Abstract
A family of oxazaborines, diazaborinones, triazaborines, and triazaborinones was prepared by reaction of polarized ethylenes, such as β-enaminoamides, with 4-methylbenzenediazonium tetraphenylborates. The reaction conditions (stirring in CH2Cl2 at room temperature (Method A) or stirring with CH3COONa in CH2Cl2 at room temperature (Method B) or refluxing in the CH2Cl2/toluene mixture (Method C)) controlled the formation and relative content of these compounds in the reaction mixtures from one to three products. Substituted oxazaborines gradually rearranged into diazaborinones at 250 °C. The prepared compounds were characterized by 1H NMR, 13C NMR, IR, and UV–Vis spectroscopy, HRMS, or microanalysis. The structure of individual compounds was confirmed by 11B NMR, 15N NMR, 1D NOESY, and X-ray analysis. The mechanism of reaction of enaminoamides with 4-methylbenzenediazonium tetraphenylborate was proposed.
1. Introduction
Activated alkenes, also known as polarized ethylenes, are molecules containing a double bond with the electron-withdrawing group (–C=O, –CN, –NO2, etc.) at one end and electron-donating groups (typically amino groups) at the other. A molecule with such an arrangement of groups is referred to as “push–pull” ethylene. Due to their versatile reactivity, they are important building blocks for the syntheses of a wide variety of different heterocyclic systems with practical usability. β-Enaminoamides 1 (Figure 1) also belong among representatives of the polarized ethylenes. The oxygen of the amide function donates the free electron pair, and the amino group in β-position of the double bond is able to withdraw it.
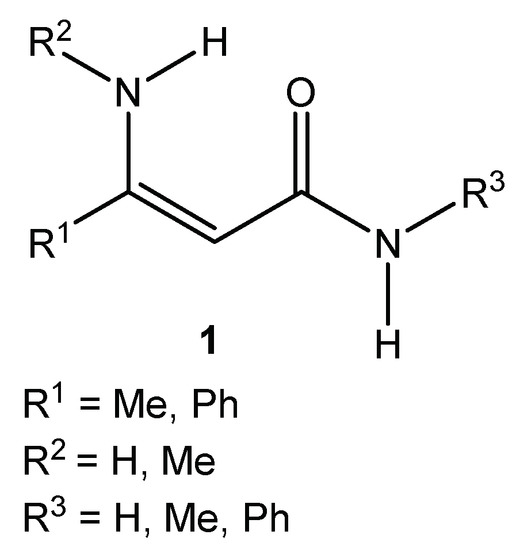
Figure 1.
General structure of simple enaminoamides.
Compounds 1 have been used for the synthesis of uracil derivatives that inhibit histone deacetylase (HDAC), and these compounds have been efficient in inhibiting human recombinant (hr) HDAC1 [1]. Compounds 1 have been also used in the synthesis of dihydropyridines [2,3,4,5,6], and these dihydropyridines have been tested in cloned human α adrenoceptors, as well as the rat L-type calcium channel [4] or platelet activating factor (PAF) antagonist activity was measured [3]. Enaminoamides 1 can be used in the synthesis of other types of heterocycles, for example, pyridines [7], pyridine-2-one [8], pyrimidines [9], pyrimidinones [10,11,12,13], spirocyclohexane [14], 1H-pyrrole-2,3-dione [15], 2-oxopyrrole [11], pyrazolone [16], and isothiazole [17]. The coordination ability of these simple compounds has not been studied yet. The first mention is from November 2017 [18,19], where the authors tested prepared oxazaborines (Scheme 1) for inhibition of the NLRP3 inflammasome in vitro and in vivo without affecting Ca2+ homeostasis.
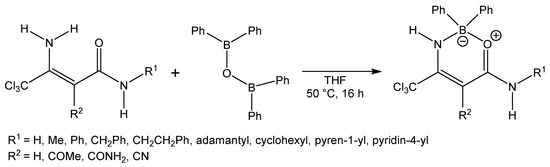
Scheme 1.
Synthesis of oxazaborines according to Baldwin et al.
We have studied the reactions of polarized ethylenes, such as β-enaminones, β-enaminonitriles, and β-enaminoamides, with substitution on the amino group, with diazonium salts, leading to heterocyclic compounds. These compounds are pyridazines [20,21,22], pyrazoles [23,24,25,26], oxazaborines, diazaborinones, and triazaborines [27,28,29,30,31]. In 2009, we published work on the reaction of β-enaminoamides 1 (Figure 1, R3 = Ph) and 4-methylbenzenediazonium tetraphenylborate to give compounds I–III (Scheme 2) [28]. The ring transformation of compound I to diazaborinones II (predominantly when R1 = Me) and triazaborines III (predominantly when R1 = H) was proceeded under reflux in N,N-dimethylformamide [28]. We decided to change the substitution at the nitrogen atom of the amide group of enaminoamides as the basis for this paper. In the new study on enaminoamide reactivity, we report on synthetic transformations leading to N–B–O, N–B–N, and newly also N–N–B–N–C(=O) heterocyclic systems using the reactions of β-enaminoamides 1 (Figure 1, R3 = H, Me) and diazonium salts.
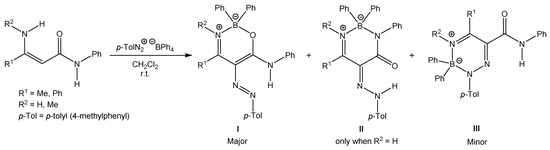
Scheme 2.
Reaction of N-phenylenaminoamides and 4-methylbenzenediazonium tetraphenylborate.
