
Construction of Non-Biaryl Atropisomeric Amide Scaffolds Bearing a C–N Axis via Enantioselective Catalysis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
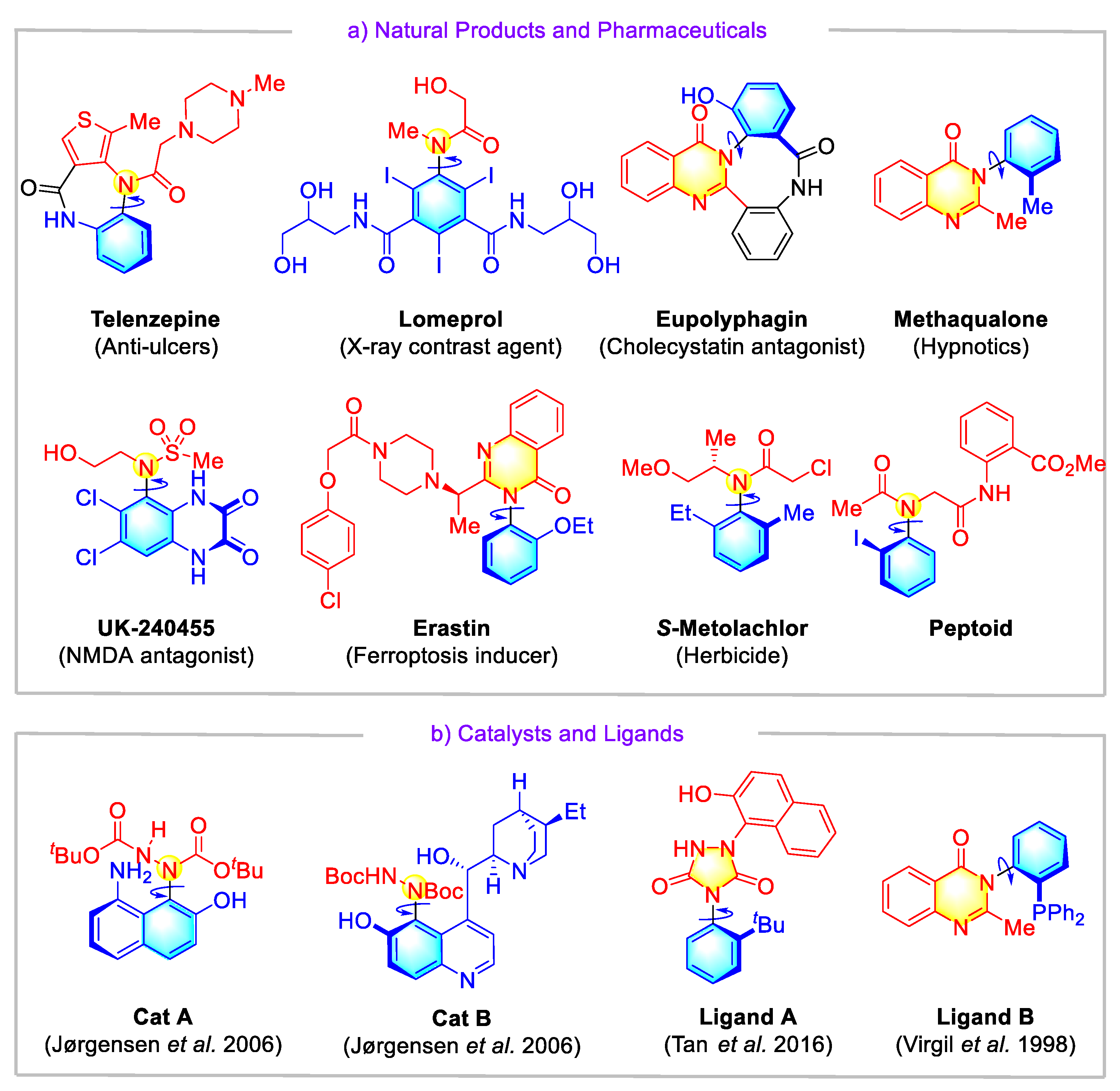
:1. Introduction
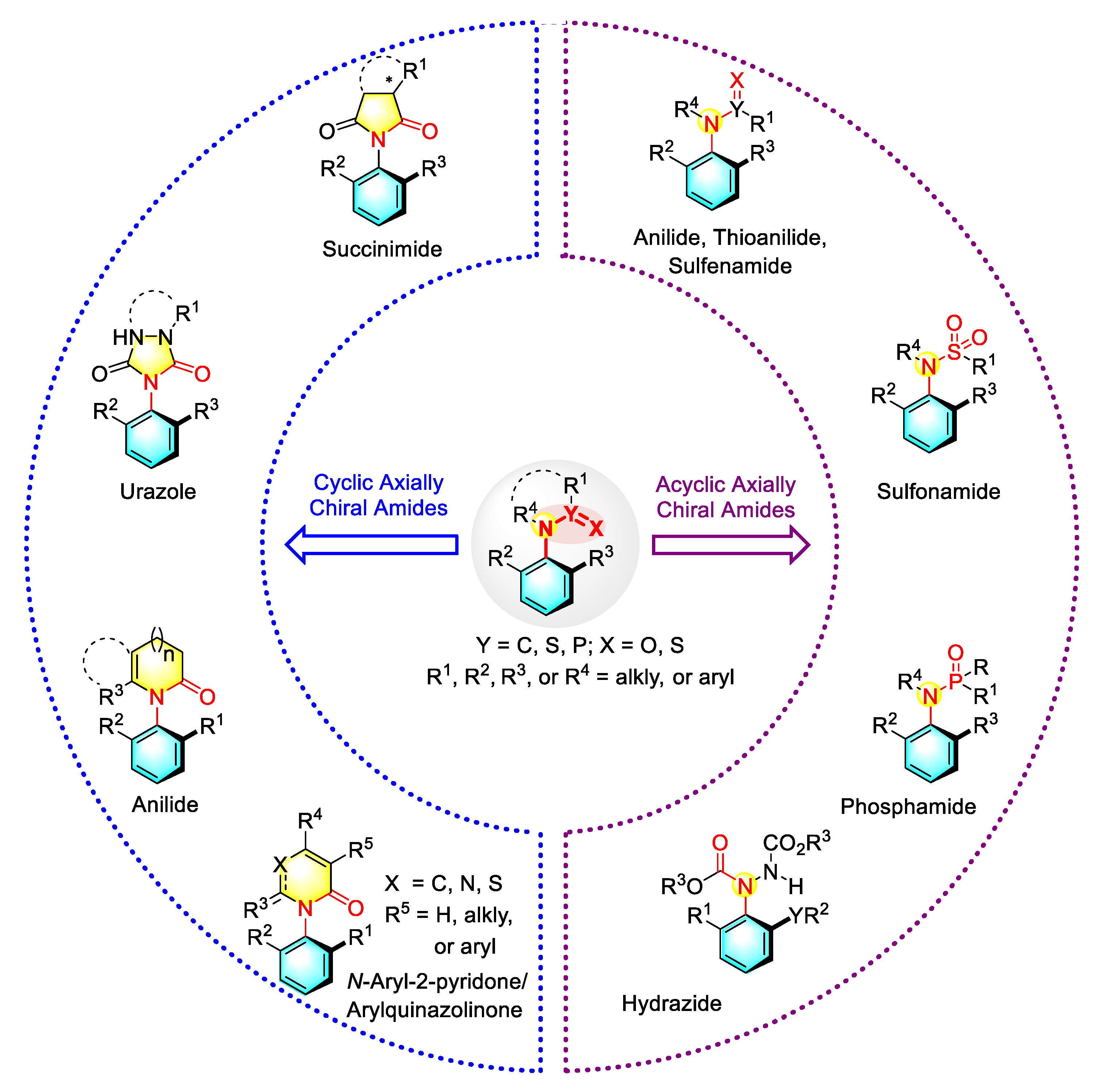
2. Enantioselective Synthesis of Acyclic C–N Axially Chiral Amides
2.1. Synthetic Strategies for Acyclic C–N Axially Chiral (Thio)anilides
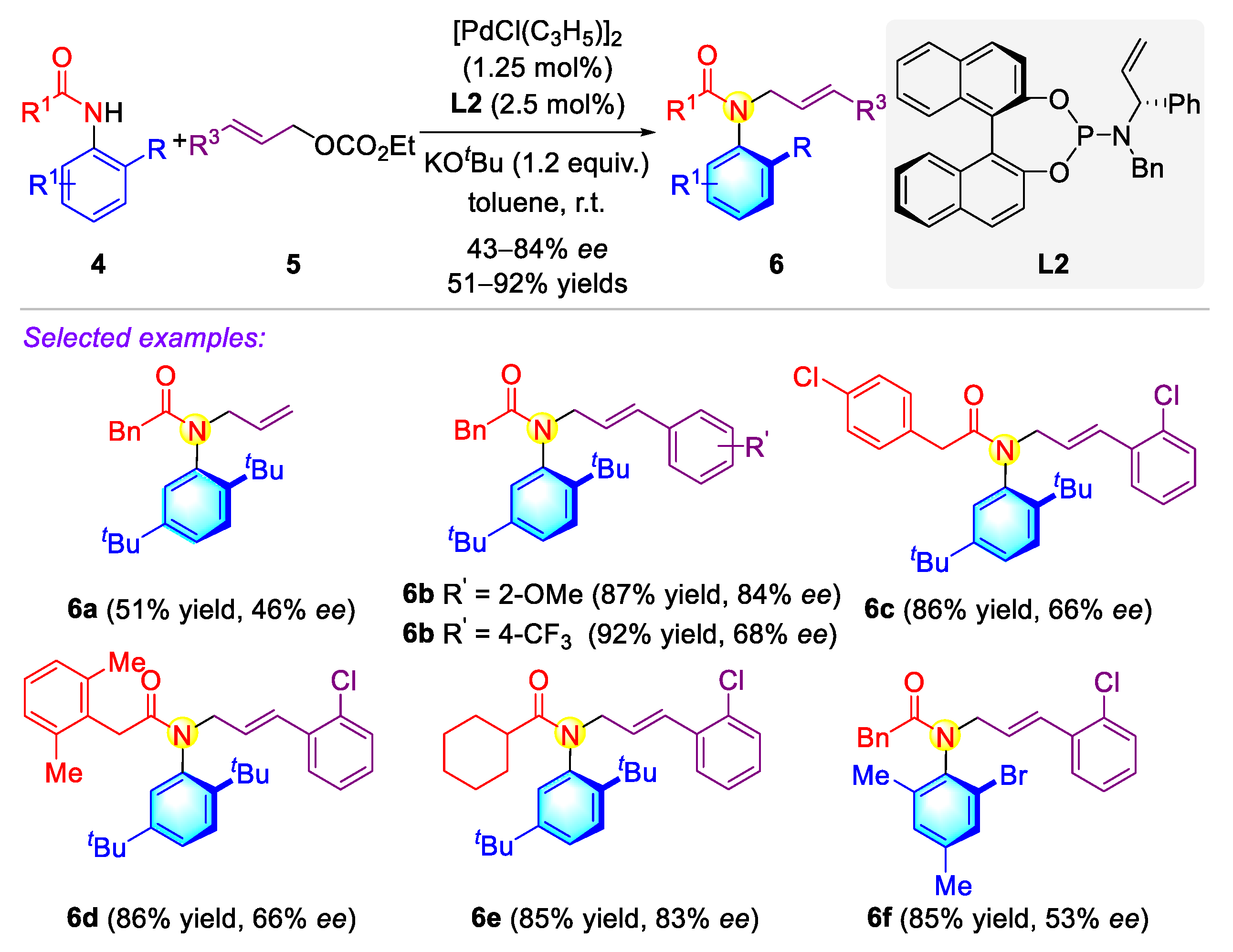
2.1.1. Atropisomeric (Thio)anilide Syntheseis via Transition-Metal Catalysis
2.1.2. Atropisomeric Anilides Syntheses via Phase-Transfer Catalysis (PTC)
2.1.3. Atropisomeric Anilides Syntheses via Organocatalysis
2.2. Synthetic Strategies for Acyclic C–N Axially Chiral Sulfonamide and Sulfinamide
2.2.1. Atropisomeric Sulfonamide Syntheses via Transition-Metal Catalysis
2.2.2. Synthetic Strategies for Acyclic C–N Axially Chiral Sulfonamide and Sulfinamide via Nucleophilic Catalysis
2.3. Synthetic Strategies for Acyclic C–N Axially Chiral Phosphamide via Nucleophilic Catalysis
2.4. Synthetic Strategies for Acyclic C–N Axially Chiral Hydrazide via Enantioselective Amination
2.4.1. Atropisomeric Hydrazide Synthesis via Asymmetric C–H Amination
2.4.2. Atropisomeric Hydrazide Synthesis via an Au-Catalyzed Cycloisomerization–Amination Cascade Reaction
3. Enantioselective Synthesis of Cyclic C–N Axially Chiral Amides
3.1. Synthetic Strategies for C–N Axially Chiral N-Aryl Succinimides via the Desymmetrization of N-Arylmaleimides
3.1.1. Synthetic Strategies for C–N Axially Chiral N-Aryl Succinimides via Transition-metal Catalyzed Desymmetrization of N-Arylmaleimides
3.1.2. Synthetic Strategies for C–N Axially Chiral N-Aryl Succinimides via the Organo-Catalyzed Desymmetrization of N-Arylmaleimides
3.1.3. Synthetic Strategies for C–N Axially Chiral N-Aryl Succinimides through Miscellaneous Strategies
3.2. Synthetic Strategies for C–N Axially Chiral Urazoles via Desymmetrization
3.3. Synthetic Strategies to Access Atropisomeric (iso-)Indolinone
3.4. Synthetic Strategies to Access Cyclic C–N Axially Chiral N-Aryl Piperidinone/Pyridones/Quinolinone/Phenanthridinone
3.4.1. Construction of Cyclic C–N Axially Chiral N-Aryl-Piperidinone via Brønsted Base Catalysis
3.4.2. Construction of Cyclic C–N Axially Chiral N-Aryl-Piperidinone via Brønsted Base Catalysis
3.4.3. Atropisomeric Quinolinone/Phenanthridinone Syntheses via Transition Metal Catalysis
3.4.4. Synthetic Strategies to Access Cyclic C–N Axially Chiral N-Aryl-Quinazolinones/Thiazines
3.5. Synthetic Strategies to Access Miscellaneous Cyclic C–N Axially Chiral N-Arylamides
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Christie, G.H.; Kenner, J. LXXI.—The molecular configurations of polynuclear aromatic compounds. Part I. The resolution of γ-6:6′-dinitro- and 4:6:4′:6′-tetranitro-diphenic acids into optically active components. J. Chem. Soc. Trans. 1922, 121, 614–620. [Google Scholar] [CrossRef]
- Eveleigh, P.; Hulme, E.C.; Schudt, C.; Birdsall, N.J.M. The existence of stable enantiomers of telenzepine and their stereoselective interaction with muscarinic receptor subtypes. Mol. Pharmacol. 1989, 35, 477–483. [Google Scholar] [PubMed]
- Clayden, J.; Moran, W.J.; Edwards, P.J.; LaPlante, S.R. The challenge of atropisomerism in drug discovery. Angew. Chem. Int. Ed. 2009, 48, 6398–6401. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, G.; Gulder, T.; Gulder, T.A.M.; Breuning, M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 2011, 111, 563–639. [Google Scholar] [CrossRef] [PubMed]
- Toenjes, S.T.; Gustafson, J.L. Atropisomerism in medicinal chemistry: Challenges and opportunities. Future Med. Chem. 2018, 10, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Sotorasib: First approval. Drugs 2021, 81, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- O’connell, P.J.; Harms, C.T.; Allen, J.R.F. Metolachlor, S-metolachlor and their role within sustainable weed-management. Crop. Prot. 1998, 17, 207–212. [Google Scholar] [CrossRef]
- Blaser, H.-U. The chiral switch of (S)-Metolachlor: A personal account of an industrial odyssey in asymmetric catalysis. Adv. Synth. Catal. 2002, 344, 17–31. [Google Scholar] [CrossRef]
