Abstract
Amylase and glucosidase enzymes are the primary harmful source in the development of the chronic condition known as diabetes mellitus. The main function of these enzymes is to break the macromolecules into simple sugar units which are directly involved in the solubility of blood, hence increasing blood glucose levels. To overcome this effect, there is a need for a potent and effective inhibitor that inhibits the conversion of macromolecules of sugar into its smaller units. In this regard, we synthesized thiazolidinone-based indole derivatives (1–20). The synthesized derivatives were evaluated for α-amylase and α-glucosidase inhibitory activity. Different substituted derivatives were found with moderate to good potentials having IC50 values ranging, for α-amylase, from 1.50 ± 0.05 to 29.60 ± 0.40 μM and, for α-glucosidase, from IC50 = 2.40 ± 0.10 to 31.50 ± 0.50 μM. Among the varied substituted compounds, the most active analogs four (1.80 ± 0.70 and 2.70 ± 0.70), five (1.50 ± 0.05 and 2.40 ± 0.10, respectively) of the series showed few folds better inhibitory activity than standard drug acarbose (IC50 = 10.20 ± 0.10 and 11.70 ± 0.10 μM, respectively). Moreover, structure–activity relationship (SAR) was established and binding interactions were analyzed for ligands and proteins (α-amylase and α-glucosidase) through a molecular docking study.
Keywords:
synthesis; α-amylase; α-glucosidase enzymes; thiazolidinone; indole; SAR; molecular docking 1. Introduction
Diabetes mellitus is a group of metabolic diseases characterized by hyperglycemia resulting from defects in insulin secretion, insulin action, or both. This metabolic disease is non-communicable involving a high mortality rate and huge healthcare costs. Approximately 387 million people were affected by diabetes mellitus and almost 4.90 million fatalities occurred in the year 2014 [1]. Postprandial hyperglycemia (PPHG), which frequently occurs after digesting a meal, is caused when the blood glucose level remains high. Diabetes mellitus is linked to secondary problems such as neuropathy, retinopathy, and cardiovascular disorders, among others [2]. There are two enzymes, Such as α-amylase, which cleaves glycosidic linkages of α-D-(1,4) of glycogen into oligosaccharide, followed by α-glucosidase which further breaks into glucose [3] The activity of these enzymes may be limited by anti-diabetic drugs that control blood glucose levels in people who consume carbohydrate-rich diets [4]. These drugs are Voglibose, Miglitol, and Acarbose, which are the current inhibitors used for the treatment of diabetic mellitus to control PPHG. All these drugs display greater potential against targeted enzymes among acarbose used to treat both enzymes while the remaining two drugs are only used against α-glucosidase.
The inhibitory profile of this drug is not sufficient due to some gastrointestinal side effects [5,6]. The majority of people in the nation are from the lower and middle classes, and he believes that such medications are far more costly. Researchers have tried to extract glucosidase and amylase inhibitors from a variety of sources, including bacteria, fungus, marine algae, and plants [7,8,9,10]. Additionally, a couple of them also researched pure substances, while others studied crude extracts (organic or aqueous) [11,12]. Due to their strong ability to interact with human sugar, glycosidases, and other protein receptors, the iminosugars, a common class of plant and microbial chemicals, have attracted significant study [13,14,15,16]. Most of the pure compounds and extracts were found to be efficient against both these enzymes, but had some complications [17,18]. Thiazolidinone is an adaptable organic molecule with a variety of biological potentials, including anti-bacterial [19], anti-fungal [20], anti-viral [21], anti-inflammatory [22], and anti-tuberculosis [23] effects, similar to other heterocyclic moieties that are utilized as anti-diabetic agents, as well as Alpha-glucosidase and Alpha-amylase inhibitors containing 4-thiazolidinone moiety [24]. The aim of this work is to report the synthetic route for synthesis of thiazolidinone-based indole derivatives and biological significances. These compounds were found with few folds more potent than acarbose against Alpha-amylase and Alpha-glucosidase (enzymes causing diabetes mellitus). Moreover, the results of the present work were compared with previous works, as shown in (Figure 1) [25,26].
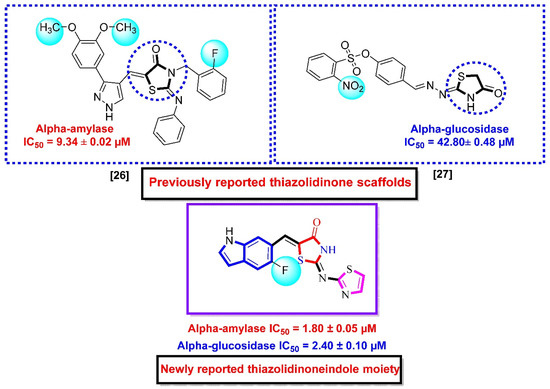
Figure 1.
Rationale of the current study.
2. Results and Discussion
2.1. Chemistry
The synthetic route was adopted for the synthesis of thiazolidinone-based indole derivatives via different stepwise reactions. Initially, 2-aminothiazole (I) and ammonium isothiocyanate were mixed and refluxed in the reaction mixture for about 4 h to obtain thiazole-bearing thiourea (II). This was further treated with chloroacetic acid in an acidified medium using acetic acid under refluxed condition for about 5 h, affording thiazole-based thiazolidinone moiety (III). Equimolar amounts of thiazole-based thiazolidinone (III) were further treated with various substituted indole-bearing aldehyde moieties in methanol, and the reaction mixture was then refluxed for about 7 h in the presence of potassium carbonate to produce thiazolidinone-based indole derivatives (1–20) (Scheme 1). All reactions were monitored at every step using thin layer chromatography (TLC). Moreover, all the synthesized derivatives were washed with n-hexane, in order to remove impurities. Fine powder was collected, and then these products were further characterized through different spectroscopic techniques such as HNMR, CNMR, and HREI-MS. Spectral analysis of the tested analogs is explained in supporting information Section 3.0.
