

Design and Synthesis of Coumarin Derivatives as Cytotoxic Agents through PI3K/AKT Signaling Pathway Inhibition in HL60 and HepG2 Cancer Cells

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
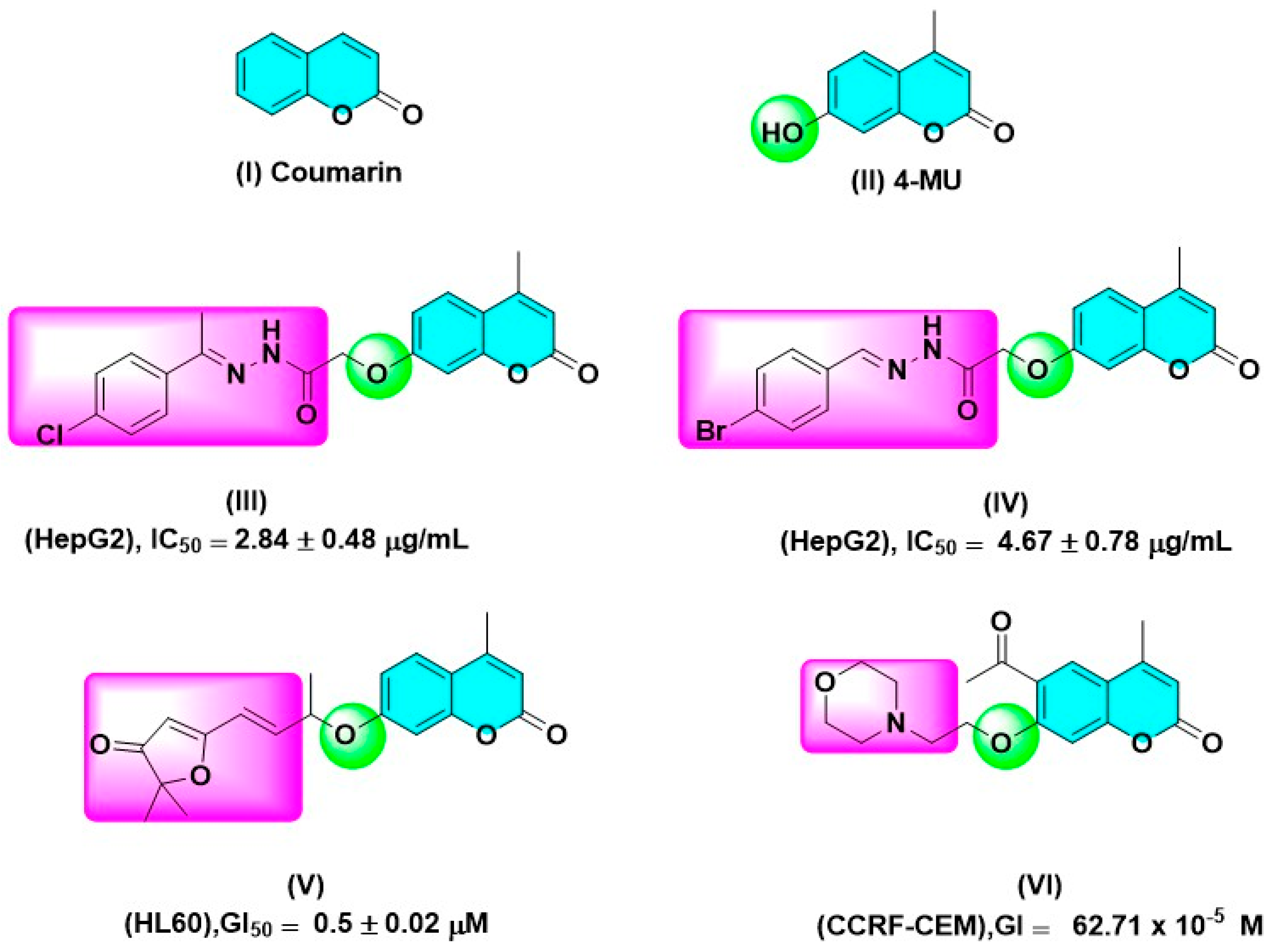
2. Results and Discussion
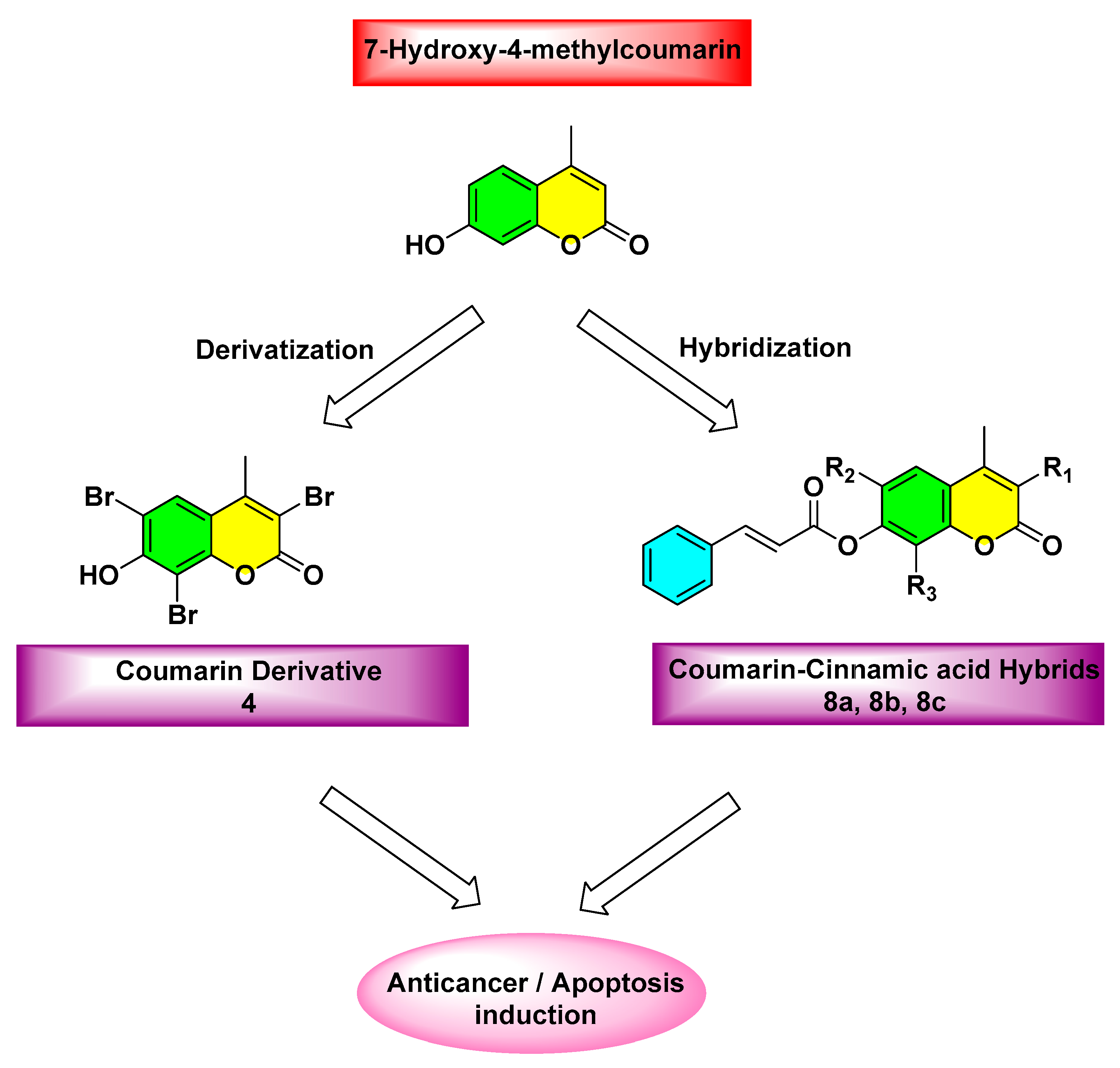
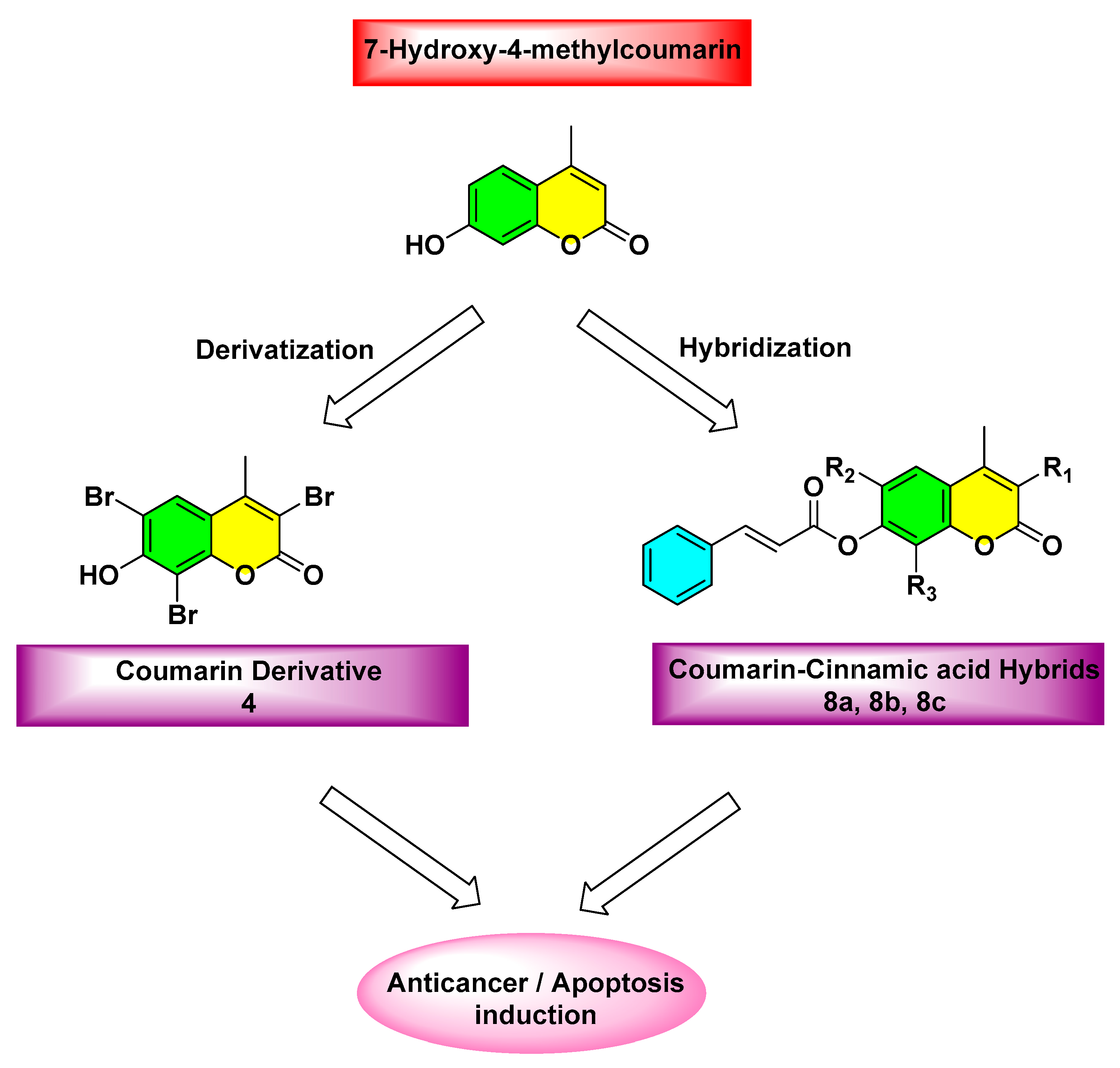
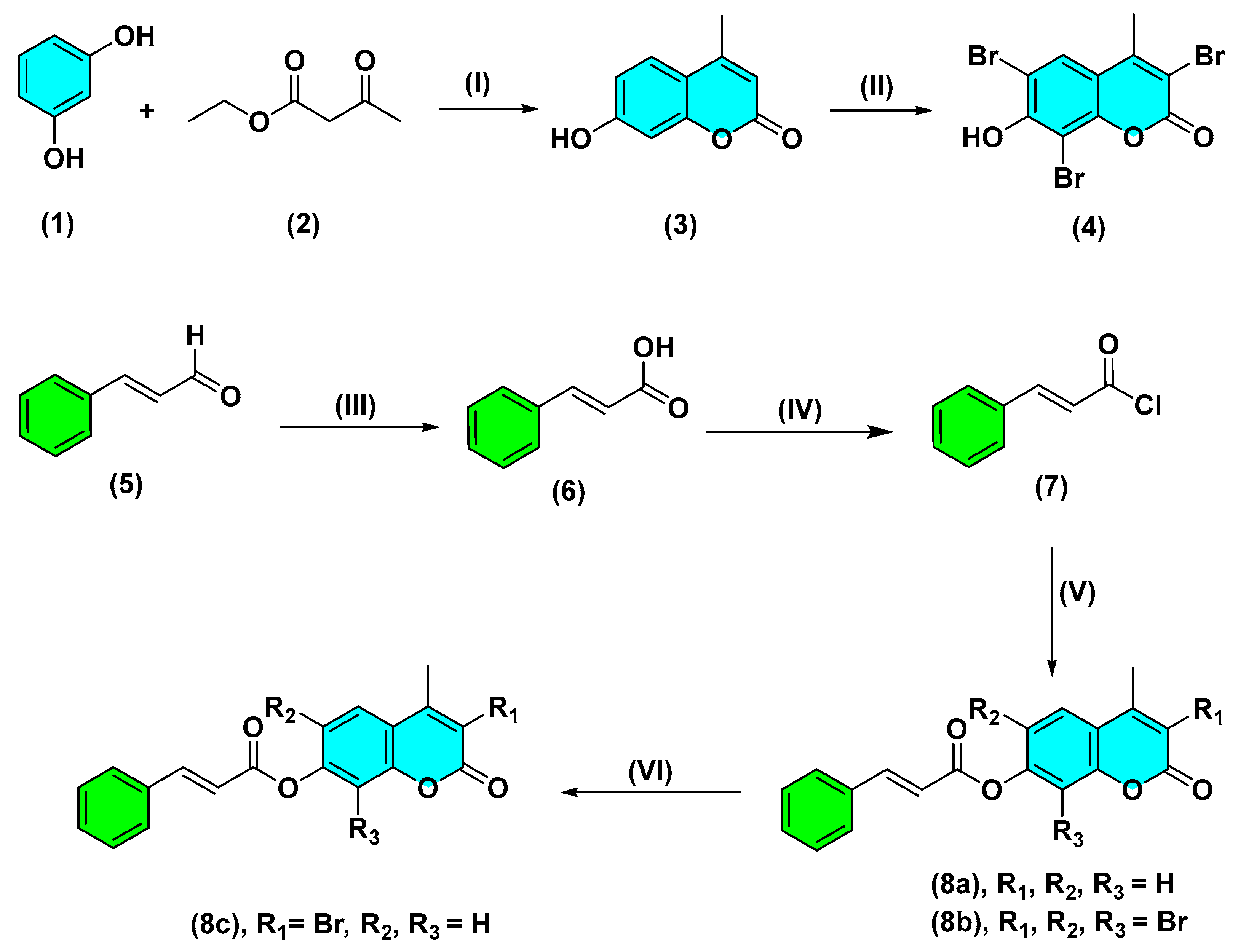
2.1. Chemistry
2.2. In Vitro Biological Evaluation
2.2.1. Cytotoxic Screening against a Panel of Cancer Cell Lines
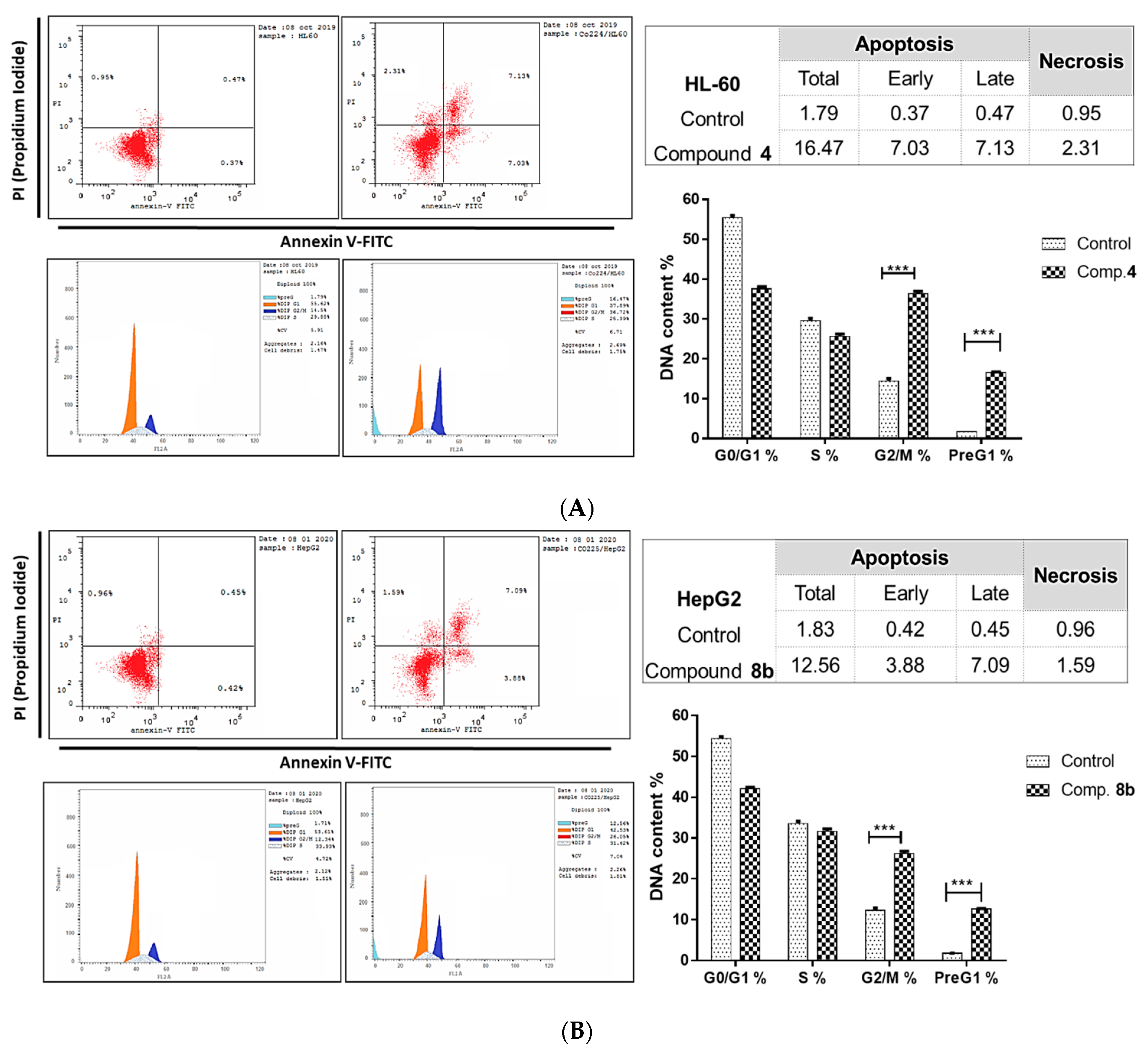
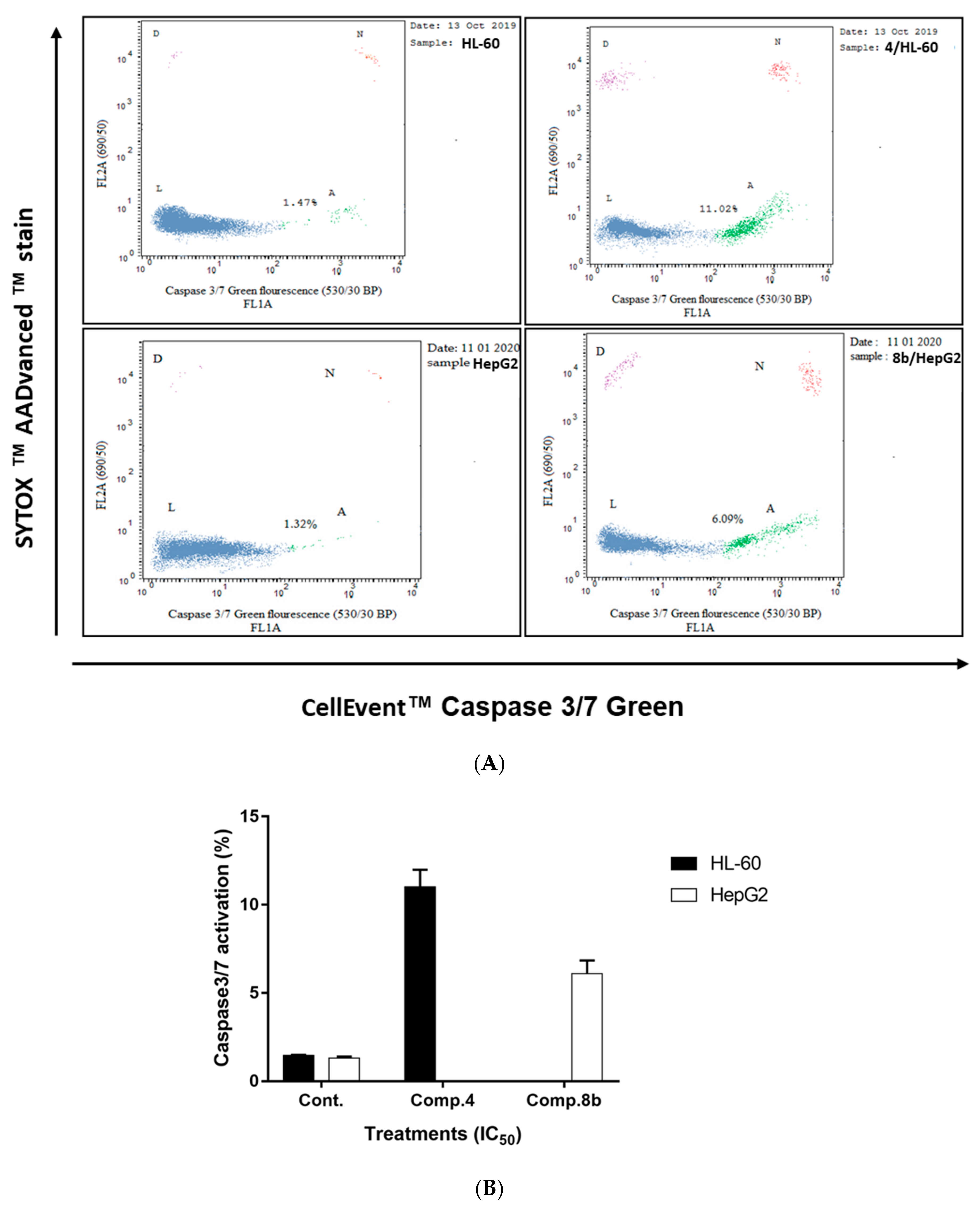
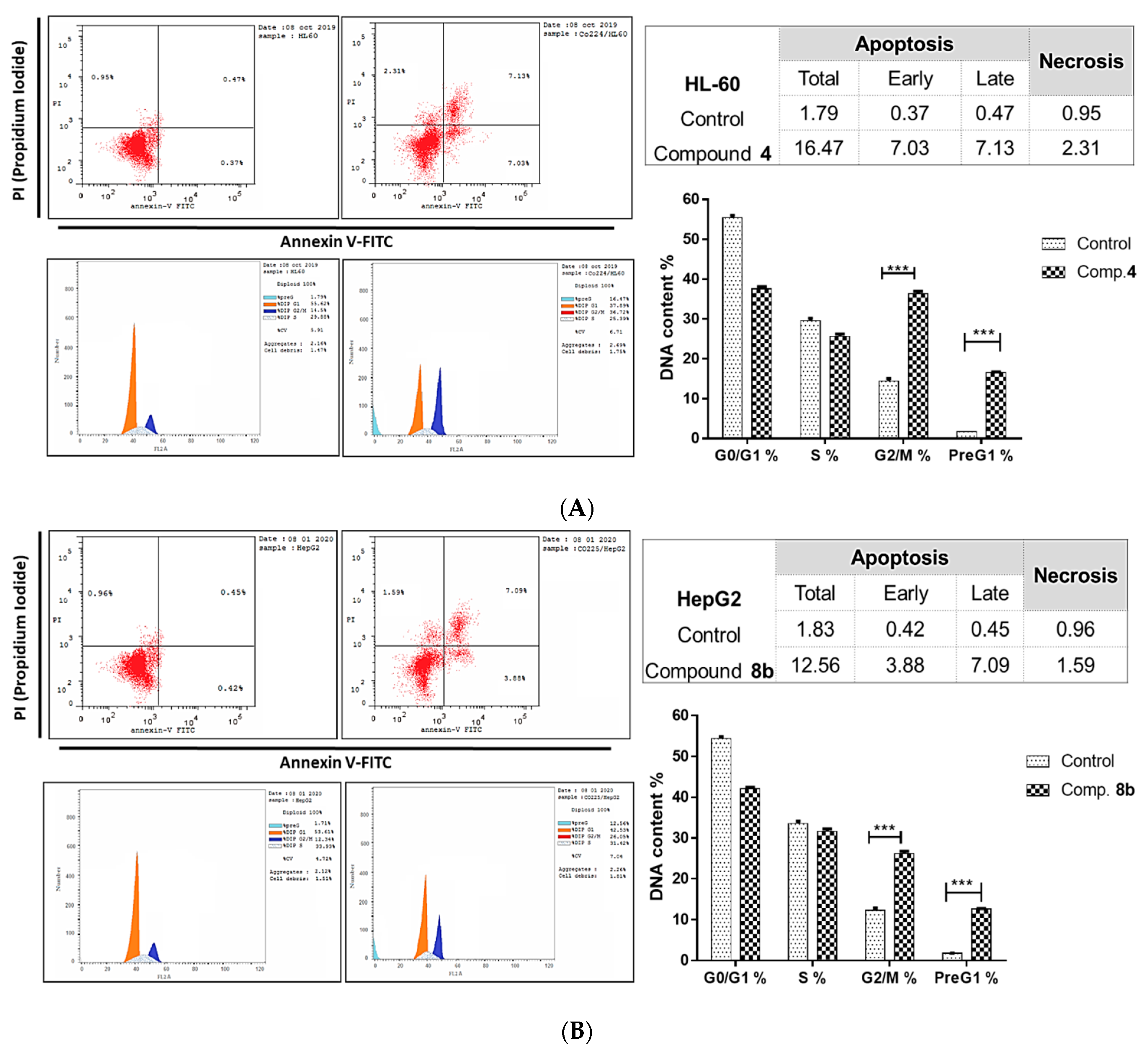
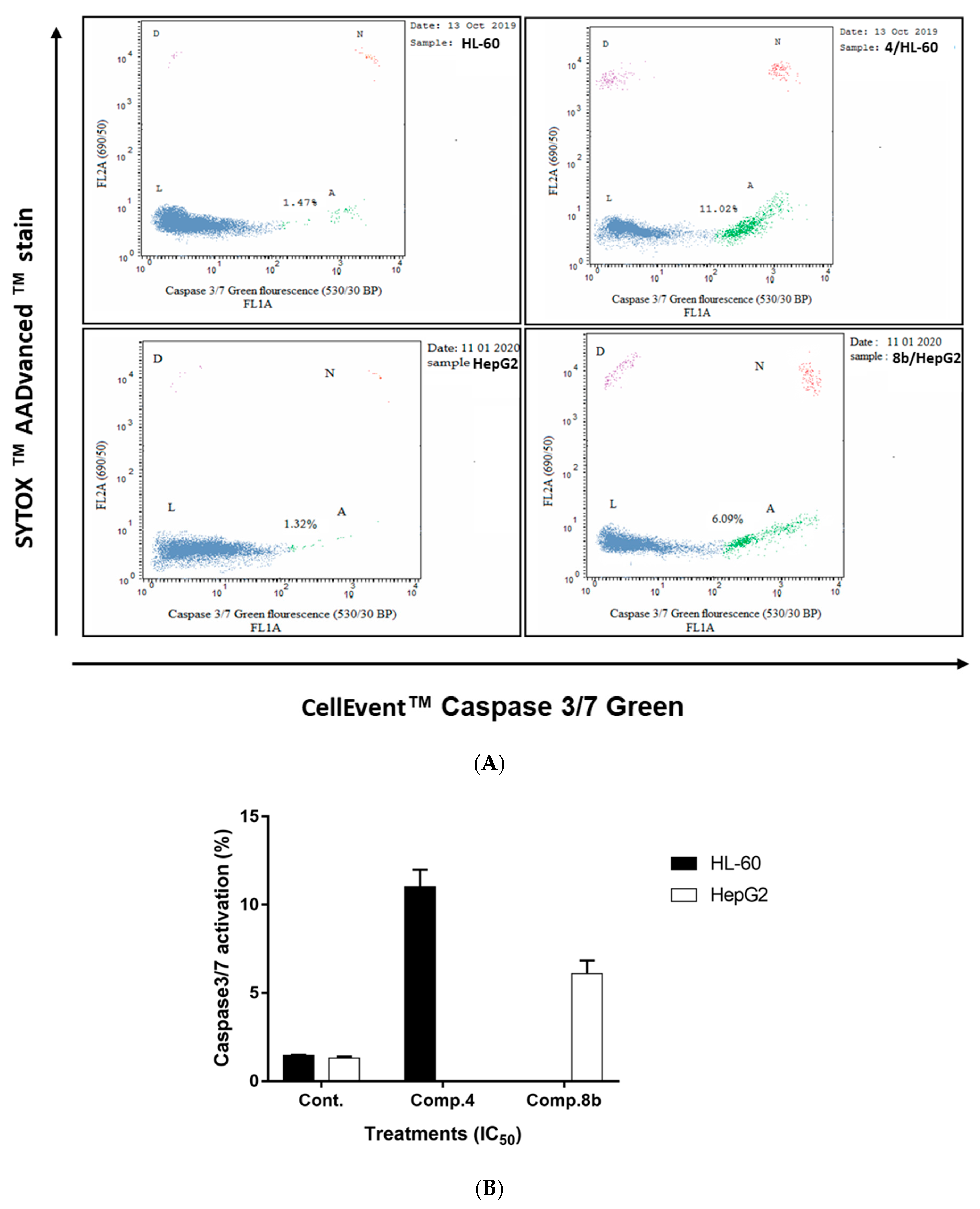
2.2.2. Investigation of Apoptotic Pathway
Flow Cytometric Analyses
Determination of Caspases 3/7 Activity
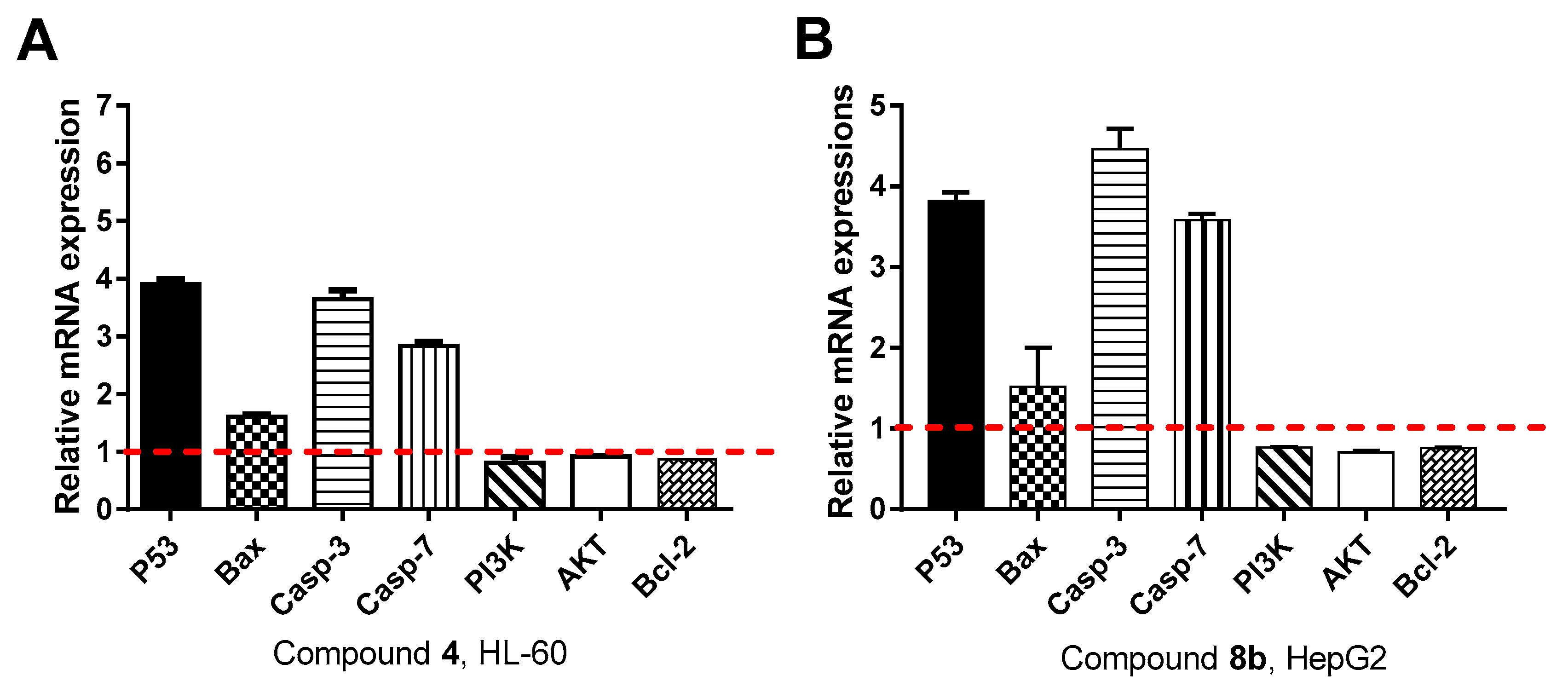
Gene Expression Analysis for the Apoptosis-Related Genes
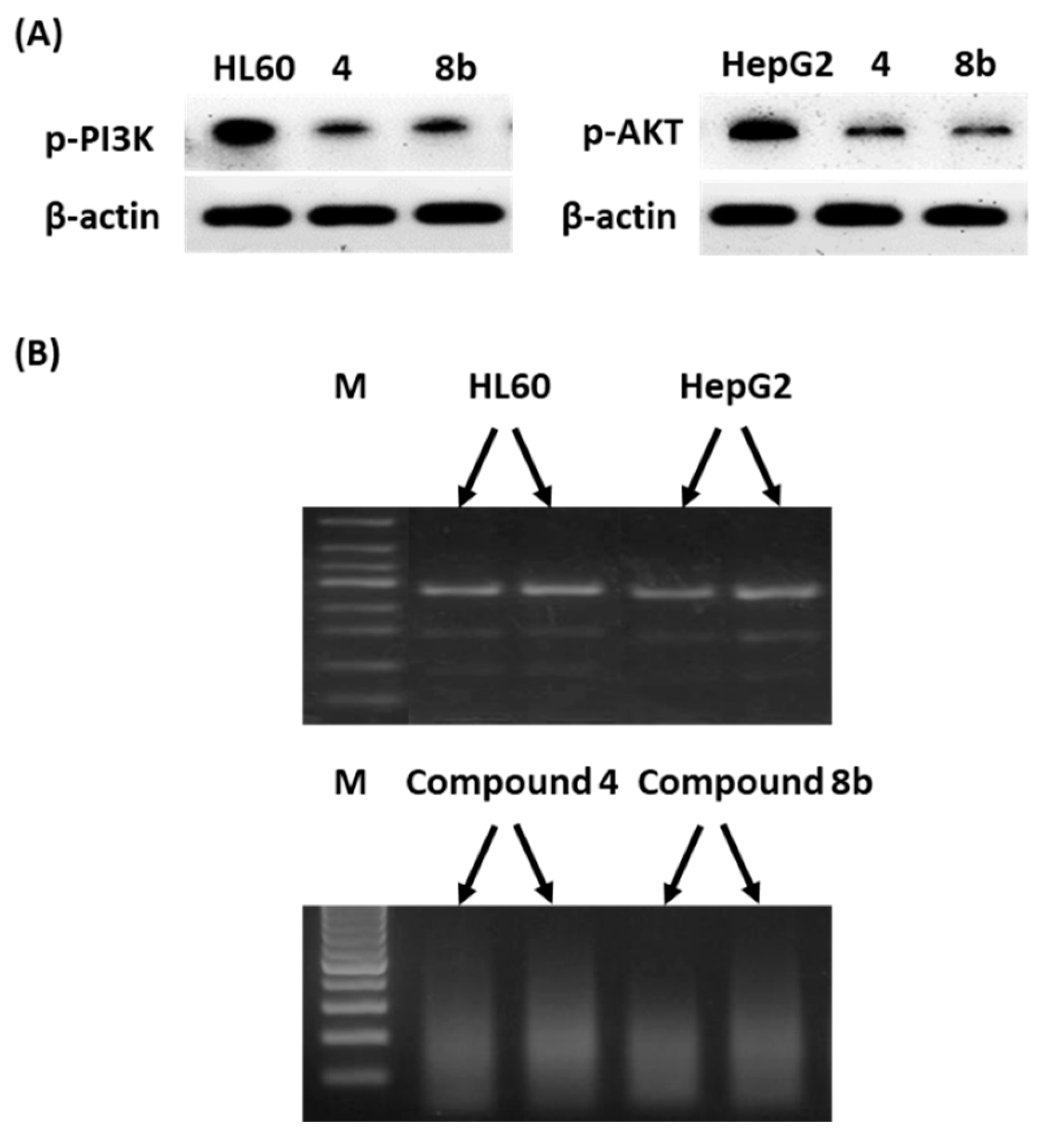
Western Blotting and DNA Fragmentation
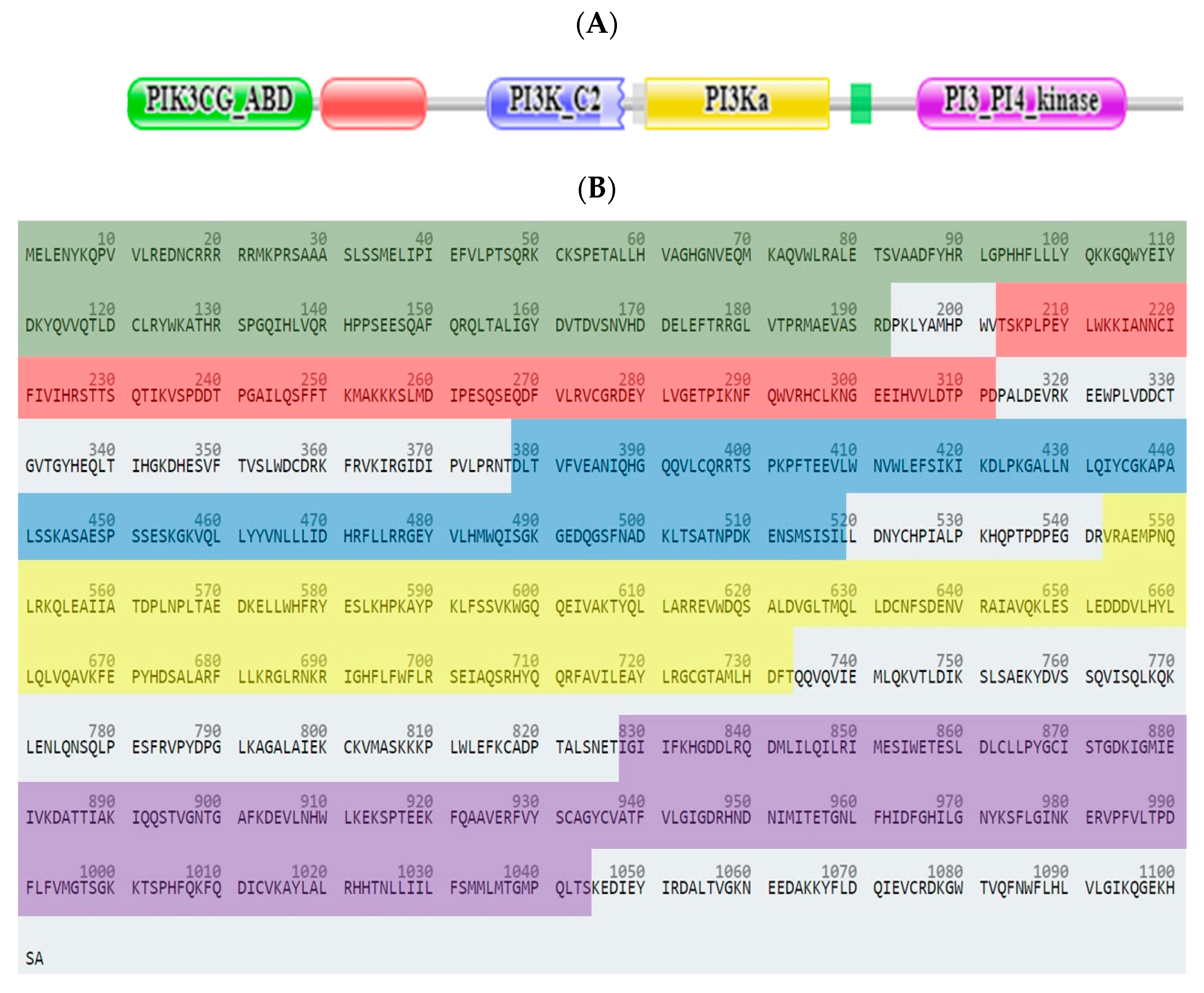
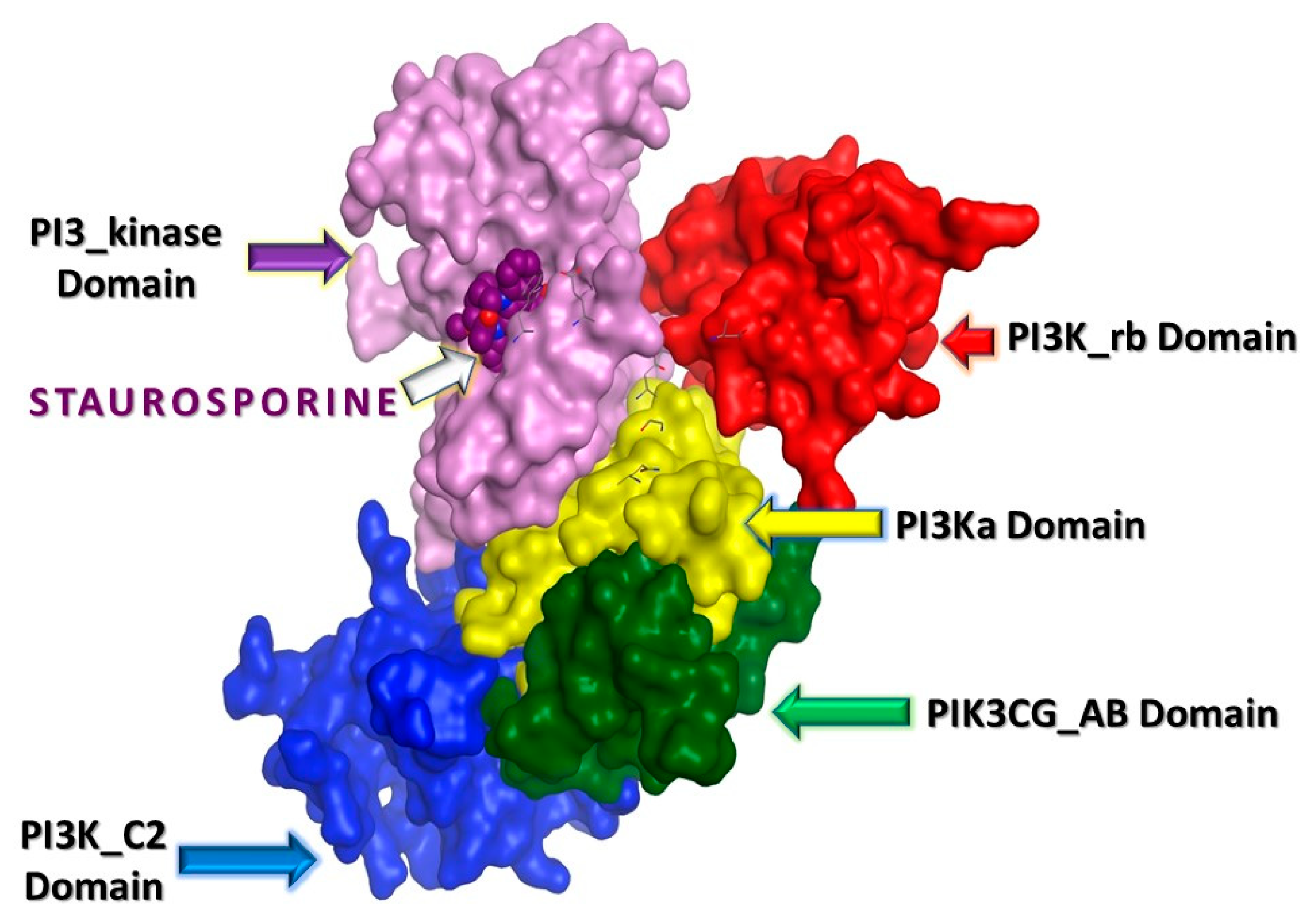
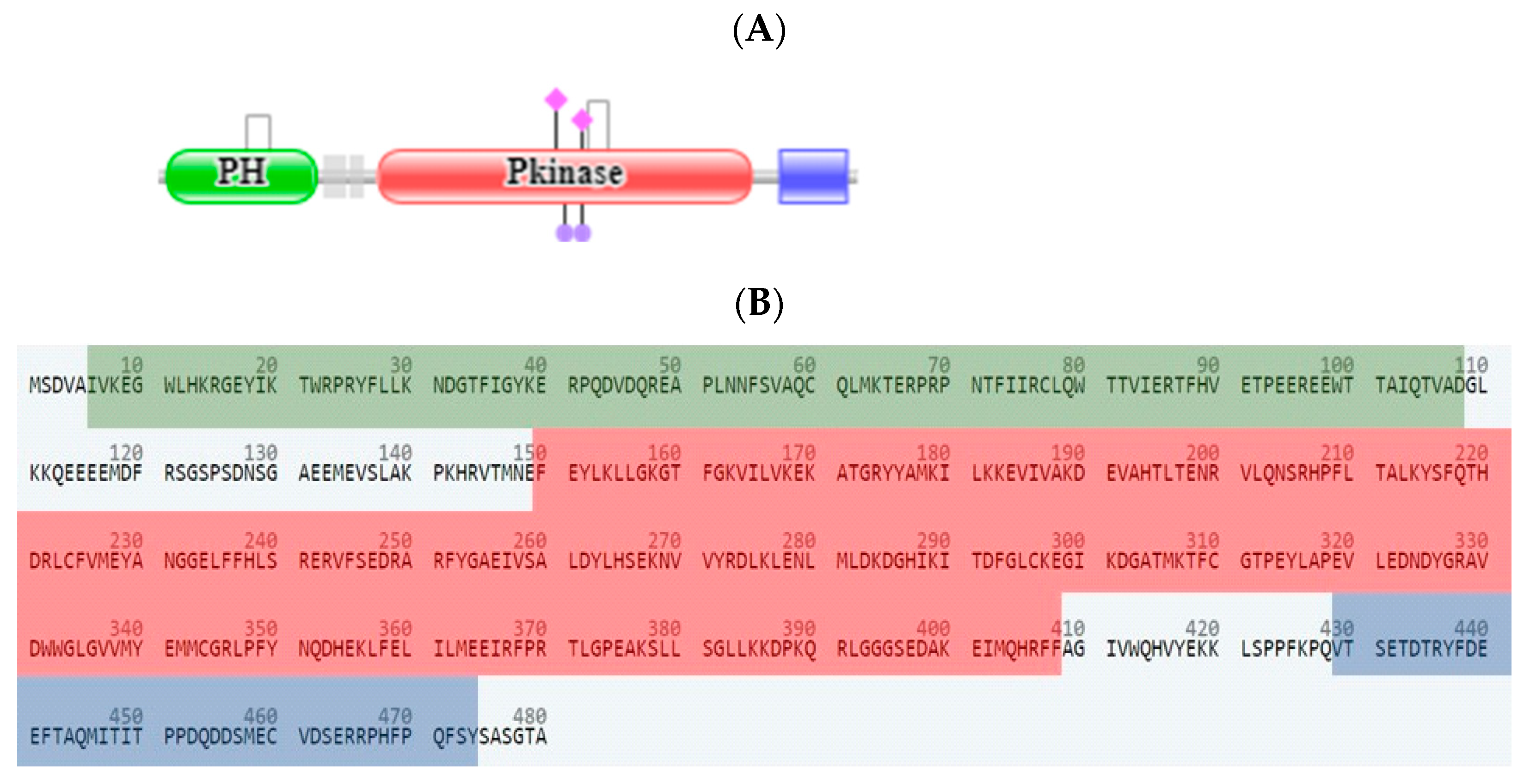
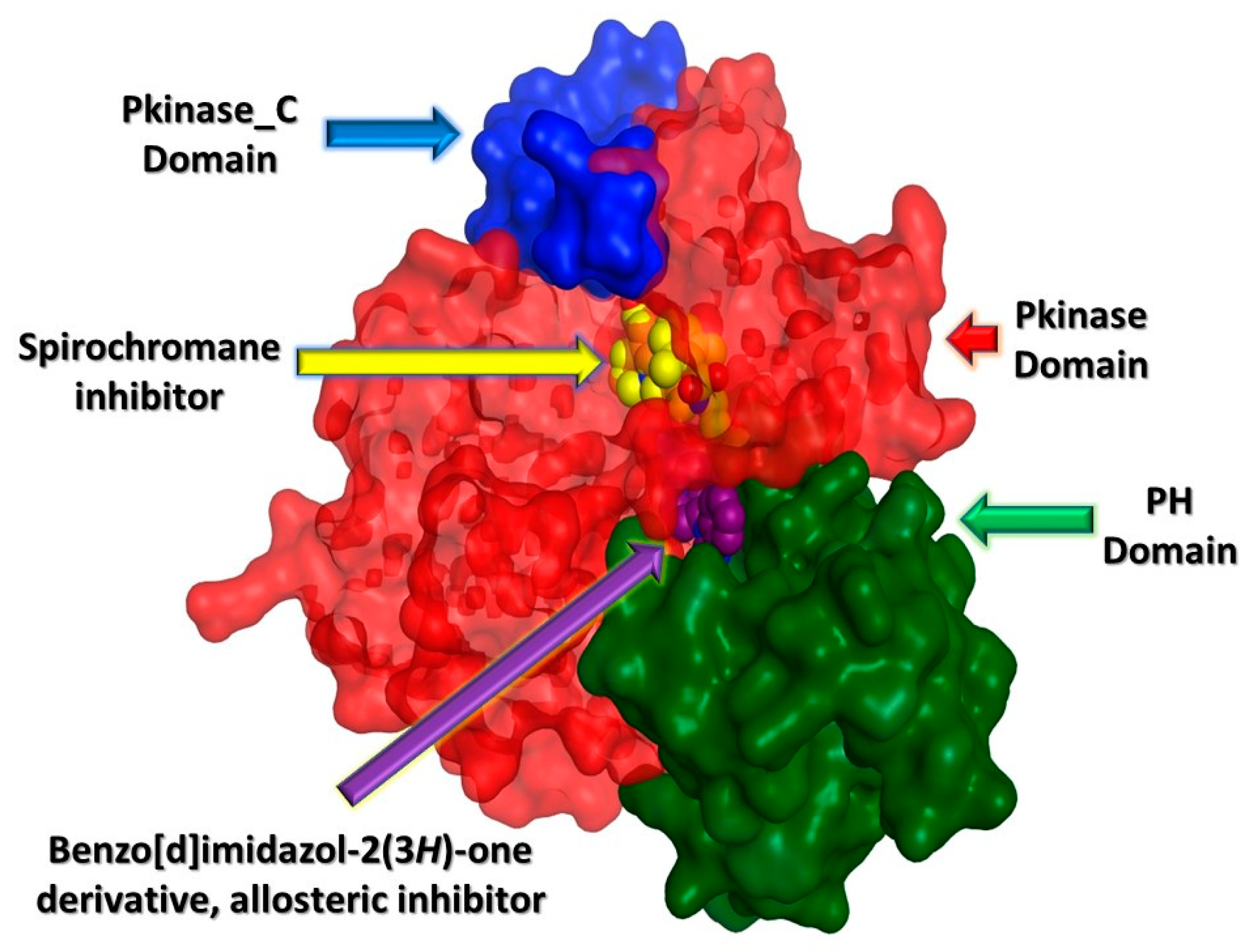
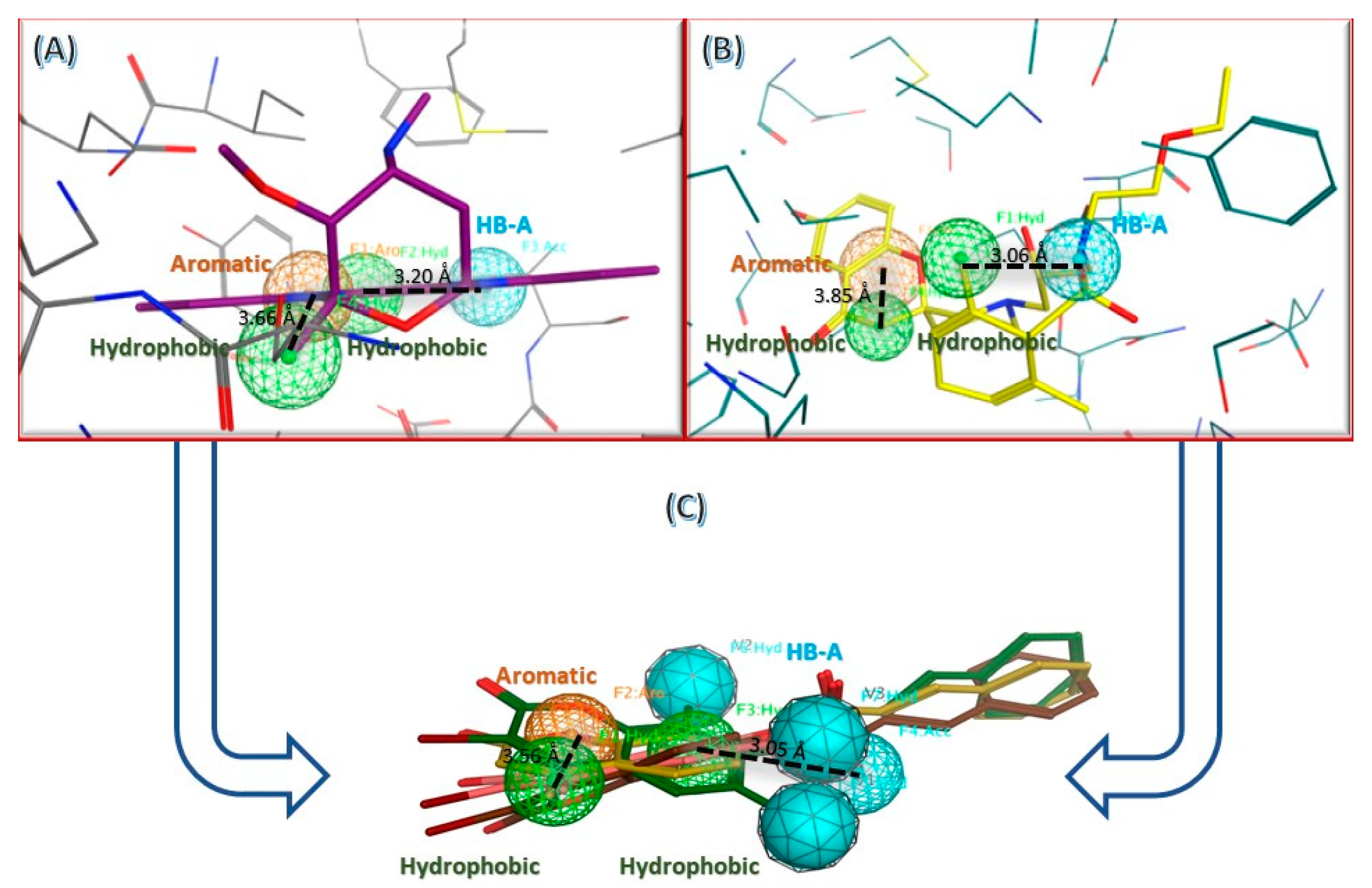
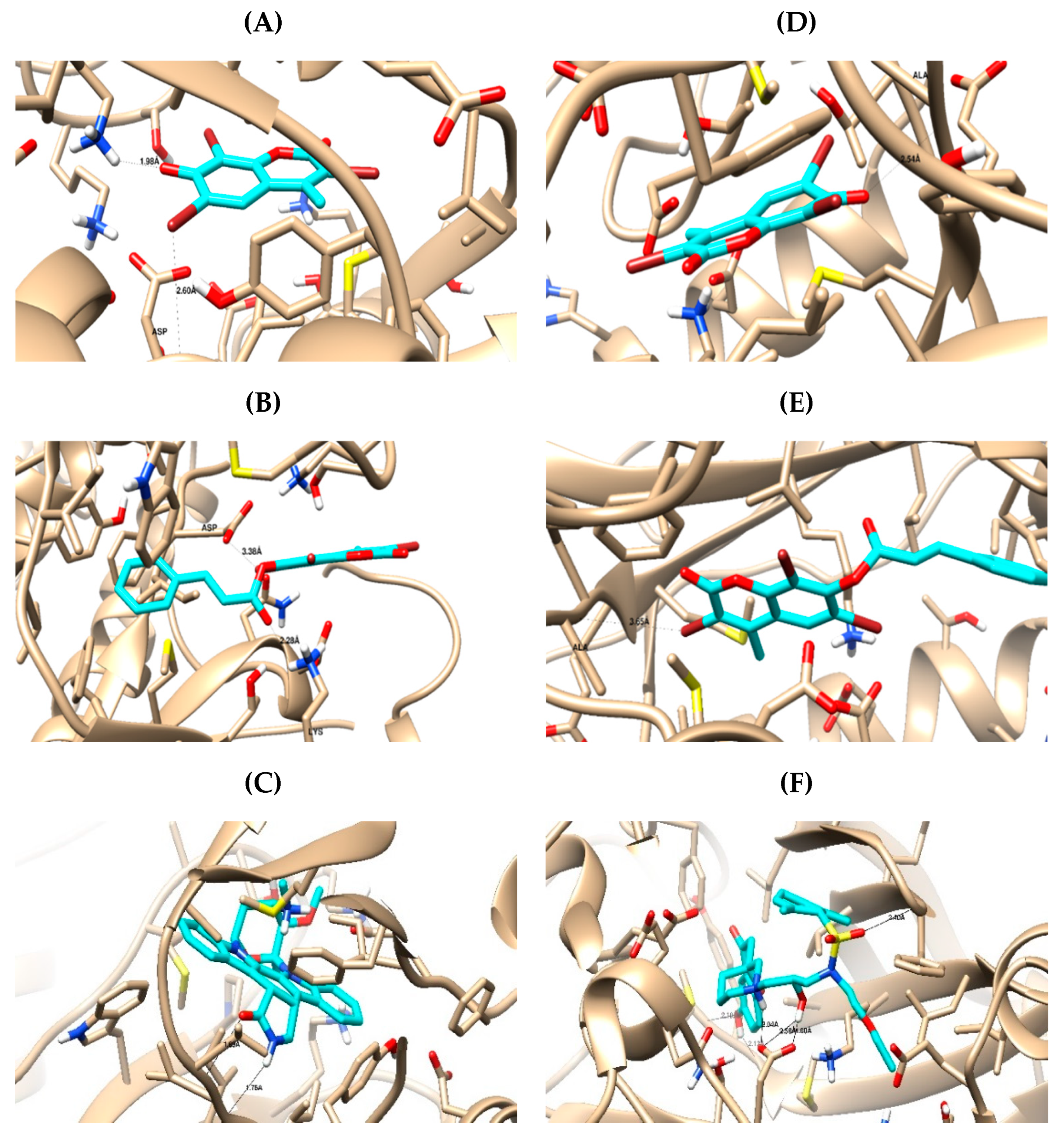
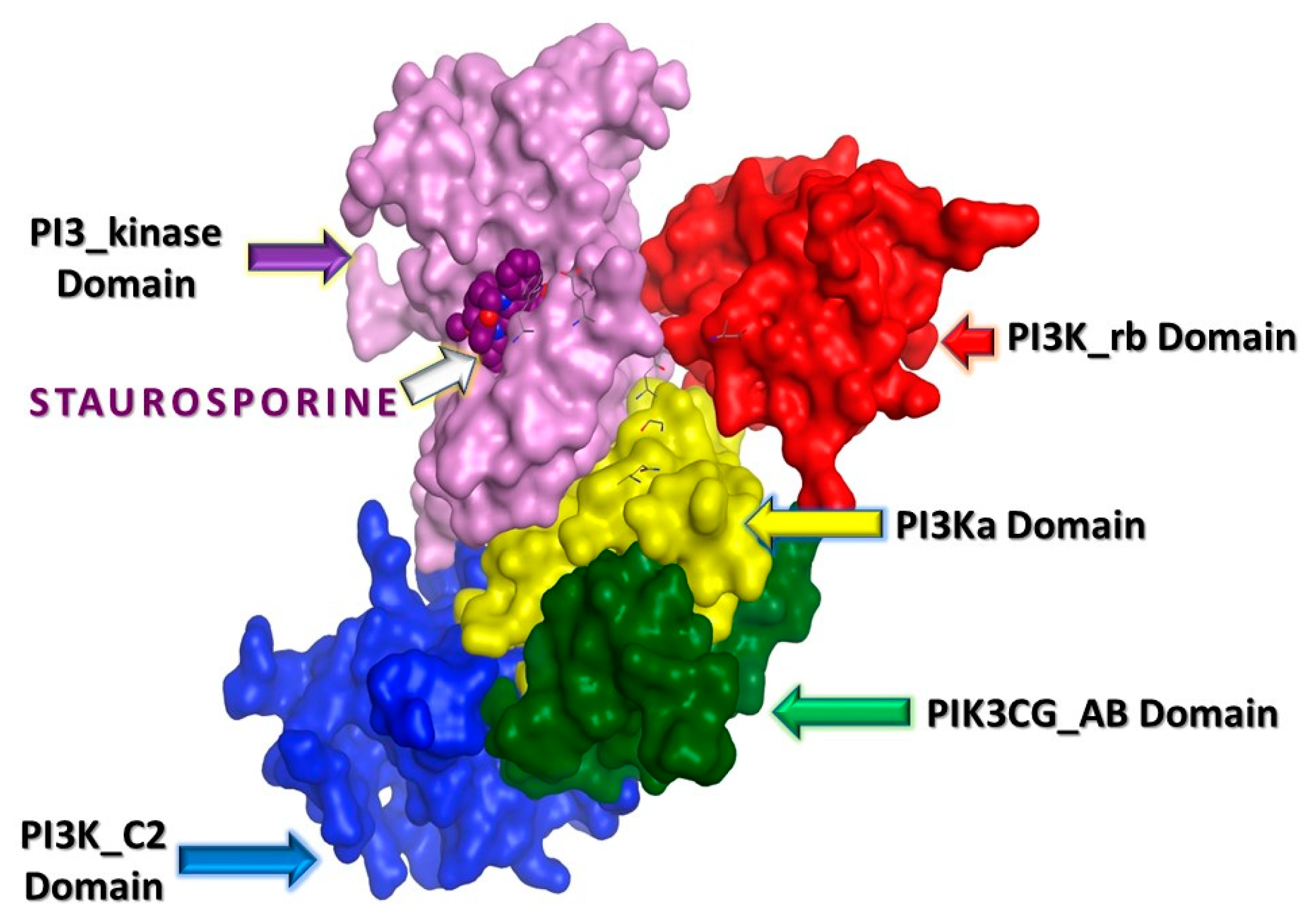
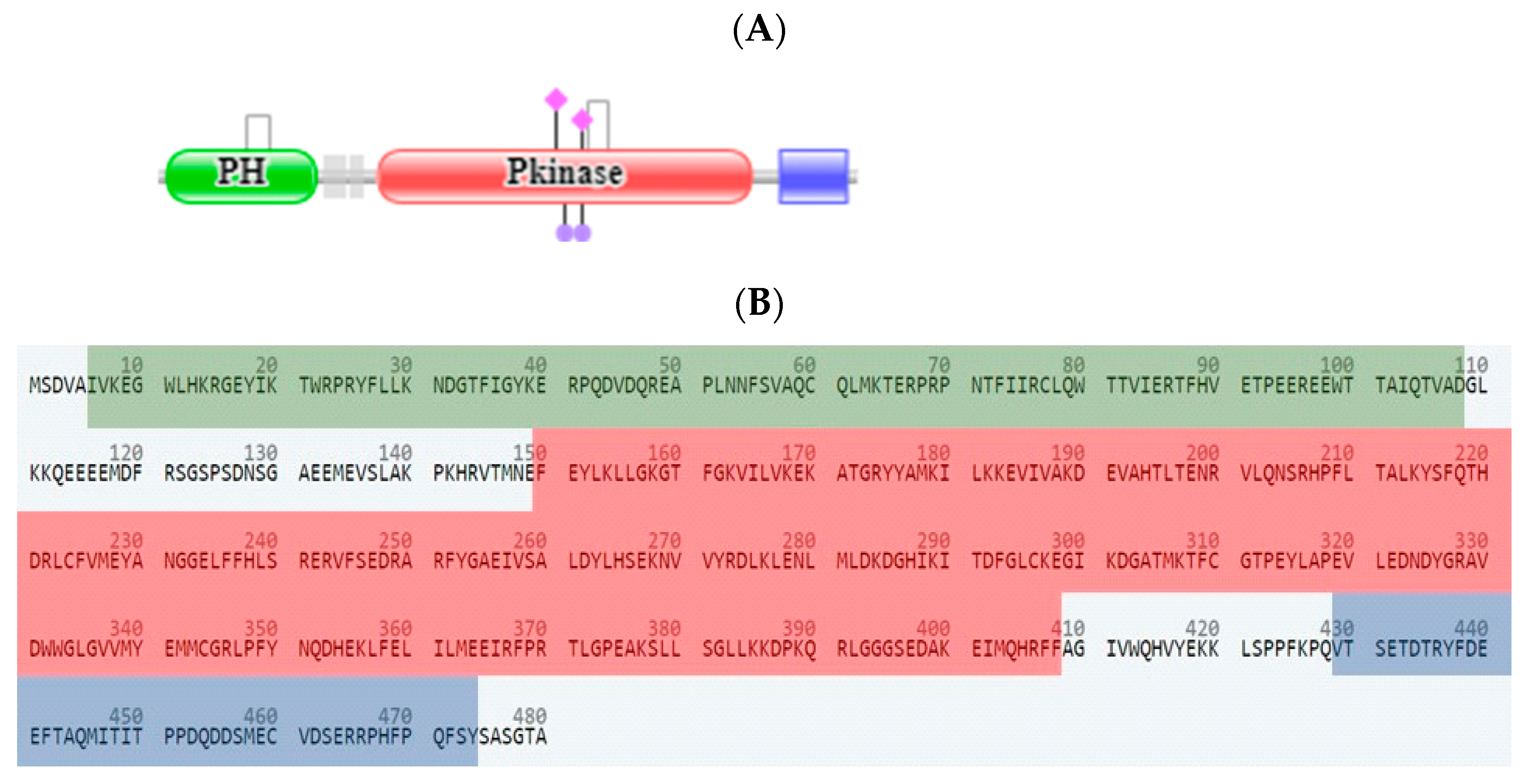
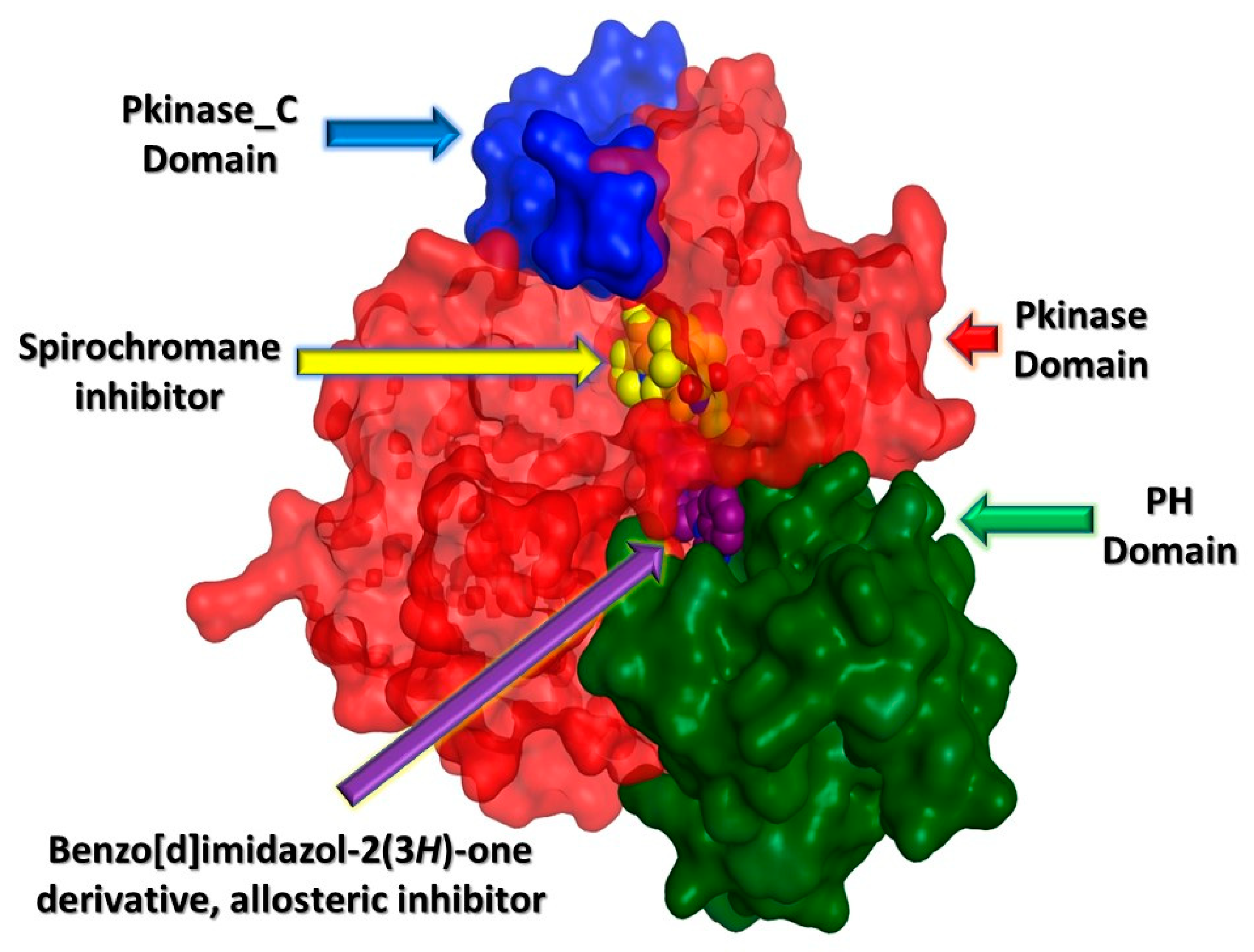
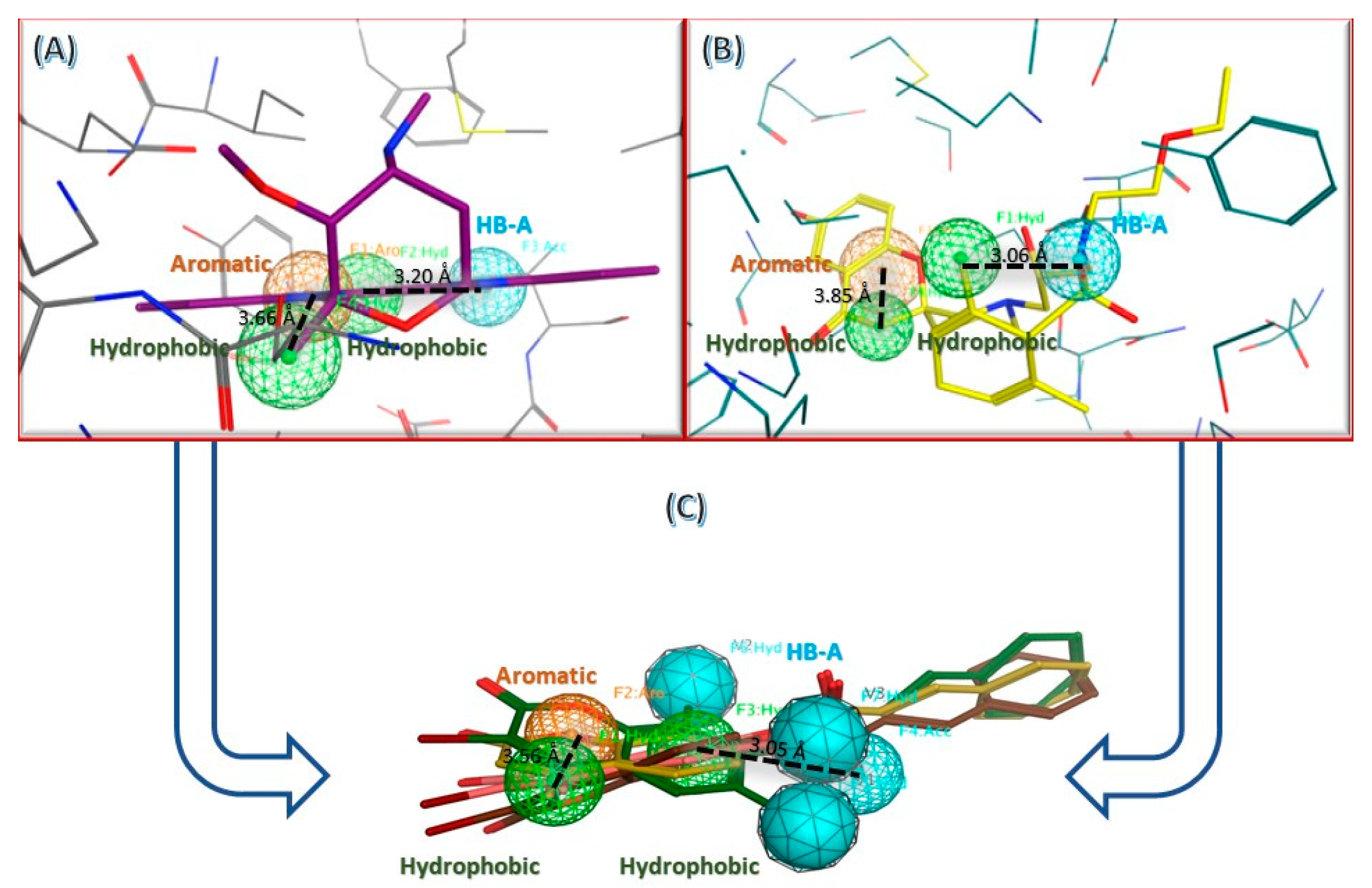
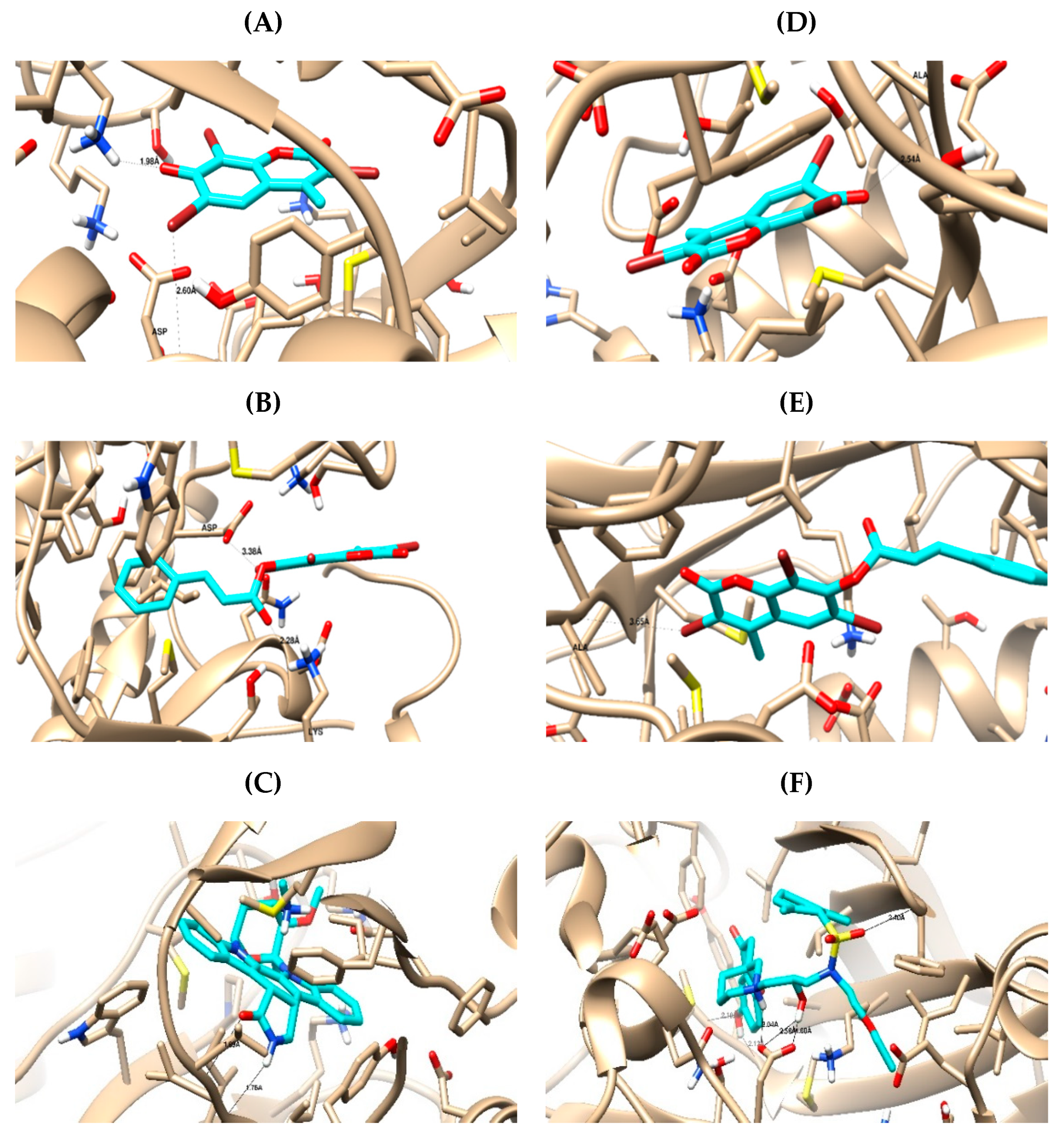
2.3. In Silico Studies
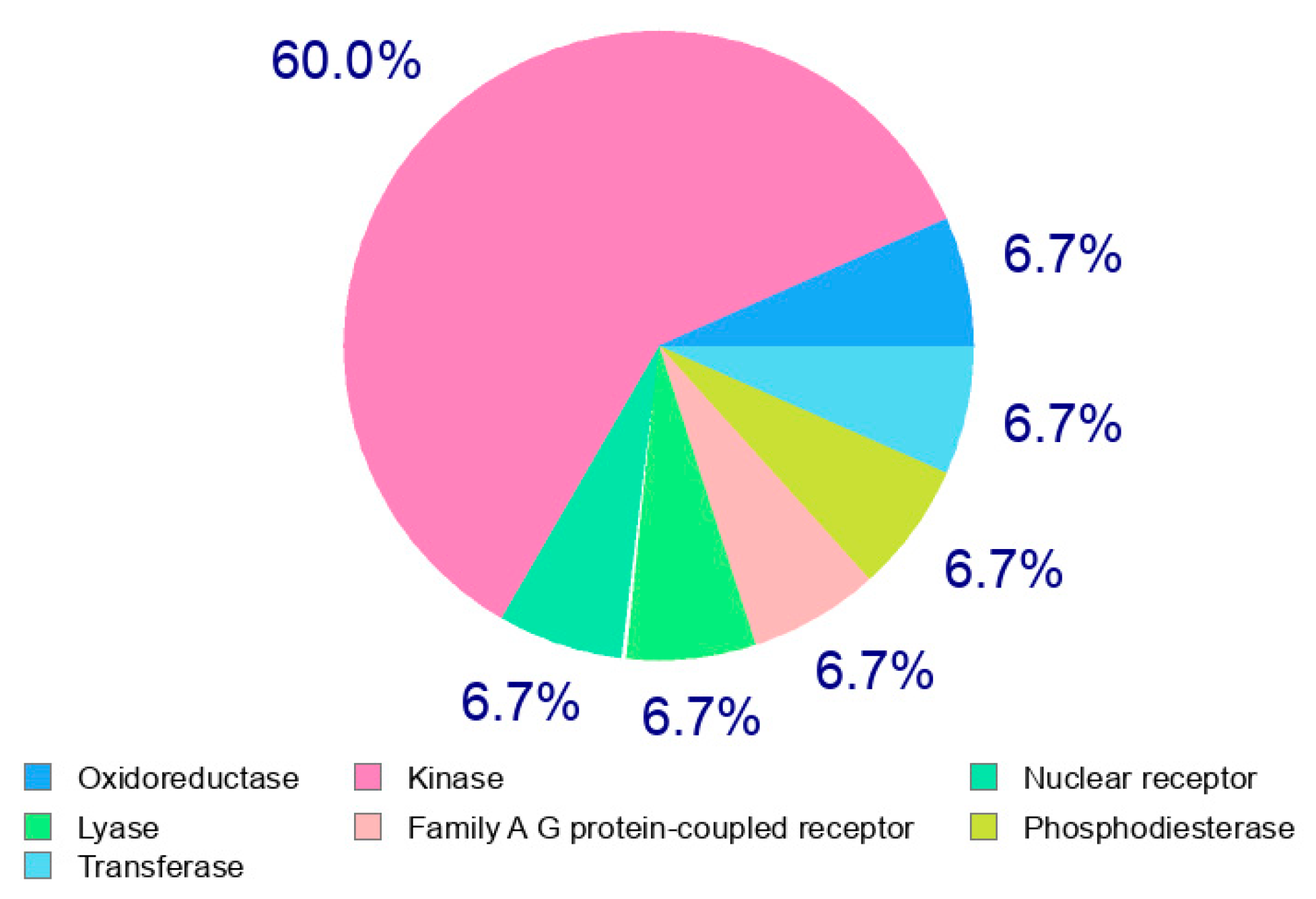
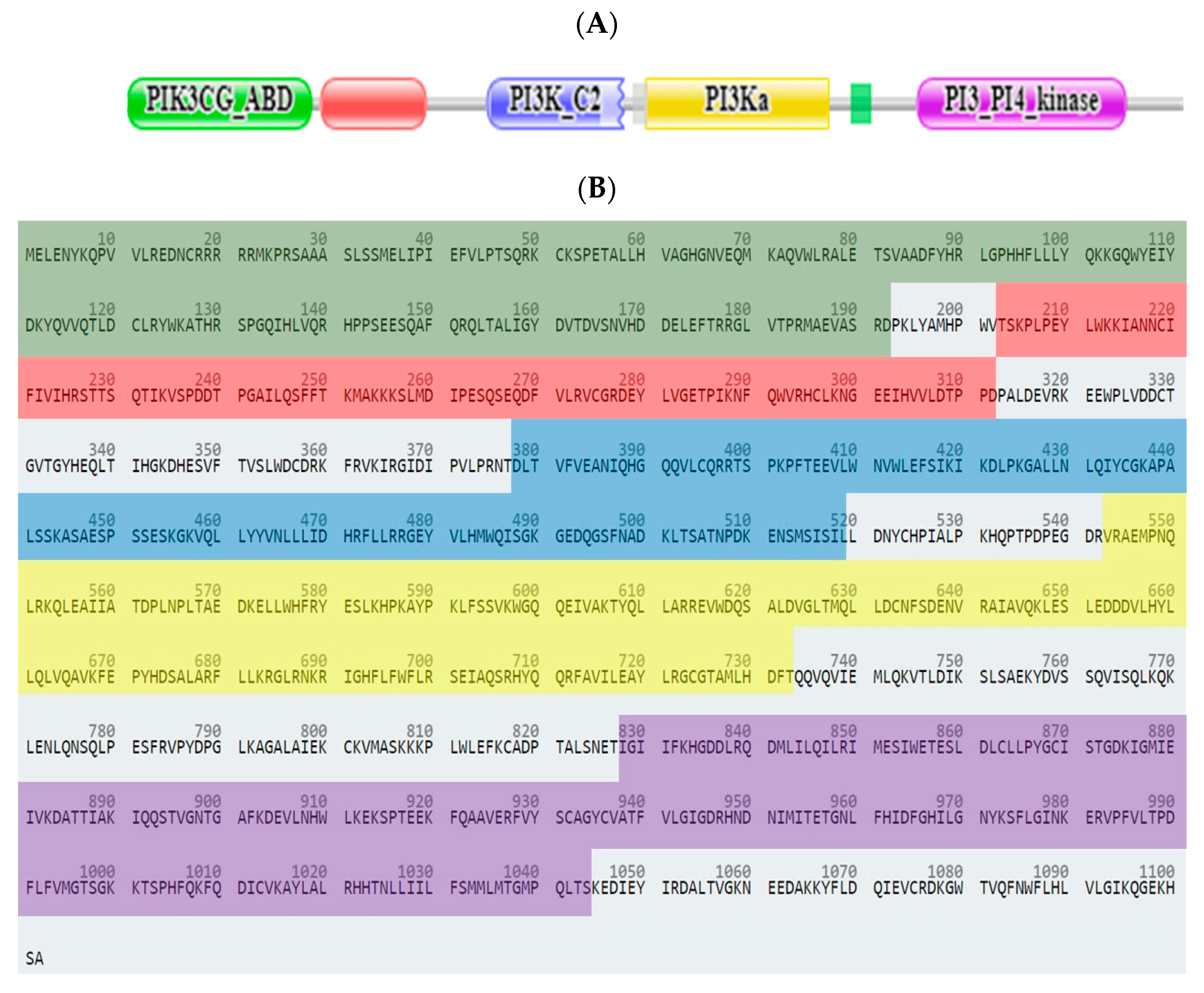
2.3.1. Computational Analysis
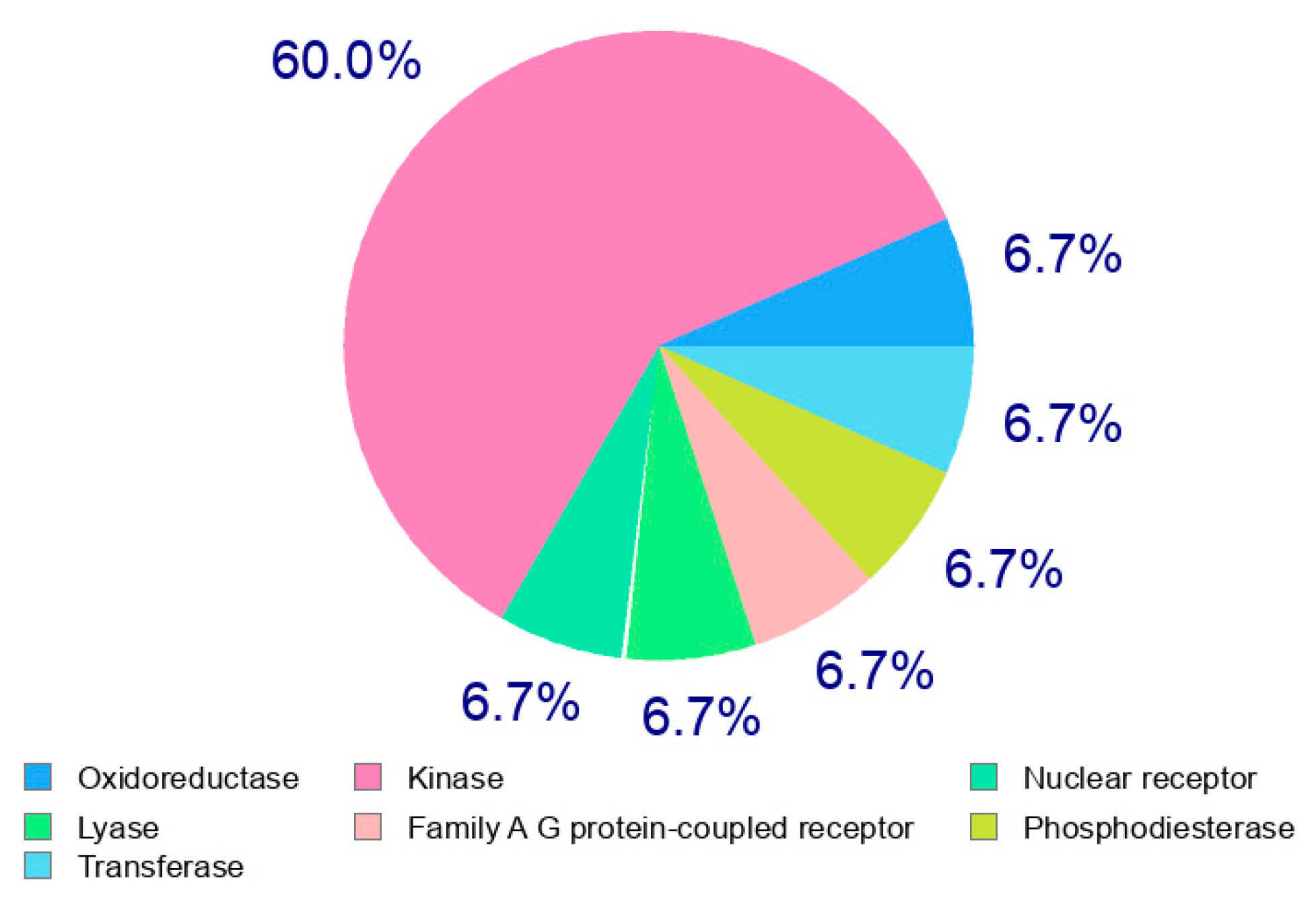
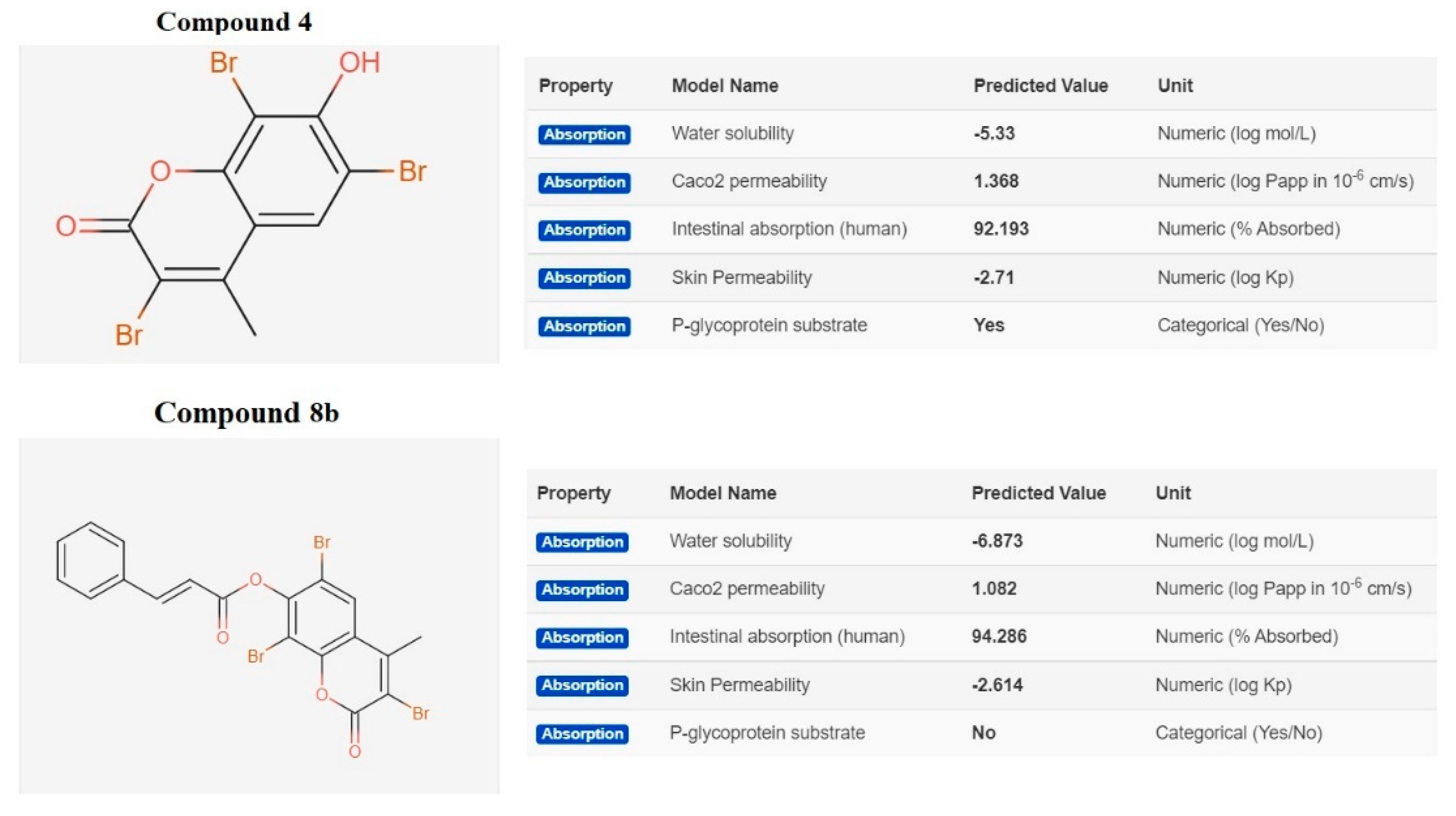
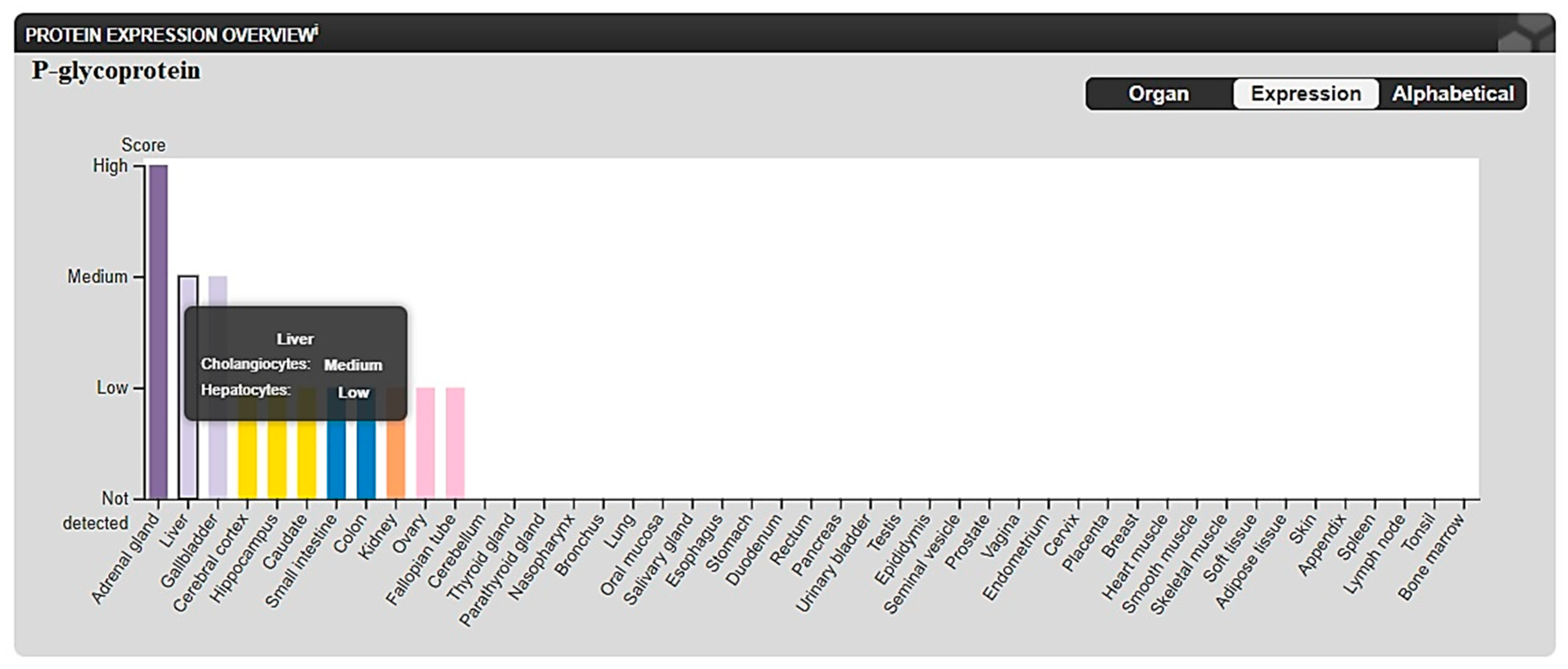
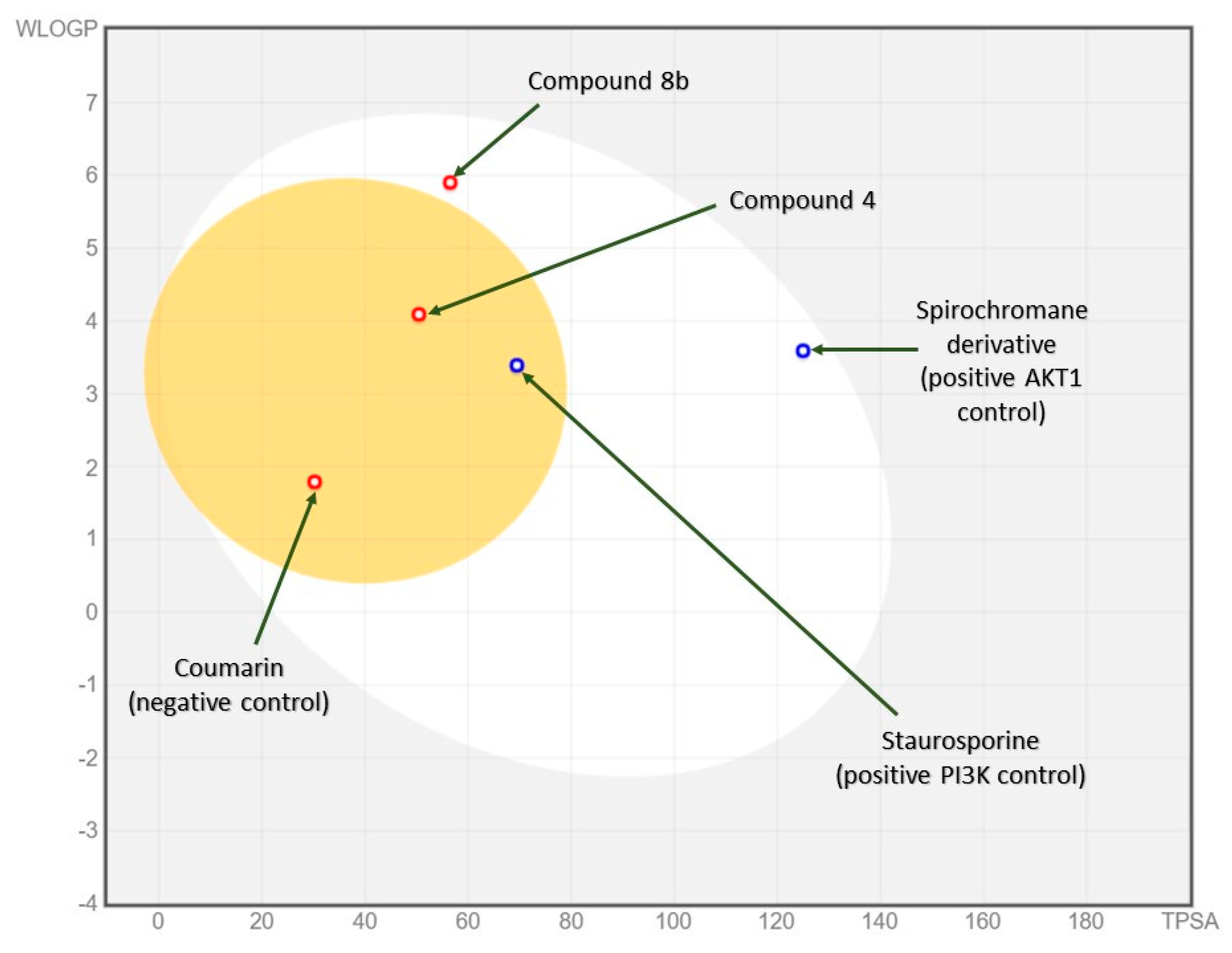
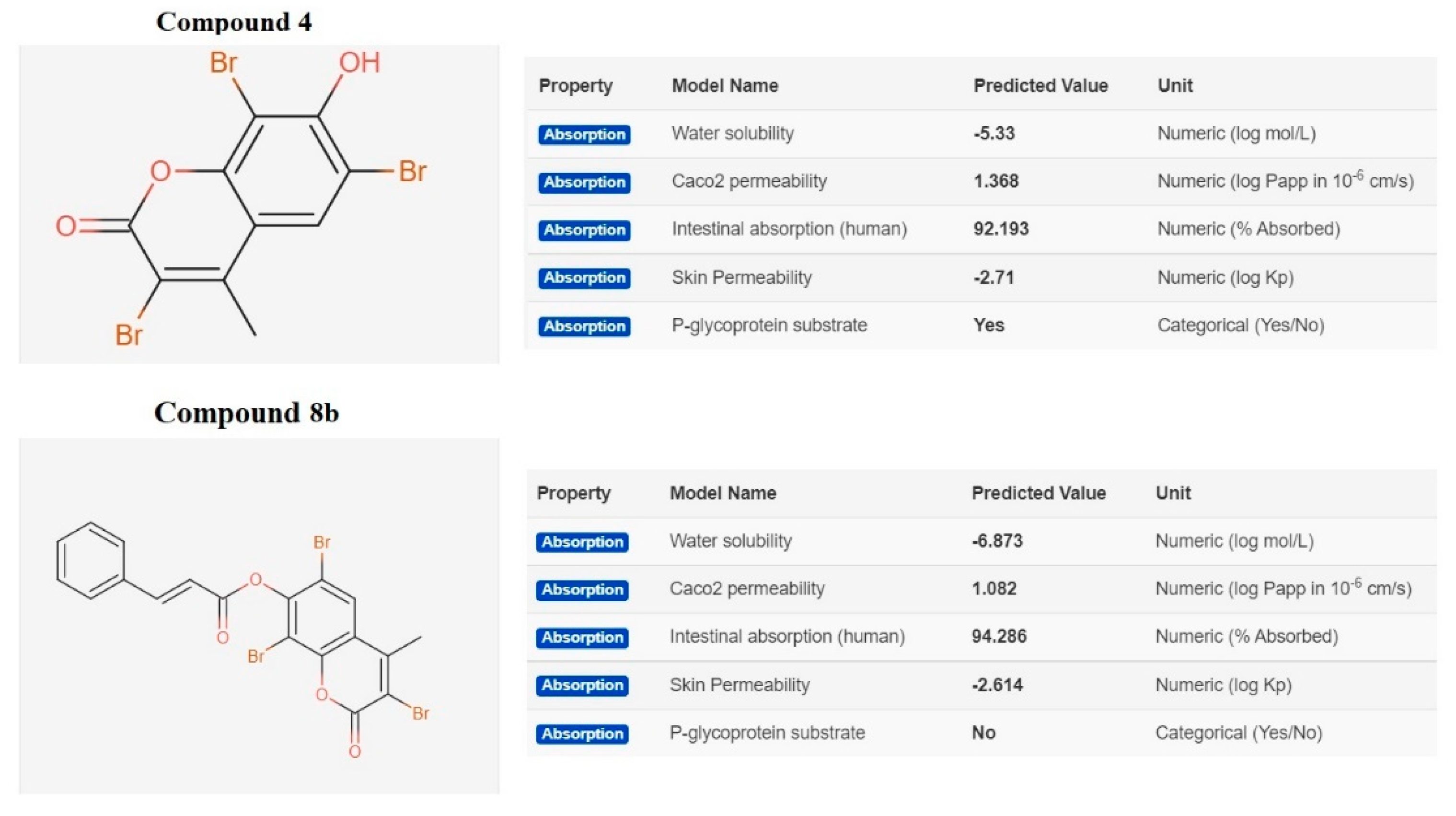
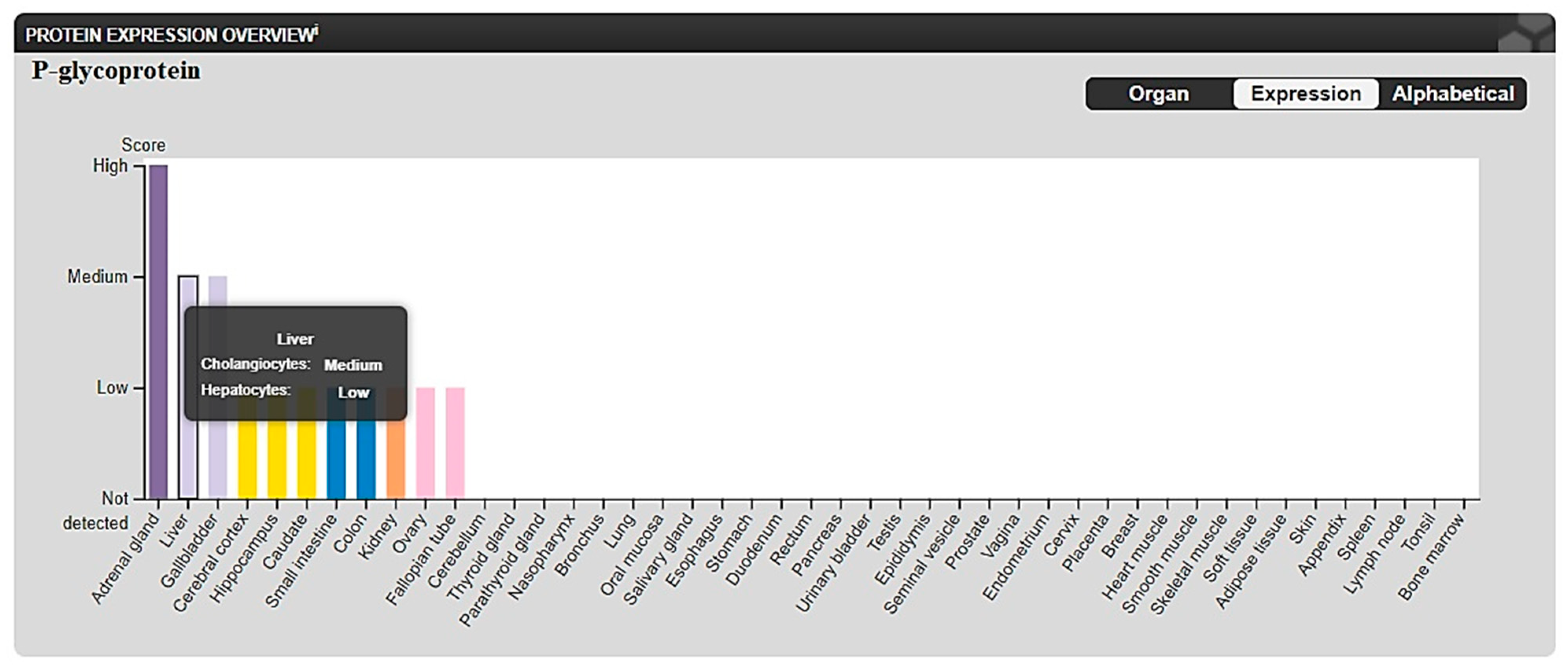
2.3.2. In Silico ADME and Bioactivity Prediction
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of 7-Hydroxy-4-methylcoumarin (3)
3.1.2. Synthesis of 7-Hydroxy-3,6,8-tribromo-4-methylcoumarin (4)
3.1.3. Isolation of cinnamaldehyde (5)
3.1.4. Preparation of cinnamic acid (6)
3.1.5. General procedure for the synthesis of compounds (8a, 8b)
(2E)-4-Methyl-2-oxo-2H-chromen-7-yl Cinnamate (8a)
(2E)-3,6,8-Tribromo-4-methyl-2-oxo-2H-chromen-7-yl Cinnamate (8b)
3.1.6. Synthesis of (2E)-3-bromo-4-methyl-2-oxo-2H-chromen-7-yl cinnamate (8c)
3.2. In Vitro Biological Assays
3.2.1. Cell culture
3.2.2. Cytotoxic Activity Using MTT Assay
3.2.3. Investigation of the Apoptotic Pathway
Flow Cytometric Analysis
Gene Expression Using RT-PCR Analysis
Western Blottting
3.3. In Silico Studies
3.3.1. Molecular Docking
3.3.2. In Silico Physicochemical Descriptors, Pharmacokinetic Properties, and Bioactivity Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer WHO: Lyon, France, 2020; Available online: http://publications.iarc.fr/586 (accessed on 18 July 2022).
- Çiftçi, G.A.; Yıldırım, Ş.U.; Altıntop, M.D.; Kaplancıklı, M.D. Induction of apoptosis in lung adenocarcinoma and glioma cells by some oxadiazole derivatives. Med. Chem. Res. 2014, 23, 3353. [Google Scholar] [CrossRef]
