
Nek2 Kinase Signaling in Malaria, Bone, Immune and Kidney Disorders to Metastatic Cancers and Drug Resistance: Progress on Nek2 Inhibitor Development

 ,
, 

Abstract
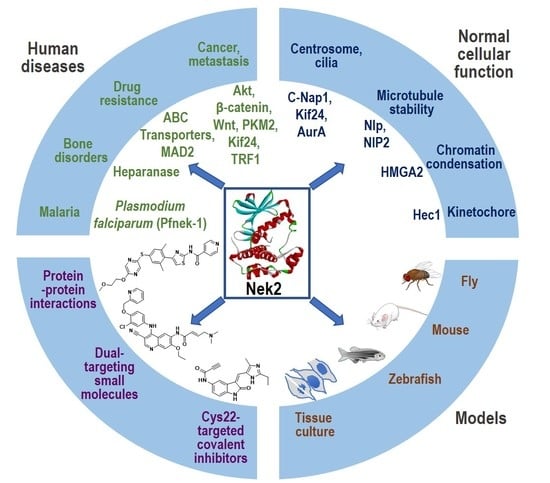
1. Introduction
2. Nek2 Signaling in Normal Human Physiology
3. The Nek2 Kinase in Polycystic Kidney Diseases (PKDs), Chromosomal Instability and Drug Resistance, Bone Destruction, and the DNA Damage Response Pathway
4. The Nek2 Kinase in Oncogenesis and Metastatic Signaling
5. Global Proteomic and Phosphoproteomic Data and Activation of the Nek2 Kinase in Cancers
6. Nek2 Ortholog in Malaria
7. Small Molecule Inhibitors of the Human Nek2 Kinase
- (a)
- (b)
- (c)
- (d)
- (e)
- (f)
- Type V: Chemotypes that occupy two distinct sites at once. This type of inhibitor has been further divided into two subcategories. These are (i) Bisubstrate analog inhibitors, which span over the ATP and substrate-binding site and (ii) Bivalent inhibitors, which span over the ATP-binding site along with any other site on the protein except at the substrate binding site [179,182].
- (g)
- Type VI: Chemotypes with a built-in electrophilic warhead that trap the accessible nucleophilic protein residue to form a covalent adduct with the target kinase [179].
8. ATP-Site Binding Inhibitors of Nek2
8.1. Pyrrole-Indoline-Based Ligand
8.2. Thiophene-Based Ligands
8.3. Viridin/Wortmannin-like Ligands
8.4. Aminopyrazine Inhibitors
8.5. Benzimidazole Inhibitors (DFG-Out)
8.6. Oxyindole-Propynamide Inhibitors
8.7. Aminopyridine Inhibitors
8.8. Quinoline-Based Inhibitors
8.9. Pyrimidine Inhibitors
8.10. Imidazo[1,2-a] Pyridine Inhibitors
8.11. Purine Inhibitors
8.12. Protein-Protein Interaction (PPI) Inhibitors
9. Expert Opinion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Janne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.J.; Krien, M.J.; Hunter, T. Never say never. The NIMA-related protein kinases in mitotic control. Trends Cell Biol. 2003, 13, 221–228. [Google Scholar] [CrossRef]
- Morris, N.R. Mitotic mutants of Aspergillus nidulans. Genet. Res. 1975, 26, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Hames, R.S.; Fry, A.M. Alternative splice variants of the human centrosome kinase Nek2 exhibit distinct patterns of expression in mitosis. Biochem. J. 2002, 361, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Fardilha, M.; Wu, W.; Sa, R.; Fidalgo, S.; Sousa, C.; Mota, C.; da Cruz e Silva, O.A.; da Cruz e Silva, E.F. Alternatively spliced protein variants as potential therapeutic targets for male infertility and contraception. Ann. N. Y. Acad. Sci. 2004, 1030, 468–478. [Google Scholar] [CrossRef]
- Hayward, D.G.; Fry, A.M. Nek2 kinase in chromosome instability and cancer. Cancer Lett. 2006, 237, 155–166. [Google Scholar] [CrossRef]
- Wu, W.; Baxter, J.E.; Wattam, S.L.; Hayward, D.G.; Fardilha, M.; Knebel, A.; Ford, E.M.; da Cruz e Silva, E.F.; Fry, A.M. Alternative splicing controls nuclear translocation of the cell cycle-regulated Nek2 kinase. J. Biol. Chem. 2007, 282, 26431–26440. [Google Scholar] [CrossRef]
- Fry, A.M.; Arnaud, L.; Nigg, E.A. Activity of the human centrosomal kinase, Nek2, depends on an unusual leucine zipper dimerization motif. J. Biol. Chem. 1999, 274, 16304–16310. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Cleveland, D.W. The role of aneuploidy in promoting and suppressing tumors. J. Cell Biol. 2009, 185, 935–937. [Google Scholar] [CrossRef]
- Bannon, J.H.; Mc Gee, M.M. Understanding the role of aneuploidy in tumorigenesis. Biochem. Soc. Trans. 2009, 37, 910–913. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Q.; Hirohashi, Y.; Du, X.; Greene, M.I.; Wang, Q. Nek2 targets the mitotic checkpoint proteins Mad2 and Cdc20: A mechanism for aneuploidy in cancer. Exp. Mol. Pathol. 2010, 88, 225–233. [Google Scholar] [CrossRef]
- Williams, B.R.; Amon, A. Aneuploidy: Cancer’s fatal flaw? Cancer Res. 2009, 69, 5289–5291. [Google Scholar] [CrossRef] [PubMed]
- Mayor, T.; Hacker, U.; Stierhof, Y.D.; Nigg, E.A. The mechanism regulating the dissociation of the centrosomal protein C-Nap1 from mitotic spindle poles. J. Cell Sci. 2002, 115, 3275–3284. [Google Scholar] [CrossRef]
- Yoo, J.C.; Chang, J.R.; Kim, S.H.; Jang, S.K.; Wolgemuth, D.J.; Kim, K.; Rhee, K. NIP1/XB51/NECAB3 is a potential substrate of Nek2, suggesting specific roles of Nek2 in Golgi. Exp. Cell Res. 2004, 292, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Graf, R. DdNek2, the first non-vertebrate homologue of human Nek2, is involved in the formation of microtubule-organizing centers. J. Cell Sci. 2002, 115, 1919–1929. [Google Scholar] [CrossRef]
- Fletcher, L.; Cerniglia, G.J.; Nigg, E.A.; Yend, T.J.; Muschel, R.J. Inhibition of centrosome separation after DNA damage: A role for Nek2. Radiat. Res. 2004, 162, 128–135. [Google Scholar] [CrossRef]
- Di Agostino, S.; Fedele, M.; Chieffi, P.; Fusco, A.; Rossi, P.; Geremia, R.; Sette, C. Phosphorylation of high-mobility group protein A2 by Nek2 kinase during the first meiotic division in mouse spermatocytes. Mol. Biol. Cell 2004, 15, 1224–1232. [Google Scholar] [CrossRef]
- Wang, S.; Li, W.; Lv, S.; Wang, Y.; Liu, Z.; Zhang, J.; Liu, T.; Niu, Y. Abnormal expression of Nek2 and beta-catenin in breast carcinoma: Clinicopathological correlations. Histopathology 2011, 59, 631–642. [Google Scholar] [CrossRef]
- Tsunoda, N.; Kokuryo, T.; Oda, K.; Senga, T.; Yokoyama, Y.; Nagino, M.; Nimura, Y.; Hamaguchi, M. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009, 100, 111–116. [Google Scholar] [CrossRef]
- Hayward, D.G.; Clarke, R.B.; Faragher, A.J.; Pillai, M.R.; Hagan, I.M.; Fry, A.M. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. 2004, 64, 7370–7376. [Google Scholar] [CrossRef]
- Li, J.; Zhan, Q. The role of centrosomal Nlp in the control of mitotic progression and tumourigenesis. Br. J. Cancer 2011, 104, 1523–1528. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Kaplan, D.D.; Deluca, J.G.; Giddings, T.H., Jr.; O’Toole, E.T.; Winey, M.; Salmon, E.D.; Casey, P.J.; Nelson, W.J.; Barth, A.I. beta-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 2008, 22, 91–105. [Google Scholar] [CrossRef]