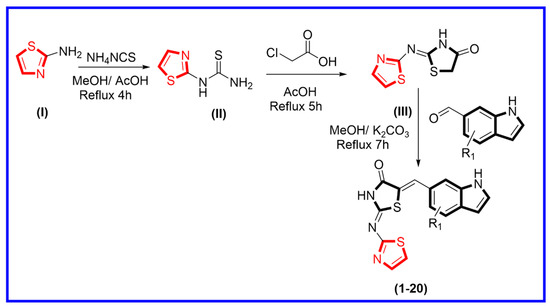
Scheme 1.
Procedure adopted for the synthesis of thiazolidinone-based indole derivatives (1–20).
2.2. In Vitro Alpha-Amylase and Alpha-Glucosidase Inhibitory Activities
Twenty analogs based on thiazolidinone-based indole (one–twenty) were synthesized and then assessed for their inhibitory potentials against targeted α-amylase and α-glucosidase (in vitro). When evaluated against a standard acarbose drug, all the synthesized thiazolidinone-based indole analogs showed varied ranges on inhibitory potentials ranging from IC50 = 1.50 ± 0.05 to 29.60 ± 0.40 μM (against α-amylase) and from IC50 = 2.40 ± 0.10 to 31.50 ± 0.50 μM (against α-glucosidase). Structure activity relationship (SAR) studies were carried out for all synthesized thiazolidinone-based indole analogs based on substitution(s) pattern around the indole ring. In order to discuss the SAR studies in a better way, the synthesized analogs were split into thiazole, thiazolidinone, and indole rings. By keeping the thiazole and thiazolidinone rings constant, the variation in inhibitory potentials for both α-amylase and α-glucosidase were observed by changing the nature, position, number(s), and electron donating or electron withdrawing groups around the indole ring (Table 1).

Table 1.
Substituted thiazolidinone-based indole derivatives along with biological profile (1–20).
Structure Activity Relationship (SAR) Studies for α-Amylase and α-Glucosidase Inhibitory Activity
The nitro group in the ortho- and meta-positions, among other substituents, on the phenyl ring, which attached to thiazolidinone moiety, as in the case of one, two, and three, has been identified and showed significant results against α-amylase and α-glucosidase, but scaffold one was found one- and three-folds better than two and three, as well as five-folds more active than standard acarbose as reference inhibitor. Among nitro-substituted analogs, scaffold one, having nitro moiety in the ortho-position, exhibited excellent potential for both α-amylase and α-glucosidase. Comparison criteria were set for those analogs bearing nitro group at meta-position two with a little bit of a decrease in α-amylase and α-glucosidase potential observed, which might be the position of substituents from ortho-position to meta-position. By this consideration, it was concluded that replacement of the nitro-group at meta-position of the phenyl ring decreases the interactive property of the molecule. The activity of analog three decreased due to the presence of chlorine moiety, which causes steric hindrance. In this stage, analog one showed better potential against α-amylase and α-glucosidase (Table 1).
Analogs four and five showed higher α-glucosidase and α-amylase performance in comparison to acarbose. This elevated α-glucosidase and α-amylase performance of analogs four and five might be due to the presence of two flouro groups that may interact better with the active site of the enzyme through hydrogen bonding. Moreover, the position of flouro moieties around the phenyl ring has a significant effect on activity. Thus, the analog five-bearing flouro group at ortho-location of the phenyl ring showed better inhibitory potentials than analog four, also bearing flouro substituent, but analog five was found with remarkable activity as compared to analog four, as well as much better potential than acarbose (Table 1).
Analogs six, seven, and eight, bearing the different number of hydroxyl moiety at ortho- (six), ortho/meta- (seven), and ortho-positions (eight), showed strong interaction against α-amylase and α-glucosidase. Two analogs were found with few-folds better activity than acarbose. By this comparative study, the better inhibitory profile was shown by analog seven when compared with acarbose drugs. Analog seven bearing two -OH groups showed superior inhibitory profile due to strong hydrogen bonding; therefore, it is ranked one in this group, while analog six bearing one -OH moiety was found with smaller interactions than analog seven, potentially due to the presence of CF3. However, analog eight was found with comparable activity with acarbose; the lower activity might be the presence of methoxy substituent which increases steric hindrance and thus reduces the biological potential (Table 1).
Analogs having either hydroxyl groups or hydroxyl groups along with a -Cl group have emerged as potent inhibitors of both Alpha-amylase and Alpha-glucosidase enzymes. Analog eleven bearing meta-OH and the ortho-Cl group was recognized as the most active analog among the hydroxyl group bearing moieties. The potency of analog eleven was reduced by the introduction of one -OH group on the meta-position instead of ortho-position, as in analog twelve, which might cause steric hindrance. This is because of the presence of a -Cl group at the ortho-site which decreases the enzymatic activity. The activity of analog eleven was further reduced due to the presence of one -Cl group which might reduce the binding interactions, as in analog thirteen, and two -Cl in the case of analog fourteen, also decreasing the biological profile of the molecule (Table 1).