- Wei, J.; Yang, Y.; Wang, Z.; Wang, Z.; Fu, C.; Zhu, J.; Shan, J.; Huang, Y.; Tang, B.; Jiang, D. A potential anti-cancer ability of 1,2-di(quinazolin-4-yl) diselane against gastric cancer cells through ROS signaling pathway. Med. Chem. Res. 2017, 26, 841. [Google Scholar] [CrossRef]
- Singh, V.; Khurana, A.; Navik, U.; Allawadhi, P.; Bharani, K.K.; Weiskirchen, R. Apoptosis and Pharmacological Therapies for Targeting Thereof for Cancer Therapeutics. Sci 2022, 4, 15. [Google Scholar] [CrossRef]
- Siedlecka-Kroplewska, K.; Wrońska, A.; Kmieć, Z. Piceatannol, a Structural Analog of Resveratrol, Is an Apoptosis Inducer and a Multidrug Resistance Modulator in HL-60 Human Acute Myeloid Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 10597. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.; Salinas-Illarena, A.; Baldauf, H.M. New strategies to treat AML: Novel insights into AML survival pathways and combination therapies. Leukemia 2021, 35, 299. [Google Scholar] [CrossRef] [PubMed]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Available online: https://www.who.int/publications/m/item/cancer-egy-2020 (accessed on 3 September 2022).
- Kang, Z.; Qiao, N.; Liu, G.; Chen, H.; Tang, Z.; Li, Y. Copper-induced apoptosis and autophagy through oxidative stress-mediated mitochondrial dysfunction in male germ cells. Toxicol. In Vitro 2019, 61, 104639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, C.; Han, X.; Yang, G.; Ge, Z.; Zhang, G. Knockdown of ADAM17 inhibits cell proliferation and increases oxaliplatin sensitivity in HCT-8 colorectal cancer through EGFR-PI3K-AKT activation. Biochem. Biophys. Res. Commun. 2018, 503, 2333. [Google Scholar] [CrossRef] [PubMed]
- Vala, R.M.; Tandon, V.; Nicely, L.G.; Guo, L.; Gu, Y.; Banerjee, S. Synthesis of a Novel Pyrano[2,3-C]pyrazole Enabling PKBβ/AKT2 Inhibitory and in Vitro Anti-Glioma Activity. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Xiong, G.; Xiaofei, L.; Chi-Tang, H.; Xiaorong, L.; Yuanyuan, Z.; Bin, L.; Zhongzheng, C. Cocoa tea (Camellia ptilophylla) induces mitochondria-dependent apoptosis in HCT116 cells via ROS generation and PI3K/Akt signaling pathway. Food Res. Int. 2020, 129, 108854. [Google Scholar]
- Luo, X.; Lin, B.; Gao, Y.; Lei, X.; Wang, X.; Li, Y.; Li, T. Genipin attenuates mitochondrial-dependent apoptosis, endoplasmic reticulum stress, and inflammation via the PI3K/AKT pathway in acute lung injury. Int. Immunopharmacol. 2019, 76, 105842. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, X.; Bai, Z.; Chi, B.; Wei, Y.; Chen, X. Curcumol induces cell cycle arrest in colon cancer cells via reactive oxygen species and Akt/GSK3β/cyclin D1 pathway. J. Ethnopharmacol. 2018, 210, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhameed, R.F.; Ibrahim, A.K.; Yamada, K.; Ahmed, S.A. Cytotoxic and anti-inflammatory compounds from Red Sea grass Thalassodendron ciliatum. Med. Chem. Res. 2018, 27, 1238. [Google Scholar] [CrossRef]