- Rapley, J.; Baxter, J.E.; Blot, J.; Wattam, S.L.; Casenghi, M.; Meraldi, P.; Nigg, E.A.; Fry, A.M. Coordinate regulation of the mother centriole component nlp by nek2 and plk1 protein kinases. Mol. Cell. Biol. 2005, 25, 1309–1324. [Google Scholar] [CrossRef]
- Frett, B.; Brown, R.V.; Ma, M.; Hu, W.; Han, H.; Li, H.Y. Therapeutic melting pot of never in mitosis gene a related kinase 2 (Nek2): A perspective on Nek2 as an oncology target and recent advancements in Nek2 small molecule inhibition. J. Med. Chem. 2014, 57, 5835–5844. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.F.; Zhang, X.W. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef]
- Fry, A.M.; O’Regan, L.; Sabir, S.R.; Bayliss, R. Cell cycle regulation y the NEK family of protein kinases. J. Cell Sci. 2012, 125, 4423–4433. [Google Scholar] [CrossRef]
- Hardy, T.; Lee, M.; Hames, R.S.; Prosser, S.L.; Cheary, D.M.; Samant, M.D.; Schultz, F.; Baxter, J.E.; Rhee, K.; Fry, A.M. Multisite phosphorylation of C-Nap1 releases it from Cep135 to trigger centrosome disjunction. J. Cell Sci. 2014, 127, 2493–2506. [Google Scholar] [CrossRef] [PubMed]
- Mardin, B.R.; Lange, C.; Baxter, J.E.; Hardy, T.; Scholz, S.R.; Fry, A.M.; Schiebel, E. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat. Cell Biol. 2010, 12, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Adamian, M.; Li, T. Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol. Biol. Cell 2006, 17, 1033–1040. [Google Scholar] [CrossRef]
- Graser, S.; Stierhof, Y.D.; Nigg, E.A. Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J. Cell Sci. 2007, 120, 4321–4331. [Google Scholar] [CrossRef]
- Helps, N.R.; Luo, X.; Barker, H.M.; Cohen, P.T. NIMA-related kinase 2 (Nek2), a cell-cycle-regulated protein kinase localized to centrosomes, is complexed to protein phosphatase 1. Biochem. J. 2000, 349, 509–518. [Google Scholar] [CrossRef]
- Schmucker, S.; Sumara, I. Molecular dynamics of PLK1 during mitosis. Mol. Cell Oncol. 2014, 1, e954507. [Google Scholar] [CrossRef]
- Liu, X. Targeting Polo-Like Kinases: A Promising Therapeutic Approach for Cancer Treatment. Transl. Oncol. 2015, 8, 185–195. [Google Scholar] [CrossRef]
- Mardin, B.R.; Agircan, F.G.; Lange, C.; Schiebel, E. Plk1 controls the Nek2A-PP1γ antagonism in centrosome disjunction. Curr. Biol. 2011, 21, 1145–1151. [Google Scholar] [CrossRef]
- Fry, A.M.; Mayor, T.; Meraldi, P.; Stierhof, Y.D.; Tanaka, K.; Nigg, E.A. C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J. Cell Biol. 1998, 141, 1563–1574. [Google Scholar] [CrossRef]
- Bahe, S.; Stierhof, Y.D.; Wilkinson, C.J.; Leiss, F.; Nigg, E.A. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J. Cell Biol. 2005, 171, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Jeong, A.L.; Lee, S.; Park, J.S.; Han, S.; Jang, C.Y.; Lim, J.S.; Lee, M.S.; Yang, Y. Cancerous inhibitor of protein phosphatase 2A (CIP2A) protein is involved in centrosome separation through the regulation of NIMA (never in mitosis gene A)-related kinase 2 (NEK2) protein activity. J. Biol. Chem. 2014, 289, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Pimanda, J.E.; Westermarck, J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res. 2013, 73, 6548–6553. [Google Scholar] [CrossRef] [PubMed]
- Harrison Pitner, M.K.; Saavedra, H.I. Cdk4 and nek2 signal binucleation and centrosome amplification in a her2+ breast cancer model. PLoS ONE 2013, 8, e65971. [Google Scholar]
- Du, J.; Cai, X.; Yao, J.; Ding, X.; Wu, Q.; Pei, S.; Jiang, K.; Zhang, Y.; Wang, W.; Shi, Y.; et al. The mitotic checkpoint kinase NEK2A regulates kinetochore microtubule attachment stability. Oncogene 2008, 27, 4107–4114. [Google Scholar] [CrossRef]
- Lou, Y.; Yao, J.; Zereshki, A.; Dou, Z.; Ahmed, K.; Wang, H.; Hu, J.; Wang, Y.; Yao, X. NEK2A interacts with MAD1 and possibly functions as a novel integrator of the spindle checkpoint signaling. J. Biol. Chem. 2004, 279, 20049–20057. [Google Scholar] [CrossRef]
- Wei, R.; Ngo, B.; Wu, G.; Lee, W.H. Phosphorylation of the Ndc80 complex protein, HEC1, by Nek2 kinase modulates chromosome alignment and signaling of the spindle assembly checkpoint. Mol. Biol. Cell 2011, 22, 3584–3594. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Riley, D.J.; Zheng, L.; Chen, P.L.; Lee, W.H. Phosphorylation of the mitotic regulator protein Hec1 by Nek2 kinase is essential for faithful chromosome segregation. J. Biol. Chem. 2002, 277, 49408–49416. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Qiu, X.L.; Zhou, L.; Zhu, J.; Chamberlin, R.; Lau, J.; Chen, P.L.; Lee, W.H. Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res. 2008, 68, 8393–8399. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wei, W.; Ye, T.; Liu, Z.; Liu, L.; Luo, Y.; Zhang, L.; Gao, C.; Wang, N.; Yu, L. Small Molecule TH-39 Potentially Targets Hec1/Nek2 Interaction and Exhibits Antitumor Efficacy in K562 Cells via G0/G1 Cell Cycle Arrest and Apoptosis Induction. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 40, 297–308. [Google Scholar] [CrossRef]
- Hu, C.M.; Zhu, J.; Guo, X.E.; Chen, W.; Qiu, X.L.; Ngo, B.; Chien, R.; Wang, Y.V.; Tsai, C.Y.; Wu, G.; et al. Novel small molecules disrupting Hec1/Nek2 interaction ablate tumor progression by triggering Nek2 degradation through a death-trap mechanism. Oncogene 2015, 34, 1220–1230. [Google Scholar] [CrossRef]
- Hames, R.S.; Crookes, R.E.; Straatman, K.R.; Merdes, A.; Hayes, M.J.; Faragher, A.J.; Fry, A.M. Dynamic recruitment of Nek2 kinase to the centrosome involves microtubules, PCM-1, and localized proteasomal degradation. Mol. Biol. Cell 2005, 16, 1711–1724. [Google Scholar] [CrossRef] [PubMed]
- Dammermann, A.; Merdes, A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 2002, 159, 255–266. [Google Scholar] [CrossRef]
- Andersen, J.S.; Wilkinson, C.J.; Mayor, T.; Mortensen, P.; Nigg, E.A.; Mann, M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003, 426, 570–574. [Google Scholar] [CrossRef]
- Casenghi, M.; Meraldi, P.; Weinhart, U.; Duncan, P.I.; Korner, R.; Nigg, E.A. Polo-like kinase 1 regulates Nlp, a centrosome protein involved in microtubule nucleation. Dev. Cell 2003, 5, 113–125. [Google Scholar] [CrossRef]