2. Results and Discussion
2.1. Synthesis
We prepared eight enaminoamides 1a–h (Supplementary Material, Scheme S1). The synthesis proceeded via an addition–elimination mechanism of the corresponding oxoamides with ammonia or methylamine. Oxoamides (except acetoacetamide (4a)) were obtained in two (compound 4b [32]) or three (compounds 4c and 4d) steps from ethyl benzoylacetate and ethyl acetoacetate, respectively. Compound 4b was prepared according to G. A. M. Giardina [32]. For the preparation of oxoamides 4c and 4d, the synthetic procedure according to B. Štefane and S. Polanc was used [33]. This method consists of activation of the carboxy group with BF3–Et2O, substitution of the alkoxy group, and hydrolysis to give oxoamide 4c and 4d, or aminolysis to give enaminoamide 1h.
Prepared enaminoamides 1a–h reacted with 4-methylbenezenediazonium tetraphenylborate under three different conditions: (1) Stirring in CH2Cl2 at room temperature (Method A), (2) Stirring with CH3COONa in CH2Cl2 at room temperature (Method B), and (3) Refluxing in the CH2Cl2/toluene mixture (Method C).
Figure 2 shows five possible product structures (A–E) for the reaction of enaminoamides 1a–h with 4-methylbenzenediazonium tetraphenylborate.
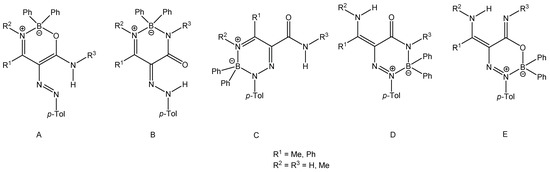
Figure 2.
Possible products of the reaction of enaminoamides 1a–h with 4-methylbenzenediazonium tetraphenylborate.
When β-enaminoamide 1c (Figure 1, R1 = R2 = Me, R3 = H) reacted with diazonium salt under heating with the mixture solvent (dichloromethane/toluene), the only product with a yield of 69% was isolated. The structure of this product was deduced on the basis of 1H, 13C, 11B, and 15N-NMR spectroscopy. In the aliphatic part of the 1H NMR spectrum of this product (Supplementary Material, Figure S12), there are three signals of three methyl groups. One of them is a doublet, and two of them are singlets. The presence of a doublet is only possible for structures D and E (Figure 2, R1 = R2 = Me, R3 = H). The 1D NOESY spectrum (Figure 3) helped us to exclude one of these suggested structures. The through-space interaction between the proton with δ = 5.52 ppm and the ortho protons of the phenyl groups on the boron atom was found after the selective excitation of this N–H proton (An analogue experiment was also carried out in the case of compounds 5a, 5b, 5g. Supplementary Material, Figures S2, S7 and S22). This interaction is not possible for structure E. The structure of triazaborinone D suggested on the basis of NMR experiments was subsequently confirmed by X-ray diffraction (Figure 4). This is the new type of heterocycle in the series of enaminoamides 1, where the substituent on the nitrogen of the amide group is not phenyl. In the case of N-phenylenaminoamides, triazaborinones were not isolated.
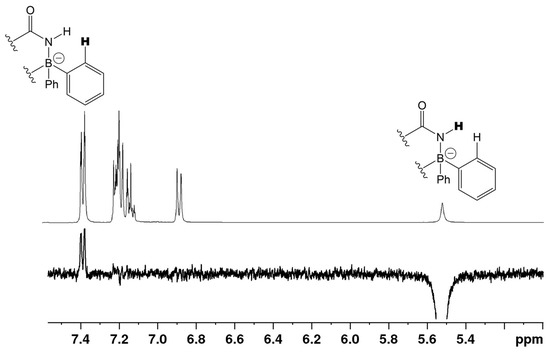
Figure 3.
The 400 MHz 1D-NOESY (mixing time d8 = 800 ms, relaxation delay d1 = 2 s) of compound 5c in CDCl3.
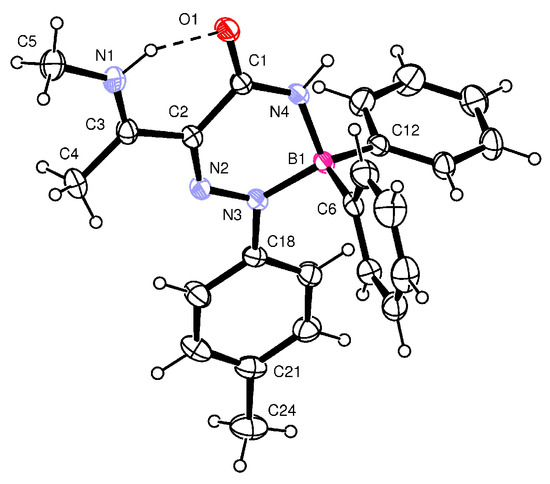
Figure 4.
ORTEP view and atom numbering scheme for 5c. Thermal ellipsoids are drawn at the 50% probability level.
We isolated the new type of boron heterocycle in six cases. When enaminoamides 1a–d, 1g, and 1h reacted with diazonium salt, the new triazaborinones 5 were obtained as major compounds together with oxazaborines 6, diazaborinones 7, or triazaborines 8 as minor products (Scheme 3). Oxazaborine 6a was also prepared from 3-aminobut-2-ennitrile and diazonium tetraphenylborate, as described in [30].