- Blaser, H.-U.; Pugin, B.; Spindler, F.; Thommen, M. From a chiral switch to a ligand portfolio for ssymmetric catalysis. Acc. Chem. Res. 2007, 40, 1240–1250. [Google Scholar] [CrossRef]
- Brandes, S.; Bella, M.; Kjærsgaard, A.; Jørgensen, K.A. Chirally aminated 2-naphthols—organocatalytic synthesis of non-biaryl atropisomers by asymmetric Friedel–Crafts amination. Angew. Chem. Int. Ed. 2006, 45, 1147–1151. [Google Scholar] [CrossRef]
- Brandes, S.; Niess, B.; Bella, M.; Prieto, A.; Overgaard, J.; Jørgensen, K.A. Non-biaryl atropisomers in organocatalysis. Chem. Eur. J. 2006, 12, 6039–6052. [Google Scholar] [CrossRef]
- Zhang, J.-W.; Xu, J.-H.; Cheng, D.-J.; Shi, C.; Liu, X.-Y.; Tan, B. Discovery and enantiocontrol of axially chiral urazoles via organocatalytic tyrosine click reaction. Nat. Commun. 2016, 7, 10677. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Wong, A.; Virgil, S.C. Discovery and enantiocontrol of axially chiral urazoles via organocatalytic tyrosine click reaction. J. Org. Chem. 1998, 63, 2597–2600. [Google Scholar] [CrossRef]
- Takahashi, I.; Suzuki, Y.; Kitagawa, O. Asymmetric synthesis of atropisomeric compounds with an N–C chiral axis. Org. Prep. Proced. Int. 2014, 46, 1–23. [Google Scholar] [CrossRef]
- Kumarasamy, E.; Ayitou, A.J.-L.; Vallavoju, N.; Raghunathan, R.; Iyer, A.; Clay, A.; Kandappa, S.K.; Sivaguru, J. Tale of twisted molecules. Atropselective photoreactions: Taming light induced asymmetric transformations through non-biaryl atropisomers. Acc. Chem. Res. 2016, 49, 2713–2724. [Google Scholar] [CrossRef]
- Kumarasamy, E.; Raghunathan, R.; Sibi, M.P.; Sivaguru, J. Nonbiaryl and heterobiaryl atropisomers: Molecular templates with promise for atropselective chemical transformations. Chem. Rev. 2015, 115, 11239–11300. [Google Scholar] [CrossRef]
- Wang, Y.-B.; Tan, B. Construction of axially chiral compounds via asymmetric organocatalysis. Acc. Chem. Res. 2018, 51, 534–547. [Google Scholar] [CrossRef]
- Kitagawa, O. Chiral Pd-catalyzed enantioselective syntheses of various N–C axially chiral compounds and their synthetic applications. Acc. Chem. Res. 2021, 54, 719–730. [Google Scholar] [CrossRef]
- Cheng, J.K.; Xiang, S.-H.; Li, S.; Ye, L.; Tan, B. Recent advances in catalytic asymmetric construction of atropisomers. Chem. Rev. 2021, 121, 4805–4902. [Google Scholar] [CrossRef]
- Mei, G.-J.; Koay, W.L.; Guan, C.-Y.; Lu, Y. Atropisomers beyond the C–C axial chirality: Advances in catalytic asymmetric synthesis. Chem 2022, 8, 1855–1893. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Liao, G.; Shi, B.-F. Stereoselective construction of atropisomers featuring a C–N chiral axis. Green Synth. Catal. 2022, 3, 117–136. [Google Scholar] [CrossRef]
- Guo, X.-X.; Gu, D.-W.; Wu, Z.; Zhang, W. Copper-catalyzed C–H functionalization reactions: Efficient synthesis of heterocycles. Chem. Rev. 2015, 115, 1622–1651. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, Q.; Yu, J.-Q. Palladium-catalyzed transformations of alkyl C–H bonds. Chem. Rev. 2017, 117, 8754–8786. [Google Scholar] [CrossRef]
- Cheng, F.; Chen, T.; Huang, Y.-Q.; Li, J.-W.; Zhou, C.; Xiao, X.; Chen, F.-E. Copper-catalyzed Ullmann-type coupling and decarboxylation cascade of arylhalides with malonates to access α-aryl esters. Org. Lett. 2022, 24, 115–120. [Google Scholar] [CrossRef]
- Xiao, X.; Huang, Y.-Q.; Tian, H.-Y.; Bai, J.; Cheng, F.; Wang, X.; Ke, M.-L.; Chen, F.-E. Robust, scalable construction of an electrophilic deuterated methylthiolating reagent: Facile access to SCD3-containing scaffolds. Chem. Commun. 2022, 58, 3015–3018. [Google Scholar] [CrossRef]
- Cheng, Q.; Tu, H.-F.; Zheng, C.; Qu, J.-P.; Helmchen, G.; You, S.-L. Iridium-catalyzed asymmetric allylic substitution reactions. Chem. Rev. 2019, 119, 1855–1969. [Google Scholar] [CrossRef]
- Woźniak, Ł.; Tan, J.-F.; Nguyen, Q.-H.; Vigné, A.M.; Smal, V.; Cao, Y.-X.; Cramer, N. Catalytic enantioselective functionalizations of C–H bonds by chiral iridium complexes. Chem. Rev. 2020, 120, 10516–10543. [Google Scholar] [CrossRef]
- Süsse, L.; Stoltz, B.M. Enantioselective formation of quaternary centers by allylic alkylation with first-row transition-metal catalysts. Chem. Rev. 2021, 121, 4084–4099. [Google Scholar] [CrossRef]