- Tandon, V.; Vala, R.M.; Chen, A.; Sah, R.L.; Patel, H.M.; Pirrung, M.C.; Banerjee, S. Syrbactin-class dual constitutive- and immuno-proteasome inhibitor TIR-199 impedes myeloma-mediated bone degeneration in vivo. Biosci Rep. 2022, 42, BSR20212721. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.K.; Batran, R.Z.; Elseginy, S.A.; Ali, M.M.; Mahmoud, A.E. Synthesis, anti-cancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis. Bioorg. Chem. 2019, 85, 253. [Google Scholar] [CrossRef] [PubMed]
- Lončarić, M.; Gašo-Sokač, D.; Jokić, S.; Molnar, M. Recent Advances in the Synthesis of Coumarin Derivatives from Different Starting Materials. Biomolecules 2020, 16, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miri, R.; Nejati, M.; Saso, L.; Khakdan, F.; Parshad, B.; Mathur, D.; Parmar, V.S.; Bracke, M.E.; Prasad, A.K.; Sharma, S.K.; et al. Structure–activity relationship studies of 4-methylcoumarin derivatives as anti-cancer agents. Pharm. Biol. 2016, 54, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lončar, M.; Jakovljević, M.; Šubarić, D.; Pavlić, M.; Buzjak Služek, V.; Cindrić, I.; Molnar, M. Coumarins in Food and Methods of Their Determination. Foods 2020, 9, 645. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.C. Trease and Evans’ Pharmacognosy, 16th ed.; Saunders Elsavier: London, UK, 2009; ISBN 9780702029332. [Google Scholar]
- Spino, C.; Dodier, M.; Sotheeswaran, S. Anti-HIV coumarins from Calophyllum seed oil. Bioorg. Med. Chem. Lett. 1998, 8, 3475. [Google Scholar] [CrossRef]
- Kempen, I.; Papapostolou, D.; Thierry, N.; Pochet, L.; Counerotte, S.; Masereel, B.; Foidart, J.-M.; Reboud-Ravaux, M.; Noël, A.; Pirotte, B. 3-Bromophenyl 6-acetoxymethyl-2-oxo-2H-1-benzopyran-3-carboxylate inhibits cancer cell invasion in vitro and tumour growth in vivo. Br. J. Cancer 2003, 88, 1111. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Rosaiah, J.N.; Kumar, A.; Bhatia, G.; Khanna, A.K. Synthesis of novel benzocoumarin derivatives as lipid lowering agents, Bioorg. Med. Chem. Lett. 2010, 20, 3065. [Google Scholar] [CrossRef] [PubMed]
- Imenshahidi, M.; Eghbal, M.; Sahebkar, A.; Iranshahi, M. Hypotensive activity of auraptene, a monoterpene coumarin from Citrus spp. Pharm. Biol. 2013, 51, 545. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.J.; Jiang, J.G. Pharmacological and Nutritional Effects of Natural Coumarins and Their Structure–Activity Relationships. Mol. Nutr. Food Res. 2018, 62, 1701073. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Q.; Huang, C.; Jia, Y.M.; Song, B.A.; Li, J.; Liu, X.H. Novel coumarin-dihydropyrazole thio-ethanone derivatives: Design, synthesis, and anti-cancer activity. Eur. J. Med. Chem. 2012, 74, 717. [Google Scholar] [CrossRef] [PubMed]