- Jeong, Y.; Lee, J.; Kim, K.; Yoo, J.C.; Rhee, K. Characterization of NIP2/centrobin, a novel substrate of Nek2, and its potential role in microtubule stabilization. J. Cell Sci. 2007, 120, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Rhee, K. NEK2 phosphorylation antagonizes the microtubule stabilizing activity of centrobin. Biochem. Biophys. Res. Commun. 2013, 431, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, J.M.; Urquhart, A.J.; Subramaniam, V.N.; Parton, R.G.; Khanna, K.K. Centrobin regulates the assembly of functional mitotic spindles. Oncogene 2010, 29, 2649–2658. [Google Scholar] [CrossRef]
- Nachury, M.V.; Mick, D.U. Establishing and regulating the composition of cilia for signal transduction. Nat. Rev. Mol. Cell Biol. 2019, 20, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Basten, S.G.; Giles, R.H. Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia 2013, 2, 6. [Google Scholar] [CrossRef]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef]
- Spalluto, C.; Wilson, D.I.; Hearn, T. Nek2 localises to the distal portion of the mother centriole/basal body and is required for timely cilium disassembly at the G2/M transition. Eur. J. Cell Biol. 2012, 91, 675–686. [Google Scholar] [CrossRef]
- Kim, S.; Lee, K.; Choi, J.H.; Ringstad, N.; Dynlacht, B.D. Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle. Nat. Commun. 2015, 6, 8087. [Google Scholar] [CrossRef] [PubMed]
- Viol, L.; Hata, S.; Pastor-Peidro, A.; Neuner, A.; Murke, F.; Wuchter, P.; Ho, A.D.; Giebel, B.; Pereira, G. Nek2 kinase displaces distal appendages from the mother centriole prior to mitosis. J. Cell Biol. 2020, 219, e201907136. [Google Scholar] [CrossRef] [PubMed]
- DeVaul, N.; Koloustroubis, K.; Wang, R.; Sperry, A.O. A novel interaction between kinase activities in regulation of cilia formation. BMC Cell Biol. 2017, 18, 33. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Mahjoub, M.R.; Trapp, M.L.; Quarmby, L.M. NIMA-related kinases defective in murine models of polycystic kidney diseases localize to primary cilia and centrosomes. J. Am. Soc. Nephrol. 2005, 16, 3485–3489. [Google Scholar] [CrossRef]
- Mahjoub, M.R.; Qasim Rasi, M.; Quarmby, L.M. A NIMA-related kinase, Fa2p, localizes to a novel site in the proximal cilia of Chlamydomonas and mouse kidney cells. Mol. Biol. Cell 2004, 15, 5172–5186. [Google Scholar] [CrossRef] [PubMed]
- DeVaul, N.; Wang, R.; Sperry, A.O. PPP1R42, a PP1 binding protein, regulates centrosome dynamics in ARPE-19 cells. Biol. Cell 2013, 105, 359–371. [Google Scholar] [CrossRef][Green Version]
- Endicott, S.J.; Basu, B.; Khokha, M.; Brueckner, M. The NIMA-like kinase Nek2 is a key switch balancing cilia biogenesis and resorption in the development of left-right asymmetry. Development 2015, 142, 4068–4079. [Google Scholar] [PubMed]
- Parker, J.D.; Bradley, B.A.; Mooers, A.O.; Quarmby, L.M. Phylogenetic analysis of the Neks reveals early diversification of ciliary-cell cycle kinases. PLoS ONE 2007, 2, e1076. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, Y.; Xia, J.; Wang, H.; Salama, M.E.; Xiong, W.; Xu, H.; Shetty, S.; Chen, T.; Zeng, Z.; et al. NEK2 Induces Drug Resistance Mainly through Activation of Efflux Drug Pumps and Is Associated with Poor Prognosis in Myeloma and Other Cancers. Cancer Cell 2013, 23, 48–62. [Google Scholar] [CrossRef]
- Das, T.K.; Dana, D.; Paroly, S.S.; Perumal, S.K.; Singh, S.; Jhun, H.; Pendse, J.; Cagan, R.L.; Talele, T.T.; Kumar, S. Centrosomal kinase Nek2 cooperates with oncogenic pathways to promote metastasis. Oncogenesis 2013, 2, e69. [Google Scholar] [CrossRef]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Choi, C.H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef][Green Version]
- Misra, S.; Ghatak, S.; Toole, B.P. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J. Biol. Chem. 2005, 280, 20310–20315. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Liu, Z.; Wei, Y.; Lin-Lee, Y.C.; Tatebe, S.; Mills, G.B.; Unate, H. Induction of human MDR1 gene expression by 2-acetylaminofluorene is mediated by effectors of the phosphoinositide 3-kinase pathway that activate NF-kappaB signaling. Oncogene 2002, 21, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, W.; Xia, J.; Gu, Z.; Wendlandt, E.; Zhan, X.; Janz, S.; Tricot, G.; Zhan, F. NEK2 mediates ALDH1A1-dependent drug resistance in multiple myeloma. Oncotarget 2014, 5, 11986–11997. [Google Scholar] [CrossRef] [PubMed]
- Marina, M.; Saavedra, H.I. Nek2 and Plk4: Prognostic markers, drivers of breast tumorigenesis and drug resistance. Front. Biosci. 2014, 19, 352–365. [Google Scholar] [CrossRef]
- Hao, M.; Franqui-Machin, R.; Xu, H.; Shaughnessy, J.; Barlogie, B.; Roodman, D.; Quelle, D.E.; Janz, S.; Tomasson, M.H.; Sanderson, R.D.; et al. NEK2 induces osteoclast differentiation and bone destruction via heparanase in multiple myeloma. 2017, 31, 1648–1650. Leukemia 2017, 31, 1648–1650. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Turgeon, M.O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8, 15. [Google Scholar] [CrossRef]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Derheimer, F.A.; Kastan, M.B. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 2010, 584, 3675–3681. [Google Scholar] [CrossRef] [PubMed]
- Pavan, I.C.B.; Peres de Oliveira, A.; Dias, P.R.F.; Basei, F.L.; Issayama, L.K.; Ferezin, C.C.; Silva, F.R.; Rodrigues de Oliveira, A.L.; Alves Dos Reis Moura, L.; Martins, M.B.; et al. On Broken Ne(c)ks and Broken DNA: The Role of Human NEKs in the DNA Damage Response. Cells 2021, 10, 507. [Google Scholar] [CrossRef]
- Fletcher, L.; Yen, T.J.; Muschel, R.J. DNA damage in HeLa cells induced arrest at a discrete point in G2 phase as defined by CENP-F localization. Radiat. Res. 2003, 159, 604–611. [Google Scholar] [CrossRef]
- Mi, J.; Guo, C.; Brautigan, D.L.; Larner, J.M. Protein phosphatase-1alpha regulates centrosome splitting through Nek2. Cancer Res. 2007, 67, 1082–1089. [Google Scholar] [CrossRef]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef]
- Seo, J.; Seong, D.; Lee, S.R.; Oh, D.B.; Song, J. Post-Translational Regulation of ARF: Perspective in CancerPost-Translational Regulation of ARF: Perspective in Cancer. Biomolecules 2020, 10, 1143. [Google Scholar] [CrossRef]