Some of the screened analogs were found with lower abilities compared to both α-glucosidase and α-amylase enzymes. When compared with standard drug acarbose (10.20 ±0.10 and 11.70 ± 0.10), these analogs displayed a varied range of inhibitory profile in both the activities, such as analogs nine (21.50 ± 0.30 and 23.20 ± 0.40), ten (23.20 ± 0.10 and 24.60 ± 0.20), fifteen (27.50 ± 0.70 and 28.40 ± 0.80), sixteen (25.20 ± 0.20 and 26.70 ± 0.30), seventeen (29.60 ± 0.40 and 31.50 ± 0.50), eighteen (22.70 ± 0.50 and 23.30 ± 0.40), nineteen (18.40 ± 0.60 and 19.80 ± 0.60), and twenty (27.60 ± 0.80 and 29.40 ± 0.90). In the case of hydroxyl groups containing analogs, especially in cases of derivatives nineteen and twenty, both having a hydroxyl group at the meta- and ortho-positions, respectively, as well as the bromo group at the ortho-position on the same ring. The presence of this bromo moiety and the decrease in activity profile were seen due to its bulky nature which congests the activity profile of the molecule (Figure 2). Thus, both the molecules were found with fewer IC50 values (Table 1).
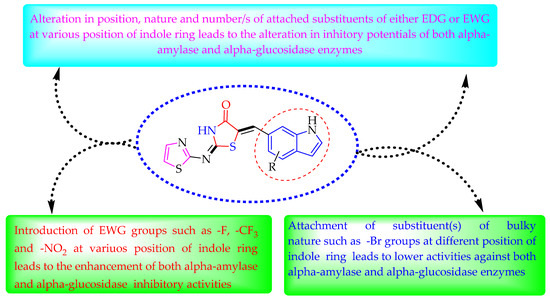
Figure 2.
Summary of SAR studies for α-amylase and α-glucosidase inhibitory activities.
2.3. Molecular Docking Study
The molecular docking study investigates the binding mode of interactions with enzyme active sites. Various pieces of software were used for this including auto Dock vina, DSV MGL tool, and Discovery Studio Visualizer [27,28,29,30]. Protein was retrieved from an online source such as a protein data bank (PDB). The designated codes for α-amylase and α-glucosidase enzymes are 1b2y and 3w37, respectively.
Molecular docking studies were conducted to explore the binding modalities between ligands and protein. Docking was performed to obtain a population of possible conformations and orientations for the ligand at the binding site. The protein was loaded in auto dock vina software, creating a PDBQT file that contained a protein structure with hydrogen in all polar residues. All bonds of ligands were set to be rotatable. All calculations for protein-fixed ligand-flexible docking were performed using the Lamarckian Genetic Algorithm (LGA) method. The docking site on protein target was defined by establishing a grid box with the dimensions of X: 38.0729, Y: 33.3208, and Z: 25.0000 Å, with a grid spacing of 0.375 Å, centered on X: 20.2892, Y: 10.3219, and Z: 32.3218 Å. The best conformation was chosen with the lowest docked energy after the docking search was completed. Nine runs with AutoDock Vina were performed in all cases per each ligand structure, and for each run the best pose was saved as shown in Table 2 and Table 3. The average affinity for best poses was taken as the final affinity value. The interactions of complex alpha-amylase and -glucosidase protein–ligand conformations, including hydrogen bonds and the bond lengths, were analyzed using DSV.
Table 2.
Represent the binding affinities and best mode RMSD of analog 4 against Alpha-amylase and Alpha-glucosidase.
Table 3.
Represent the binding affinities and best mode RMSD of analog 5 against Alpha-amylase and Alpha-glucosidase.
A molecular docking study revealed the interactive residues with active sites of ligands. The interactions are generally obtained between protein and ligand interest. The target enzymes Alpha-amylase and Alpha-glucosidase were attained from the online source www.rcsb.org. The potential of ligands against targeted enzymes might be the presence of attached substituents. Here, the nature of substituents is important for binding interactions. Thus, the electron donating group activates the ring by a negative charge, which dominantly increases the interaction toward the positive center or deficient species. In this study, analogs (four, five, six, and seven) bearing flouro moieties, which are electron withdrawing but have the property to make hydrogen bonds, were found with better interactions, so they were considered as ranked first. They were followed by the interaction procedure of analogs bearing hydroxyl and trifluoro methyl moieties, which also exhibited superposed surface complex structures, as illustrated in (Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10, Figure 11, Figure 12, Figure 13 and Figure 14). In addition, these ligands (four, five, six, and seven) were found with varied binding modalities (Table 2, Table 3, Table 4 and Table 5) against targeted enzymes, which demonstrated excellent potency in vitro study and were found with the best potential (in silico). The best mode of analogs four and five was observed when compared with the acarbose moiety, and their binding affinity and RMSD value were found lower than that of standard drugs.
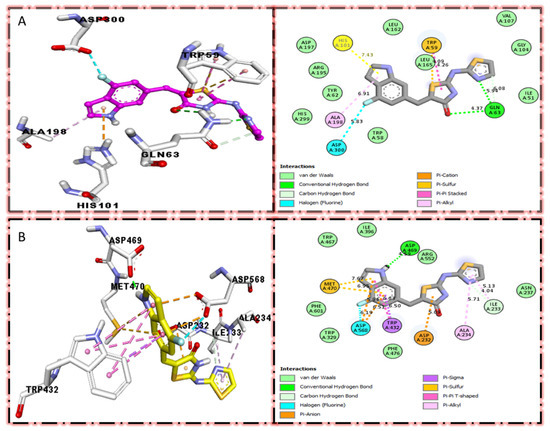
Figure 3.