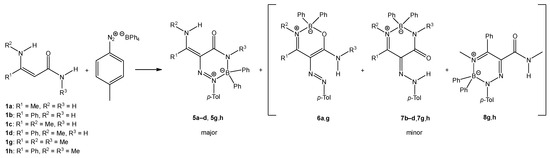
Scheme 3.
The reaction of enaminoamides 1a–d, 1g, and 1h.
When enaminoamides 1e and 1f reacted with diazonium tetraphenylborate, only oxazaborines and diazaborinones were isolated (Scheme 4). Triazaborine 8e was also isolated when enaminoamide 1e and diazonium salt were refluxed after stirring 30 min at room temperature. The yield of 8e was 26%.
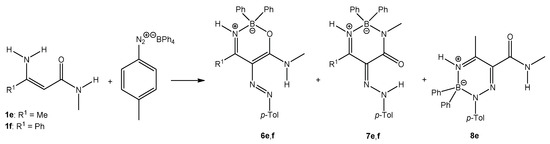
Scheme 4.
The reaction of enaminoamides 1e and 1f.
The yields of the prepared heterocyclic boron compounds are shown in Table 1, and their values are given either after crystallization or after washing with an appropriate solvent.

Table 1.
Yields of boron compounds prepared under three different reaction conditions.
We suggested the mechanism for the reaction of enaminones with diazonium tetraphenylborates [27]. In the first step, the azocoupling reaction proceeds in which tetraphenylborate acts as a base and during which the protodeboronation of Ph3B––[C6H6]+ occurs to give triphenylborane and benzene [27]. Triphenylborane is able to coordinate to the nitrogen atom of the amine group of enaminones followed by further protodeboronation and cyclization. In the case of enaminoamides, two nitrogen atoms are possible for coordination (amine group and amide group) of triphenylborane. Our previously studied enaminoamides having phenyl at amidic nitrogen (Figure 1, R3 = Ph) gave products of coordination of triphenylborane with the amine group of enaminoamides (Scheme 5, Path B). Enaminoamides 1 described in this paper gave different results. Triphenylborane likely bonded to amidic nitrogen (Scheme 5, Path A), and after protodeboronation and cyclization, the new type of heterocyclic compounds formed. Triazaborinones 5 are the main products in most cases. The by-product was mainly diazaborinone. Exceptions are reactions of enaminoamides 1e and 1f, having –CONHMe, and the primary amine group. The behavior of these compounds is similar to that of enaminoamides having –CONHPh studied earlier. Oxazaborines 6e and 6f, diazaborinones 7e and 7f, and triazaborine 8e were isolated when these enaminoamides were used.
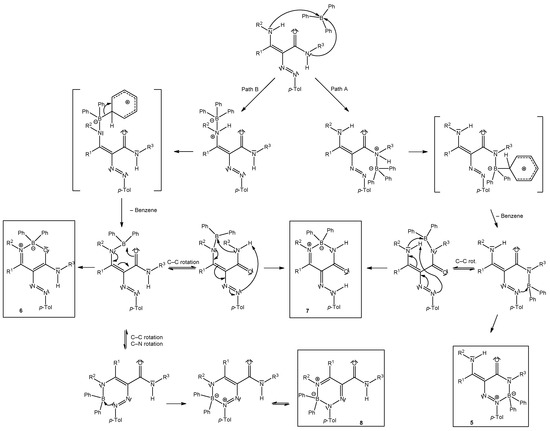
Scheme 5.
Suggested mechanism for the reaction of enaminoamides 1a–h with 4-methylbenzenediazonium tetraphenylborate.
Additionally, the products of double coordination of BPh3 (9g and 9h, Figure 5) were isolated in the case of the reaction of enaminoamides 1g and 1h with diazonium tetraphenylborate carried out in dichloromethane at room temperature (Method A) for 72 h and 96 h, respectively. The yield was 4% for 9g and 15% for 9h. Compound 9h was also obtained using the Method C (refluxing the mixture for 3 h), but the yield was only 3%. Chemical shifts of 11B and 15N are similar to those for oxazaborine 6h and triazaborinone 5h. The study of this type of structure will be the subject of further investigation.
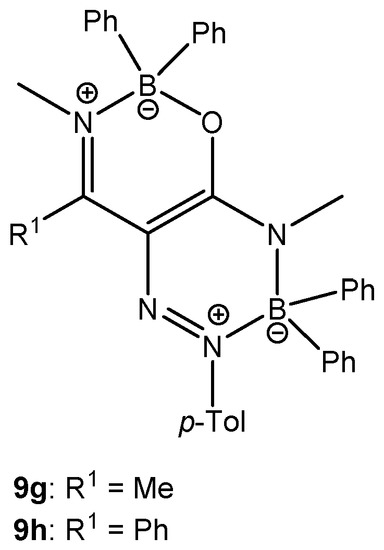
Figure 5.
Oxazaborine–triazaborines 9g and 9h.
When oxazaborines 6a and 6e–h were heated at 250 °C without solvent (Scheme 6), a ring transformation occurred to give the corresponding diazaborinones in good yields (49–82%). Diazaborinone 7a was not obtained from enaminoamide 1a and diazonium salt, while recyclization gave compound 7a in 82% yield. Here is the difference between the previously prepared oxazaborines I (Scheme 2) with the N-phenyl group and those in this publication, since only diazaborinones were isolated, and it does not matter what the substitution of the amino group of the starting enaminoamide is.