- Kitagawa, O.; Kohriyama, M.; Taguchi, T. Catalytic asymmetria synthesis of optically active atropisomeric anilides through enantioselective N-allylation with chiral Pd-tol-BINAP catalyst. J. Org. Chem. 2002, 67, 8682–8684. [Google Scholar] [CrossRef]
- Terauchi, J.; Curran, D.P. N-Allylation of anilides with chiral palladium catalysts: The first catalytic asymmetric synthesis of axially chiral anilides. Tetrahedron Asymmetry 2003, 14, 587–592. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, X.; Du, H. Asymmetric synthesis of axially chiral anilides by Pd-catalyzed allylic substitutions with P/Olefin ligands. Org. Biomol. Chem. 2015, 13, 125–132. [Google Scholar] [CrossRef]
- Kitagawa, O.; Takahashi, M.; Yoshikawa, M.; Taguchi, T. Efficient synthesis of optically active atropisomeric anilides through catalytic asymmetric N-arylation reaction. J. Am. Chem. Soc. 2005, 127, 3676–3677. [Google Scholar] [CrossRef]
- Kitagawa, O.; Yoshikawa, M.; Tanabe, H.; Morita, T.; Takahashi, M.; Dobashi, Y.; Taguchi, T. Highly enantioselective synthesis of atropisomeric anilide derivatives through catalytic asymmetric N-arylation: Conformational analysis and application to asymmetric enolate chemistry. J. Am. Chem. Soc. 2006, 128, 12923–12931. [Google Scholar] [CrossRef]
- Tanaka, K.; Nishida, G.; Wada, A.; Noguchi, K. Enantioselective synthesis of axially chiral phthalides through cationic [RhI(H8-binap)]-catalyzed cross alkyne cyclotrimerization. Angew. Chem. Int. Ed. 2004, 43, 6510–6512. [Google Scholar] [CrossRef]
- Tanaka, K.; Nishida, G.; Ogino, M.; Hirano, M.; Noguchi, K. Enantioselective synthesis of axially chiral biaryls through rhodium-catalyzed complete intermolecular cross-cyclotrimerization of internal alkynes. Org. Lett. 2005, 7, 3119–3121. [Google Scholar] [CrossRef]
- Tanaka, K.; Wada, A.; Noguchi, K. Rhodium-catalyzed chemo-, regio-, and enantioselective [2+2+2] cycloaddition of alkynes with isocyanates. Org. Lett. 2005, 7, 4737–4739. [Google Scholar] [CrossRef]
- Nishida, G.; Suzuki, N.; Noguchi, K.; Tanaka, K. Enantioselective synthesis of tetra-ortho-substituted axially chiral biaryls through rhodium-catalyzed double [2+2+2] cycloaddition. Org. Lett. 2006, 8, 3489–3492. [Google Scholar] [CrossRef]
- Tanaka, K.; Suda, T.; Noguchi, K.; Hirano, M. Catalytic [2+2+2] and thermal [4+2] cycloaddition of 1,2-bis(arylpropiolyl)benzenes. J. Org. Chem. 2007, 72, 2243–2246. [Google Scholar] [CrossRef]
- Nishida, G.; Noguchi, K.; Hirano, M.; Tanaka, K. Asymmetric assembly of aromatic rings to produce tetra-ortho-substituted axially chiral biaryl phosphorus compounds. Angew. Chem. Int. Ed. 2007, 46, 3951–3954. [Google Scholar] [CrossRef]
- Tanaka, K.; Otake, Y.; Wada, A.; Hirano, M. Cationic Rh(I)/modified-BINAP-catalyzed reactions of carbonyl compounds with 1,6-diynes leading to dienones and ortho-functionalized aryl ketones. Org. Lett. 2007, 9, 2203–2206. [Google Scholar] [CrossRef]
- Tanaka, K.; Takeishi, K.; Noguchi, K. Enantioselective synthesis of axially chiral anilides through Rhodium-catalyzed [2+2+2] cycloaddition of 1,6-Diynes with trimethylsilylynamides. J. Am. Chem. Soc. 2006, 128, 4586–4587. [Google Scholar] [CrossRef]
- Tanaka, K.; Takeishi, K. Preparation of enantioenriched axially chiral anilides via [2+2+2] cycloaddition of 1,6-Diynes with trimethylsilylynamides. Synthesis 2007, 18, 2920–2923. [Google Scholar] [CrossRef]
- Giri, R.; Shi, B.-F.; Engle, K.M.; Maugel, N.; Yu, J.-Q. Transition metal-catalyzed C–H activation reactions: Diastereoselectivity and enantioselectivity. Chem. Soc. Rev. 2009, 38, 3242–3272. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Recent development of direct asymmetric functionalization of inert C–H bonds. RSC Adv. 2014, 4, 6173–6214. [Google Scholar] [CrossRef]
- Newton, C.G.; Wang, S.-G.; Oliveira, C.C.; Cramer, N. Catalytic enantioselective transformations involving C–H bond cleavage by transition-metal complexes. Chem. Rev. 2017, 117, 8908–8976. [Google Scholar] [CrossRef]