Protein–ligand interaction profile for active analog against the Alpha-amylase and Alpha-glucosidase represents the surface of the corresponding enzyme. (A,B) indicate the PLI profile for compound 4.
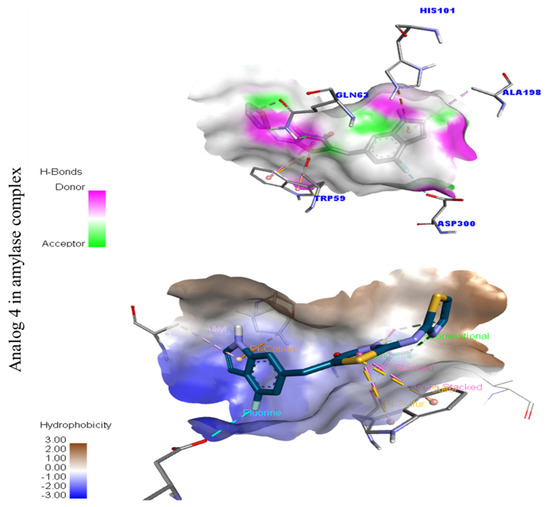
Figure 4.
Represent hydrogen bond and hydrophobic character against Alpha-amylase indicate the PLI profile for compound 4.
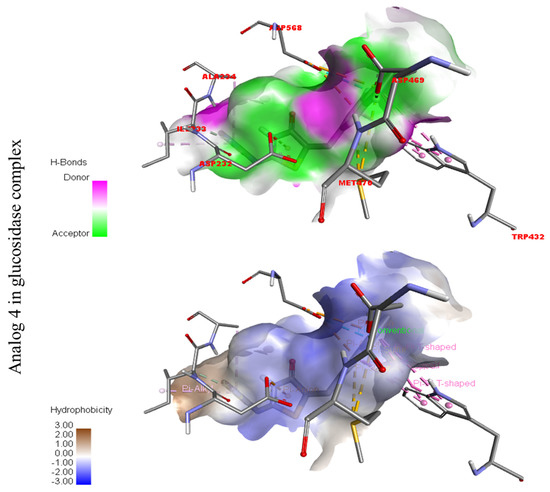
Figure 5.
Represent hydrogen bond and hydrophobic character against Alpha-glucosidase indicate the PLI profile for compound 4.
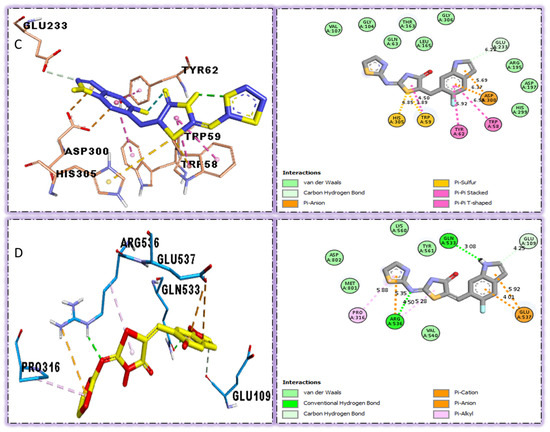
Figure 6.
The surfaces of the relevant enzymes are shown by the PLI profiles for powerful drugs against Alpha-amylase and Alpha-glucosidase (C,D). The PLI profile for chemical 5 is shown by the letters (C,D).
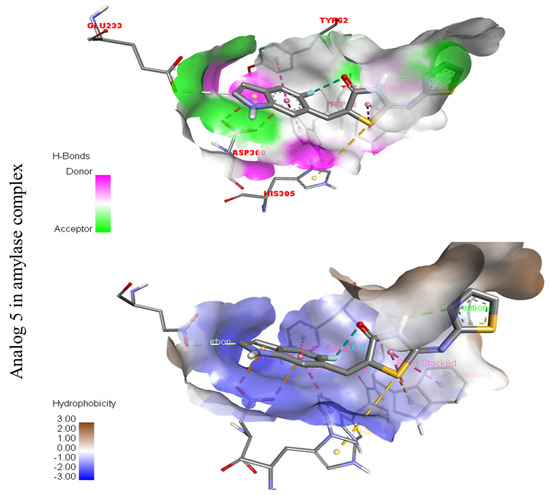
Figure 7.
Represent hydrogen bond and hydrophobic character against Alpha-amylase indicate the PLI profile for compound 5.
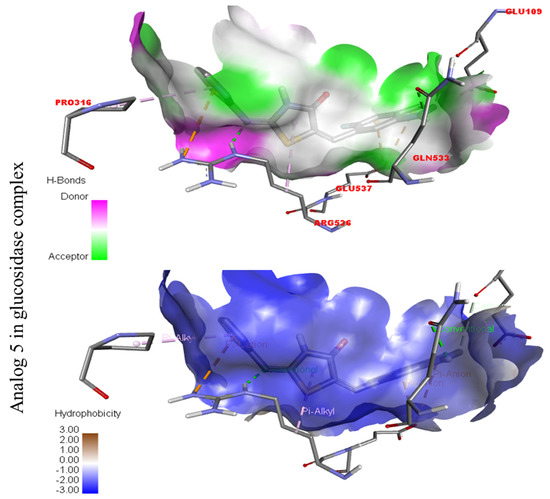
Figure 8.
Represent hydrogen bond and hydrophobic character against Alpha-glucosidase indicate the PLI profile for compound 5.