- Vianna, D.R.; Hamerski, L.; Figueiró, F.; Bernardi, A.; Visentin, L.C.; Pires, E.N.S.; Teixeira, H.F.; Salbego, C.G.; Eifler-Lima, V.L.; Battastini, A.M.O.; et al. Selective cytotoxicity and apoptosis induction in glioma cell lines by 5-oxygenated-6,7-methylenedioxycoumarins from Pterocaulon species. Eur. J. Med. Chem. 2012, 57, 268. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, H.R.; Liu, H.S.; Cheng, M.; Xia, P.; Qian, K.; Wu, P.C.; Lai, C.Y.; Xia, Y.; Yang, Z.Y.; et al. Antitumor agents 292. Design, synthesis, and pharmacological study of S- and O-substituted 7-mercapto- or hydroxy-coumarins and chromones as potent cytotoxic agents. Eur. J. Med. Chem. 2012, 49, 74. [Google Scholar] [CrossRef]
- Molaverdi, F.; Khoobi, M.; Emami, S.; Alipour, M.; Firuzi, O.; Foroumadi, A.; Dehghan, G.; Miri, R.; Shaki, F.; Jafarpour, L.; et al. Polyoxygenated cinnamoylcoumarins as conformationally constrained analogs of cytotoxic diarylpentanoids: Synthesis and biological activity. Eur. J. Med. Chem. 2013, 68, 103. [Google Scholar] [CrossRef]
- Vázquez, R.; Riveiro, M.E.; Vermeulen, M.; Alonso, E.; Mondillo, C.; Facorro, G.; Piehl, L.; Gómez, N.; Moglioni, A.; Fernández, N.; et al. Structure-anti-leukemic activity relationship study of ortho-dihydroxycoumarins in U-937 cells: Key role of the δ-lactone ring in determining differentiation-inducing potency and selective pro-apoptotic action. Bioorg. Med. Chem. 2012, 20, 5537. [Google Scholar] [CrossRef]
- García-Vilas, J.A.; Quesada, A.R.; Medina, M.Á. 4-Methylumbelliferone Inhibits Angiogenesis in Vitro and in Vivo. J. Agric. Food Chem. 2013, 61, 4063. [Google Scholar] [CrossRef]
- Blahová, j.; Svobodová, Z. Assessment of coumarin levels in ground cinnamon available in the Czech retail market. Sci. World J. 2012, 2012, 26385. [Google Scholar] [CrossRef] [PubMed]
- Nagy, N.; Kuipers, H.F.; Frymoyer, A.R.; Ishak, H.D.; Bollyky, J.B.; Wight, T.N.; Bollyky, P.L. 4-Methylumbelliferone Treatment and Hyaluronan Inhibition as a Therapeutic Strategy in Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2015, 6, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.S.; Paul, S.; Mandal, S.K.; Banerjee, A.; Boujedaini, N.; Khuda-Bukhsh, A.R. A synthetic coumarin (4-Methyl-7 hydroxy coumarin) has anti-cancer potentials against DMBA-induced skin cancer in mice. Eur. J. Pharmacol. 2009, 614, 128. [Google Scholar] [CrossRef] [PubMed]
- Duangdee, N.; Mahavorasirikul, W.; Prateeptongkum, S. Design synthesis and antiproliferative activity of some new coumarin substituted hydrazide–hydrazone derivatives. J. Chem. Sci. 2020, 132, 66. [Google Scholar] [CrossRef]
- Chimichi, S.; Boccalini, M.; Salvador, A.; Dall’Acqua, F.; Basso, G.; Viola, G. Synthesis and Biological Evaluation of New Geiparvarin Derivatives. Chem. Med. Chem. 2009, 4, 769. [Google Scholar] [CrossRef] [PubMed]
- Trykowska Konc, J.; Hejchman, E.; Kruszewska, H.; Wolska, I.; Maciejewska, D. Synthesis and pharmacological activity of O-aminoalkyl derivatives of 7-hydroxycoumarin. Eur. J. Med. Chem. 2011, 46, 2252. [Google Scholar] [CrossRef] [PubMed]
- Hamidpour, R.; Hamidpour, M.; Hamidpour, S.; Shahlari, A.R. Cinnamon from the selection of traditional applications to its novel effects on the inhibition of angiogenesis in cancer cells and prevention of Alzheimer’s disease, and a series of functions such as antioxidant, anticholesterol, antidiabetes, antibacterial. J. Tradit. Complement. Med. 2015, 5, 66. [Google Scholar] [PubMed] [Green Version]