- Saint-Denis, T.G.; Zhu, R.-Y.; Chen, G.; Wu, Q.-F.; Yu, J.-Q. Enantioselective C(sp3)–H bond activation by chiral transition metal catalysts. Science 2018, 359, eaao4798. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shi, B.-F. From reactivity and regioselectivity to stereoselectivity: An odyssey of designing PIP amine and related directing groups for C–H activation. Chin. J. Chem. 2019, 37, 647–656. [Google Scholar] [CrossRef]
- Loup, J.; Dhawa, U.; Pesciaioli, F.; Wencel-Delord, J.; Ackermann, L. Enantioselective C–H activation with earth-abundant 3d transition metals. Angew. Chem. Int. Ed. 2019, 58, 12803–12818. [Google Scholar] [CrossRef]
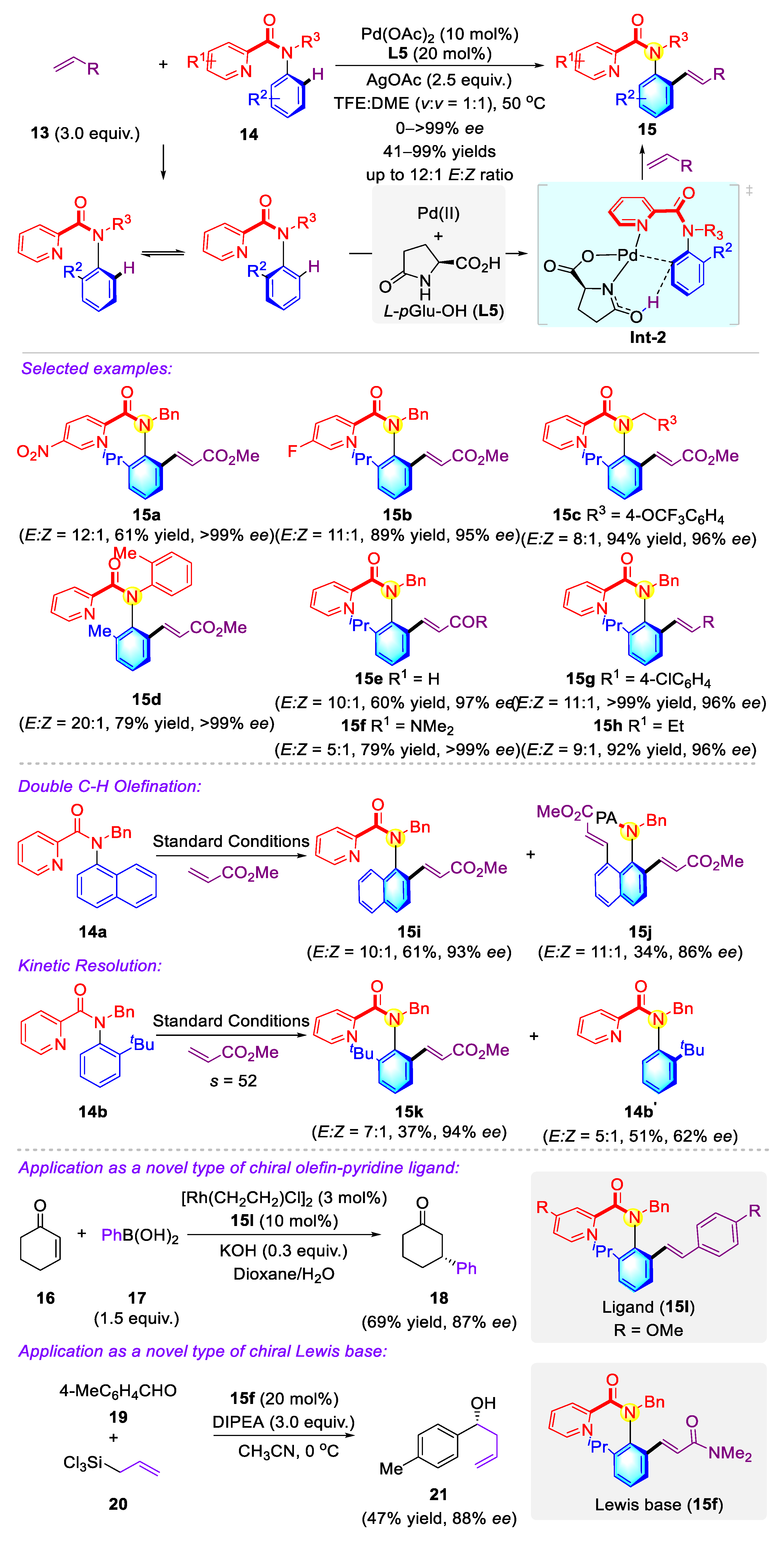
- Yao, Q.-J.; Xie, P.-P.; Wu, Y.-J.; Feng, Y.-L.; Teng, M.-Y.; Hong, X.; Shi, B.-F. Enantioselective synthesis of atropisomeric anilides via Pd(II)-catalyzed asymmetric C–H olefination. J. Am. Chem. Soc. 2020, 142, 18266–18276. [Google Scholar] [CrossRef]
- Jia, Z.-S.; Wu, Y.-J.; Yao, Q.-J.; Xu, X.-T.; Zhang, K.; Shi, B.-F. Pd(II)-catalyzed atroposelective C–H allylation: Synthesis of enantioenriched N-Aryl peptoid atropisomers. Org. Lett. 2022, 24, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-J.; Xie, P.-P.; Zhou, G.; Yao, Q.-J.; Hong, X.; Shi, B.-F. Atroposelective synthesis of N-Aryl peptoid atropisomers via a Palladium(II)-catalyzed asymmetric C–H alkynylation strategy. Chem. Sci. 2021, 12, 9391–9397. [Google Scholar] [CrossRef]
- Vedejs, E.; Jure, M. Efficiency in nonenzymatic kinetic resolution. Angew. Chem. Int. Ed. 2005, 44, 3974–4001. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-J.; Geng, R.-L.; Wei, J.-H.; Gong, L.-Z. Atroposelective sp3 C–H coupling for kinetic resolution of thioanilide atropisomers. Chin. J. Chem. 2021, 39, 3269–3276. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent development and application of chiral phase-transfer catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Maruoka, K. Recent advances in asymmetric phase-transfer catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef]
- Lygo, B.; Andrews, B.I. Asymmetric phase-transfer catalysis utilizing chiral quaternary ammonium salts: Asymmetric alkylation of glycine imines. Acc. Chem. Res. 2004, 37, 518–525. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.J. The Enantioselective synthesis of α-amino acids by phase-transfer catalysis with achiral schiff base esters. Acc. Chem. Res. 2004, 37, 506–517. [Google Scholar] [CrossRef]
- Ooi, T.; Maruoka, K. Asymmetric organocatalysis of structurally well-defined chiral quaternary ammonium fluorides. Acc. Chem. Res. 2004, 37, 526–533. [Google Scholar] [CrossRef]
- Shirakawa, S.; Liu, K.; Maruoka, K. Catalytic asymmetric synthesis of axially chiral o-iodoanilides by phase-transfer catalyzed alkylations. J. Am. Chem. Soc. 2012, 134, 916–919. [Google Scholar] [CrossRef]
- Liu, K.; Wu, X.; Kan, S.; Shirakawa, S.; Maruoka, K. Phase-transfer-catalyzed asymmetric synthesis of axially chiral anilides. Chem. Asian J. 2013, 8, 3214–3221. [Google Scholar] [CrossRef]
- Xiao, X.; Shao, B.-X.; Lu, Y.-J.; Cao, Q.-Q.; Xia, C.-N.; Chen, F.-E. Recent advances in asymmetric organomulticatalysis. Adv. Synth. Catal. 2021, 363, 352–387. [Google Scholar] [CrossRef]
- Wang, N.; Wu, Z.; Wang, J.; Ullah, N.; Lu, Y. Recent applications of asymmetric organocatalytic annulation reactions in natural products synthesis. Chem. Soc. Rev. 2021, 50, 9766–9793. [Google Scholar] [CrossRef]
- Brak, K.; Jacobsen, E.N. Asymmetric ion-pairing catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef] [Green Version]
- Doyle, A.G.; Jacobsen, E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar]
- Xiao, X.; Tian, H.-Y.; Huang, Y.-Q.; Lu, Y.-J.; Fang, J.-J.; Zhou, G.-J.; Chen, F.-E. Atom- and step-economic 1,3-thiosulfonylation of activated allenes with thiosulfonates to access vinyl sulfones/sulfides. Chem. Commun. 2022, 58, 6756–6768. [Google Scholar] [CrossRef]
- Wang, N.; Lang, Y.; Wang, J.; Wu, Z.; Lu, Y. Phosphine-catalyzed sequential [3+2]/[3+2] annulation between allenoates and arylidenemalononitriles for the enantioselective construction of bicyclo[3,3,0]octenes and cyclopenta[c]quinolinones. Org. Lett. 2022, 24, 3712–3716. [Google Scholar] [CrossRef]
- Catalytic asymmetric inverse-electron-demand aza-Diels–Alder reaction of 1,3-diazadienes with 3-vinylindoles. Chem. Commun. 2022, 58, 7515–7518. [CrossRef]