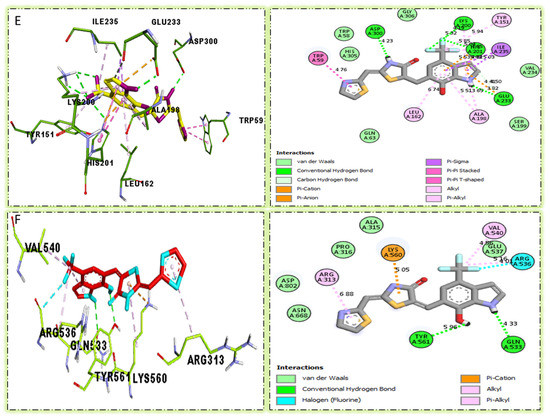
Figure 9.
The surface of the appropriate enzyme is shown by the PLI profile for drugs that are effective against Alpha-amylase and Alpha-glucosidase. The PLI profile for chemical 6 is shown by (E,F).
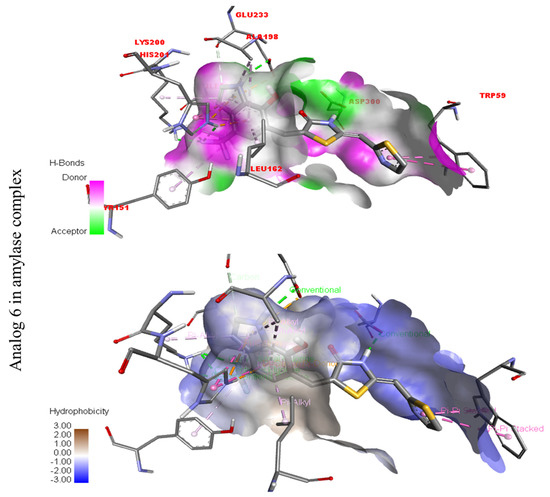
Figure 10.
Represent hydrogen bond and hydrophobic character against Alpha-amylase indicate the PLI profile for compound 6.
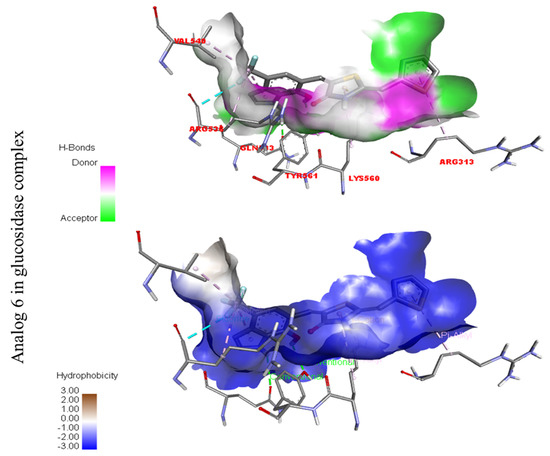
Figure 11.
Represent hydrogen bond and hydrophobic character against Alpha-glucosidase indicate the PLI profile for compound 6.
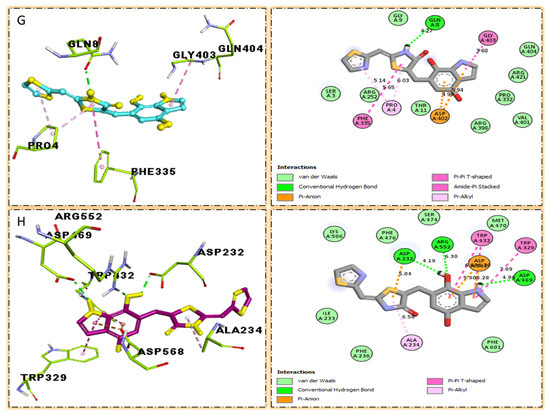
Figure 12.
The surface of the relevant enzyme is represented by the PLI profile for drugs that are effective against Alpha-amylase and Alpha-glucosidase. The PLI profile for chemical 7 is shown by (G,H).
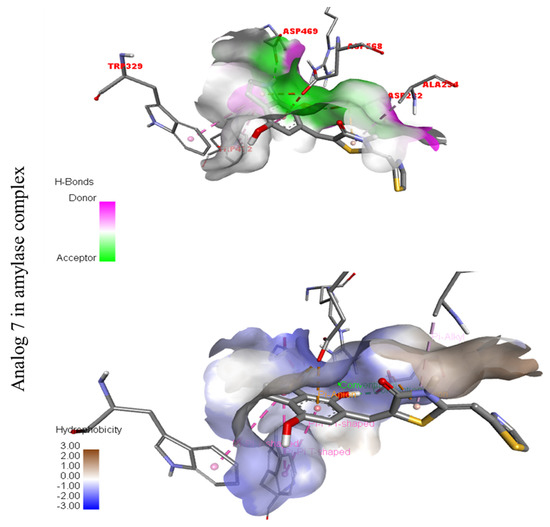
Figure 13.
Represent hydrogen bond and hydrophobic character against Alpha-amylase indicate the PLI profile for compound 7.
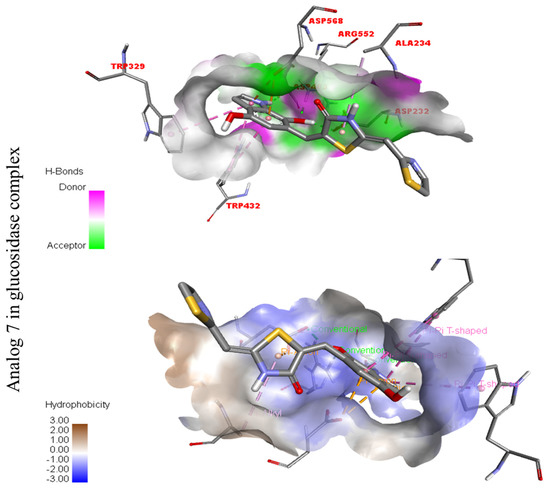
Figure 14.