- Lee, J.; Gollahon, L. Mitotic perturbations induced by Nek2 overexpression require interaction with TRF1 in breast cancer cells. Cell Cycle 2013, 12, 3599–3614. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prime, G.; Markie, D. The telomere repeat binding protein Trf1 interacts with the spindle checkpoint protein Mad1 and Nek2 mitotic kinase. Cell Cycle 2005, 4, 121–124. [Google Scholar] [CrossRef]
- Walker, J.R.; Zhu, X.D. Post-translational modifications of TRF1 and TRF2 and their roles in telomere maintenance. Mech. Ageing Dev. 2012, 133, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Linardopoulos, S.; Turner, N.C. Tumour selective targeting of cell cycle kinases for cancer treatment. Curr. Opin. Pharm. 2013, 13, 529–535. [Google Scholar] [CrossRef]
- Camidge, D.R. Cell cycle-associated kinases as targets for therapy in lung cancer. J. Thorac. Oncol. 2010, 5, S461–S462. [Google Scholar] [CrossRef]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jang, C.; Lee, K.A. Polo-like kinases (plks), a key regulator of cell cycle and new potential target for cancer therapy. Dev. Reprod. 2014, 18, 65–71. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, G.D. Targeting serine/threonine protein kinase B/Akt and cell-cycle checkpoint kinases for treating cancer. Curr. Top. Med. Chem. 2002, 2, 939–971. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle kinases in cancer. Curr. Opin. Genet. Dev. 2007, 17, 60–65. [Google Scholar] [CrossRef]
- Moniz, L.; Dutt, P.; Haider, N.; Stambolic, V. Nek family of kinases in cell cycle, checkpoint control and cancer. Cell Div. 2011, 6, 18. [Google Scholar] [CrossRef]
- Pitts, T.M.; Davis, S.L.; Eckhardt, S.G.; Bradshaw-Pierce, E.L. Targeting nuclear kinases in cancer: Development of cell cycle kinase inhibitors. Pharmacol. Ther. 2014, 142, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.; Sutherland, R.L.; Musgrove, E.A. Inhibitors of cell cycle kinases: Recent advances and future prospects as cancer therapeutics. Crit. Rev. Oncog. 2012, 17, 175–198. [Google Scholar] [CrossRef]
- Fry, A.M.; Bayliss, R.; Roig, J. Mitotic Regulation by NEK Kinase Networks. Front. Cell Dev. Biol. 2017, 5, 102. [Google Scholar]
- Fry, A.M. The Nek2 protein kinase: A novel regulator of centrosome structure. Oncogene 2002, 21, 6184–6194. [Google Scholar] [CrossRef]
- Fry, A.M.; Meraldi, P.; Nigg, E.A. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998, 17, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Feng, L.; Zhu, M.; Shen, Q.; Fang, Y.; Liu, X.; Zhang, X. NEK2 promotes proliferation, migration and tumor growth of gastric cancer cells via regulating KDM5B/H3K4me3. Am. J. Cancer Res. 2019, 9, 2364–2378. [Google Scholar]
- Rivera-Rivera, Y.; Marina, M.; Jusino, S.; Lee, M.; Velazquez, J.V.; Chardon-Colon, C.; Vargas, G.; Padmanabhan, J.; Chellappan, S.P.; Saavedra, H.I. The Nek2 centrosome-mitotic kinase contributes to the mesenchymal state, cell invasion, and migration of triple-negative breast cancer cells. Sci. Rep. 2021, 11, 9016. [Google Scholar] [CrossRef]
- Stricker, T.P.; Henriksen, K.J.; Tonsgard, J.H.; Montag, A.G.; Krausz, T.N.; Pytel, P. Expression profiling of 519 kinase genes in matched malignant peripheral nerve sheath tumor/plexiform neurofibroma samples is discriminatory and identifies mitotic regulators BUB1B, PBK and NEK2 as overexpressed with transformation. Mod. Pathol. 2013, 26, 930–943. [Google Scholar] [CrossRef]
- Ning, Z.; Wang, A.; Liang, J.; Liu, J.; Zhou, T.; Yan, Q.; Wang, Z. Abnormal expression of Nek2 in pancreatic ductal adenocarcinoma: A novel marker for prognosis. Int. J. Clin. Exp. Pathol. 2014, 7, 2462–2469. [Google Scholar]
- Barbagallo, F.; Paronetto, M.P.; Franco, R.; Chieffi, P.; Dolci, S.; Fry, A.M.; Geremia, R.; Sette, C. Increased expression and nuclear localization of the centrosomal kinase Nek2 in human testicular seminomas. J. Pathol. 2009, 217, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Iwaya, T.; Sawada, G.; Kurashige, J.; Matsumura, T.; Uchi, R.; Ueo, H.; Takano, Y.; Eguchi, H.; Sudo, T.; et al. Up-regulation of NEK2 by microRNA-128 methylation is associated with poor prognosis in colorectal cancer. Ann. Surg. Oncol. 2014, 21, 205–212. [Google Scholar] [CrossRef]
- Wai, D.H.; Schaefer, K.L.; Schramm, A.; Korsching, E.; Van Valen, F.; Ozaki, T.; Boecker, W.; Schweigerer, L.; Dockhorn-Dworniczak, B.; Poremba, C. Expression analysis of pediatric solid tumor cell lines using oligonucleotide microarrays. Int. J. Oncol. 2002, 20, 441–451. [Google Scholar] [CrossRef][Green Version]
- Liu, X.; Gao, Y.; Lu, Y.; Zhang, J.; Li, L.; Yin, F. Upregulation of NEK2 is associated with drug resistance in ovarian cancer. Oncol. Rep. 2014, 31, 745–754. [Google Scholar] [CrossRef]
- Kokuryo, T.; Senga, T.; Yokoyama, Y.; Nagino, M.; Nimura, Y.; Hamaguchi, M. Nek2 as an effective target for inhibition of tumorigenic growth and peritoneal dissemination of cholangiocarcinoma. Cancer Res. 2007, 67, 9637–9642. [Google Scholar] [CrossRef] [PubMed]
- Kokuryo, T.; Yokoyama, Y.; Yamaguchi, J.; Tsunoda, N.; Ebata, T.; Nagino, M. NEK2 Is an Effective Target for Cancer Therapy With Potential to Induce Regression of Multiple Human Malignancies. Anticancer Res. 2019, 39, 2251–2258. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Guan, X.; Dong, Q.; Yang, S.; Liu, W.; Zhang, L. Examining Nek2 as a better proliferation marker in non-small cell lung cancer prognosis. Tumour Biol. 2014, 35, 7155–7162. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Guan, X.; Liu, W.; Zhang, L. Aberrant expression of NEK2 and its clinical significance in non-small cell lung cancer. Oncol. Lett. 2014, 8, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Zhou, G.X.; Li, X.Y.; Zhang, Q.; Zhao, K.; Zhang, C.P.; Xue, C.H.; Yang, K.; Tian, Z.B. Effects of the hippo signaling pathway in human gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 5199–5205. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Dong, J. The Hippo Signaling Pathway in Drug Resistance in Cancer. Cancers 2021, 13, 318. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Weber, U.; Mlodzik, M. APC/C(Fzr/Cdh1)-Dependent Regulation of Planar Cell Polarity Establishment via Nek2 Kinase Acting on Dishevelled. Dev. Cell 2017, 40, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.; Meghini, F.; Florio, F.; Kimata, Y. The APC/C Coordinates Retinal Differentiation with G1 Arrest through the Nek2-Dependent Modulation of Wingless Signaling. Dev. Cell 2017, 40, 67–80. [Google Scholar] [CrossRef]