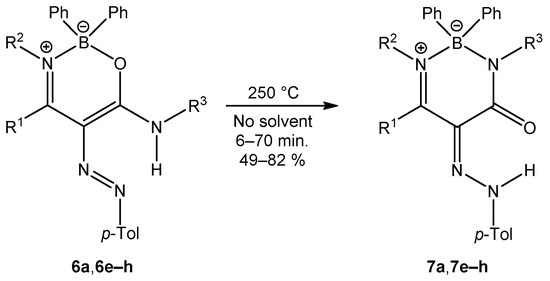
Scheme 6.
Ring transformation of oxazaborines 6.
NMR parameters such as 15N and 11B NMR chemical shifts (δ) and coupling constants (J) for heterocyclic compounds 5–9 are shown in Table 2.
Table 2.
NMR parameters for prepared boron heterocyclic compounds 5–9.
2.2. UV–Visible Spectroscopy
UV–Vis analyses of the compounds 5g, 6g, and 7a in various solvents (cyclohexane, diethyl ether, dichloromethane, tetrahydrofurane, acetonitrile, methanol, formamide) are shown in Figure 6 (5g, 7a) and Figure S112 (6g). All absorption spectra for prepared compounds in dichloromethane are listed in the Supplementary Materials (Figures S113–S124). The results of UV–Vis absorption data as an evaluated molar absorption coefficient (ε) for individual compounds in the solvents used are presented in Tables S1–S4. The dependence of the maximum absorption spectra (λMAX) on the solvent properties is not apparent from the above measurements. The possible solvatochromic effects are therefore rather insignificant.
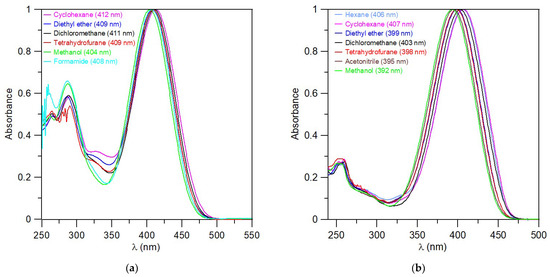
Figure 6.
Normalized UV–Vis spectra of triazaborinone 5g (a); and diazaborinone 7a (b).
2.3. IR Spectroscopy
The set of measured IR spectra for triazaborinones 5 is depicted in Figure 7, for oxazaborines 6 in Figure 8, for diazaborinones 7 in Figure 9, and for triazaborines 8 and oxazaborine–triazaborines 9 in Figure 10. Weak stretching vibrations of the C–H bonds of aromatic benzene rings at 3080–3000 cm−1 are evident in all spectra. Next, it is possible to mention the adjacent H–H out-of-plane deformation at the para-substituted ring at 820 cm−1 and several strong vibrations of the monosubstituted benzene ring, the out-of-plane ring bending at 706 cm−1, and the adjacent H–H out-of-plane deformation at 746 cm−1.
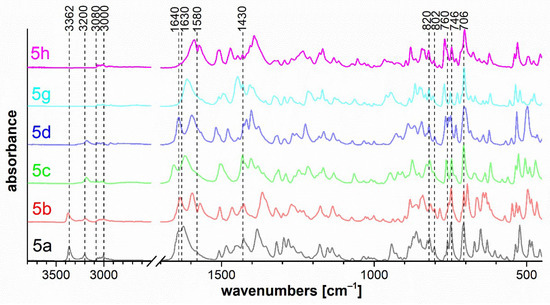
Figure 7.
FTIR ATR spectra of triazaborinones 5a–d, 5g, 5h.
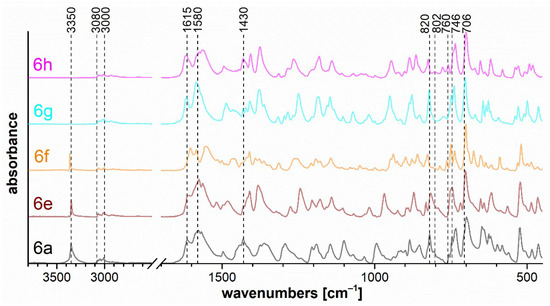
Figure 8.
FTIR ATR spectra of oxazaborines 6a, 6e–h.
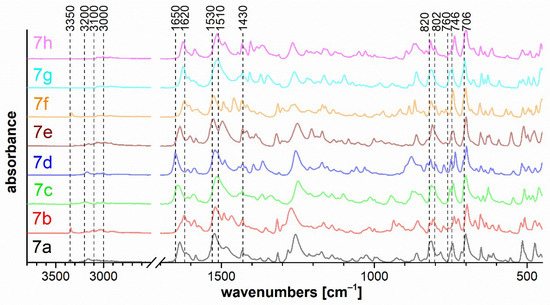
Figure 9.
FTIR ATR spectra of diazaborinones 7a–h.
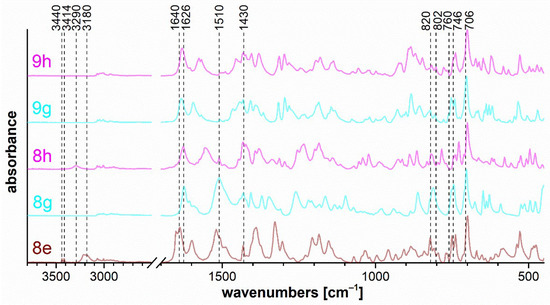
Figure 10.
FTIR ATR spectra of triazaborines 8e, 8g, and 8h, and oxazaborine–triazaborines 9g and 9h.