Represent hydrogen bond and hydrophobic character against Alpha-glucosidase indicate the PLI profile for compound 7.
Table 4.
Represent the binding affinities and best mode RMSD of analog 6 against Alpha-amylase and Alpha-glucosidase.
Table 5.
Represent the binding affinities and best mode RMSD of analog 7 against Alpha-amylase and Alpha-glucosidase.
In the case of flouro-substituted moiety four and its protein–ligand interaction profile exhibited varied interactive residues for Alpha-amylase (A) with distances from 4.09–7.43 A, as shown in (Figure 3). Such residues are HIS-A-101(π-cation), LEU-A-165 (VW), TRP-A-59(π-S), GLN-A-63(H-B), ASP-A-300(H-F), and ALA-A-300(π-R). Similarly, for Alpha-glucosidase (B), the interactive residues, with distances from 4.04–7.67 A, are MET-A-470(π-anion), ASP-A-469(H-B), ILE-A-233(π-X), ALA-A-234(π-R), ASP-A-232(π-anion), TRP-A-432(π-σ), and ASP-A-568(H-F), as shown in (Figure 3).
The strongest molecule in the series, number five, had a fantastic interactions profile with the targeted enzymes. Flouro-substituted analog five was found with different interactions against Alpha-amylase (C) with distances from 4.50–6.85 A, such as HIS-A-305(π-S), TRP-A-59(π-S), TYR-A-62(π-π-stacked), TRP-A-58(π-π-T-shaped), and ASP-A-300(π-anion), as shown in (Figure 6). While those against Alpha-glucosidase (D) had distances from 3.08–5.92 A, including Pro-a-316(π-R), ARG-A-536(H-B), GLU-A-537(π-anion), GLU-A-537(π-cation), GLN-A-533(H-B), etc., as shown in (Figure 6).
Compound six in the series exhibited a significant PLI profile. Analog six containing the CF3-group at the meta- and-OH at the ortho-positions on the phenyl moiety showed better interactions against Alpha-amylase (E) with distances from 3.82–6.74 A, such as TRP-A-59(π-π-stacked), ASP-A-300(H-B), LYS-A-300(H-B), ASP-A-201(H-B), and TYR-A-151(R), ILE-A-235(π-sigma), GLU-A-233(H-B), ALA-A198(π-R), LEU-A-162(π-R), etc., as shown in (Figure 9). While those against Alpha-glucosidase (F) had distances from 4.33–6.88 A, including ARG-A-313(π-R), LYS-A-560(π-cation), VAL-A-540(R), ARG-A-536(H-F) GLN-A-533(H-B), TYR-A-561(H-B), etc., as shown in (Figure 9).
The third most potent compound seven in the series illustrated comparable activity with analogs four and five having better interaction profiles. Analog seven contains nitro moiety which showed different interactions against Alpha-amylase (G) with distance from 3.68–5.94 A, such as PHE-A-335(π-π-stacked), PRO-A-4(π-R), ASP-A-402(π-anion), GLY-A-403(π-π-T-shaped), GLN-A-8(H-B), etc., as shown in (Figure 12). While those against Alpha-glucosidase (H) had distances from 4.19–7.09 A, including ASP-A-232(H-B), ARG-A-552(H-B), TRP-A-432(π-π-T-shaped), TRP-A-329(π-π-T-shaped), ASP-A-469(π-anion), ALA-A-234(π-R), etc., as shown in (Figure 12).
Resultantly, it was observed that analogs four and five were found to have excellent potential against targeted enzymes. It might be that the presence of electrons withdrawing and donating groups, respectively, increased the binding modalities with different interactions such as pi-anion, pi-cation, pi-sulfur, hydrogen bonding, and pi–pi interaction. The interactive ability of these scaffolds is due to the presence of benzene rings and different heteroatoms.
3. Experimental Section
3.1. Material and Methods
For the synthesis of desired compounds, all the chemicals and reagents were purchased from the USA (Sigma Aldrich). Thin layer chromatography (TLC) was performed on pre-coated silica gel aluminum plates (Kieselgel 60,254, E. Merck, Germany). Chromatograms were visualized by UV at 254 and 365 nm. High-resolution electron impact mass spectra (HREI-MS) were recorded on a Finnigan MAT-311A (Germany) mass spectrometer. NMR experiments were performed on an Avance Bruker AM 600MHz machine.
3.2. General Procedure for the Synthesis of Thiazolidinone-Based Indole Derivatives (1–20)
The mixture of 2-aminothiazole (I, 1 equivalent) and ammonium isothiocyanate (1 equivalent) was mixed in methanol (10 mL), and the reaction mixture was refluxed in the presence of glacial acetic acid to yield thiazole-based thiourea analog (II), which was then treated with chloroacetic acid (1 equivalent) under reflux in acidic medium and afforded thiazole-based thiazolidinone derivatives (III). In the next step, compound (III, 1 equivalent) was further treated with a different substituted indole-bearing aldehyde (1 equivalent) in methanol (10 mL) and the reaction mixture was refluxed in the presence of potassium carbonate (0.75 equivalents) to yield thiazolidinone-based indole derivatives (1–20) in appropriate yield.