- Kim, W.; Cho, Y.S.; Wang, X.; Park, O.; Ma, X.; Kim, H.; Gan, W.; Jho, E.H.; Cha, B.; Jeung, Y.J.; et al. Hippo signaling is intrinsically regulated during cell cycle progression by APC/C(Cdh1). Proc. Natl. Acad. Sci. USA 2019, 116, 9423–9432. [Google Scholar] [CrossRef]
- Nelson, W.J.; Nusse, R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.D.; Meigs, T.E.; Kelly, P.; Casey, P.J. Identification of a role for beta-catenin in the establishment of a bipolar mitotic spindle. J. Biol. Chem. 2004, 279, 10829–10832. [Google Scholar] [CrossRef]
- Kolligs, F.T.; Bommer, G.; Goke, B. Wnt/beta-catenin/tcf signaling: A critical pathway in gastrointestinal tumorigenesis. Digestion 2002, 66, 131–144. [Google Scholar] [CrossRef]
- Leung, J.Y.; Kolligs, F.T.; Wu, R.; Zhai, Y.; Kuick, R.; Hanash, S.; Cho, K.R.; Fearon, E.R. Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J. Biol. Chem. 2002, 277, 21657–21665. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Wu, R.; Schwartz, D.R.; Darrah, D.; Reed, H.; Kolligs, F.T.; Nieman, M.T.; Fearon, E.R.; Cho, K.R. Role of beta-catenin/T-cell factor-regulated genes in ovarian endometrioid adenocarcinomas. Am. J. Pathol. 2002, 160, 1229–1238. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Mbom, B.C.; Siemers, K.A.; Ostrowski, M.A.; Nelson, W.J.; Barth, A.I. Nek2 phosphorylates and stabilizes β-catenin at mitotic centrosomes downstream of Plk1. Mol. Biol. Cell 2014, 25, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zeng, Y.; Shi, L.; Yang, Q.; Chen, Y.; Wu, G.; Li, G.; Xu, S. Targeting NEK2 impairs oncogenesis and radioresistance via inhibiting the Wnt1/beta-catenin signaling pathway in cervical cancer. J. Exp. Clin. Cancer Res. 2020, 39, 183. [Google Scholar] [CrossRef]
- Neal, C.P.; Fry, A.M.; Moreman, C.; McGregor, A.; Garcea, G.; Berry, D.P.; Manson, M.M. Overexpression of the Nek2 kinase in colorectal cancer correlates with beta-catenin relocalization and shortened cancer-specific survival. J. Surg. Oncol. 2014, 110, 828–838. [Google Scholar] [CrossRef]
- Kaowinn, S.; Oh, S.; Moon, J.; Yoo, A.Y.; Kang, H.Y.; Lee, M.R.; Kim, J.E.; Hwang, D.Y.; Youn, S.E.; Koh, S.S.; et al. CGK062, a small chemical molecule, inhibits cancer upregulated gene 2induced oncogenesis through NEK2 and betacatenin. Int. J. Oncol. 2019, 54, 1295–1305. [Google Scholar] [PubMed]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Esernio, J.; Cagan, R.L. Restraining Network Response to Targeted Cancer Therapies Improves Efficacy and Reduces Cellular Resistance. Cancer Res. 2018, 78, 4344–4359. [Google Scholar] [CrossRef]
- Rellos, P.; Ivins, F.J.; Baxter, J.E.; Pike, A.; Nott, T.J.; Parkinson, D.M.; Das, S.; Howell, S.; Fedorov, O.; Shen, Q.Y.; et al. Structure and regulation of the human Nek2 centrosomal kinase. J. Biol. Chem. 2007, 282, 6833–6842. [Google Scholar] [CrossRef]
- Emmitte, K.A.; Adjabeng, G.M.; Andrews, C.W.; Alberti, J.G.; Bambal, R.; Chamberlain, S.D.; Davis-Ward, R.G.; Dickson, H.D.; Hassler, D.F.; Hornberger, K.R.; et al. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorg. Med. Chem. Lett. 2009, 19, 1694–1697. [Google Scholar] [CrossRef]
- Hayward, D.G.; Newbatt, Y.; Pickard, L.; Byrne, E.; Mao, G.; Burns, S.; Sahota, N.K.; Workman, P.; Collins, I.; Aherne, W.; et al. Identification by high-throughput screening of viridin analogs as biochemical and cell-based inhibitors of the cell cycle-regulated nek2 kinase. J. Biomol. Screen 2010, 15, 918–927. [Google Scholar] [CrossRef]
- Whelligan, D.K.; Solanki, S.; Taylor, D.; Thomson, D.W.; Cheung, K.M.; Boxall, K.; Mas-Droux, C.; Barillari, C.; Burns, S.; Grummitt, C.G.; et al. Aminopyrazine inhibitors binding to an unusual inactive conformation of the mitotic kinase Nek2: SAR and structural characterization. J. Med. Chem. 2010, 53, 7682–7698. [Google Scholar] [CrossRef] [PubMed]
- Solanki, S.; Innocenti, P.; Mas-Droux, C.; Boxall, K.; Barillari, C.; van Montfort, R.L.; Aherne, G.W.; Bayliss, R.; Hoelder, S. Benzimidazole inhibitors induce a DFG-out conformation of never in mitosis gene A-related kinase 2 (Nek2) without binding to the back pocket and reveal a nonlinear structure-activity relationship. J. Med. Chem. 2011, 54, 1626–1639. [Google Scholar] [CrossRef] [PubMed]
- Henise, J.C.; Taunton, J. Irreversible Nek2 kinase inhibitors with cellular activity. J. Med. Chem. 2011, 54, 4133–4146. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, P.; Cheung, K.M.; Solanki, S.; Mas-Droux, C.; Rowan, F.; Yeoh, S.; Boxall, K.; Westlake, M.; Pickard, L.; Hardy, T.; et al. Design of potent and selective hybrid inhibitors of the mitotic kinase Nek2: Structure-activity relationship, structural biology, and cellular activity. J. Med. Chem. 2012, 55, 3228–3241. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Carpenter, K.; Mollard, A.; Vankayalapati, H.; Warner, S.L.; Sharma, S.; Tricot, G.; Zhan, F.; Bearss, D.J. Inhibition of Nek2 by small molecules affects proteasome activity. BioMed Res. Int. 2014, 2014, 273180. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Kong, Y.; Xi, J.; Zhu, M.; Zhu, T.; Jiang, T.; Hu, W.; Ma, M.; Zhang, X. Preclinical activity of MBM-5 in gastrointestinal cancer by inhibiting NEK2 kinase activity. Oncotarget 2016, 7, 79327–79341. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, P.; Pavlyukov, M.S.; Yu, H.; Zhang, Z.; Kim, S.H.; Minata, M.; Mohyeldin, A.; Xie, W.; Chen, D.; et al. Targeting NEK2 attenuates glioblastoma growth and radioresistance by destabilizing histone methyltransferase EZH2. J. Clin. Investig. 2017, 127, 3075–3089. [Google Scholar] [CrossRef]
- Coxon, C.R.; Wong, C.; Bayliss, R.; Boxall, K.; Carr, K.H.; Fry, A.M.; Hardcastle, I.R.; Matheson, C.J.; Newell, D.R.; Sivaprakasam, M.; et al. Structure-guided design of purine-based probes for selective Nek2 inhibition. Oncotarget 2017, 8, 19089–19124. [Google Scholar] [CrossRef]