- Xiao, X.; Shao, B.; Li, J.; Yang, Z.; Lu, Y.-J.; Ling, F.; Zhong, W. Enantioselective synthesis of functionalized 1,4-dihydropyrazolo[4′,3′:5,6]pyrano[2,3-b]quinolines through ferrocenyl-phosphine-catalyzed annulation of modified MBH carbonates and pyrazolones. Chem. Commun. 2021, 57, 4690–4693. [Google Scholar] [CrossRef]
- Li, S.-L.; Yang, C.; Wu, Q.; Zheng, H.-L.; Li, X.; Cheng, J.-P. Atroposelective catalytic asymmetric allylic alkylation reaction for axially chiral anilides with achiral Morita–Baylis–Hillman carbonates. J. Am. Chem. Soc. 2018, 140, 12836–12843. [Google Scholar] [CrossRef]
- Lu, S.; Ng, S.V.H.; Lovato, K.; Ong, J.-Y.; Poh, S.B.; Ng, X.Q.; Kürti, L.; Zhao, Y. Practical access to axially chiral sulfonamides and biaryl amino phenols via organocatalytic atroposelective N-alkylation. Nat. Commun. 2019, 10, 3061–3069. [Google Scholar] [CrossRef]
- Xiao, X.; Lu, Y.-J.; Tian, H.-Y.; Zhou, H.-J.; Li, J.-W.; Yao, Y.-P.; Ke, M.-L.; Chen, F.-E. Organocatalytic atroposelective N-alkylation: Divergent synthesis of axially chiral sulfonamides and biaryl amino phenols. Org. Chem. Front. 2022, 9, 2830–2839. [Google Scholar] [CrossRef]
- Zheng, G.; Li, X.; Cheng, J.-P. Access to axially and centrally chiral culfinamides via asymmetric allylic alkylation. Org. Lett. 2021, 23, 3997–4001. [Google Scholar] [CrossRef]
- Yang, G.-H.; Zheng, H.; Li, X.; Cheng, J.-P. Asymmetric synthesis of axially chiral phosphamides via atroposelective N-allylic alkylation. ACS Catal. 2020, 10, 2324–2333. [Google Scholar] [CrossRef]
- Li, D.; Wang, G.S.; Dong, S.; Feng, X. Asymmetric synthesis of axially chiral anilides via organocatalytic atroposelective N-acylation. Org. Lett. 2020, 22, 5331–5336. [Google Scholar] [CrossRef]
- Ong, J.-Y.; Ng, X.Q.; Lu, S.; Zhao, Y. Isothiourea-catalyzed atroposelective N-acylation of sulfonamides. Org. Lett. 2020, 22, 6447–6451. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Nakamura, C.; Matsuoka, M.; Asami, R.; Kitagawa, O. Catalytic enantioselective synthesis of N–C axially chiral sulfonamides through chiral palladium-catalyzed N-allylation. J. Org. Chem. 2019, 84, 8112–8120. [Google Scholar] [CrossRef]
- Gao, Z.; Yan, C.-X.; Qian, J.; Yang, H.; Zhou, P.; Zhang, J.; Jiang, G. Enantioselective synthesis of axially chiral sulfonamides via atroposelective hydroamination of allenes. ACS Catal. 2021, 11, 6931–6938. [Google Scholar] [CrossRef]
- Ma, S. Typical advances in the synthetic applications of allenes. Chem. Rev. 2005, 105, 2829–2872. [Google Scholar]
- Yu, S.; Ma, S. How easy are the syntheses of allenes? Chem. Commun. 2011, 47, 5384–5418. [Google Scholar] [CrossRef] [PubMed]
- He, F.-S.; Bao, P.; Yu, F.; Zeng, L.-H.; Deng, W.-P.; Wu, J. Copper-catalyzed regioselective 1,4-selenosulfonylation of 1,3-enynes to access cyanoalkylsulfonylated allenes. Org. Lett. 2021, 23, 7472–7476. [Google Scholar] [CrossRef] [PubMed]
- Han, T.-J.; Zhang, Z.-X.; Wang, M.-C.; Xu, L.-P.; Mei, G.-J. The rational design and atroposelective synthesis of axially chiral C2-arylpyrrole-derived amino alcohols. Angew. Chem. Int. Ed. 2022, 61, e202207517. [Google Scholar] [CrossRef] [PubMed]
- Mei, G.-J.; Wong, J.J.; Zheng, W.; Nangia, A.A.; Houk, K.N.; Lu, Y. Rational design and atroposelective synthesis of N–N axially chiral compounds. Chem 2021, 7, 2743–2757. [Google Scholar] [CrossRef]
- Bai, H.-Y.; Tan, F.-X.; Liu, T.-Q.; Zhu, G.-D.; Tian, J.-M.; Ding, T.-M.; Chen, Z.-M.; Zhang, S.-Y. Highly atroposelective synthesis of nonbiaryl naphthalene-1,2-diamine N-C atropisomers through direct enantioselective C–H amination. Nat. Commun. 2019, 10, 3063. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Li, K.-N.; Liu, B.; Zhu, H.-J.; Fan, Y.-M.; Gong, L.-Z. Asymmetric synthesis of heteroaryl atropisomers via a gold-catalyzed cycloisomerization–amination cascade reaction. Chem. Commun. 2014, 50, 5451–5454. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.-L.; Imazaki, Y.; Shintani, R.; Hayashi, T. Asymmetric construction of chiral C–N axes through rhodium-catalyzed 1,4-addition. Tetrahedron 2007, 63, 8529–8536. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Lin, L.; Yao, Q.; Liu, X.; Feng, X. Catalytic asymmetric desymmetrization of N-arylmaleimides: Efficient construction of both atom chirality and axial chirality. Chem. Commun. 2015, 51, 10554. [Google Scholar] [CrossRef]