- Ranasinghe, P.; Pigera, S.; Premakumara, G.A.S.; Galappaththy, P.; Constantine, G.R.; Katulanda, P. Medicinal properties of ‘true’ cinnamon (Cinnamomum zeylanicum): A systematic review. BMC Complement. Altern. Med. 2013, 13, 275. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Chakraborty, A. Cinnamon in Anticancer Armamentarium: A Molecular Approach. J. Toxicol. 2018, 2018, 8978731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.H.; Ismail, I.A.; Kang, S.M.; Han, D.C.; Kwon, B.M. Cinnamaldehydes in Cancer Chemotherapy. Phyther. Res. 2016, 30, 754. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Baltas, M.; Bedos-Belval, F. Cinnamic Acid Derivatives as Anticancer Agents-A Review. Curr. Med. Chem. 2011, 18, 1672. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.S.; Cheng, J.B.; Su, W.Q.; Li, H.Z.; Xiao, T.; Chen, D.A.; Zhang, Z.L. Cinnamic acid hybrids as anticancer agents: A mini review. Arch. Pharm. 2022, 355, e2200052. [Google Scholar] [CrossRef]
- Nasr, T.; Bondock, S.; Youns, M. Anti-cancer activity of new coumarin substituted hydrazide-hydrazone derivatives. Eur. J. Med. Chem. 2014, 76, 539. [Google Scholar] [CrossRef] [PubMed]
- Erşatır, M.; Yıldırım, M.; Giray, E.S.; Yalın, S. Synthesis and antiproliferative evaluation of novel biheterocycles based on coumarin and 2-aminoselenophene-3-carbonitrile unit. Monatshefte Chem. Chem. Mon. 2020, 151, 625. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Reddy, J.J.; Lakshmi, P.S.; Krishna, P.R. An efficient ZrCl4 catalyzed one-pot solvent free protocol for the synthesis of 4-substituted coumarins. Tetrahedron Lett. 2005, 46, 6119. [Google Scholar] [CrossRef]
- Deshmukh, M.; Pawar, P.; Joseph, M.; Phalgune, U.; Kashalkar, R. Efficacy of 4-methyl-7-hydroxy coumarin derivatives against vectors Aedes aegypti and Culex quinquefasciatus. Indian J. Exp. Biol. 2008, 46, 788. [Google Scholar] [PubMed]
- Al-Bayati, F.A.; Mohammed, M.J. Isolation, identification, and purification of cinnamaldehyde from Cinnamomum zeylanicum bark oil. An antibacterial study. Pharm. Biol. 2009, 47, 61. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S. Synthesis of Cinnamaldehyde Hydroxyl Sulfonic Sodium and its Anti-Fungal Activity. Adv. Mater. Res. 2011, 183–185, 1942. [Google Scholar] [CrossRef]
- Utchariyakiat, I.; Surassmo, S.; Jaturanpinyo, M.; Khuntayaporn, P.; Chomnawang, M.T. Efficacy of cinnamon bark oil and cinnamaldehyde on anti-multidrug resistant Pseudomonas aeruginosa and the synergistic effects in combination with other antimicrobial agents. BMC Complement. Altern. Med. 2016, 16, 158. [Google Scholar] [CrossRef] [Green Version]
- Moss, E.; Debeuckelaere, C.; Berl, V.; Elbayed, K.; Moussallieh, F.M.; Namer, I.J.; Lepoittevin, J.P. In Situ Metabolism of Cinnamyl Alcohol in Reconstructed Human Epidermis: New Insights into the Activation of This Fragrance Skin Sensitizer. Chem. Res. Toxicol. 2016, 29, 1172. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Gowda, R.R.; Malik, P. Silver nitrate-catalyzed oxidation of aldehydes to carboxylic acids by H2O2. Tetrahedron Lett. 2009, 50, 6553. [Google Scholar] [CrossRef]