- Matheson, C.J.; Coxon, C.R.; Bayliss, R.; Boxall, K.; Carbain, B.; Fry, A.M.; Hardcastle, I.R.; Harnor, S.J.; Mas-Droux, C.; Newell, D.R.; et al. 2-Arylamino-6-ethynylpurines are cysteine-targeting irreversible inhibitors of Nek2 kinase. RSC Med. Chem. 2020, 11, 707–731. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhu, Y.; Ran, K.; Liu, Z.; Wang, N.; Feng, Q.; Zeng, J.; Zhang, L.; He, B.; Ye, T.; et al. Synthesis and biological evaluation of N-(4-phenylthiazol-2-yl)cinnamamide derivatives as novel potential anti-tumor agents. Med. Chem. Commun. 2015, 6, 1036–1042. [Google Scholar] [CrossRef]
- Huang, L.Y.; Lee, Y.S.; Huang, J.J.; Chang, C.C.; Chang, J.M.; Chuang, S.H.; Kao, K.J.; Tsai, Y.J.; Tsai, P.Y.; Liu, C.W.; et al. Characterization of the biological activity of a potent small molecule Hec1 inhibitor TAI-1. J. Exp. Clin. Cancer Res. 2014, 33, 6. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Chuang, S.H.; Huang, L.Y.; Lai, C.L.; Lin, Y.H.; Yang, J.Y.; Liu, C.W.; Yang, S.C.; Lin, H.S.; Chang, C.C.; et al. Discovery of 4-aryl-N-arylcarbonyl-2-aminothiazoles as Hec1/Nek2 inhibitors. Part I: Optimization of in vitro potencies and pharmacokinetic properties. J. Med. Chem. 2014, 57, 4098–4110. [Google Scholar] [CrossRef]
- Huang, L.Y.; Chang, C.C.; Lee, Y.S.; Chang, J.M.; Huang, J.J.; Chuang, S.H.; Kao, K.J.; Lau, G.M.; Tsai, P.Y.; Liu, C.W.; et al. Activity of a novel Hec1-targeted anticancer compound against breast cancer cell lines in vitro and in vivo. Mol. Cancer Ther. 2014, 13, 1419–1430. [Google Scholar] [CrossRef]
- Huang, L.Y.; Chang, C.C.; Lee, Y.S.; Huang, J.J.; Chuang, S.H.; Chang, J.M.; Kao, K.J.; Lau, G.M.; Tsai, P.Y.; Liu, C.W.; et al. Inhibition of Hec1 as a novel approach for treatment of primary liver cancer. Cancer Chemother. Pharmacol. 2014, 74, 511–520. [Google Scholar] [CrossRef]
- Chuang, S.-H.; Lee, Y.-S.E.; Huang, L.Y.L.; Chen, C.-K.; Lai, C.-L.; Lin, Y.-H.; Yang, J.-Y.; Yang, S.-C.; Chang, L.-H.; Chen, C.-H.; et al. Discovery of T-1101 tosylate as a first-in-class clinical candidate for Hec1/Nek2 inhibition in cancer therapy. Eur. J. Med. Chem. 2020, 191, 112118. [Google Scholar] [CrossRef] [PubMed]
- Deb, B.; Sengupta, P.; Sambath, J.; Kumar, P. Bioinformatics Analysis of Global Proteomic and Phosphoproteomic Data Sets Revealed Activation of NEK2 and AURKA in Cancers. Biomolecules 2020, 10, 237. [Google Scholar] [CrossRef]
- World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019; Licence: CC BY-NC-SA 3.0 IGO.
- White, N.J. Antimalarial drug resistance. J. Clin. Investig. 2004, 113, 1084–1092. [Google Scholar] [CrossRef]
- Yeung, S.; Pongtavornpinyo, W.; Hastings, I.M.; Mills, A.J.; White, N.J. Antimalarial drug resistance, artemisinin-based combination therapy, and the contribution of modeling to elucidating policy choices. Am. J. Trop. Med. Hyg. 2004, 71, 179–186. [Google Scholar] [CrossRef]
- Conrad, M.D.; Rosenthal, P.J. Antimalarial drug resistance in Africa: The calm before the storm? Lancet Infect. Dis. 2019, 19, e338–e351. [Google Scholar] [CrossRef]
- Plowe, C.V. Antimalarial drug resistance in Africa: Strategies for monitoring and deterrence. Curr. Top. Microbiol. Immunol. 2005, 295, 55–79. [Google Scholar] [PubMed]
- Takala-Harrison, S.; Laufer, M.K. Antimalarial drug resistance in Africa: Key lessons for the future. Ann. N. Y. Acad. Sci. 2015, 1342, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Solyakov, L.; Halbert, J.; Alam, M.M.; Semblat, J.P.; Dorin-Semblat, D.; Reininger, L.; Bottrill, A.R.; Mistry, S.; Abdi, A.; Fennell, C.; et al. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2011, 2, 565. [Google Scholar] [CrossRef] [PubMed]
- Doerig, C.; Billker, O.; Haystead, T.; Sharma, P.; Tobin, A.B.; Waters, N.C. Protein kinases of malaria parasites: An update. Trends Parasitol. 2008, 24, 570–577. [Google Scholar] [CrossRef]
- Doerig, C.D. Stopping malaria parasites dead in their tracks. Nat. Chem. Biol. 2008, 4, 334–335. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, J.M.; Doerig, C. The protein-phosphatome of the human malaria parasite Plasmodium falciparum. BMC Genom. 2008, 9, 412. [Google Scholar] [CrossRef]
- Ward, P.; Equinet, L.; Packer, J.; Doerig, C. Protein kinases of the human malaria parasite Plasmodium falciparum: The kinome of a divergent eukaryote. BMC Genom. 2004, 5, 79. [Google Scholar] [CrossRef]
- Le Roch, K.G.; Zhou, Y.; Blair, P.L.; Grainger, M.; Moch, J.K.; Haynes, J.D.; De La Vega, P.; Holder, A.A.; Batalov, S.; Carucci, D.J.; et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 2003, 301, 1503–1508. [Google Scholar] [CrossRef]
- Dorin-Semblat, D.; Schmitt, S.; Semblat, J.P.; Sicard, A.; Reininger, L.; Goldring, D.; Patterson, S.; Quashie, N.; Chakrabarti, D.; Meijer, L.; et al. Plasmodium falciparum NIMA-related kinase Pfnek-1: Sex specificity and assessment of essentiality for the erythrocytic asexual cycle. Microbiology 2011, 157, 2785–2794. [Google Scholar] [CrossRef]
- Reininger, L.; Garcia, M.; Tomlins, A.; Muller, S.; Doerig, C. The Plasmodium falciparum. Nima-related kinase Pfnek-4: A marker for asexual parasites committed to sexual differentiation. Malar. J. 2012, 11, 250. [Google Scholar] [CrossRef] [PubMed]
- Dorin, D.; Le Roch, K.; Sallicandro, P.; Alano, P.; Parzy, D.; Poullet, P.; Meijer, L.; Doerig, C. Pfnek-1, a NIMA-related kinase from the human malaria parasite Plasmodium falciparum Biochemical properties and possible involvement in MAPK regulation. Eur. J. Biochem. FEBS 2001, 268, 2600–2608. [Google Scholar] [CrossRef] [PubMed]
- Laurent, D.; Jullian, V.; Parenty, A.; Knibiehler, M.; Dorin, D.; Schmitt, S.; Lozach, O.; Lebouvier, N.; Frostin, M.; Alby, F.; et al. Antimalarial potential of xestoquinone, a protein kinase inhibitor isolated from a Vanuatu marine sponge Xestospongia sp. Bioorg. Med. Chem. 2006, 14, 4477–4482. [Google Scholar] [CrossRef] [PubMed]
- Desoubzdanne, D.; Marcourt, L.; Raux, R.; Chevalley, S.; Dorin, D.; Doerig, C.; Valentin, A.; Ausseil, F.; Debitus, C. Alisiaquinones and alisiaquinol, dual inhibitors of Plasmodium falciparum enzyme targets from a New Caledonian deep water sponge. J. Nat. Prod. 2008, 71, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Tamaoki, T.; Nomoto, H.; Takahashi, I.; Kato, Y.; Morimoto, M.; Tomita, F. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem. Biophys. Res. Commun. 1986, 135, 397–402. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “gatekeeper door”: Exploiting the active kinase conformation. J. Med. Chem. 2010, 53, 2681–2694. [Google Scholar] [CrossRef]