4. Conclusions
Thiazolidinone-based indole derivatives (one–twenty) were synthesized successfully and confirmed through different spectroscopic techniques such as NMR and HR-MS. The biological profile of all the derivatives was screened against α-amylase and α-glucosidase to investigate the better potential of the scaffolds. Among them, few derivatives were found with moderate to good activity, while the most potent behavior was shown in this study in scaffolds four (2Z,5E)-5-((4-fluoro-1H-indol-6-yl)methylene)-2-(thiazol-2-ylimino)thiazolidin-4-one(1.80 ± 0.70 and 2.70 ± 0.70) and five (2Z,5E)-5-((5-fluoro-1H-indol-6-yl)methylene)-2-(thiazol-2-ylimino)hiazolidine-4-one (1.50 ± 0.05 and 2.40 ± 0.10, respectively) of the series, showing to have few folds better inhibitory activity than standard drug acarbose (IC50 = 10.20 ±0.10 and 11.70 ± 0.10 μM, respectively) The interaction of these scaffolds might be due to the presence of flouro moieties forming a strong hydrogen bond with enzyme active sites. The potential of the scaffold depends upon the nature of the substituent, either small in size or bulky group, which increases or decreases the binding interactions. A molecular docking study also revealed the significant binding modalities of subjected candidates. This study identifies a new class of thiazolidinone-based indole derivatives as α-amylase and α-glucosidase inhibitors.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27196564/s1, Section S3: Spectral analysis. Refs. [31,32] are cited in the supplementary materials.
Author Contributions
Conceptualization, writing-original draft Preparation, S.K. and S.I.; methodology, F.R.; software, M.S.; validation, project administration, critical revision, R.H., H.A. and W.R.; writing review and editing, resources, funding, A.A.D., L.R. and H.H.S.; investigation, writing-original draft preparation, R.A.P., E.A. and A.-E.F. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant Code: (22UQU4320141DSR49). This work was supported by King Khalid University through a grant (KKU/RCAMS/22) under the Research Center for Advanced Materials Science (RCAMS) at King Khalid University, Saudi Arabia.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Atlas, D. International Diabetes Federation. IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015; p. 33. [Google Scholar]
- Gin, H.; Rigalleau, V. Post-prandial hyperglycemia. post-prandial hyperglycemia and diabetes. Diabetes Metab. 2000, 26, 265–272. [Google Scholar] [PubMed]
- Lordan, S.; Smyth, T.J.; Soler-Vila, A.; Stanton, C.; Ross, R.P. The α-amylase and α glucosidase inhibitory effects of Irish seaweed extracts. Food Chem. 2013, 141, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, H.E. α-glucosidase inhibitors. Endocrinol. Metab. Clin. N. Am. 1997, 26, 539–551. [Google Scholar] [CrossRef]
- Van de Laar, F.A. Alpha-glucosidase inhibitors in the early treatment of type 2 diabetes. Vasc. Health Risk Manag. 2008, 4, 1189. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, U.; de la Garza, A.L.; Campión, J.; Martínez, J.A.; Milagro, F.I. Antidiabetic effects of natural plant extracts via inhibition of carbohydrate hydrolysis enzymes with emphasis on pancreatic alpha amylase. Expert Opin. Ther. Targets 2012, 16, 269–297. [Google Scholar] [CrossRef] [PubMed]
- Fatmawati, S.; Shimizu, K.; Kondo, R. Ganoderol B: A potent α-glucosidase inhibitor isolated from the fruiting body of Ganoderma lucidum. Phytomedicine 2011, 18, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Kawamura-Konishi, Y.; Watanabe, N.; Saito, M.; Nakajima, N.; Sakaki, T.; Katayama, T.; Enomoto, T. Isolation of a new phlorotannin, a potent inhibitor of carbohydrate-hydrolyzing enzymes, from the brown alga Sargassum patens. J. Agric. Food Chem. 2012, 60, 5565–5570. [Google Scholar] [CrossRef]
- Orhan, N.; Aslan, M.; Şüküroğlu, M.; Orhan, D.D. In vivo and in vitro antidiabetic effect of Cistus laurifolius L. and detection of major phenolic compounds by UPLC–TOF-MS analysis. J. Ethnopharmacol. 2013, 146, 859–865. [Google Scholar] [CrossRef]
- Panwar, H.; Calderwood, D.; Grant, I.R.; Grover, S.; Green, B.D. Lactobacillus strains isolated from infant faeces possess potent inhibitory activity against intestinal alpha-and beta-glucosidases suggesting anti-diabetic potential. Eur. J. Nutr. 2014, 53, 1465–1474. [Google Scholar] [CrossRef]
- Ali, R.B.; Atangwho, I.J.; Kuar, N.; Ahmad, M.; Mahmud, R.; Asmawi, M.Z. In vitro and in vivo effects of standardized extract and fractions of Phaleria macrocarpa fruits pericarp on lead carbohydrate digesting enzymes. BMC Complement. Altern. Med. 2013, 13, 39. [Google Scholar] [CrossRef]