- Zaczek, A.J.; Korter, T.M. Polymorphism in cis–trans Muconic Acid Crystals and the Role of C–H···O Hydrogen Bonds. Cryst. Growth Des. 2017, 17, 4458–4466. [Google Scholar] [CrossRef]
- Peperidou, A.; Kapoukranidou, D.; Kontogiorgis, C.; Hadjipavlou-Litina, D. Multitarget Molecular Hybrids of Cinnamic Acids. Molecules 2014, 19, 20197. [Google Scholar] [CrossRef] [PubMed]
- Kattan, S.W.; Nafie, M.S.; Elmgeed, G.A.; Alelwani, W.; Badar, M.; Tantawy, M.A. Molecular docking, antiproliferative activity and induction of apoptosis in human liver cancer cells treated with androstane derivatives: Implication of PI3K/AKT/mTOR pathway. J. Steroid Biochem. Mol. Biol. 2020, 198, 105604. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, A.A.M.; Boraei, A.T.A.; Barakat, A.; Nafie, M.S. Discovery of hydrazide-based pyridazino[4,5-b] indole scaffold as a new phosphoinositide 3-kinase (PI3K) inhibitor for breast cancer therapy. RSC Adv. 2020, 10, 19534–19541. [Google Scholar] [CrossRef] [PubMed]
- Di Yang, M.; Shen, X.B.; Hu, Y.S.; Chen, Y.Y.; Liu, X.H. Novel naphthalene-enoates: Design and anti-cancer activity through regulation cell autophagy. Biomed. Pharmacother. 2020, 113, 108747. [Google Scholar] [CrossRef] [PubMed]
- Uniprot. Available online: https://www.uniprot.org/uniprotkb/P48736/entry (accessed on 15 July 2020).
- EMBL-EBI. Available online: http://pfam.xfam.org/protein/P48736 (accessed on 15 July 2020).
- UniProt. Available online: https://www.uniprot.org/uniprotkb/P31749/entry (accessed on 15 July 2020).
- EMBL-EBI. Available online: http://pfam.xfam.org/protein/P31749 (accessed on 15 July 2020).
- Abdelnaby, R.M.; Rateb, H.S.; Ali, O.; Saad, A.S.; Nadeem, R.I.; Abou-Seri, S.M.; Amin, K.M.; Younis, N.S.; Abdelhady, R. Dual PI3K/Akt Inhibitors Bearing Coumarin-Thiazolidine Pharmacophores as Potential Apoptosis Inducers in MCF-7 Cells. Pharmaceuticals 2022, 15, 428. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.C.; Liu, Z.P. Design and Synthesis of Coumarin Derivatives as Novel PI3K Inhibitors. Anticancer Agents Med. Chem. 2017, 17, 395. [Google Scholar] [CrossRef] [PubMed]
- Long, H.-Z.; Cheng, Y.; Zhou, Z.-W.; Luo, H.-Y.; Wen, D.-D.; Gao, L.-C. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer’s Disease and Parkinson’s Disease. Front. Pharmacol. 2021, 12, 648636. [Google Scholar] [CrossRef] [PubMed]
- Bin, Z.; Jiaying, Q.; Yakun, Y.; Li, L.; Yu, L.; Xue, H.; Weizhong, Q.; Li, C. Mechanisms of cinnamic aldehyde against myocardial ischemia/hypoxia injury in vivo and in vitro: Involvement of regulating PI3K/AKT signaling pathway. Biomed. Pharmacother. 2022, 147, 753. [Google Scholar]