- Liu, H.-C.; Tao, H.-Y.; Cong, H.; Wang, C.-J. Silver(I)-catalyzed atroposelective desymmetrization of N-arylmaleimide via 1,3-dipolar cycloaddition of azomethine ylides: Access to octahydropyrrolo[3,4-c]pyrrole derivatives. J. Org. Chem. 2016, 81, 3752–3760. [Google Scholar] [CrossRef]
- Gu, X.-W.; Sun, Y.-L.; Xie, J.-L.; Wang, X.-B.; Xu, Z.; Yin, G.-W.; Li, L.; Yang, K.-F.; Xu, L.-W. Stereospecific Si-C coupling and remote control of axial chirality by enantioselective palladiumcatalyzed hydrosilylation of maleimides. Nat. Commun. 2020, 11, 2904. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Kong, L.; Wang, F.; Lan, Y.; Li, X. Enantioselective and diastereoselective C–H alkylation of benzamides: Synergized axial and central chirality via a single stereodetermining step. ACS Catal. 2021, 11, 9151–9158. [Google Scholar] [CrossRef]
- Sun, F.; Wang, T.; Cheng, G.-J.; Fang, X. Enantioselective nickel-catalyzed hydrocyanative desymmetrization of norbornene derivatives. ACS Catal. 2021, 11, 7578–7583. [Google Scholar] [CrossRef]
- Iorio, N.D.; Crotti, S.; Bencivenni, G. Organocatalytic desymmetrization reactions for the synthesis of axially chiral compounds. Chem. Rec. 2019, 19, 2095–2104. [Google Scholar] [CrossRef]
- Iorio, N.; Righi, P.; Mazzanti, A.; Mancinelli, M.; Ciogli, A.; Bencivenni, G. Remote control of axial chirality: Aminocatalytic desymmetrization of N-arylmaleimides via vinylogous Michael addition. J. Am. Chem. Soc. 2014, 136, 10250–10253. [Google Scholar] [CrossRef]
- Eudier, F.; Righi, P.; Mazzanti, A.; Ciogli, A.; Bencivenni, G. Organocatalytic atroposelective formal Diels–Alder desymmetrization of N-arylmaleimides. Org. Lett. 2015, 17, 1728–1731. [Google Scholar] [CrossRef]
- Di Iorio, N.; Champavert, F.; Erice, A.; Righi, P.; Mazzanti, A.; Bencivenni, G. Targeting remote axial chirality control of N-(2-tert-butylphenyl)succinimides by means of michael addition type reactions. Tetrahedron 2016, 72, 5191–5201. [Google Scholar] [CrossRef]
- Di Iorio, N.; Soprani, L.; Crotti, S.; Marotta, E.; Mazzanti, A.; Righi, P.; Bencivenni, G. Michael addition of oxindoles to N-(2-tert-butylphenyl)maleimides: Efficient desymmetrization for the synthesis of atropisomeric succinimides with quaternary and tertiary stereocenters. Synthesis 2017, 49, 1519–1530. [Google Scholar]
- Song, R.; Jin, Z.; Chi, Y.R. NHC-catalyzed covalent activation of heteroatoms for enantioselective reactions. Chem. Sci. 2021, 12, 5037–5043. [Google Scholar] [CrossRef]
- Chen, X.; Wang, H.; Jin, Z.; Chi, Y.R. N-Heterocyclic carbene organocatalysis: Activation modes and typical reactive intermediates. Chin. J. Chem. 2020, 38, 1167–1202. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic reactions enabled by N-heterocyclic carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [Green Version]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef]
- Nelson, D.J.; Nolan, S.P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, X.; Glorius, F. Organocatalytic umpolung: N-heterocyclic carbenes and beyond. Chem. Soc. Rev. 2012, 41, 3511–3522. [Google Scholar] [CrossRef] [PubMed]
- Barik, S.; Shee, S.; Das, S.; Gonnade, R.G.; Jindal, G.; Mukherjee, S.; Biju, A.T. NHC-Catalyzed desymmetrization of N-aryl maleimides leading to the atroposelective synthesis of N-aryl succinimides. Angew. Chem. Int. Ed. 2021, 60, 12264–12268. [Google Scholar] [CrossRef] [PubMed]
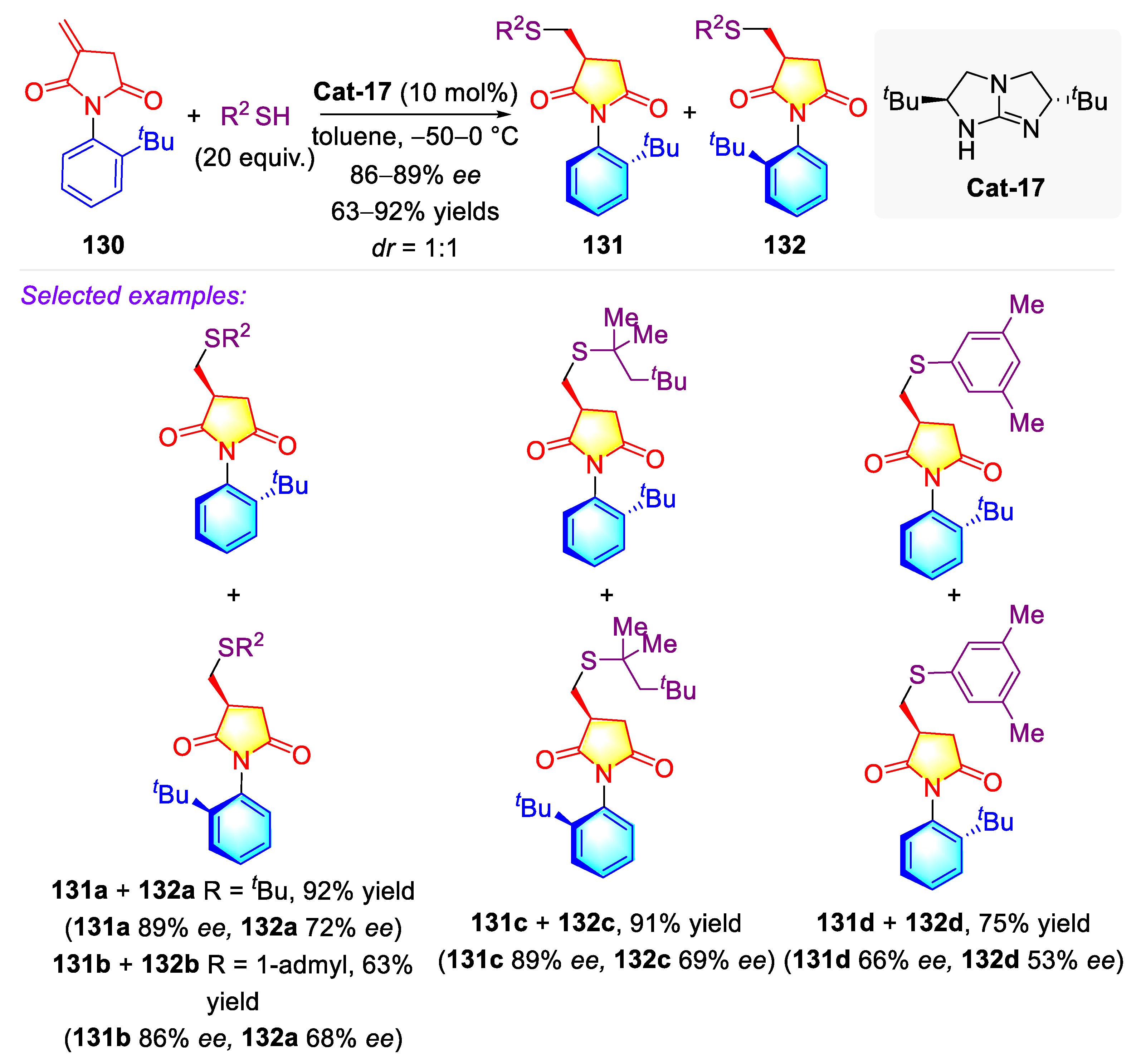
- Lin, S.; Leow, D.; Huang, K.-W.; Tan, C.-H. Enantioselective protonation of itaconimides with thiols and the rotational kinetics of the axially chiral C–N bond. Chem. Asian J. 2009, 4, 1741–1744. [Google Scholar] [CrossRef]
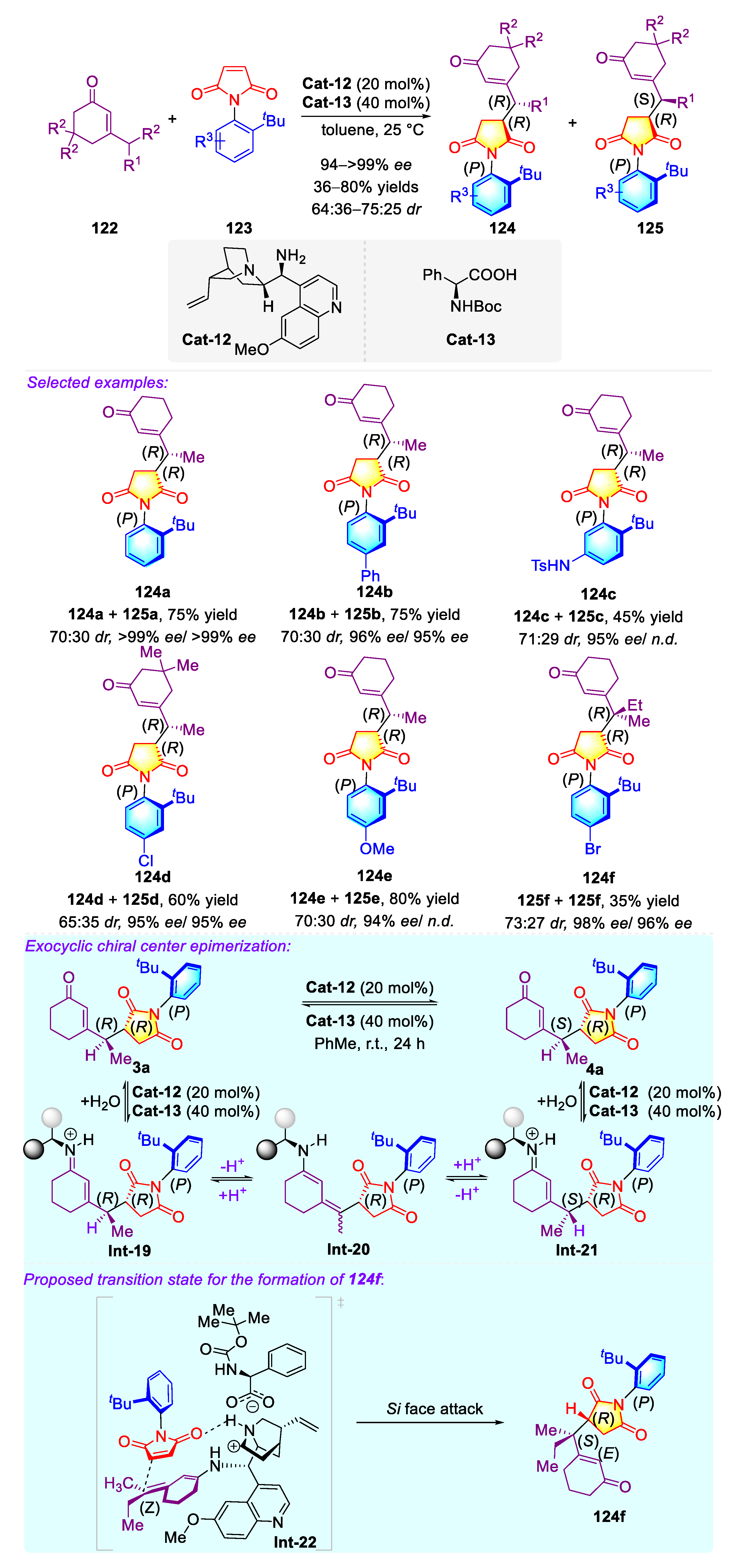
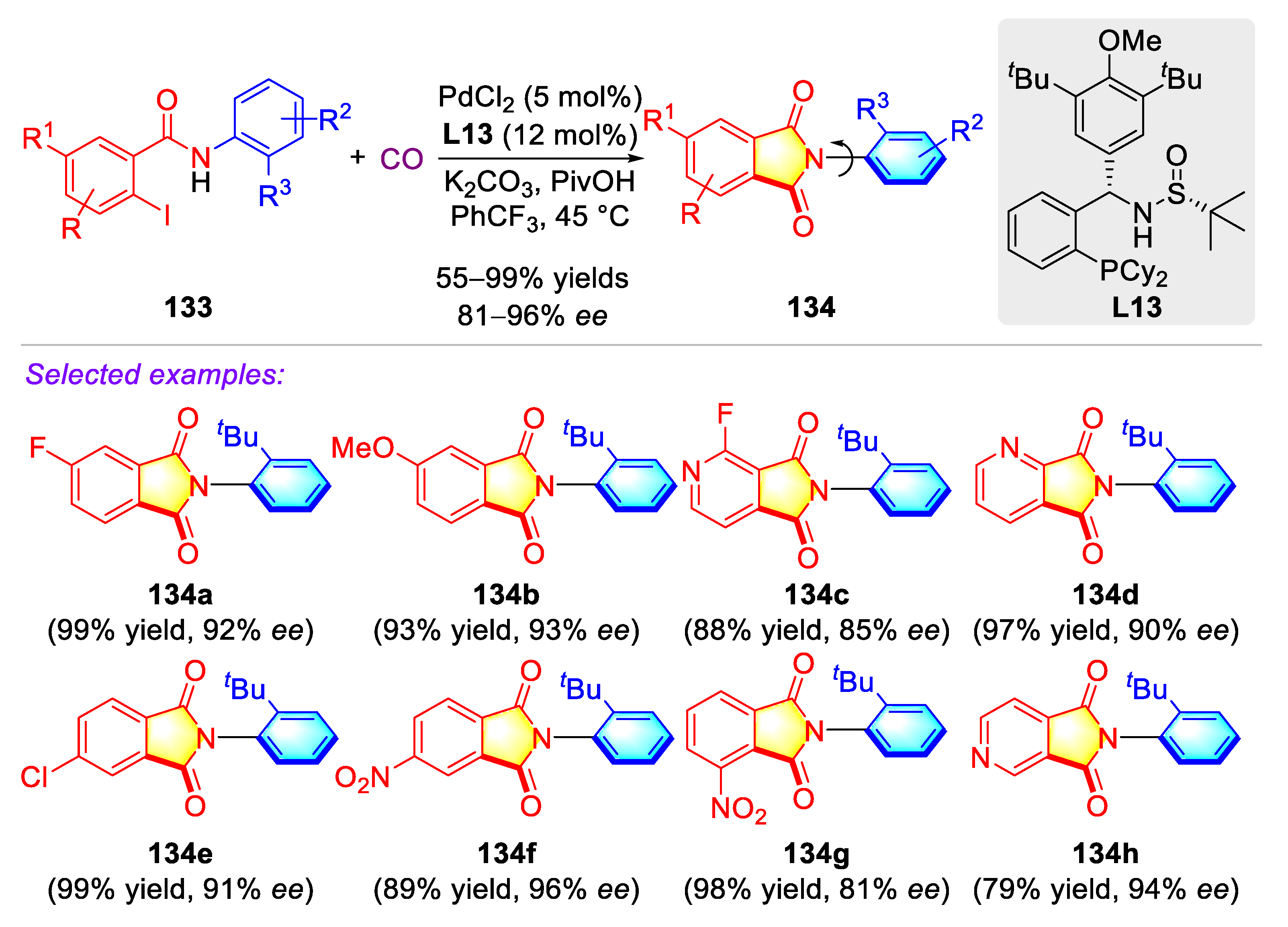
- Chen, L.-P.; Chen, J.-F.; Zhang, Y.-J.; He, X.-Y.; Han, Y.-F.; Xiao, Y.-T.; Lv, G.; Lu, X.; Teng, F.; Sun, Q.; et al. Atroposelective carbonylation of aryl iodides with amides: Facile synthesis of enantioenriched cyclic and acyclic amides. Org. Chem. Front. 2021, 8, 6067–6073. [Google Scholar] [CrossRef]
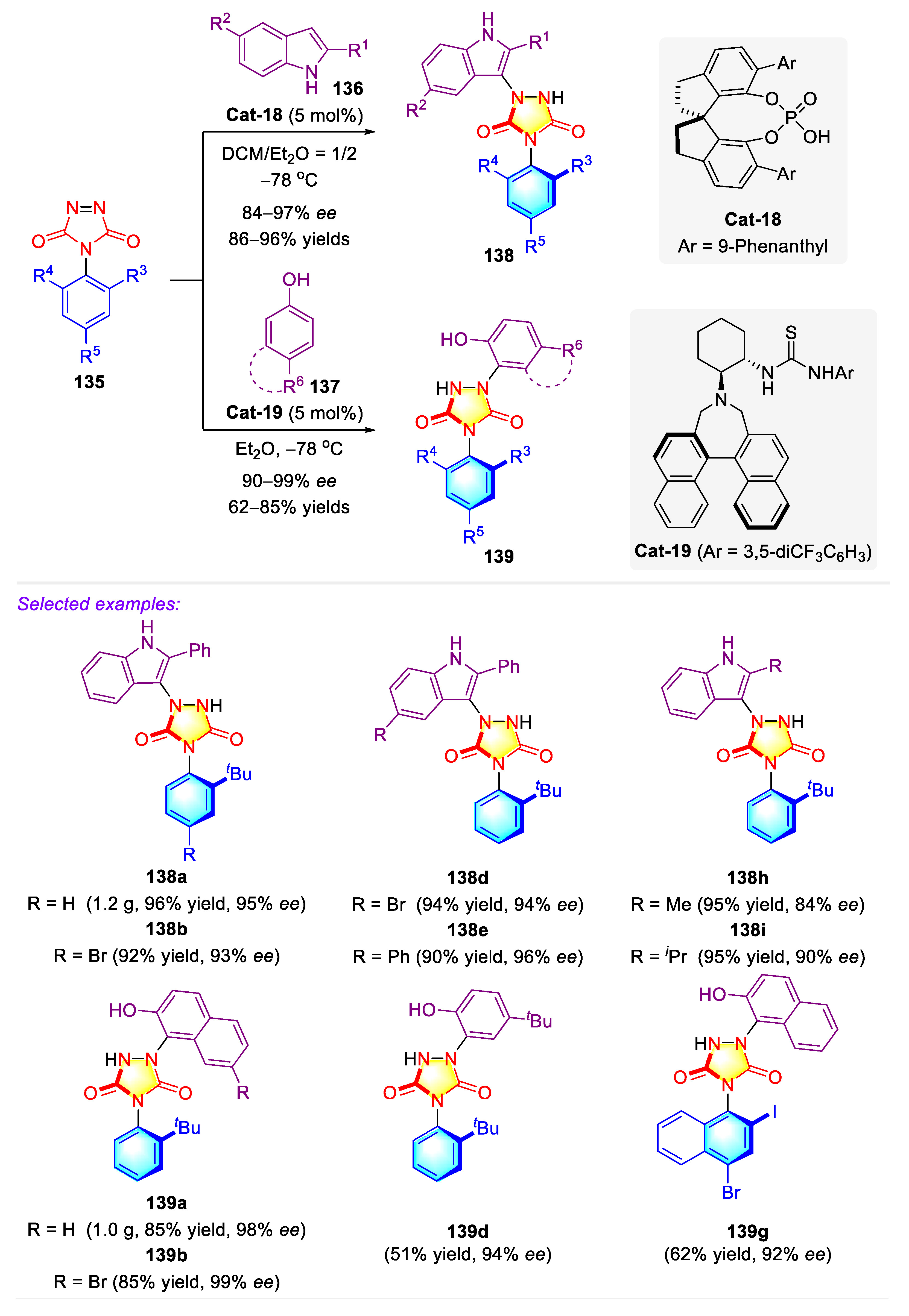