IR spectra 5a and 5b show the stretching vibration of the primary amino group at 3362 cm−1. The stretching vibration of the N–H and CO groups in the cis configuration is evident for spectra 5a–d at 3200 and 1640 cm−1, respectively. Strong stretching vibrations of the CO of α,β-unsaturated β-amino-substituted imide are located in the 1630–1580 cm−1 region. In this case, the effect of substitution on the double bond is evident. The methyl-substituted (R1 = Me) compounds 5a, 5c, and 5g are shifted to higher wavenumbers than the phenyl-substituted (R1 = Ph) compounds 5b, 5d, and 5h. The sharp band caused by the vibration of the ring at 1430 cm−1 is characteristic of compounds containing a B–phenyl bond. The ring C–H out-of-plane deformation for phenyl-substituted boron at 760 cm−1 is present as a shoulder. Two phenyl groups should appear as a doublet with a 20 cm−1 separation. These bands can be overlayed by the adjacent H–H out-of-plane deformation at 746 cm−1, as mentioned above.
Figure 8 shows the band at 3350 cm−1, which corresponds to the free N–H stretching of the imine bound to the oxazaborine ring. This is consistent with the fact that this vibration occurs only for compounds 6a, 6e, and 6f. In the case of 6a, this band is broadened due to the stretching vibration of the primary amino group.
The bands of the N–H stretching vibrations of the imine bonded to the diazaborinone ring at 3350 cm−1 for 7a, 7b, 7e, and 7f are shown in Figure 9. The stretching vibration of the N–H (spectra 7a–d) and CO (spectra 7a–h) groups in the cis configuration is evident in the range of 3200–3100 cm−1 and 1650–1620 cm−1, respectively. The spectra of all diazaborinones 7a–h contain very strong bands in the 1530–1510 cm−1 region, corresponding to an arrangement of atoms in a hydrazoketones (R1–CO–CR2N–NH–). For oxazaborines 6, Figure 8 shows two strong absorption bands—one at approximately 1615 cm−1 and another more intense near 1580 cm−1—due to vibrations of the conjugated system of the unsaturated double bond with the aromatic diazo group.
Triazaborine 8e (Figure 10) shows bands at 3440 and 3414 cm−1, which correspond to the free N–H stretching of imine bonded to the triazaborine ring. The stretching vibration of the N–H and CO groups in the cis configuration is evident for spectra 8e and 8h in the range of 3290–3180 cm−1 and 1640–1626 cm−1, respectively. The position of these bands is affected by methyl- or phenyl-substitution adjacent to the amide. The sharp band caused by the ring vibration at 1430 cm−1 is characteristic of compounds containing a B–phenyl bond and is evident in all cases in the Figure 10. The spectra of triazaborines 8g, 8e, and 8h contain very strong bands at 1510 cm−1, which corresponds to an arrangement of atoms in a hydrazoketones (R1–CO–CR2N–NH–).
2.4. X-ray
For compounds 5c, 5d, 5g, 6a, 6e, and 7f, we were able to prepare crystals suitable for single-crystal X-ray diffraction. The results are summarized in Table S5.
Triazaborinone 5c contains two types of hydrogen bonds. The first intramolecular bond is located between the N1 and O1 atoms. The other two intermolecular hydrogen bonds between N4 from the first molecule and O1 from the second molecule and vice versa bind the dimer-forming molecules. See Figure 11a for details. The plane passing through the atoms forming these intermolecular hydrogen bonds is shown in this Figure. The center of gravity of all dimer atoms is in the center of the hydrogen bond system. The point is marked as a centroid. The periodic arrangement of these basic dimers in the crystal is shown in Figures S125–S127 in the Supplementary Material. The view in the direction of the crystallographic axis a and c, and the reciprocal cell axis b* is shown. The conjugate system of three double bonds (between O1 and C1, C2 and N2, and N1 and C3 in one molecule) lies in a plane. The analogous plane from the second molecule of the dimer is parallel to the first plane and is shifted by 0.128 Å, as can be seen in Figure S128. Thus, all of these double bonds in the dimer lie practically in the plane. These bonds were assigned as doubles on the basis of their lengths, and the fact that they lie in a plane supports the indicated structure with a lower application of conjugation between the triazaborinone ring and the aromatic ring. The aromatic benzene rings are deflected out of these planes as well as boron atoms.
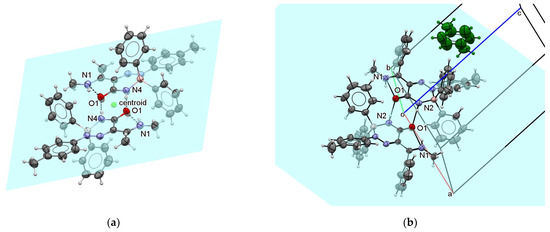
Figure 11.
ORTEP diagram of the dimer forming the basis of the triazaborinone 5c crystal (a); and ORTEP diagram of the triazaborinone 5d crystal (b).
Triazaborinone 5d forms dimers such as 5c. The structure of the dimer is shown in Figure 11b. As in the previous case, the dimer molecule is symmetric according to the center of symmetry, which in this case is at the beginning of the coordinates of the unit cell. By replacing the methyl group bound to carbon C3 with a phenyl group, hydrophobic cavities are formed in the structure of triazaborinone 5d in which toluene is enclosed as a solvent, one molecule of toluene stoichiometrically per dimer 5d. The toluene molecule is captured in the middle of the bc side of the crystallographic unit cell and is green-colored for clarity. The toluene molecule is located freely in the cavity. The presence of a toluene methyl group on two sides is a disorder for statistical reasons. Similar host–guest structures have been previously described by us for 3-amino-2-(4-dimethylaminophenyldiazenyl)-1-phenylbut-2-en-1-one (reaction of 3-amino-1-phenylbut-2-en-1-one with 4-dimethylaminobenzenediazonium tetrafluoroborate) [34]. The arrangement of the molecules in the crystal is shown in Figures S129 and S130. The hydrogen bonds that form the dimers are again shown in the figures for easier orientation.