- Dotolo, S.; Cervellera, C.; Russo, M.; Russo, G.L.; Facchiano, A. Virtual Screening of Natural Compounds as Potential PI3K-AKT1 Signaling Pathway Inhibitors and Experimental Validation. Molecules 2021, 26, 492. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Hasan, S.E.; Abu Khalaf, R.; Bardaweel, S.K.; Hajjo, R.; Alqaisi, K.M.; Sweidan, K.A.; Al-Zuheiri, A.M. Molecular Modeling, Synthesis and Biological Evaluation of N-Phenyl-4-Hydroxy-6-Methyl-2-Quinolone-3-CarboxAmides as Anticancer Agents. Molecules 2020, 25, 5348. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Eric, A.B.; Paul, S. Understanding the Behavior of Halogens as Hydrogen Bond Acceptors. Crystal Growth Design 2001, 1, 277–290. [Google Scholar]
- Riley, K.E.; Tran, K.-A. Strength and Character of R–X···π Interactions Involving Aromatic Amino Acid Sidechains in Protein-Ligand Complexes Derived from Crystal Structures in the Protein Data Bank. Crystals 2017, 7, 273. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural Determinants of Phosphoinositide 3-Kinase Inhibition by Wortmannin, LY294002, Quercetin, Myricetin, and Staurosporine. Mol. Cell. 2000, 6, 909. [Google Scholar] [CrossRef]
- Kallan, N.C.; Spencer, K.L.; Blake, J.F.; Xu, R.; Heizer, J.; Bencsik, J.R.; Mitchell, I.S.; Gloor, S.L.; Martinson, M.; Risom, T.; et al. Discovery and SAR of spirochromane Akt inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2410. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Voegtli, W.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. Chem. Med. Chem. 2016, 11, 1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- pkCSM. Available online: https://biosig.lab.uq.edu.au/pkcsm/ (accessed on 25 August 2022).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000085563-ABCB1/tissue (accessed on 25 August 2022).
- Freshney, R.I. Culture of tumor cells. In Culture of Animal Cells: A Manual of Basic Technique; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; ISBN 978-0-470-64936-7. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55. [Google Scholar] [CrossRef]
- Sharma, M.G.; Pandya, J.; Patel, D.M.; Vala, R.M.; Ramkumar, V.; Subramanian, R.; Gupta, V.K.; Gardas, R.L.; Dhanasekaran, A.; Patel, H.M. One-Pot Assembly for Synthesis of 1,4-Dihydropyridine Scaffold and Their Biological Applications. Polycyclic Aromat. Compd. 2021, 41, 1495. [Google Scholar] [CrossRef]
- Swiss TargetPrediction. Available online: http://www.swisstargetprediction.ch/ (accessed on 3 August 2020).
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455. [Google Scholar] [CrossRef] [PubMed]
- Center of Computational Structural Biology (CCSB). AutoDock Vina Documentation. Release 1.2.0; Center of Computational Structural Biology (CCSB): La Jolla, CA, USA, 2022. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605. [Google Scholar] [CrossRef]
- Swissadme. Available online: http://www.swissadme.ch/ (accessed on 20 August 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | IC50 (µM) ± SEM *# | |||||
|---|---|---|---|---|---|---|
| Leukemia Cancer HL60 | Liver Cancer HepG2 | Breast Cancer MCF-7 | Lung Cancer A549 | Normal Liver THLE2 | Normal WISH Cells | |
| 3 | 42.55 ± 0.22 | 95.65 ± 1.13 | 29.3 ± 1.02 | 16.5 ± 1.98 | - | - |