- Zhang, L.-L.; Zhang, J.-W.; Xiang, S.-H.; Guo, Z.; Tan, B. Remote control of axial chirality: Synthesis of spirooxindole–urazoles via desymmetrization of ATAD. Org. Lett. 2018, 20, 6022–6026. [Google Scholar] [CrossRef]
- Jin, J.; Huang, X.; Xu, J.; Li, T.; Peng, X.; Zhu, X.; Zhang, J.; Jin, Z.; Chi, Y.R. Carbene-catalyzed atroposelective annulation and desymmetrization of urazoles. Org. Lett. 2021, 23, 3991–3996. [Google Scholar] [CrossRef]
- Min, C.; Lin, Y.; Seidel, D. Catalytic enantioselective synthesis of Mariline A and related isoindolinones through a biomimetic approach. Angew. Chem. Int. Ed. 2017, 56, 15353–15357. [Google Scholar] [CrossRef]
- Li, H.; Yan, X.; Zhang, J.; Guo, W.; Jiang, J.; Wang, J. Enantioselective synthesis of C–N axially chiral N-aryloxindoles by asymmetric rhodium-catalyzed dual C–H activation. Angew. Chem. Int. Ed. 2019, 58, 6732–6736. [Google Scholar] [CrossRef]
- Wei, S.; Le, S.; Lei, Z.; Zhou, L.; Zhang, Z.; Dolbier, W.R., Jr. Difluoromethylarylation of α,β-unsaturated amides via a photocatalytic radical smiles rearrangement. Org. Biomol. Chem. 2022, 20, 2064–2068. [Google Scholar] [CrossRef]
- Liu, H.; Feng, W.; Kee, C.; Leow, D.; Loh, W.-T.; Tan, C.-H. Brønsted base-catalyzed tandem isomerization–Michael reactions of alkynes: Synthesis of oxacycles and azacycles. Adv. Synth. Catal. 2010, 352, 3373–3379. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Chan, B.K. Methodology for the synthesis of pyridines and pyridones: Development and applications. Heterocycles 2007, 74, 101–124. [Google Scholar] [CrossRef]
- Mercedes, T.; Salvador, G.; Margarita, P. New synthetic methods to 2-pyridone rings. Curr. Org. Chem. 2005, 9, 1757–1779. [Google Scholar]
- Tanaka, K.; Takahashi, Y.; Suda, T.; Hirano, M. Synthesis of enantioenriched N-aryl-2-pyridones with chiral C–N axes by rhodium-catalyzed [2+2+2] cycloaddition of alkynes with isocyanates. Synlett 2008, 11, 1724–1728. [Google Scholar] [CrossRef]
- Onodera, G.; Suto, M.; Takeuchi, R. Iridium-catalyzed [2+2+2] cycloaddition of α,ω-diynes with isocyanates. J. Org. Chem. 2012, 77, 908–920. [Google Scholar] [CrossRef]
- Hirata, T.; Takahashi, I.; Suzuki, Y.; Yoshida, H.; Hasegawa, H.; Kitagawa, O. Catalytic Enantioselective Synthesis of N-C Axially Chiral Phenanthridin-6-one Derivatives. J. Org. Chem. 2016, 81, 318–323. [Google Scholar] [CrossRef]
- Zhou, F.; Cai, Q. Recent advances in copper-catalyzed asymmetric coupling reactions. Beilstein J. Org. Chem. 2015, 11, 2600–2615. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Chen, Y.; Ma, D. Enantioselective arylation of 2-methylacetoacetates catalyzed by CuI/trans-4-hydroxy-L-proline at low reaction temperatures. J. Am. Chem. Soc. 2006, 128, 16050–16051. [Google Scholar] [CrossRef]
- Zhou, F.; Guo, J.; Liu, J.; Ding, K.; Yu, S.; Cai, Q. Copper catalyzed desymmetric intramolecular Ullmann C-N coupling: An enantioselective preparation of indolines. J. Am. Chem. Soc. 2012, 134, 14326–14329. [Google Scholar] [CrossRef]
- Liu, J.; Tian, Y.; Shi, J.; Zhang, S.; Cai, Q. An enantioselective synthesis of spirobilactams through copper-catalyzed intramolecular double N-arylation and phase separation. Angew. Chem. Int. Ed. 2015, 54, 10917–10920. [Google Scholar] [CrossRef]
- Rae, J.; Frey, J.; Jerhaoui, S.; Choppin, S.; Wencel-Delord, J.; Colobert, F. Synthesis of axially chiral C-N scaffolds via asymmetric coupling with enantiopure sulfinyl iodanes. ACS Catal. 2018, 8, 2805–2809. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, X.; Li, C.; Gu, Z. Enantioselective Atropisomeric Anilides Synthesis via Cu-Catalyzed Intramolecular Adjacent C–N Coupling. ACS Catal. 2019, 9, 2286–2291. [Google Scholar] [CrossRef]
- Liu, Z.-S.; Xie, P.-P.; Hua, Y.; Wu, C.; Ma, Y.; Chen, J.; Cheng, H.-G.; Hong, X.; Zhou, Q. An Axial-to-Axial Chirality Transfer Strategy for Atroposelective Construction of C–N Axial Chirality. Chem 2021, 7, 1917–1932. [Google Scholar] [CrossRef]
- Dong, Y.; Liu, R.; Wang, W. Catalytic asymmetric Catellani-type reaction: A powerful tool for axial chirality construction. Green Synth. Catal. 2020, 1, 83–85. [Google Scholar] [CrossRef]