Triazaborinone 5g has a similar arrangement of double bonds lying in the plane as triazaborinone 5c. The ORTEP diagram is shown in Figure 12a. Because of the substitution of imide hydrogen on the triazaborinone ring with a methyl group, triazaborinone 5g cannot form intramolecular hydrogen bonds leading to dimers. In this case, the crystal structure is formed by individual molecules that are bound together by weak nonbonding interactions, e.g., between aromatic rings. This arrangement is shown in Figure S131. The interaction mentioned can be seen in the middle of an indicated crystallographic unit cell. These phenyl groups attached to the boron atom form graphite-like π–π stacking with a distance between these rings of 3.293 Å. The chains formed by this layering in the direction of the crystallographic axis b are repeated regularly in the directions of the crystallographic axes a and c. The planes indicated in the figure are those in which the conjugated double bonds between the atoms C1O1, C2N2, and C3N4 lie. All atoms of the triazaborinone ring of compound 5g lie in the plane from which the ring of the p-tolyl group is slightly turned out.
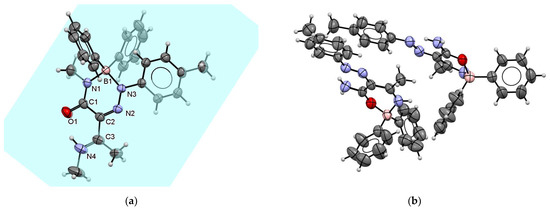
Figure 12.
ORTEP diagram of the triazaborinone 5g crystal (a); and ORTEP diagram of the oxazaborine 6a crystal (b).
Oxazaborine 6a, which was previously prepared from 3-aminobut-2-ennitrile as the starting compound, was characterized in our previous publication [30]. Unlike our earlier publication, the prepared single crystal 6a was now measured at room temperature. The structure obtained is identical, and the newly estimated values are given in Table S5. The new parameters slightly differ from those previously published due to the measurement temperature, and the resulting statistics are slightly better. Unlike other prepared single crystals at this work, only oxazaborine 6a needs two molecules to form an asymmetric unit (see Figure 12b or Figure S132). In addition, in the case of this substance, three conjugated double bonds lie in the plane. They are N3N4, C1C2, and C3N2. In this case, the boron atoms are completely deflected out of these planes, and the N–B–O angles are 103.69° and 104.13°. Sizes of such angles are not common but are described in the literature.
Oxazaborine 6e differs from previously published 6a by substituting the hydrogen atom of the amide with a methyl group. Although the structure of oxazaborine 6a consists of dimers due to hydrogen bonds similar to the structure of triazaborinone 5c described above, the formation of such dimers in the structure of oxazaborine 6e is not possible. The oxygen atom O1 in compound 6e does not participate in the formation of any hydrogen bond. In this case, the entire oxazaborine ring lies in the plane, as well as the p-tolyl group bonded to nitrogen N4. Benzene rings attached to a boron atom outside this plane form a roughly tetragonal system. The ORTEP diagram of this compound shows Figure 13a. The crystal structure of oxazaborine 6e is formed by chains of individual molecules bound to each other by π–π interactions. These interactions in the direction of the crystallographic axis c are shown in Figure S133. Figure S134 shows the alternation of these chains in the direction of the crystallographic axes a and c.
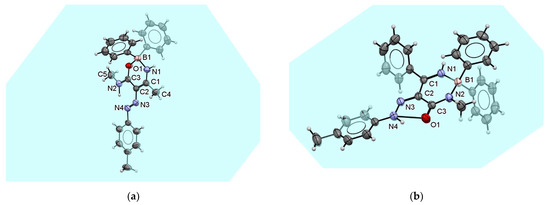
Figure 13.
ORTEP diagram of the oxazaborine 6e crystal (a); and ORTEP diagram of the diazaborinone 7f crystal (b).
The ORTEP diagram of the diazaborinone 7f is shown in Figure 13b. Two similar diazaborinones have been previously published [28]. The first differs by the substitutions on the C1 and N2 atoms, where the substituents are exchanged. The second differs by the substitution on the N2 atom, where the phenyl group is replaced by a methyl group. In the structures described previously, the formation of chains of individual molecules was observed due to the short distance interaction between the oxygen atom O1 and the imine hydrogen bonded to the nitrogen atom N1 of the second molecule. In the case of the newly described structure of diazaborinone 7f, such an intermolecular hydrogen bond does not occur, and only weak interactions occur between the molecules in a crystal. The crystal packing in the direction of the crystallographic axes a and b is shown in Figures S135 and S136. The first interaction is a π–π stacking between the p-tolyl groups. These interactions lie on the ab side of the crystallographic unit cell. The second interaction is between phenyl groups bonded to boron atoms. These groups form layers that are nested inside each other. These interactions lie in the center plane of the crystallographic unit cell in one-half of the edge length c.
2.5. Fluorescence
Some substances showed visible fluorescence when irradiated with a UV lamp (366 nm). This was found during thin-layer chromatography with UV detection. Therefore, steady-state photoluminescence measurements in the solid state were performed for a series of triazaborinones 5a–d, 5g, and 5h; diazaborinones 7a and 7f–g; triazaborine 8h; and several previously prepared substances. The measured substances were excited by radiation at a wavelength of 360 nm, and the corresponding spectra are shown in Figure 14 and Figure 15.