- Kim, K.T.; Rioux, L.E.; Turgeon, S.L. Alpha-amylase and alpha-glucosidase inhibition is differentially modulated by fucoidan obtained from Fucus vesiculosus and Ascophyllum nodosum. Phytochemistry 2014, 98, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Nash, R.J.; Kato, A.; Yu, C.-Y.; Fleet, G.W.J. Iminosugars as therapeutic agents: Recent advances and promising trends. Future Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Chennaiah, A.; Dahiya, A.; Dubbu, S.; Vankar, Y.D. A Stereoselective Synthesis of an IminoGlycal: Application in the Synthesis of (−)-1-Epi -Adenophorine and a Homoiminosugar. Eur. J. Org. Chem. 2018, 2018, 6574–6581. [Google Scholar] [CrossRef]
- Chennaiah, A.; Bhowmick, S.; Vankar, Y.D. Conversion of glycals into vicinal-1,2-diazides and 1,2-(or 2,1)-azidoacetates using hypervalent iodine reagents and Me3SiN3. Application in the synthesis of N-glycopeptides, pseudo-trisaccharides and an iminosugar. RSC Adv. 2017, 7, 41755–41762. [Google Scholar] [CrossRef]
- Horne, G.; Wilson, F.X.; Tinsley, J.; Williams, D.H.; Storer, R. Iminosugars past, present and future: Medicines for tomorrow. Drug Dis. Today 2011, 16, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.A.H.; Siddiqui, M.J.A.; Ang, L.F.; Sadikun, A.; Chan, S.H.; Tan, S.C.; Asmawi, M.Z.; Yam, M.F. Potent α-glucosidase and α-amylase inhibitory activities of standardized 50% ethanolic extracts and sinensetin from Orthosiphon stamineus Benth as anti-diabetic mechanism. BMC Complement. Altern. Med. 2012, 12, 176. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gutierrez, R.M.; Damian-Guzman, M. Meliacinolin: A potent α-glucosidase and α-amylase inhibitor isolated from Azadirachta indica leaves and in vivo antidiabetic property in streptozotocin-nicotinamide-induced type 2 diabetes in mice. Biol. Pharm. Bull. 2012, 35, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Kumari, P.; Patel, N. Synthesis and characterization of some new thiazolidinones containing coumarin moiety and their antimicrobial study. Arch. Appl. Sci. Res. 2010, 2, 68–75. [Google Scholar]
- Mishra, R.; Tomar, I.; Singhal, S.; Jha, K.K. Facile synthesis of thiazolidinones bearing thiophene nucleus as antimicrobial agents. Der Pharma Chem. 2012, 4, 489–496. [Google Scholar]
- Rao, A.; Carbone, A.; Chimirri, A.; De Clercq, E.; Monforte, A.M.; Monforte, P.; Pannecouque, C.; Zappalà, M. Synthesis and anti-HIV activity of 2, 3-diaryl-1, 3-thiazolidin-4-ones. Il Farm. 2003, 58, 115–120. [Google Scholar] [CrossRef]
- Jain, A.K.; Vaidya, A.; Ravichandran, V. SK kashaw, RK Agrawal. Bioorg. Med. Chem. 2012, 20, 3378–3395. [Google Scholar] [CrossRef] [PubMed]
- Küçükgüzel, Ş.G.; Oruç, E.E.; Rollas, S.; Şahin, F.; Özbek, A. Synthesis, characterisation and biological activity of novel 4-thiazolidinones, 1, 3, 4-oxadiazoles and some related compounds. Eur. J. Med. Chem. 2002, 37, 197–206. [Google Scholar] [CrossRef]
- Rahim, F.; Zaman, K.; Ullah, H.; Taha, M.; Wadood, A.; Javed, M.T.; Rehman, W.; Ashraf, M.W.; Uddin, R.; Uddin, I.; et al. Synthesis of 4-thiazolidinone analogs as potent in vitro anti-urease agents. Bioorg. Chem. 2015, 63, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Ganavi, D.; Ramu, R.; Kumar, V.; Patil, S.M.; Martiz, R.M.; Shirahatti, P.S.; Sathyanarayana, R.; Poojary, B.; Holl, B.S.; Poojary, V.; et al. In vitro and in silico studies of fluorinated 2, 3-disubstituted thiazolidinone-pyrazoles as potential α-amylase inhibitors and antioxidant agents. Arch. Pharm. 2022, 355, 2100342. [Google Scholar] [CrossRef]
- Kaur, R.; Kumar, R.; Dogra, N.; Yadav, A.K. Design, synthesis, biological evaluations and in silico studies of sulfonate ester derivatives of 2-(2-benzylidenehydrazono) thiazolidin-4-one as potential α-glucosidase inhibitors. J. Mol. Struct. 2022, 1247, 131266. [Google Scholar] [CrossRef]
- Kharb, M.; Jat, R.K.; Parjapati, G.; Gupta, A. Introduction to molecular docking software technique in medicinal chemistry. Int. J. Drug Res. Technol. 2012, 2, 189–197. [Google Scholar]
- Li, Z.; Gu, J.; Zhuang, H.; Kang, L.; Zhao, X.; Guo, Q. Adaptive molecular docking method based on information entropy genetic algorithm. Appl. Soft Comput. 2015, 26, 299–302. [Google Scholar] [CrossRef]
- Rao, C.M.M.P.; Naidu, N.; Priya, J.; Rao, K.P.C.; Ranjith, K.; Shobha, S.; Chowdary, B.S.; Siddiraju, S. Molecular docking and dynamic simulations of benzimidazoles with beta-tubulins. Bioinformation 2021, 17, 404. [Google Scholar]
- Khan, S.; Ullah, H.; Rahim, F.; Nawaz, M.; Hussain, R.; Rasheed, L. Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. J. Mol. Struct. 2022, 1269, 133812. [Google Scholar] [CrossRef]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar]



































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).