- Sun, C.; Qi, X.; Min, X.-L.; Bai, X.-D.; Liu, P.; He, Y. Asymmetric allylic substitution-isomerization to axially chiral enamides via hydrogen-bonding assisted central-to-axial chirality transfer. Chem. Sci. 2020, 11, 10119–10126. [Google Scholar] [CrossRef]
- Wu, Y.-X.; Liu, Q.; Zhang, Q.; Ye, Z.; He, Y. Asymmetric allylic substitution-isomerization for accessing axially chiral vinylindoles by intramolecular π…π stacking interactions. Cell Rep. Phys. Sci. 2022, 3, 101005. [Google Scholar] [CrossRef]
- Hirai, M.; Terada, S.; Yoshida, H.; Ebine, K.; Hirata, T.; Kitagawa, O. Catalytic enantioselective synthesis of N–C axially chiral mebroqualone and its derivatives through reductive asymmetric desymmetrization. Org. Lett. 2016, 18, 5700–5703. [Google Scholar] [CrossRef]
- Wang, Y.-B.; Zheng, S.-C.; Hu, Y.-M.; Tan, B. Brønsted acid-catalysed enantioselective construction of axially chiral arylquinazolinones. Nat. Commun. 2017, 8, 15489–15497. [Google Scholar] [CrossRef] [Green Version]
- Diener, M.E.; Metrano, A.J.; Kusano, S.; Miller, S.J. Enantioselective synthesis of 3-arylquinazolin-4(3H)-ones via peptide-catalyzed atroposelective bromination. J. Am. Chem. Soc. 2015, 137, 12369–12377. [Google Scholar] [CrossRef] [Green Version]
- Crawford, J.M.; Stone, E.A.; Metrano, A.J.; Miller, S.J.; Sigman, M.S. Parameterization and analysis of peptide-based catalysts for the atroposelective bromination of 3-arylquinazolin-4(3H)-ones. J. Am. Chem. Soc. 2018, 140, 868–871. [Google Scholar] [CrossRef] [Green Version]
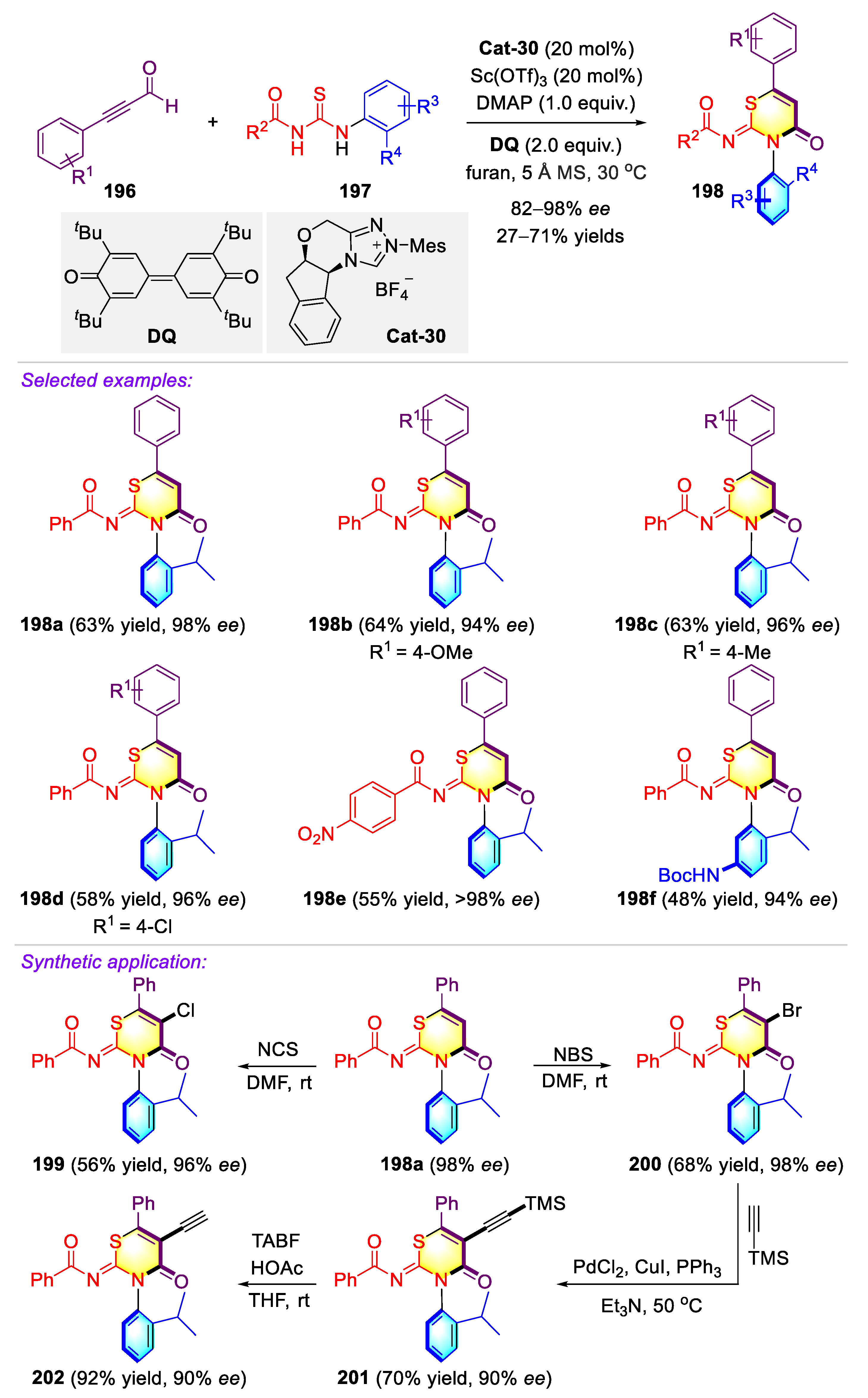
- Li, T.; Mou, C.; Qi, P.; Peng, X.; Jiang, S.; Hao, G.; Xue, W.; Yang, S.; Hao, L.; Chi, Y.R.; et al. N-Heterocyclic carbene-catalyzed atroposelective annulation for access to thiazine derivatives with C-N axial chirality. Angew. Chem. Int. Ed. 2021, 60, 9362–9367. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, J.; Hsung, R.; Figueroa, R.; Johnson, W. Stereochemical control of both C–C and C–N axial chirality in the synthesis of chiral N,O-biaryls. Org. Lett. 2007, 9, 3969–3972. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, S.; Blîmel, M.; Philipps, A.R.; Wang, A.; Puttreddy, R.; Rissanen, K.; Enders, D. Asymmetric synthesis of spirobenzazepinones with atroposelectivity and spiro-1,2-diazepinones by NHC-catalyzed [3+4] annulation reactions. Angew. Chem. Int. Ed. 2016, 55, 11110–11114. [Google Scholar] [CrossRef] [PubMed]




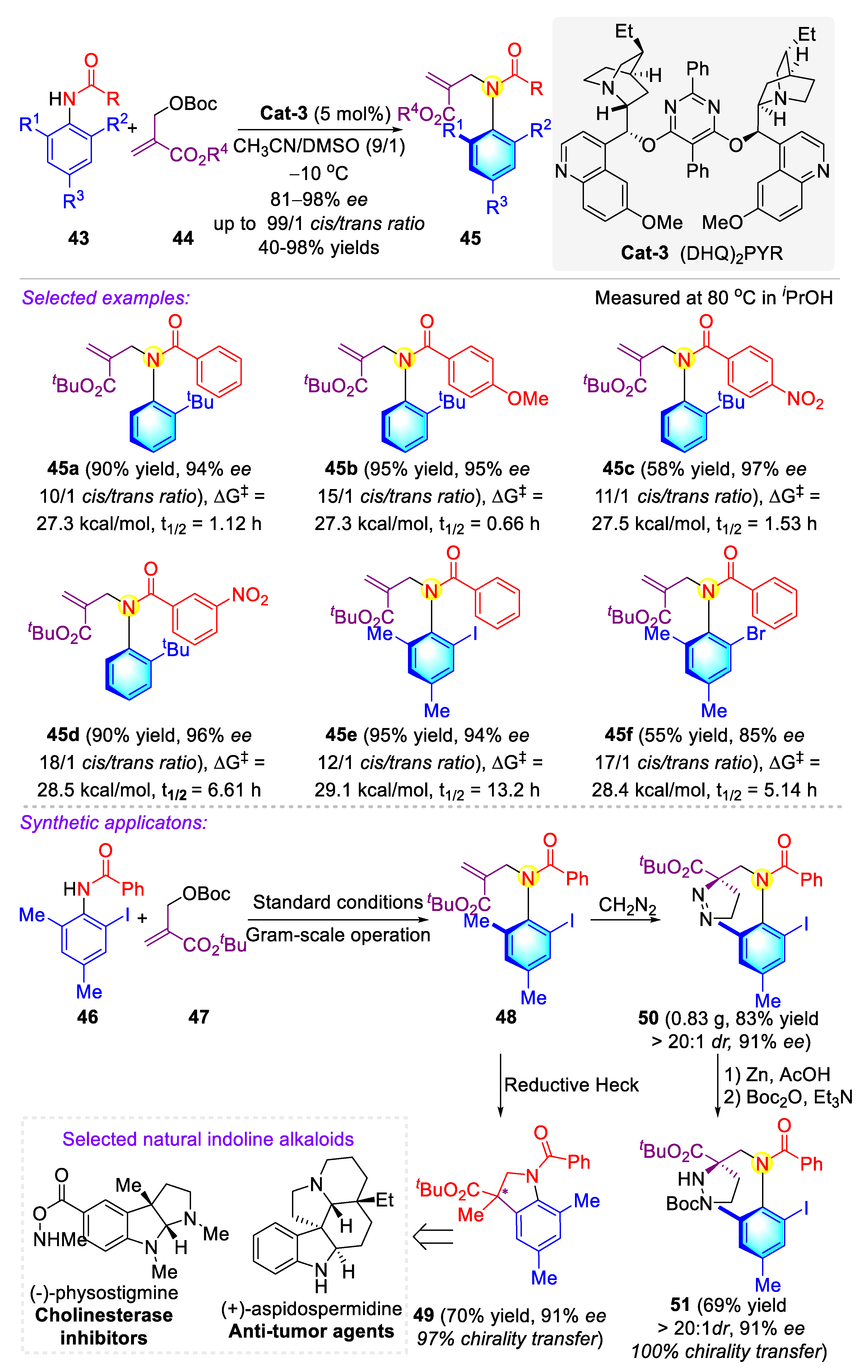
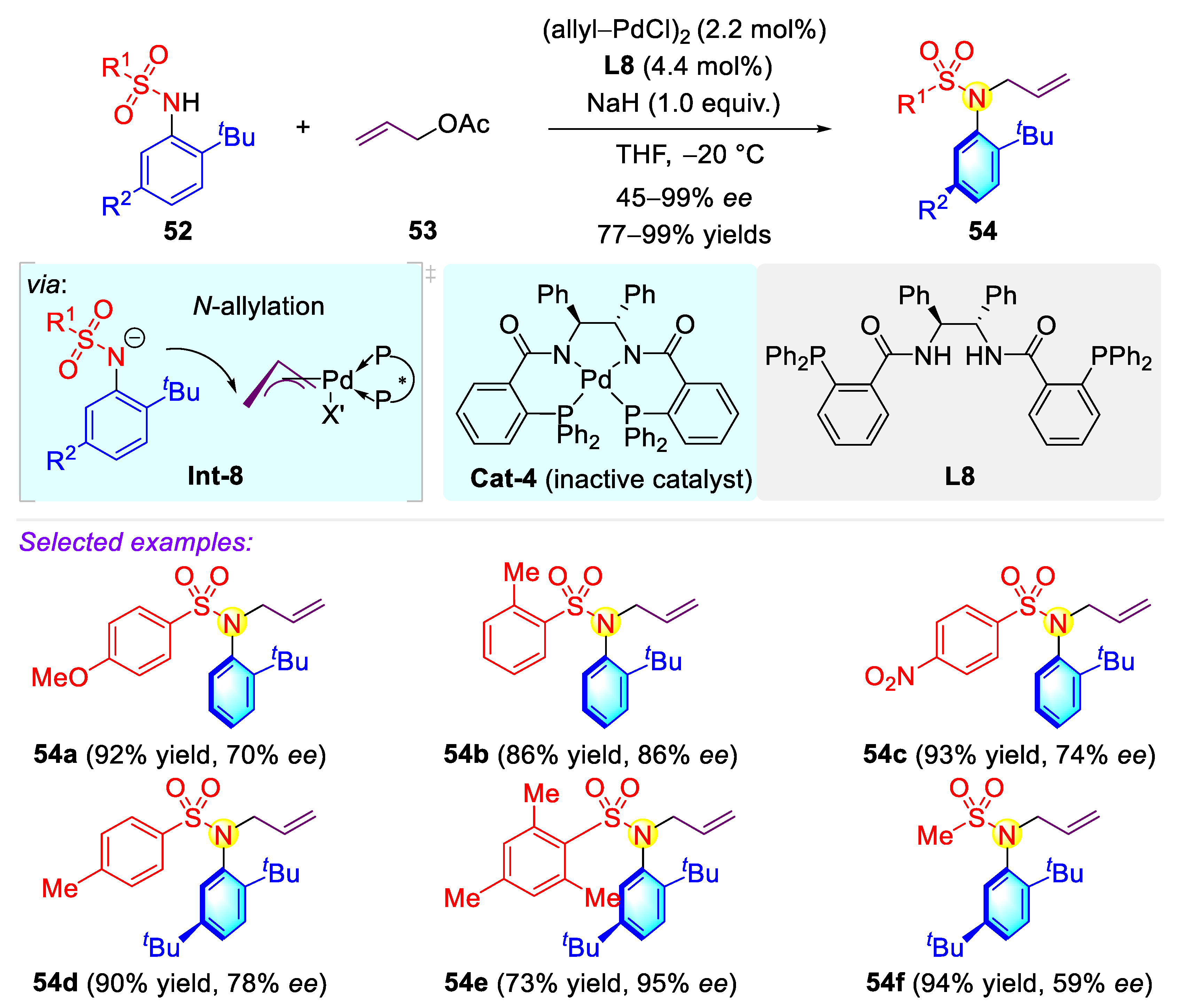
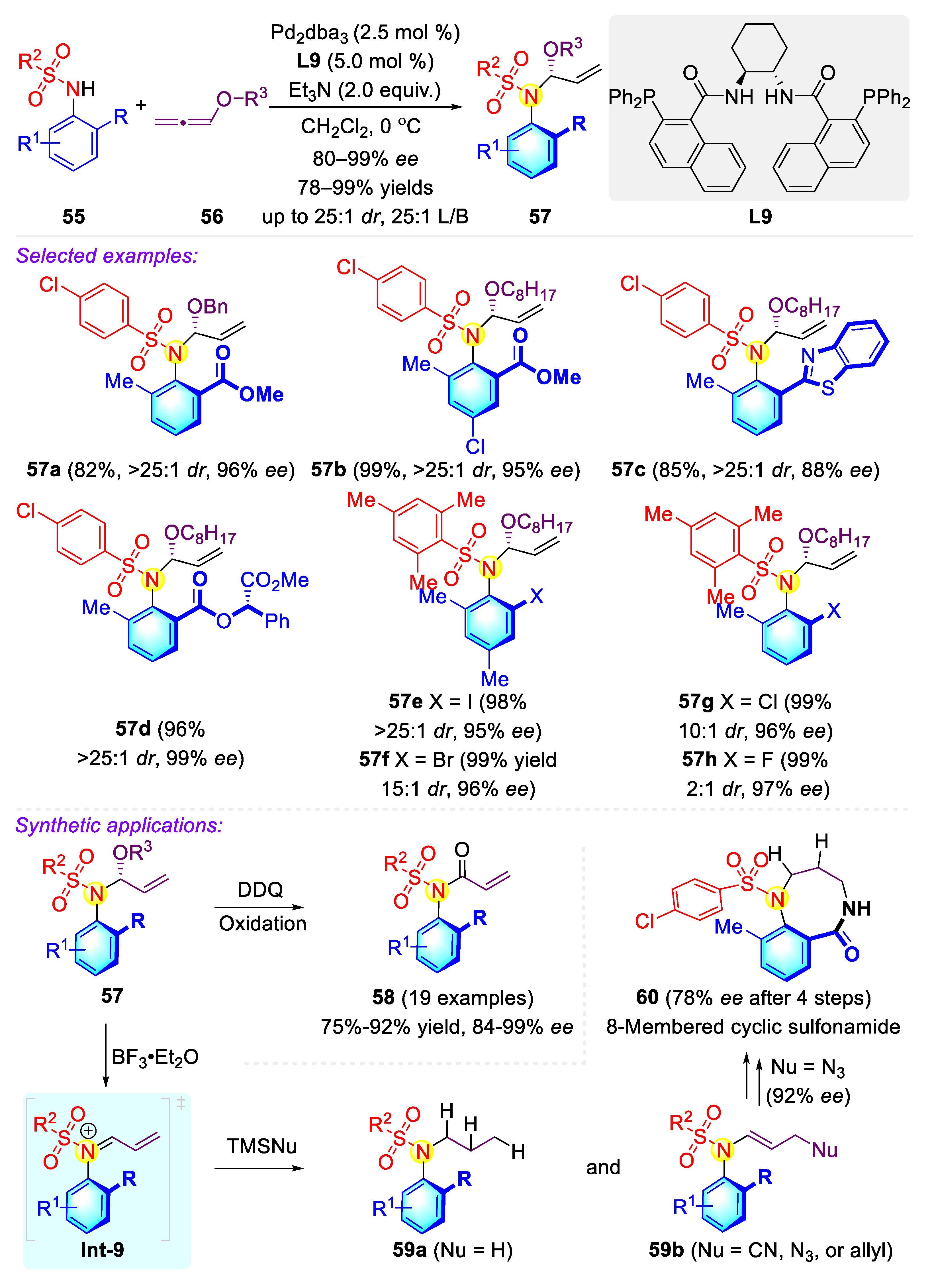
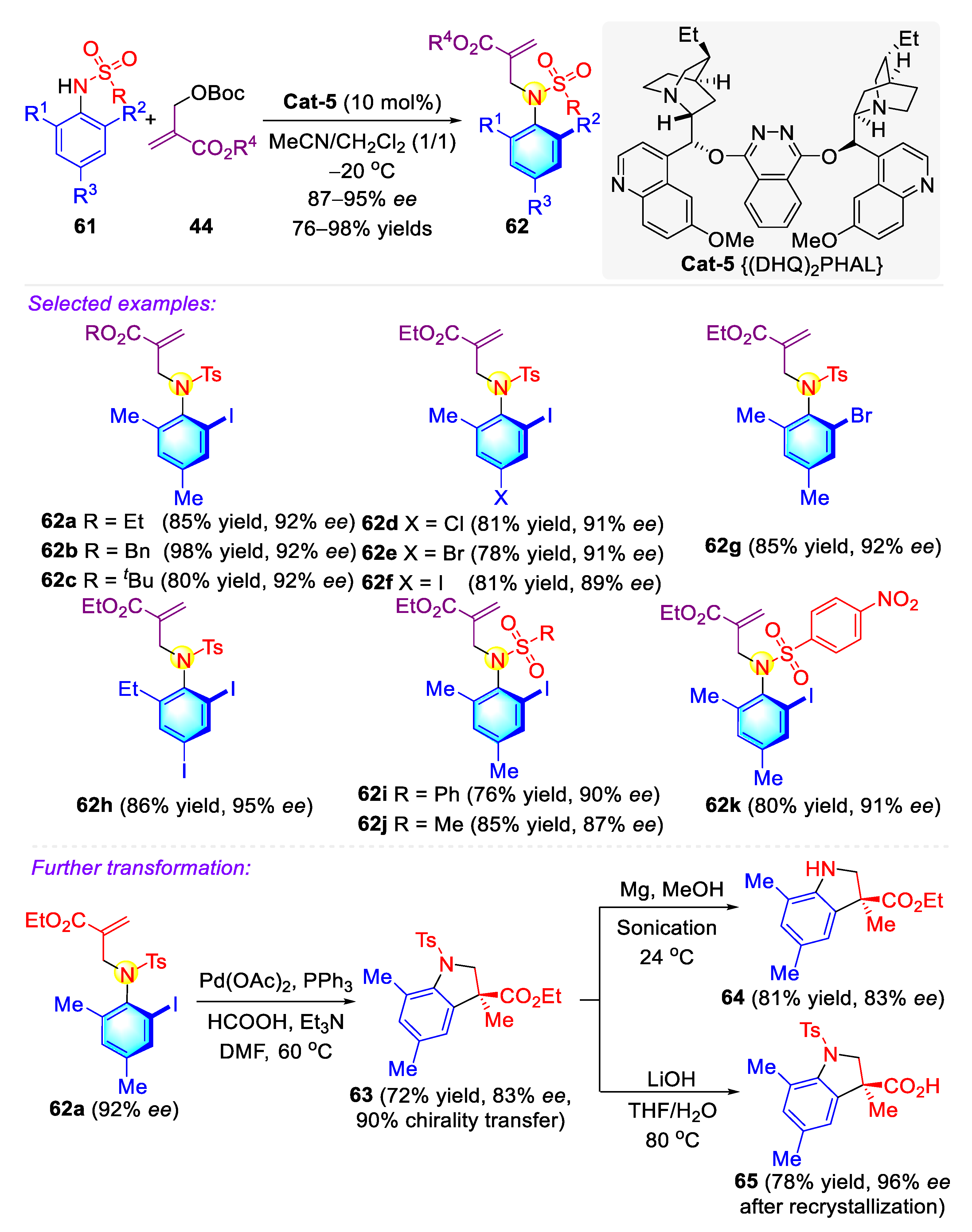

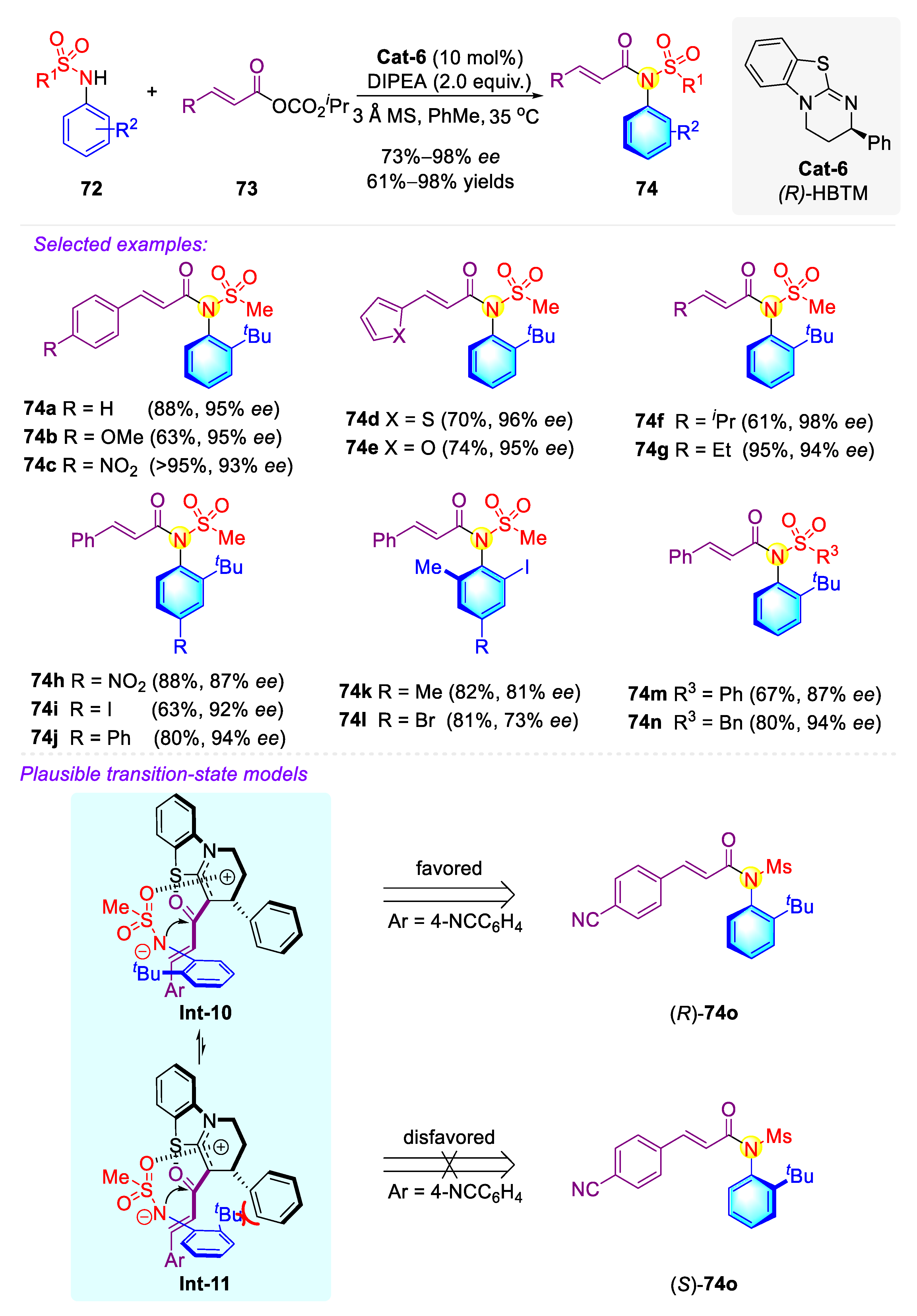

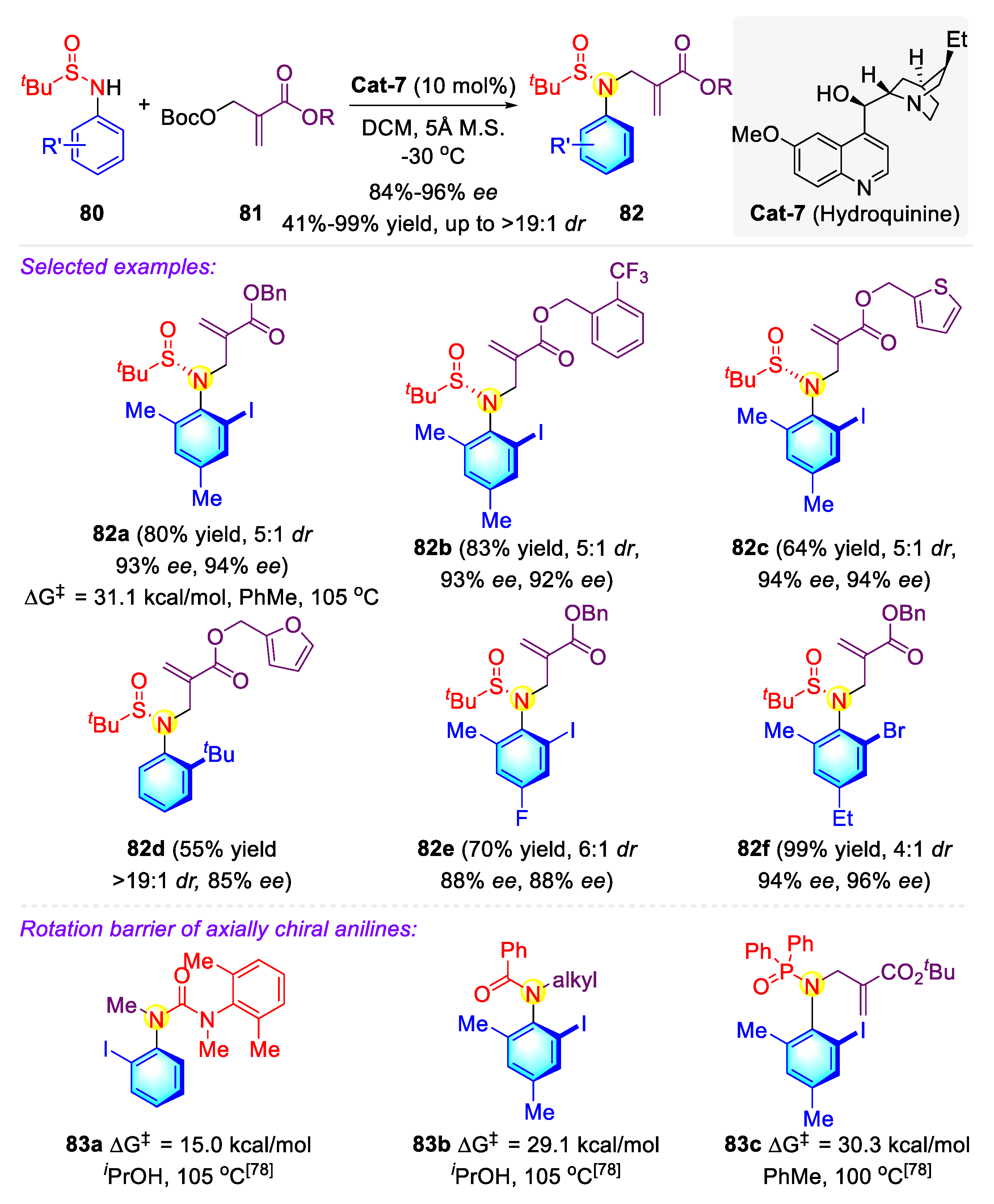



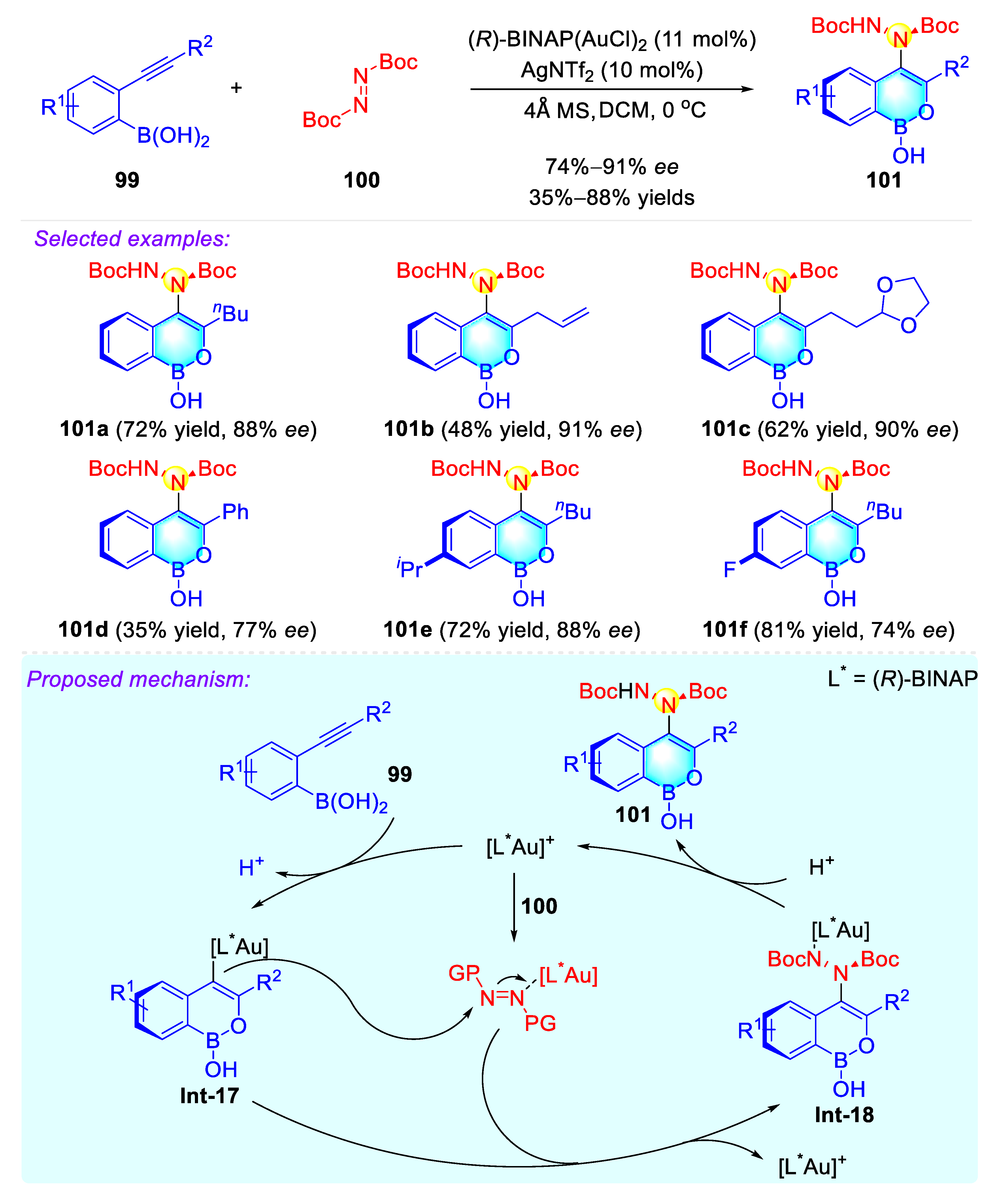
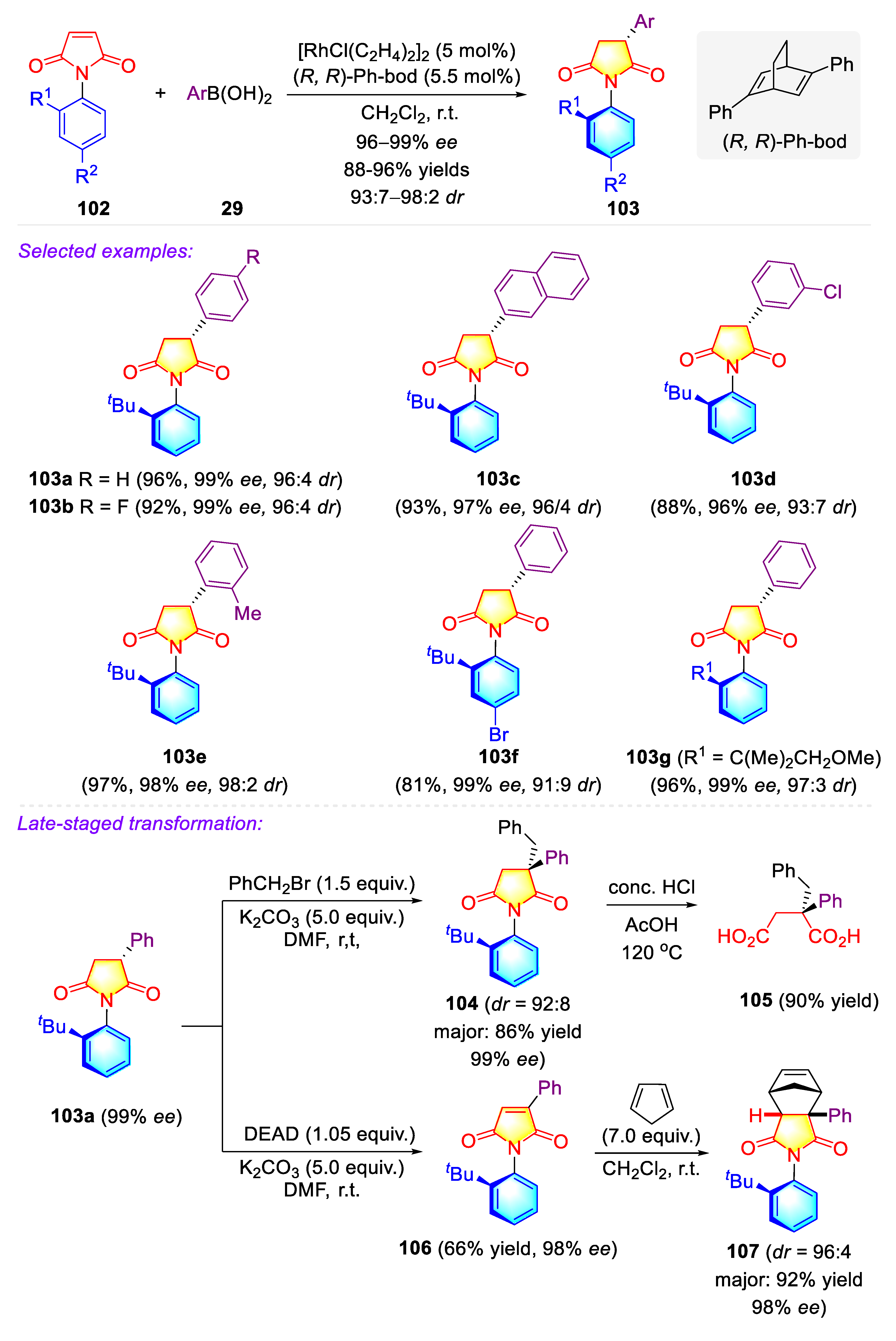

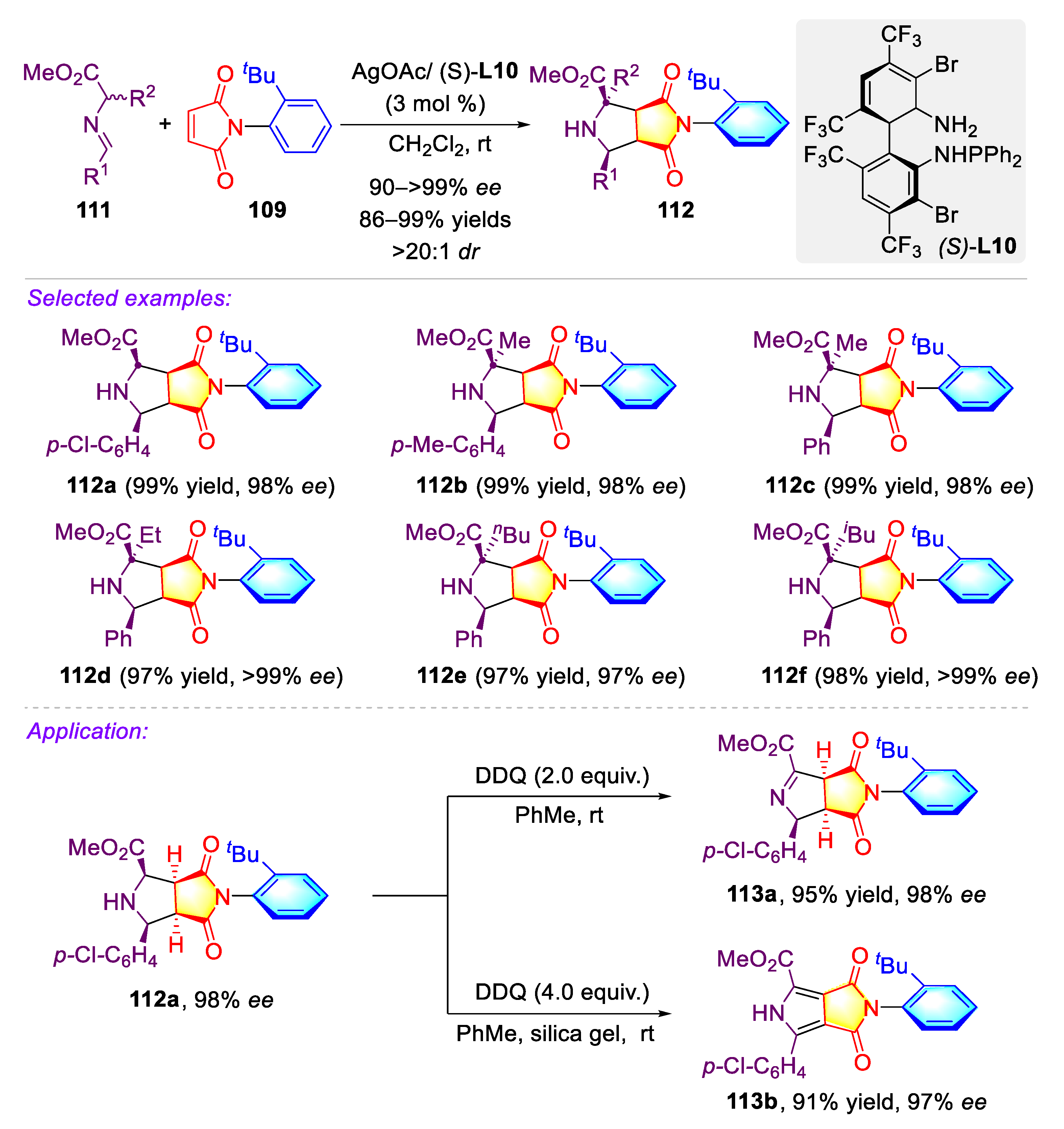
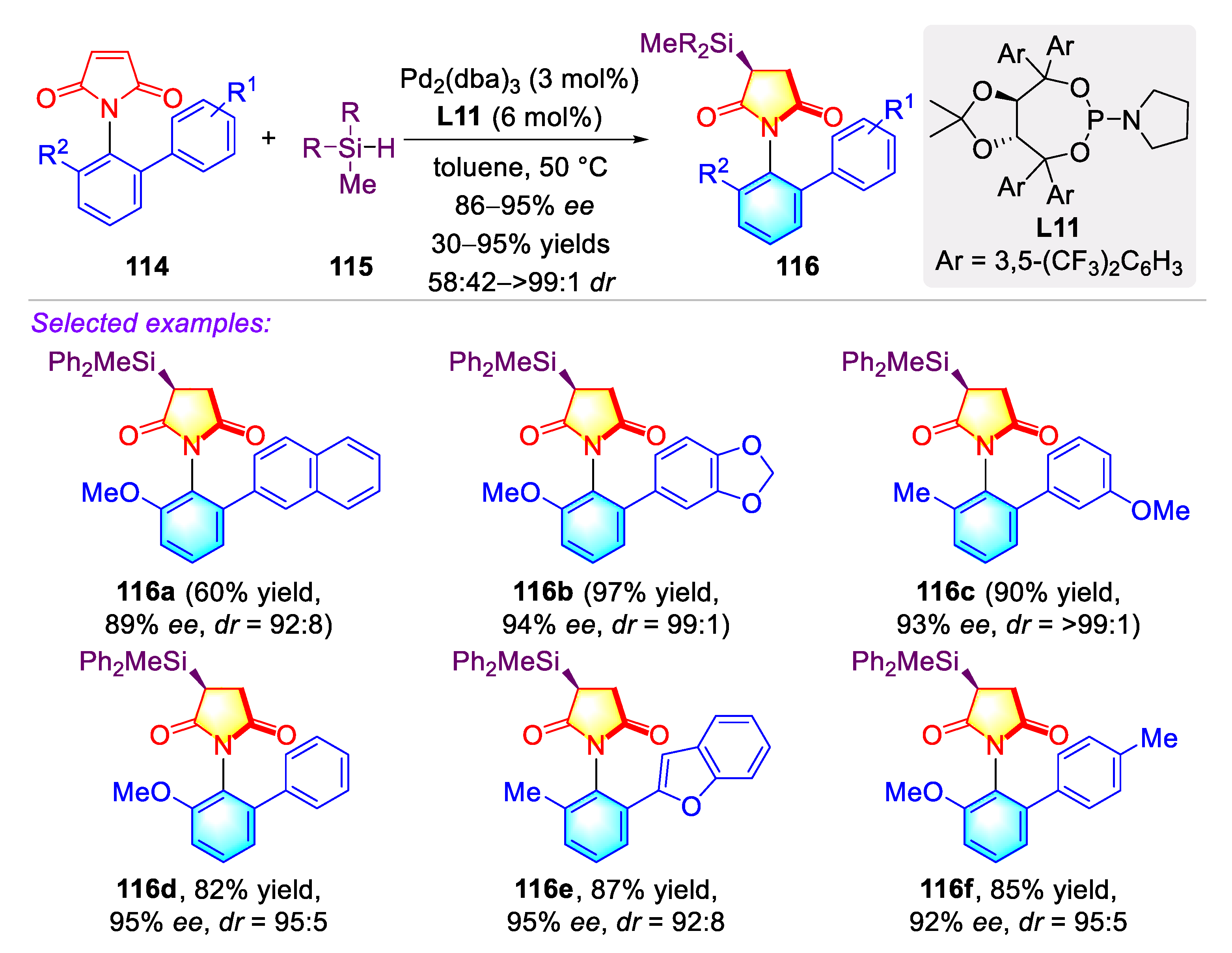

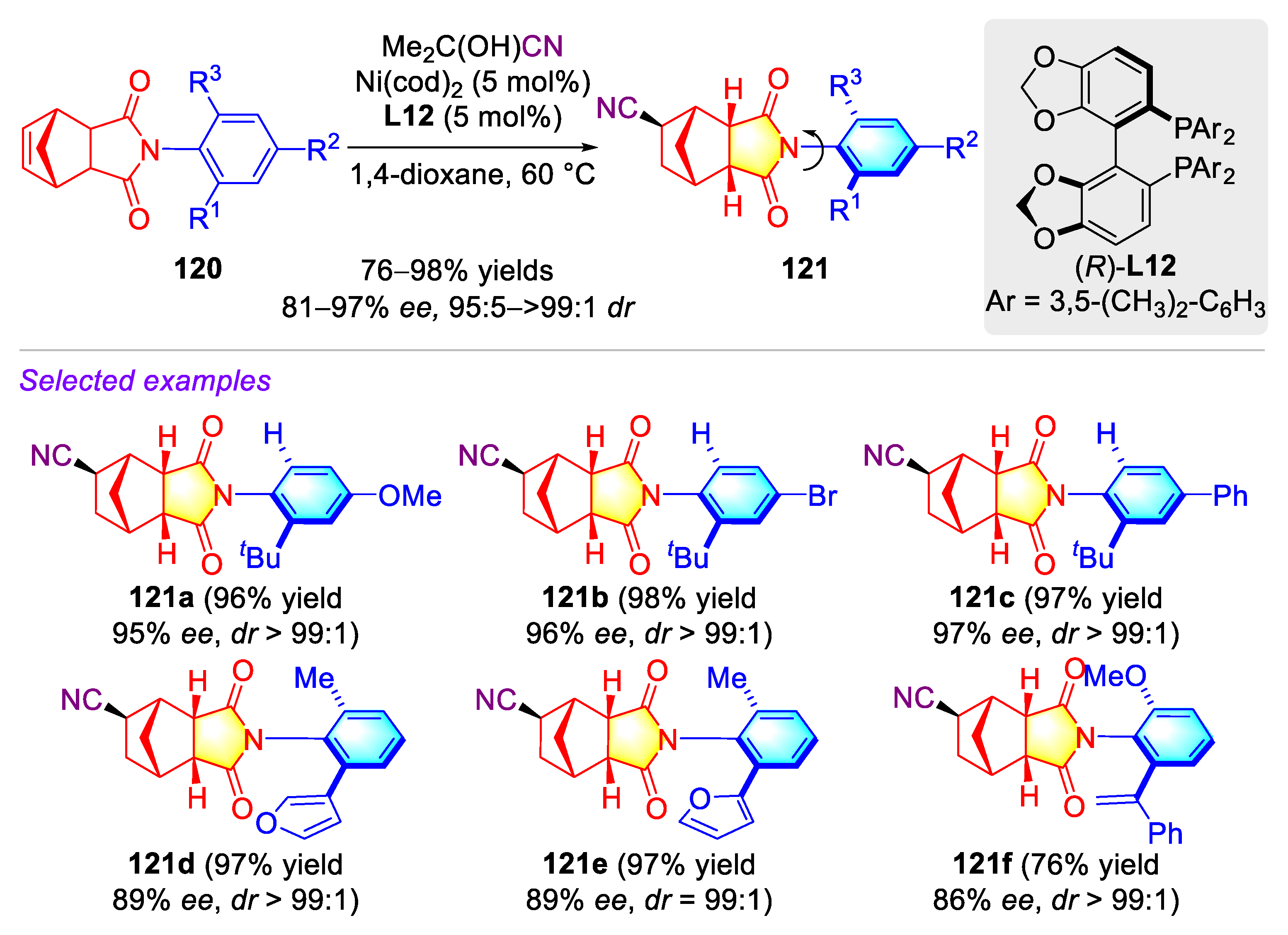
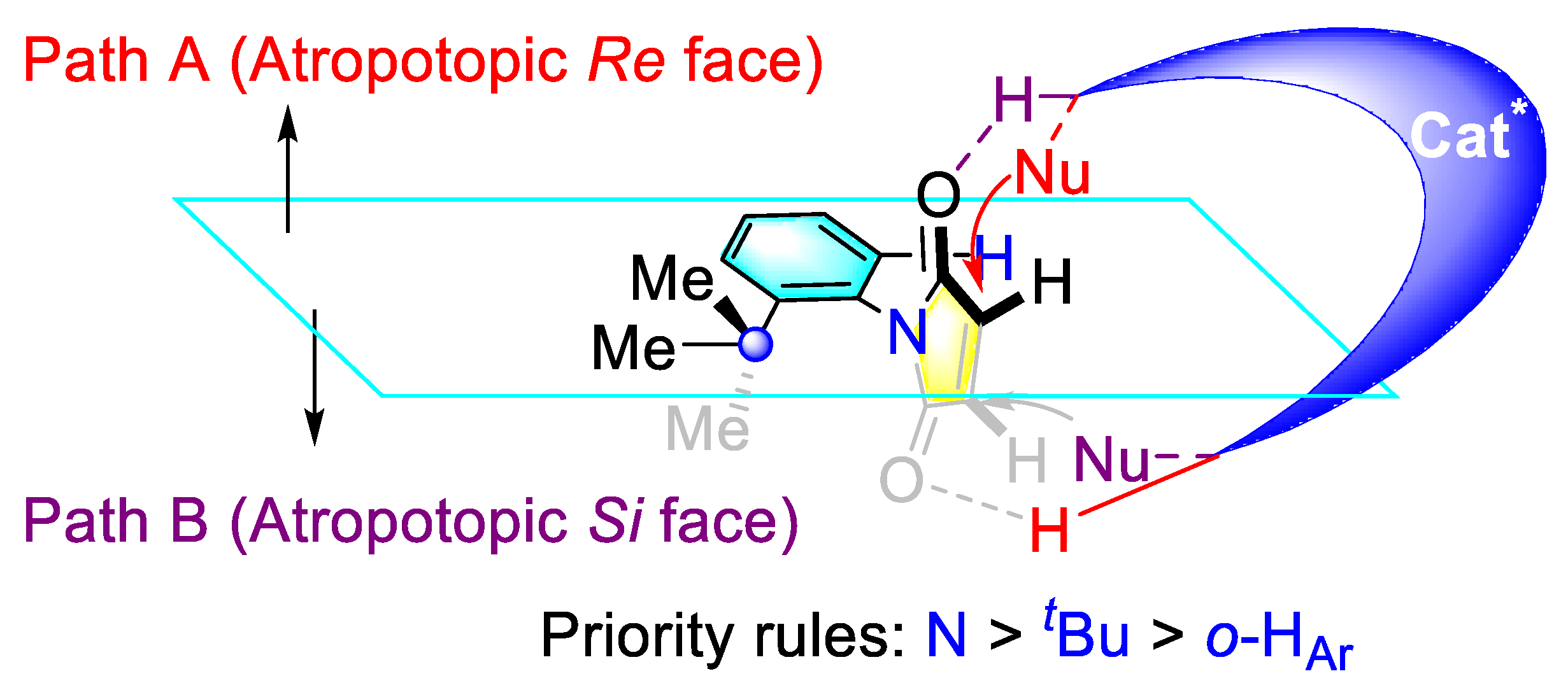


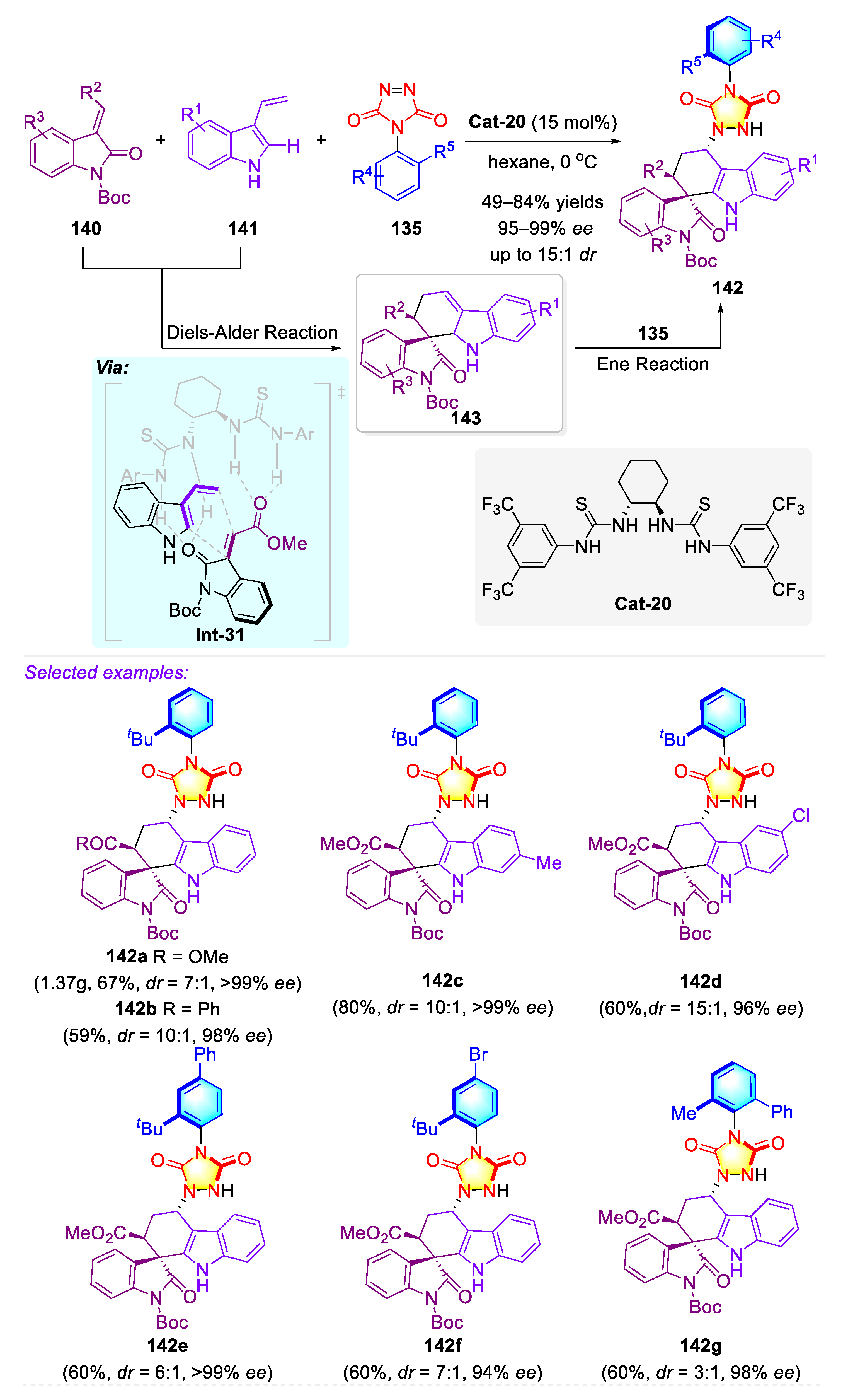
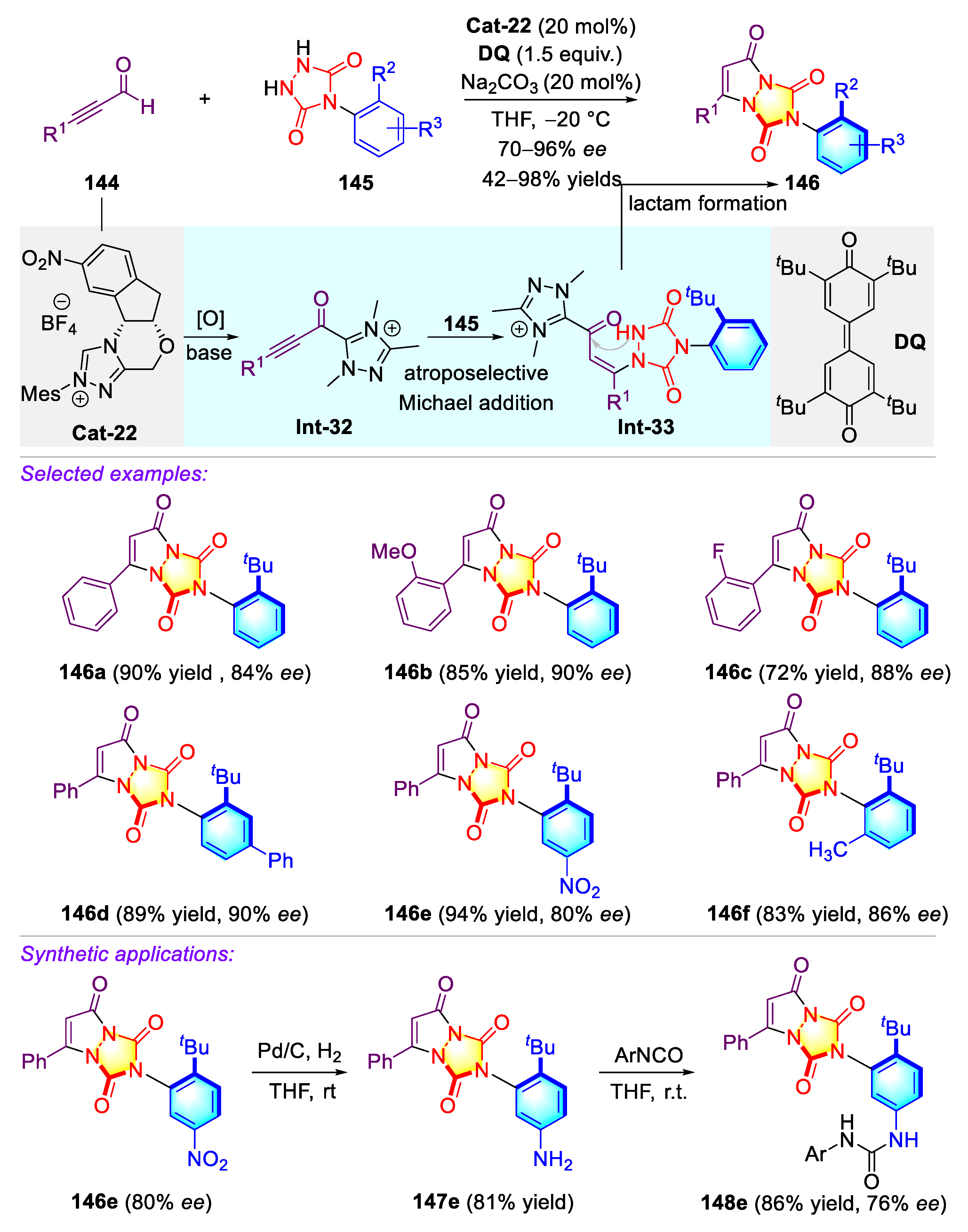

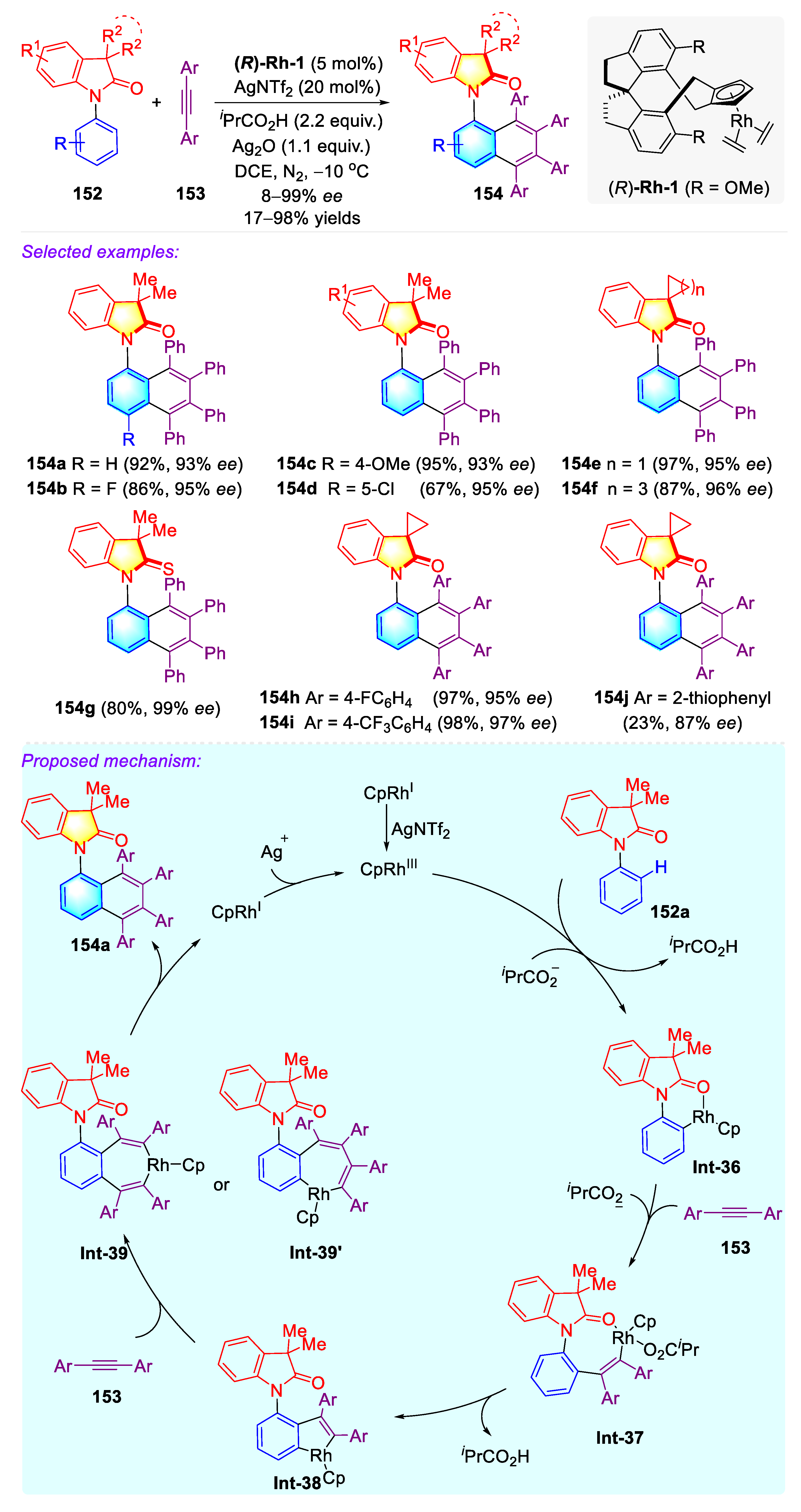

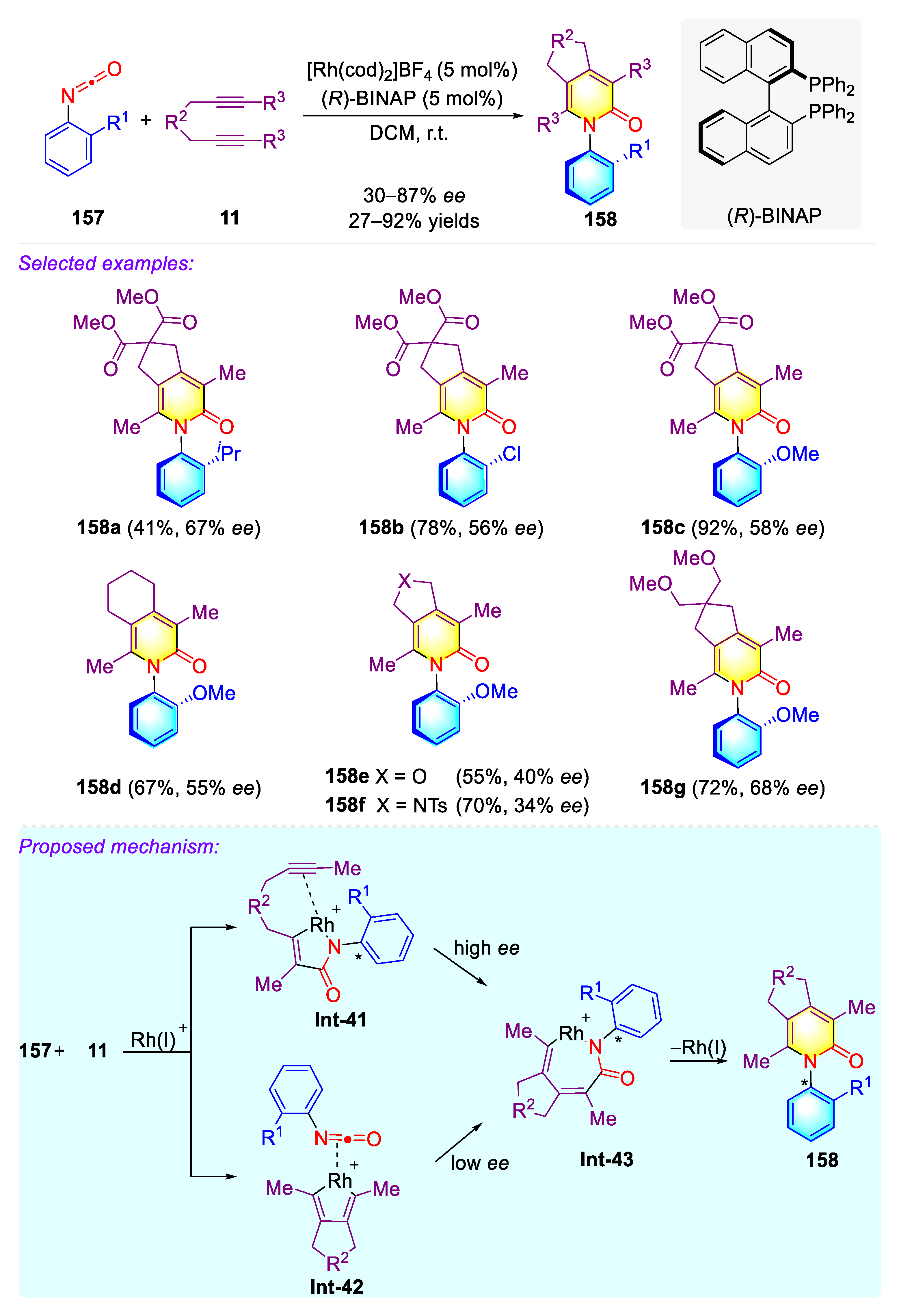
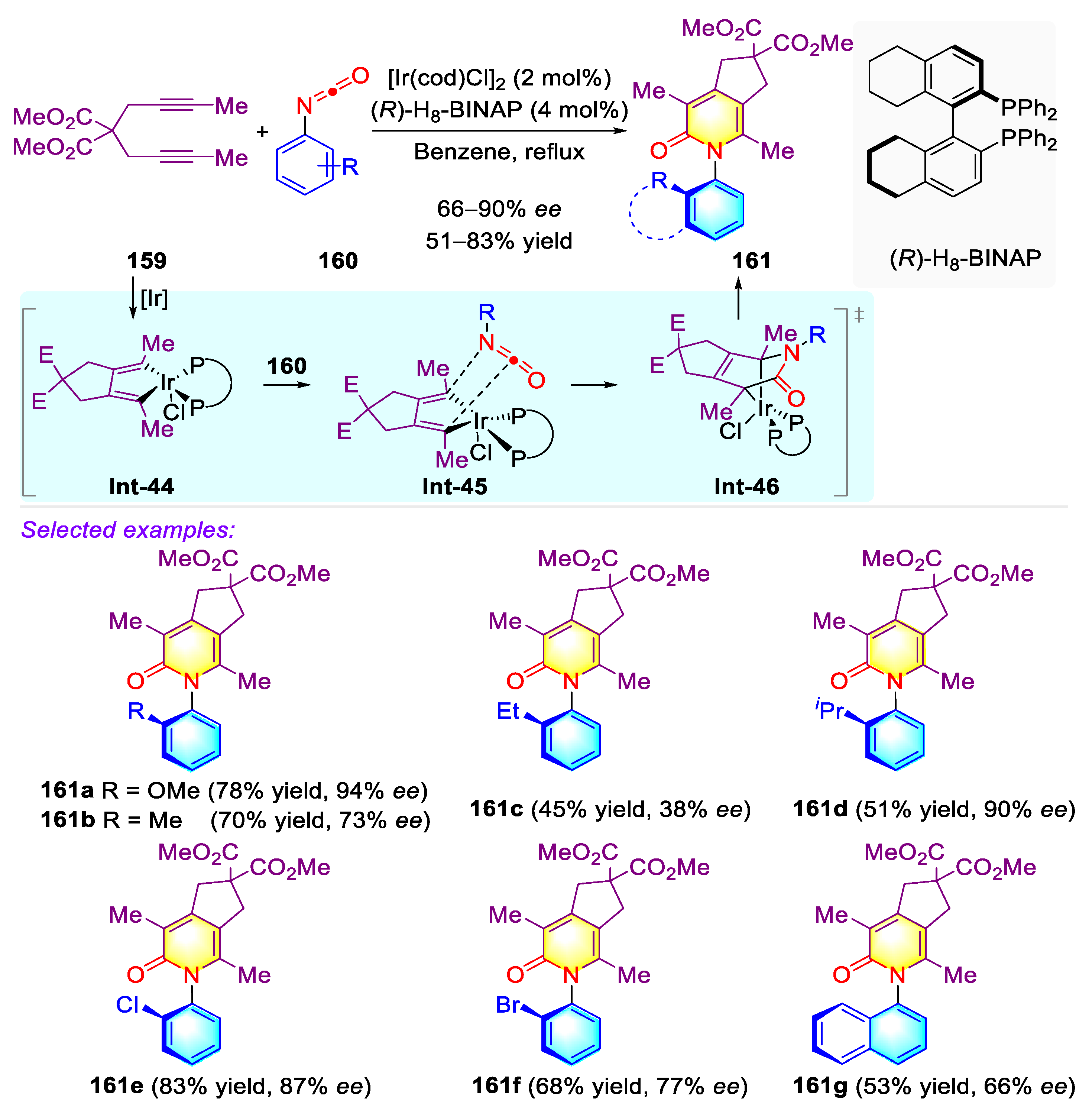

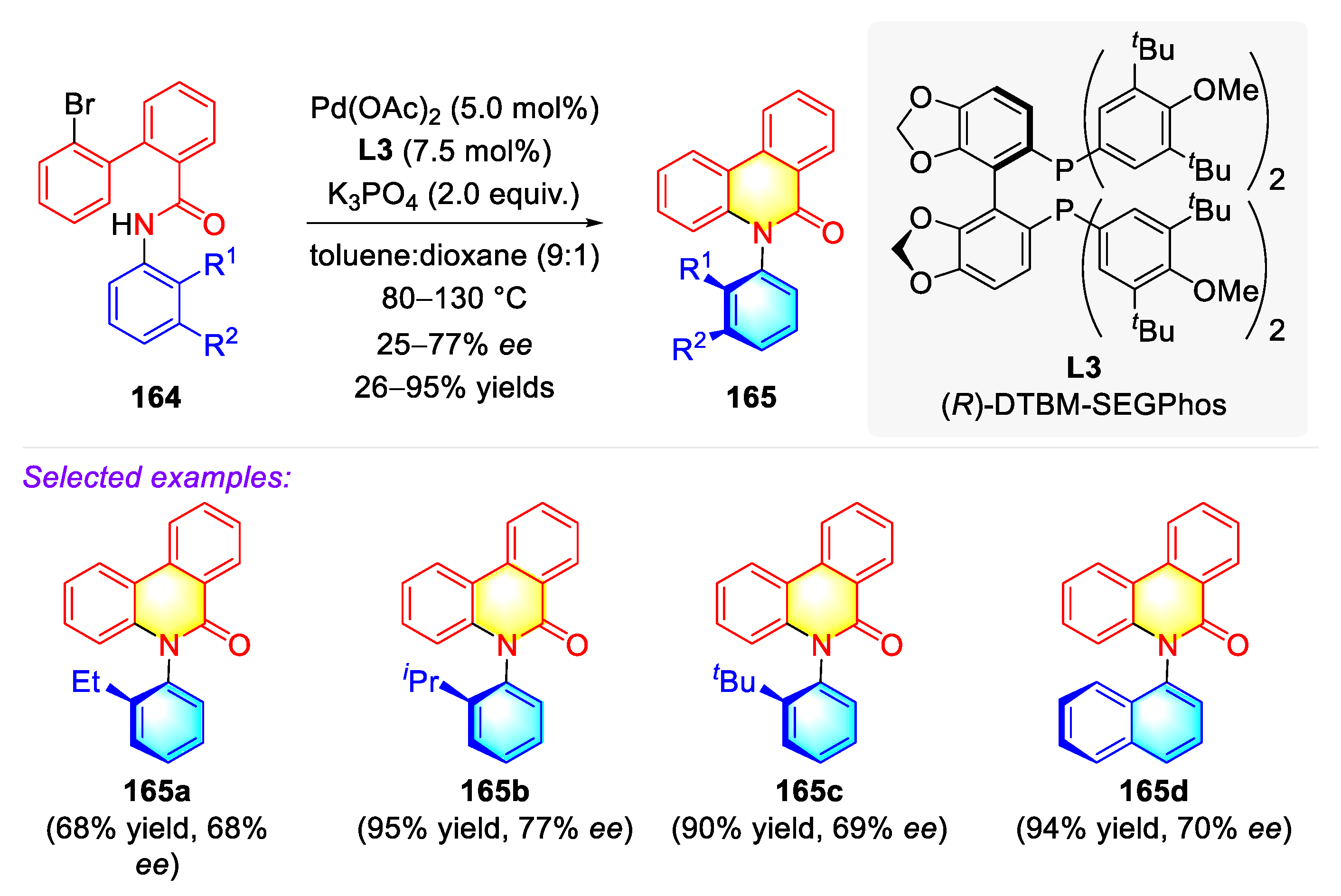
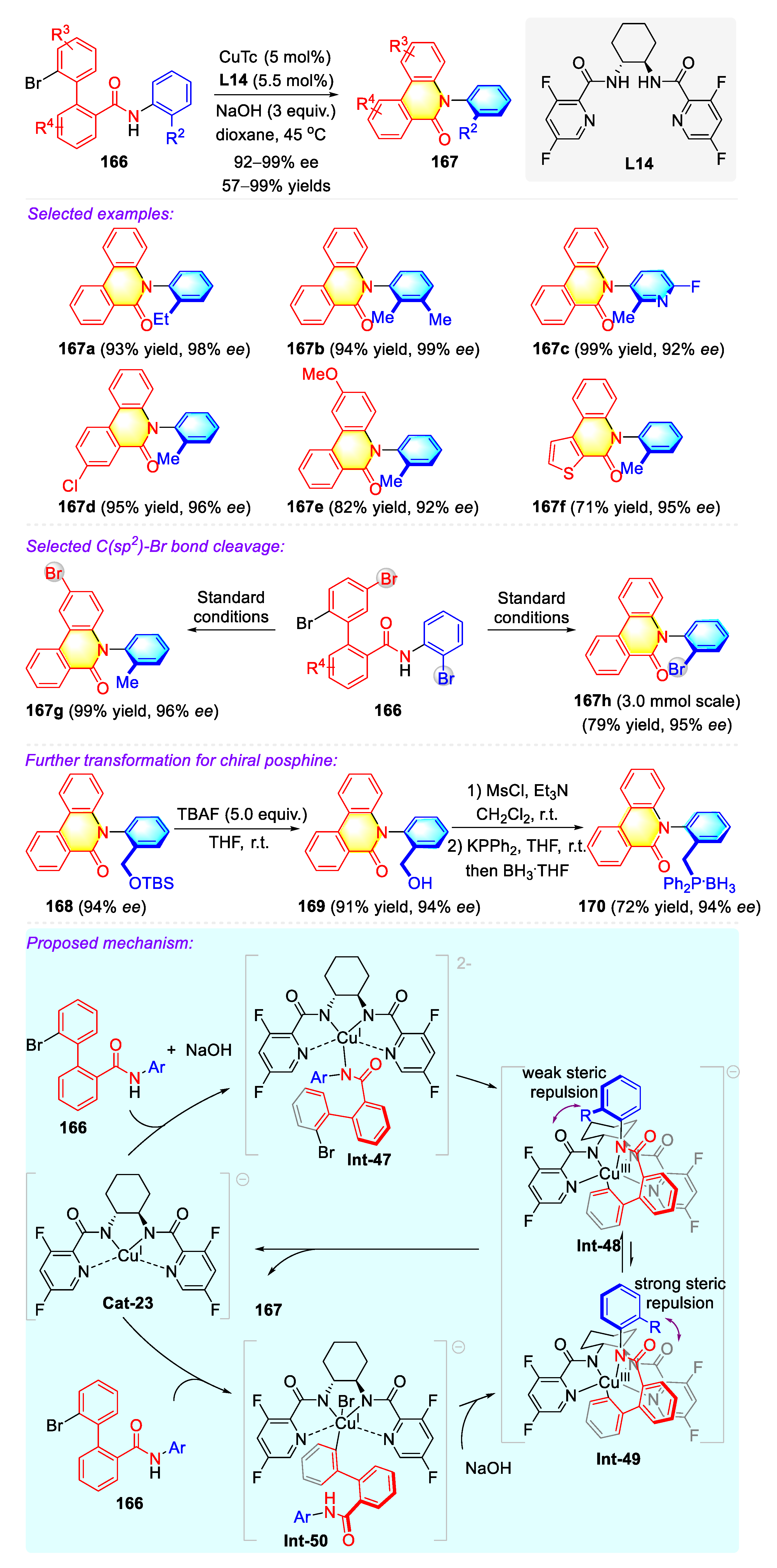
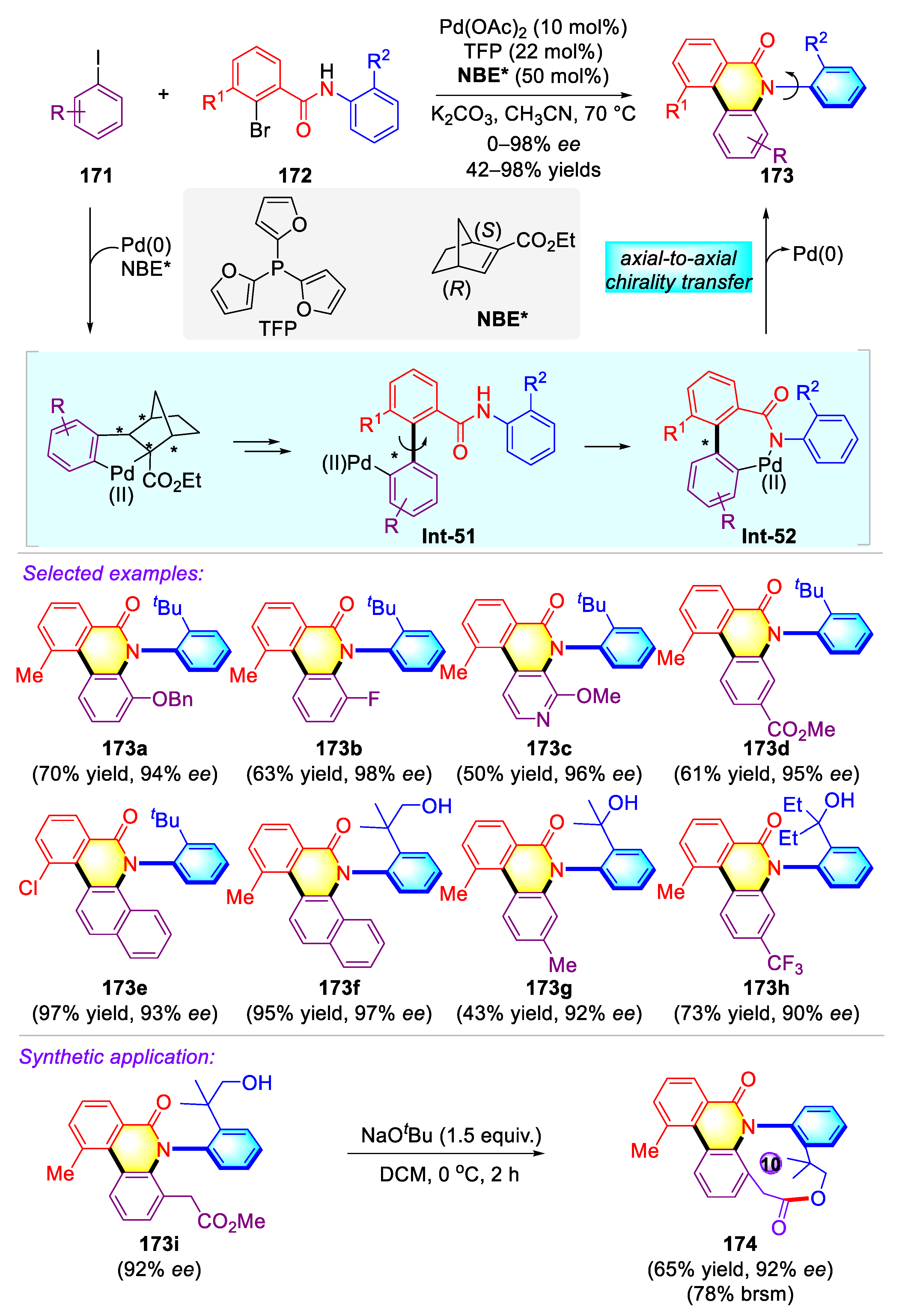
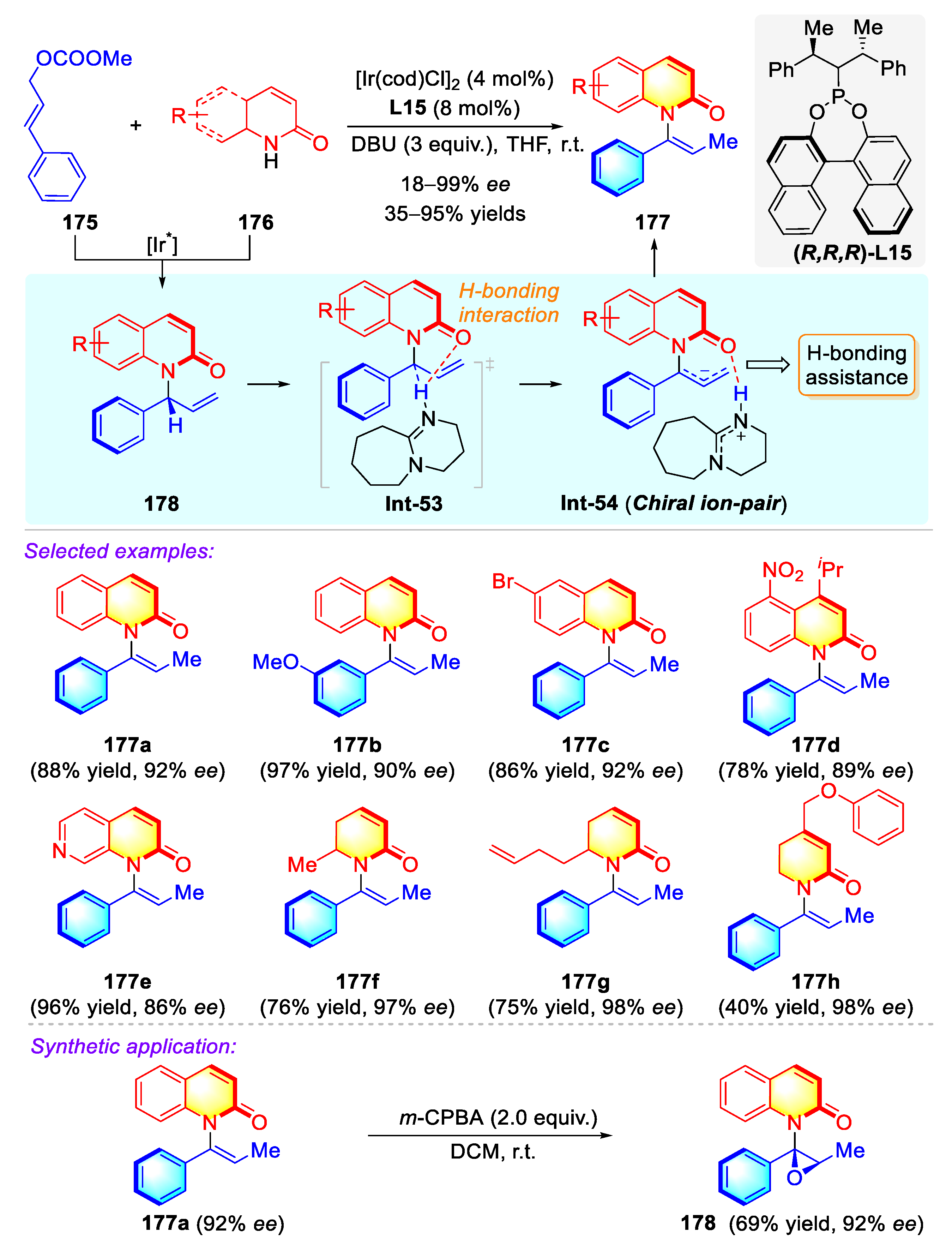
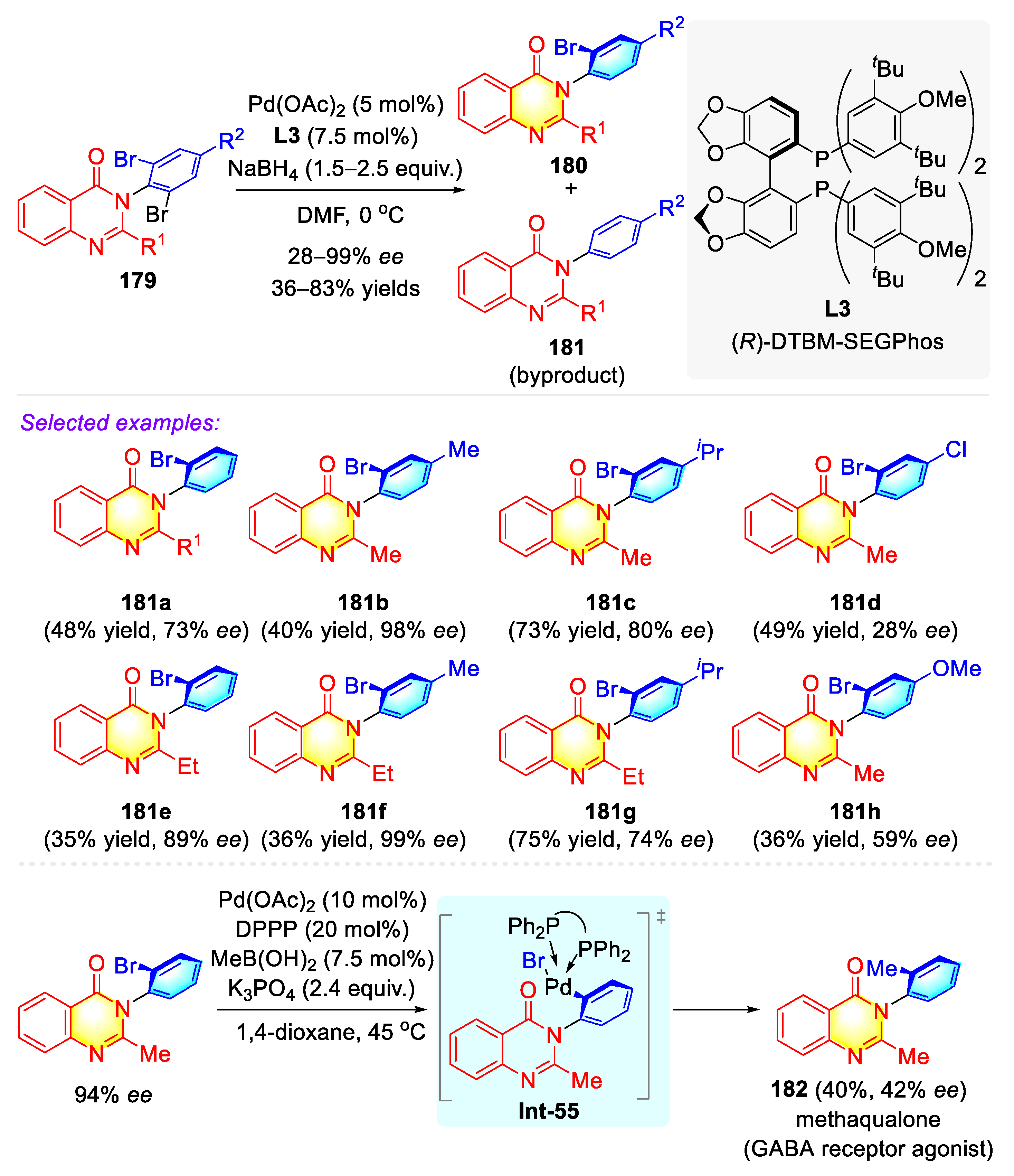
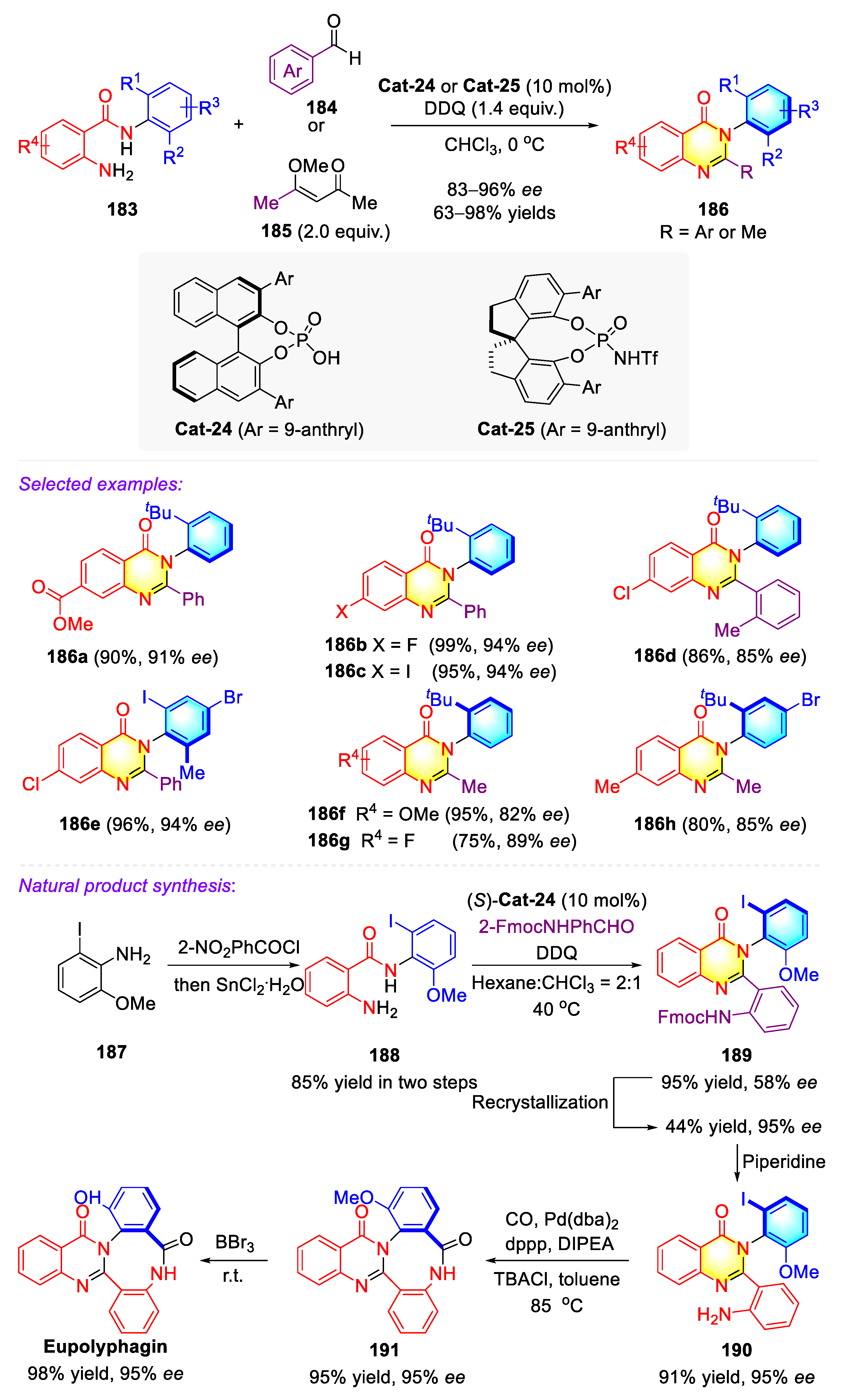
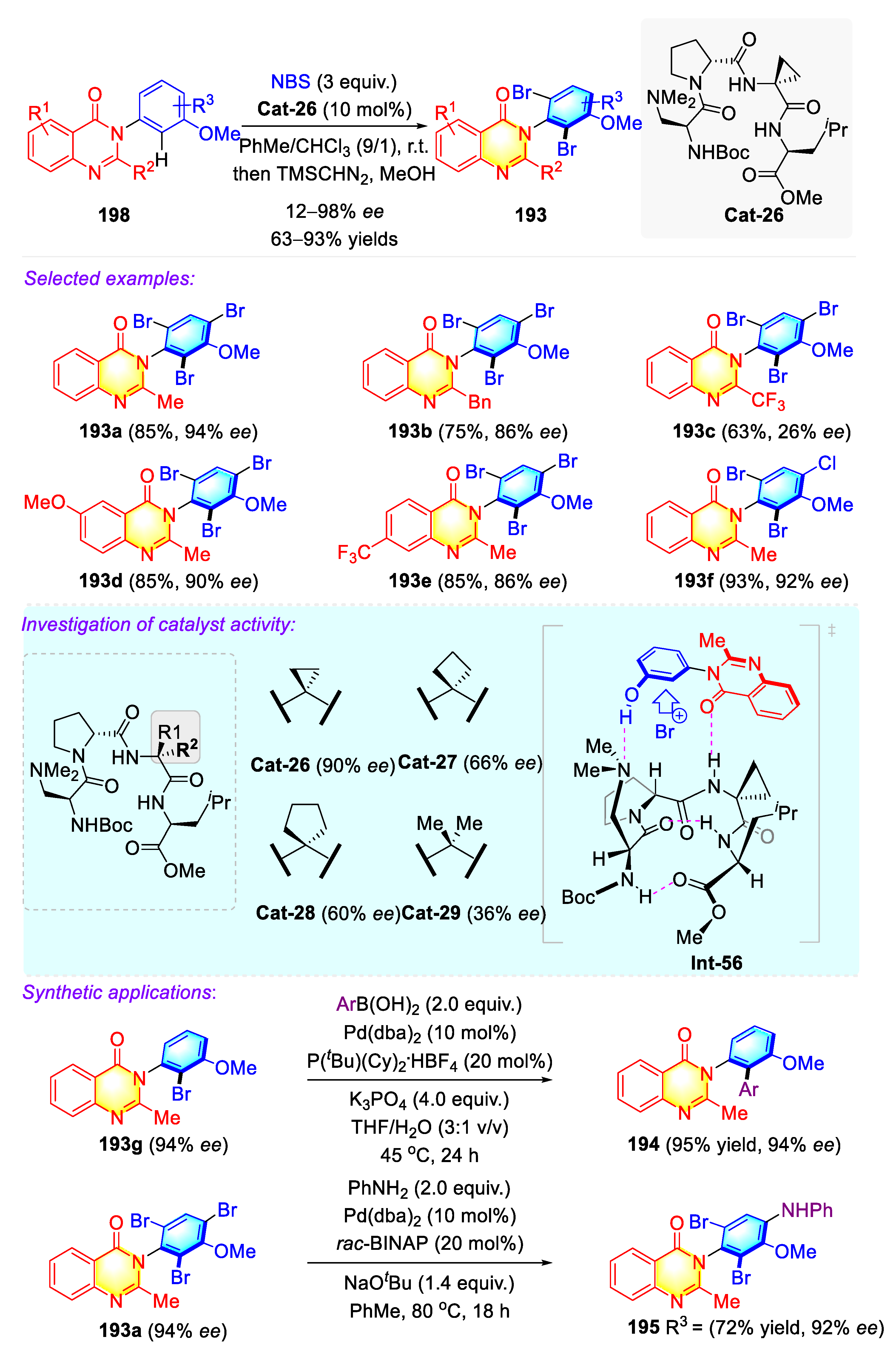


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, X.; Chen, B.; Yao, Y.-P.; Zhou, H.-J.; Wang, X.; Wang, N.-Z.; Chen, F.-E. Construction of Non-Biaryl Atropisomeric Amide Scaffolds Bearing a C–N Axis via Enantioselective Catalysis. Molecules 2022, 27, 6583. https://doi.org/10.3390/molecules27196583
Xiao X, Chen B, Yao Y-P, Zhou H-J, Wang X, Wang N-Z, Chen F-E. Construction of Non-Biaryl Atropisomeric Amide Scaffolds Bearing a C–N Axis via Enantioselective Catalysis. Molecules. 2022; 27(19):6583. https://doi.org/10.3390/molecules27196583
Chicago/Turabian StyleXiao, Xiao, Biao Chen, Yi-Ping Yao, Hai-Jie Zhou, Xu Wang, Neng-Zhong Wang, and Fen-Er Chen. 2022. "Construction of Non-Biaryl Atropisomeric Amide Scaffolds Bearing a C–N Axis via Enantioselective Catalysis" Molecules 27, no. 19: 6583. https://doi.org/10.3390/molecules27196583