- Gavrin, L.K.; Saiah, E. Approaches to discover non-ATP site kinase inhibitors. Med. Chem. Commun. 2013, 4, 41–51. [Google Scholar] [CrossRef]
- Lamba, V.; Ghosh, I. New directions in targeting protein kinases: Focusing upon true allosteric and bivalent inhibitors. Curr. Pharm. Des. 2012, 18, 2936–2945. [Google Scholar] [CrossRef]
- Heryanto, B.; Lipson, K.E.; Rogers, P.A. Effect of angiogenesis inhibitors on oestrogen-mediated endometrial endothelial cell proliferation in the ovariectomized mouse. Reproduction 2003, 125, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liang, C.; Shirazian, S.; Zhou, Y.; Miller, T.; Cui, J.; Fukuda, J.Y.; Chu, J.Y.; Nematalla, A.; Wang, X.; et al. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J. Med. Chem. 2003, 46, 1116–1119. [Google Scholar] [PubMed]
- Liao, A.T.; Chien, M.B.; Shenoy, N.; Mendel, D.B.; McMahon, G.; Cherrington, J.M.; London, C.A. Inhibition of constitutively active forms of mutant kit by multitargeted indolinone tyrosine kinase inhibitors. Blood 2002, 100, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.A.; Biggs, W.H., 3rd; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- McInnes, C.; Mezna, M.; Fischer, P.M. Progress in the discovery of polo-like kinase inhibitors. Curr. Top. Med. Chem. 2005, 5, 181–197. [Google Scholar] [CrossRef]
- Liu, Y.; Shreder, K.R.; Gai, W.; Corral, S.; Ferris, D.K.; Rosenblum, J.S. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem. Biol. 2005, 12, 99–107. [Google Scholar] [CrossRef]
- Wipf, P.; Halter, R.J. Chemistry and biology of wortmannin. Org. Biomol. Chem. 2005, 3, 2053–2061. [Google Scholar] [CrossRef]
- Meraldi, P.; Nigg, E.A. Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J. Cell Sci. 2001, 114, 3749–3757. [Google Scholar] [CrossRef]
- Liu, R.; Yue, Z.; Tsai, C.C.; Shen, J. Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J. Am. Chem. Soc. 2019, 141, 6553–6560. [Google Scholar] [CrossRef]
- Wymann, M.P.; Bulgarelli-Leva, G.; Zvelebil, M.J.; Pirola, L.; Vanhaesebroeck, B.; Waterfield, M.D.; Panayotou, G. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol. Cell. Biol. 1996, 16, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Singla, P.; Luxami, V.; Paul, K. Benzimidazole-biologically attractive scaffold for protein kinase inhibitors. RSC Adv. 2014, 4, 12422–12440. [Google Scholar] [CrossRef]
- Singh, J.; Dobrusin, E.M.; Fry, D.W.; Haske, T.; Whitty, A.; McNamara, D.J. Structure-based design of a potent, selective, and irreversible inhibitor of the catalytic domain of the erbB receptor subfamily of protein tyrosine kinases. J. Med. Chem. 1997, 40, 1130–1135. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Kluge, A.F. Targeted covalent drugs of the kinase family. Curr. Opin. Chem. Biol. 2010, 14, 475–480. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef]
- Leproult, E.; Barluenga, S.; Moras, D.; Wurtz, J.M.; Winssinger, N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: Application to the design of selective covalent inhibitors. J. Med. Chem. 2011, 54, 1347–1355. [Google Scholar] [CrossRef]
- Cohen, M.S.; Zhang, C.; Shokat, K.M.; Taunton, J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005, 308, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Hadjivassiliou, H.; Taunton, J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat. Chem. Biol. 2007, 3, 156–160. [Google Scholar] [CrossRef]
- Zhou, W.; Hur, W.; McDermott, U.; Dutt, A.; Xian, W.; Ficarro, S.B.; Zhang, J.; Sharma, S.V.; Brugge, J.; Meyerson, M.; et al. A structure-guided approach to creating covalent FGFR inhibitors. Chem. Biol. 2010, 17, 285–295. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef]
- Doehn, U.; Hauge, C.; Frank, S.R.; Jensen, C.J.; Duda, K.; Nielsen, J.V.; Cohen, M.S.; Johansen, J.V.; Winther, B.R.; Lund, L.R.; et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol. Cell 2009, 35, 511–522. [Google Scholar] [CrossRef]
- Franqui-Machin, R.; Hao, M.; Bai, H.; Gu, Z.; Zhan, X.; Habelhah, H.; Jethava, Y.; Qiu, L.; Frech, I.; Tricot, G.; et al. Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J. Clin. Investig. 2018, 128, 2877–2893. [Google Scholar] [CrossRef] [PubMed]
- Lebraud, H.; Coxon, C.R.; Archard, V.S.; Bawn, C.M.; Carbain, B.; Matheson, C.J.; Turner, D.M.; Cano, C.; Griffin, R.J.; Hardcastle, I.R.; et al. Model system for irreversible inhibition of Nek2: Thiol addition to ethynylpurines and related substituted heterocycles. Org. Biomol. Chem. 2014, 12, 141–148. [Google Scholar] [CrossRef]
- Qiu, X.L.; Li, G.; Wu, G.; Zhu, J.; Zhou, L.; Chen, P.L.; Chamberlin, A.R.; Lee, W.H. Synthesis and biological evaluation of a series of novel inhibitor of Nek2/Hec1 analogues. J. Med. Chem. 2009, 52, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Hames, R.S.; Wattam, S.L.; Yamano, H.; Bacchieri, R.; Fry, A.M. APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 2001, 20, 7117–7127. [Google Scholar] [CrossRef] [PubMed]
- Serafimova, I.M.; Pufall, M.A.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frodin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. [Google Scholar] [CrossRef]
- Pei, H.; Peng, Y.; Zhao, Q.; Chen, Y. Small molecule PROTACs: An emerging technology for targeted therapy in drug discovery. Rsc. Adv. 2019, 9, 16967–16976. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- Dar, A.C.; Das, T.K.; Shokat, K.M.; Cagan, R.L. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 2012, 486, 80–84. [Google Scholar] [CrossRef]
- Pattni, B.S.; Torchilin, V.P. Targeted Drug Delivery Systems: Strategies and Challenges. In Targeted Drug Delivery: Concepts and Design; Devarajan, P.V., Jain, S., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–38. [Google Scholar]
- Lanning, B.R.; Whitby, L.R.; Dix, M.M.; Douhan, J.; Gilbert, A.M.; Hett, E.C.; Johnson, T.O.; Joslyn, C.; Kath, J.C.; Niessen, S.; et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 2014, 10, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Cagan, R.L. Non-mammalian models of multiple endocrine neoplasia type 2. Endocr. Relat. Cancer 2018, 25, T91–T104. [Google Scholar] [CrossRef]
