen, Y.; et al. Harnessing the E3 Ligase KEAP1 for Targeted Protein Degradation. J. Am. Chem. Soc. 2021, 143, 15073–15083. [Google Scholar] [CrossRef]
- Pei, J.; Xiao, Y.; Liu, X.; Hu, W.; Sobh, A.; Yuan, Y.; Zhou, S.; Hua, N.; Mackintosh, S.G.; Zhang, X.; et al. Identification of Piperlongumine (PL) as a new E3 ligase ligand to induce targeted protein degradation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Donovan, K.A.; Ferguson, F.M.; Bushman, J.W.; Eleuteri, N.A.; Bhunia, D.; Ryu, S.; Tan, L.; Shi, K.; Yue, H.; Liu, X.; et al. Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 2020, 183, 1714–1731. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Lee, Y.; Jung, Y.M.; Park, J.H.; Yoo, H.S.; Park, J. Discovery of E3 Ligase Ligands for Target Protein Degradation. Molecules 2022, 27, 6515. https://doi.org/10.3390/molecules27196515
Lee J, Lee Y, Jung YM, Park JH, Yoo HS, Park J. Discovery of E3 Ligase Ligands for Target Protein Degradation. Molecules. 2022; 27(19):6515. https://doi.org/10.3390/molecules27196515
Chicago/Turabian StyleLee, Jaeseok, Youngjun Lee, Young Mee Jung, Ju Hyun Park, Hyuk Sang Yoo, and Jongmin Park. 2022. "Discovery of E3 Ligase Ligands for Target Protein Degradation" Molecules 27, no. 19: 6515. https://doi.org/10.3390/molecules27196515
APA StyleLee, J., Lee, Y., Jung, Y. M., Park, J. H., Yoo, H. S., & Park, J. (2022). Discovery of E3 Ligase Ligands for Target Protein Degradation. Molecules, 27(19), 6515. https://doi.org/10.3390/molecules27196515