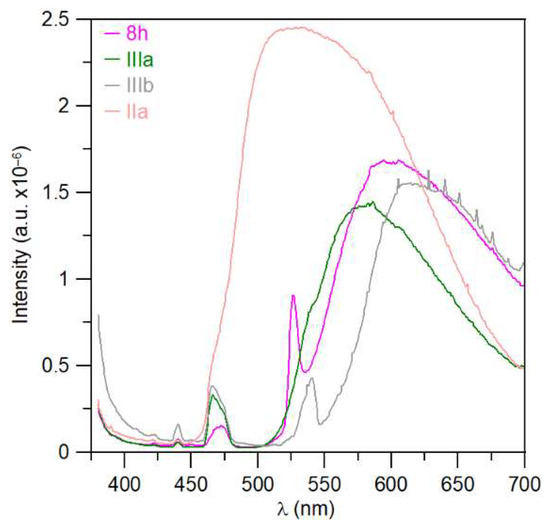
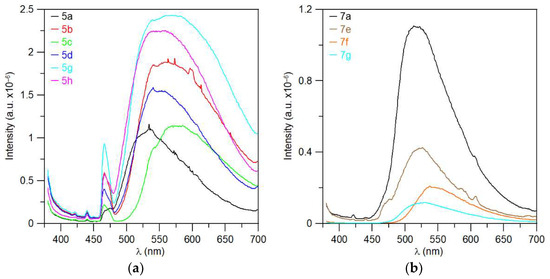
Figure 15.
Fluorescence spectrum: (a) triazaborines 5a–d, 5g, and 5h; and (b) diazaborinones 7a, 7f–g.
Time-resolved photoluminescence measurements were also performed, and quantum yields were evaluated. Unfortunately, measurable quantum yields were observed for only three substances. Fluorescence quantum yields values are not very high. The quantum yield for triazaborinone 5h is 3.84% ± 1%, for triazaborinone 5g 4.08% ± 1%, and for diazaborinone 7a 14.62% ± 1%. The absorption and emission spectra for these substances are shown in Figures S137–139.
3. Experimental
3.1. Materials and Methods
All chemicals and dried dichloromethane except those mentioned below were purchased from commercial suppliers (Acros Organics (Part of Thermo Fisher Scientific, Geel, Belgium)), Sigma-Aldrich (Merck, Darmstadt, Germany), or Fluorochem (Hadfield, Derbyshire, UK). Toluene and diethylether were dried over sodium.
NMR spectra were measured in CDCl3 or in DMSO-d6 using a Bruker AVANCE III 400 spectrometer (Ettlingen, Germany) operating at 400 MHz (1H) and 100 MHz (13C) or using a Bruker Ascend 500 spectrometer (Ettlingen, Germany) operating at 500 MHz (1H), 125 MHz (13C), 160 MHz (11B), and 50 MHz (15N). All of the pulse sequences were taken from the Bruker software library. The 13C NMR spectra were measured in a standard way and by means of the APT pulse sequence. The data are reported as follows: chemical shift in ppm (δ), multiplicity (s = singlet, d = doublet, q = quartet, m = multiplet, br s = broad singlet, br q = broad quartet). The coupling constants J are reported in Hertz (Hz). TMS was used as an internal standard for 1H NMR in CDCl3 (δ 0.00). CDCl3 was used as an internal standard for 13C NMR (the middle signal, δ 77.16). B(OMe)3 was used as an external standard for 11B NMR (δ 18.1). Nitromethane was used as an external standard for 15N NMR (δ 0.00). DMSO-d6 was used as an internal standard for both 1H NMR (the middle signal δ 2.55) and 13C NMR (the middle signal δ 39.6).
Elemental analyses were performed on a Flash 2000 CHNS Elemental Analyzer (Thermo Fisher Scientific, Milan, Italy).
Melting points were measured on a Kofler Boetius PHMK 80/2644 hot-stage microscope and were not corrected.
High-resolution mass spectra were recorded on a MALDI LTQ Orbitrap XL (Thermo Fisher Scientific, Bremen, Italy) equipped with a nitrogen UV laser (337 nm, 60 Hz, 8–20 μJ) in the positive ion mode. For the CID experiment using the linear trap quadrupole (LTQ) helium was used as the collision gas and 2,5-dihydroxybenzoic acid (DHB) as the MALDI matrix.
UV−Vis spectra were recorded on a UV–Vis spectrophotometer Hewlett-Packard 8453 (Waldbronn, Germany). IR spectra were recorded on a Nicolet iS50 (Madison, Wisconsin, USA) equipped with an ATR diamond crystal (neat solid samples). The wavenumber range 2500–1700 cm−1 was excluded due to diamond absorption. The spectra were processed with the SPECTRAGRYPH 1.2.15 (Dr. Friedrich Menges, Oberstdorf, Germany).
Steady-state and time-resolved PL measurements were performed based on a multifunctional spectrometer with L-geometry of excitation-detection pathways (FluoTime 300, PicoQuant, Berlin, Germany). A Xenon arc lamp and a PMT were used as the excitation sources and detectors for PL experiments. The PL-QY value could be obtained based on the spectrometer incorporated with an integrating sphere. Excitation wavelength: 360 nm. Range: 340–700 nm.