- Giacomotto, J.; Segalat, L. High-throughput screening and small animal models, where are we? Br. J. Pharmacol. 2010, 160, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Rudrapatna, V.A.; Cagan, R.L.; Das, T.K. Drosophila cancer models. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2012, 241, 107–118. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry # | Chemical Structure | Inhibition Profile with the Compound I.D. (In Parenthesis) | Special Notes and Reference | Inhibitor Type |
|---|---|---|---|---|
| 1 |  | IC50 (SU11652): 8 µM IC50 (SU11248): 12 µM | The first reported non-selective and cell-permeable chemotype that binds to the inactive conformation of Nek2 [140]. | Type II |
| 2 |  | IC50 (2a): 21 µM IC50 (2b): 25 µM | Thiophene-based inhibitors, originally developed for targeting PLK1, emerged as potential Nek2 inhibitory scaffold [141]. | NA |
| 3 |  | IC50 (3a): 1.4 µM IC50 (3b): 4.4 µM | Cell-active Nek2 inhibitors that showed significant Nek2 selectivity when compared to Nek6 and Nek7 but not toward Aurora A, Plk 1, and Cdk1. Affected centrosome separation in Nek2-inducible human tumor cells [142]. | Type VI $ |
| 4 |  | IC50 (4): 0.23 µM | Inhibits Nek2 by inducing notable conformational changes, “Tyr-down”, in protein. Non-selective and suffers from poor cell-permeability [143]. | NA |
| 5 |  | IC50 (5): 0.36 µM | The first benzimidazole-type inhibitor to induce DFG-out conformation in Nek2 kinase. Although it exhibited remarkable selectivity over Plk1, it failed to maintain the selectivity when tested against other kinases. It also suffered from the lack of cellular potency [144]. | Type II |
| 6 |  | IC50 (6): 0.77 µM | The first developed irreversible inhibitor of Nek2 with cellular activity which covalently modifies the protein by trapping Cys22 [145]. | Type VI |
| 7 |  | IC50 (7): 0.022 µM | A cell permeable Nek2 inhibitor with improved selectivity that induces a DFG-out conformation of protein [146]. | Type II |
| 8 |  | IC50: (Pelitinib/EKB-569): 661 nM IC50: (Neratinib/HKI-272): 247 nM | Using a whole-animal-based Nek2 overexpression model in flies, two cell active EGFR inhibitors were found to be active against Nek2 kinase [69]. | NA |
| 9 |  | IC50 (HCI-2389): 0.016 µM | Highly potent and cell-active Nek2 inhibitor that effectively sensitized bortezomib-resistant multiple myeloma cells [147]. | Type VI |
| 10 |  | IC50 (MBM-5): 0.34 µM | Effectively inhibited Nek2 kinase in leukemia and gastric and colorectal cancer cell lines and retained its efficacy while being evaluated in vivo using MGC-803 gastric and HCT-116 xenografts in mouse models. Although promising, MBM-5 suffered from unimpressive pharmacokinetic profile [148]. | NA |
| 11 |  | IC50 (CMP3a): 0.082 µM | Disrupts the Nek2-EZH2 nexus in glioma stem cells and silenced tumor in xenotransplanted mouse. Poor pharmacokinetic profile of CMP3a only allowed it to be used as a chemical tool and not as a therapeutic modality [149]. | NA |
| 12. |  | IC50 (12a): 0.62 µM IC50 (12b): 0.27 µM | First inhibitor of its kind to invoke DFG-in configuration in Nek2 kinase [150]. Most potent analog of this series. Despite notable potency, it failed to become a therapeutic candidate due to instability reasons under physiological conditions. | Type I 1/2 |
| 13 |  | IC50 (13): 0.062 µM | A cell-active irreversible inhibitor of Nek2 kinase that showed modest selectivity profile over several other kinases; however, it lacked a desirable pharmacokinetic (PK) profile [151]. | Type VI |
| 14 |  | IC50 (INH1): NA | First discovered 2-aminothiazole-based PPI inhibitor of Hec1/Nek2 axis with cellular activity [45]. | NA |
| 15 |  | IC50 (15): NA | Second generation of cell-permeable thiazole derivatives that affected Hec1/Nek2 activity [47]. | NA |
| 16 |  | IC50 (INH154): NA | Third generation thiazole-derivative with little or no toxicity that suppressed tumor growth effectively in mouse xenograft model upon peritoneal administration [47]. | NA |
| 17 |  | IC50 (TH-39): NA | A cell-active 2-aminothiazole derivative that exhibited the hallmark of Hec1/Nek2 inhibition [46,152]. | NA |
| 18 |  | IC50 (TAI-1): NA | The first orally administered Hec1/Nek2 inhibitor—also effective when administered intravenously—with little or no adverse effect. It showed tremendous promise in suppressing tumor growth in mouse model [153]. | NA |
| 19 |  | IC50 (19): NA | Extremely potent Hec1/Nek2 disruptor that presented a high AUC when administered in SD rats [154]. | NA |
| 20 |   | IC50 (TAI-95): NA IC50 (T-1101 tosylate): NA | A very potent HEC1/Nek2 inhibitor with impressive pharmacological profile that is currently being evaluated in clinical trials [155,156,157]. | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dana, D.; Das, T.; Choi, A.; Bhuiyan, A.I.; Das, T.K.; Talele, T.T.; Pathak, S.K. Nek2 Kinase Signaling in Malaria, Bone, Immune and Kidney Disorders to Metastatic Cancers and Drug Resistance: Progress on Nek2 Inhibitor Development. Molecules 2022, 27, 347. https://doi.org/10.3390/molecules27020347
Dana D, Das T, Choi A, Bhuiyan AI, Das TK, Talele TT, Pathak SK. Nek2 Kinase Signaling in Malaria, Bone, Immune and Kidney Disorders to Metastatic Cancers and Drug Resistance: Progress on Nek2 Inhibitor Development. Molecules. 2022; 27(2):347. https://doi.org/10.3390/molecules27020347
Chicago/Turabian StyleDana, Dibyendu, Tuhin Das, Athena Choi, Ashif I. Bhuiyan, Tirtha K. Das, Tanaji T. Talele, and Sanjai K. Pathak. 2022. "Nek2 Kinase Signaling in Malaria, Bone, Immune and Kidney Disorders to Metastatic Cancers and Drug Resistance: Progress on Nek2 Inhibitor Development" Molecules 27, no. 2: 347. https://doi.org/10.3390/molecules27020347
APA StyleDana, D., Das, T., Choi, A., Bhuiyan, A. I., Das, T. K., Talele, T. T., & Pathak, S. K. (2022). Nek2 Kinase Signaling in Malaria, Bone, Immune and Kidney Disorders to Metastatic Cancers and Drug Resistance: Progress on Nek2 Inhibitor Development. Molecules, 27(2), 347. https://doi.org/10.3390/molecules27020347