| 4 | 8.09 ± 0.13 | 71.3 ± 1.52 | 3.26 ± 0.12 | 9.34 ± 0.34 | 116.6 ± 3.34 | 89.6 ± 2.34 |
| 6 | 88.21 ± 0.51 | 36.58 ± 0.19 | 28.6 ± 0.91 | 27.6 ± 1.95 | - | - |
| 8a | 34.8 ± 0.64 | 75.2 ± 1.39 | 39.3 ± 1.69 | 19.6 ± 0.89 | - | - |
| 8b | 68.95 ± 2.14 | 13.14 ± 0.22 | 7.35 ± 0.67 | 4.63 ± 0.16 | 86.5 ± 2.16 | 96.8 ± 3.21 |
| 8c | 48.1 ± 0.88 | 17.4 ± 0.19 | 67.3 ± 2.03 | 24.3 ± 0.37 | 124 ± 2.12 | - |
| Staurosporine | 7.48 ± 0.11 | 10.24 ± 0.16 | 3.06 ± 0.42 | 3.7 ± 0.09 | 73.2 ± 2.8 | 69.7 ± 2.4 |
| Compound | (4) | (8b) | Staurosporine (Positive PI3K Control) | Spirochromane Derivative (Positive AKT1 Control) | Coumarin (Negative Control) |
|---|---|---|---|---|---|
| Heavy atoms | 16 | 26 | 35 | 38 | 11 |
| Rotatable bonds | 0 | 4 | 2 | 10 | 0 |
| H-bond donors | 1 | 0 | 2 | 2 | 0 |
| H-bond acceptors | 3 | 4 | 4 | 9 | 2 |
| Fraction Csp3 | 0.10 | 0.05 | 0.32 | 0.61 | 0.00 |
| Silicos-IT | −5.89 | −8.62 | −7.59 | −5.08 | −3.59 |
| XLogP3 | 3.75 | 5.94 | 3.24 | 1.66 | 1.39 |
| Molar refractivity | 72.57 | 111.66 | 139.39 | 150.15 | 42.48 |
| Gene | Forward | Reverse |
|---|---|---|
| P53 | 5′-CCCCTCCTGGCCCCTGTCATCTTC-3′ | 5′-GCAGCGCCTCACAACCTCCGTCAT-3′ |
| BAX | 5′-GTTTCATCCAGGATCGAGCAG-3′ | 5′-CATCTTCTTCCAGATGGTGA-3′ |
| CASP3 | 5′-TGGCCCTGAAATACGAAGTC-3′ | 5′-GGCAGTAGTCGACTCTGAAG-3′ |
| CASP9 | 5′-CGAACTAACAGGCAAGCAGC-3′ | 5′-ACCTCACCAAATCCTCCAGAAC-3′ |
| PI3K | 5′-CTCTCCTGTGCTGGCTACTGT-3′ | 5′-GCTCTCGGTTGATTCCAAACT-3′ |
| AKT | 5′-GGACAAGGACGGGCACATTA-3′ | 5′-CGACCGCACATCATCTCGTA-3′ |
| BCL2 | 5′-CCTGTGGATGACTGAGTACC-3′ | 5′-GAGACAGCCAGGAGAAATCA-3′ |
| β-actin | 5′-GTGACATCCACACCCAGAGG-3′ | 5′-ACAGGATGTCAAAACTGCCC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kishk, S.M.; Eltamany, E.E.; Nafie, M.S.; Khinkar, R.M.; Hareeri, R.H.; Elhady, S.S.; Yassen, A.S.A. Design and Synthesis of Coumarin Derivatives as Cytotoxic Agents through PI3K/AKT Signaling Pathway Inhibition in HL60 and HepG2 Cancer Cells. Molecules 2022, 27, 6709. https://doi.org/10.3390/molecules27196709
Kishk SM, Eltamany EE, Nafie MS, Khinkar RM, Hareeri RH, Elhady SS, Yassen ASA. Design and Synthesis of Coumarin Derivatives as Cytotoxic Agents through PI3K/AKT Signaling Pathway Inhibition in HL60 and HepG2 Cancer Cells. Molecules. 2022; 27(19):6709. https://doi.org/10.3390/molecules27196709
Chicago/Turabian StyleKishk, Safaa M., Enas E. Eltamany, Mohamed S. Nafie, Roaa M. Khinkar, Rawan H. Hareeri, Sameh S. Elhady, and Asmaa S. A. Yassen. 2022. "Design and Synthesis of Coumarin Derivatives as Cytotoxic Agents through PI3K/AKT Signaling Pathway Inhibition in HL60 and HepG2 Cancer Cells" Molecules 27, no. 19: 6709. https://doi.org/10.3390/molecules27196709
APA StyleKishk, S. M., Eltamany, E. E., Nafie, M. S., Khinkar, R. M., Hareeri, R. H., Elhady, S. S., & Yassen, A. S. A. (2022). Design and Synthesis of Coumarin Derivatives as Cytotoxic Agents through PI3K/AKT Signaling Pathway Inhibition in HL60 and HepG2 Cancer Cells. Molecules, 27(19), 6709. https://doi.org/10.3390/molecules27196709