Crystal data for all compounds were collected at 295 K using a Nonius Kappa CCD diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Ǻ). The data sets were integrated with the Denzo SMN package [35] and corrected for Lorentz, polarization, and absorption effects (SORTAV) [36]. Structures were solved by direct methods using the SIR97 [37] system of programs and refined anisotropically by using full-matrix least-squares for all non-hydrogen atoms and hydrogen atoms included on their calculated positions, riding on their carrier atoms, except the N–H hydrogens forming intramolecular hydrogen bonds, which were refined isotropically. All calculations were performed by using SHELXL 2014/6 [38], PARST [39] and PLATON [40] implemented in the WINGX [41] system of programs. Visualization of structures was done with MERCURY 2020.2.0.
3.2. Synthetic Procedure
3-Aminobut-2-enamide (1a). The dry ammonia gas was bubbled into a mixture of 3-oxobutanamide (4a) (10.1 g, 100 mmol) and diethyl ether (20 mL) over a period of 1 h. Then liquid ammonia (15 mL) was added and the mixture was stirred at room temperature overnight. The crystals were filtered off and boiled in chloroform (30 mL) for 5 min. After cooling, the white crystals were filtered off. Yield 7.13 g (71%), m.p. 95.5–99 °C (Ref. [42] 98–100 °C). 1H-NMR (DMSO-d6, 500 MHz): δ 7.31 (br s, 2H), 6.45 (br s, 1H), 6.09 (br s, 1H), 4.35 (s, 1H), 1.76 (s, 3H); 13C-NMR (DMSO-d6, 125 MHz): δ 172.6, 157.0, 85.4, 25.7, 21.8.
3-Amino-3-phenylprop-2-enamide (1b). The solution of ethyl 3-oxo-3-phenylpropanoate (22.1 g, 115 mmol) and 25% aq. ammonia (22.7 mL, 300 mmol) was heated with stirring in an Ace pressure tube at 130 °C (oil bath) for 4.5 h. After cooling, the solid product was filtered off. Yield 6.6 g. The filtrate was concentrated and then aq. ammonia (16 mL) was added and heated in an Ace pressure tube at 130 °C for 3 h. After cooling, the solid product was filtered off. Yield 1.5 g. Yellowish crystal, total yield 8.1 g (43%), m.p. 162–164.5 °C (Ref. [16] 164 °C). 1H-NMR (DMSO-d6, 400 MHz): δ 7.58–7.60 (m, 2H), 7.46–7.48 (m, 3H), 6.85 (br s, 1H), 6.26 (br s, 1H), 4.91 (s, 1H); 13C-NMR (DMSO-d6, 100 MHz,): δ 172.2, 157.1, 137.9, 129.6, 128.6, 126.1, 85.9.
3-(Methylamino)but-2-enamide (1c). 3-Oxobutanamide (4a) (5 g, 50 mmol) and methylamine solution (33 wt.% in absolute ethanol, 18.5 mL, 150 mmol) were heated in an Ace pressure tube for 4 h at 130–140 °C. After cooling to room temperature, ethanol was evaporated, and the oil was cooled. Ethyl acetate (5 mL) was added, and the mixture was stirred for 5 min at cooling. The crystals were filtered off, and the filtrate was evaporated. Ethyl acetate (3 mL) was added to the filtrate and the mixture was stirred for 5 min at cooling. The crystals were filtered off. This compound must be stored in the refrigerator. Total yield 3.33 g (58%), m.p. 70–74 °C. 1H-NMR (CDCl3, 400 MHz): δ 8.99 (br s, 1H), 5.01 (br s, 2H), 4.37 (s, 1H), 2.87 (d, J = 5.3 Hz, 3H), 1.87 (s, 3H); 13C-NMR (CDCl3, 100 MHz): δ 173.0, 161.0, 83.1, 29.3, 19.1. HR-MS (MALDI), cald. for C5H11N2O: [M + H]+ 115.0866, found: 115.0867 [M + H]+.
3-(Methylamino)-3-phenylprop-2-enamide (1d). The solution of 3-oxo-3-phenylprop-2-enamide (4b) (3.2 g, 19.6 mmol) and 33 wt% methylamine in ethanol (7.4 mL, 58.8 mmol) was heated with stirring in an Ace pressure tube at 135–140 °C. After heating for 4 h, ethanol was evaporated under reduced pressure. Ethyl acetate (5 mL) was added to the viscous oil and the mixture was heated for 2 min. After cooling, the light-yellow crystals were filtered off. Yield 2.8 g (81%), m.p. 93.5–96 °C. 1H-NMR (CDCl3, 500 MHz): δ 9.01 (br s, 1H), 7.38–7.39 (m, 3H), 7.32–7.34 (m, 2H), 5.09 (br s, 2H), 4.48 (s, 1H), 2.71 (d, J = 5.2 Hz, 3H); 13C-NMR (CDCl3, 125 MHz): δ 172.7, 164.2, 136.3, 129.0, 128.3, 127.9, 86.1, 31.3. For elemental analysis, the product was recrystallized from the cyclohexane/toluene mixture. Anal. Calc. for C10H12N2O: C, 68.16; H, 6.86; N, 15.90. Found: C, 68.40; H, 6.90; N, 15.87%.
3-Amino-N-methylbut-2-enamide (1e). The title compound was prepared according to a modified published procedure [43]. Dry ammonia gas was bubbled into a mixture of N-methyl-3-oxobutanamide (4c) (2.26 g, 19.6 mmol), dry toluene (10 mL), and molecular sieve (1 g) over a period of 2 h at 10 °C. Then ethanol (30 mL) was added. The solid was dissolved, the mixture was filtered off, and the filtrate was evaporated to dryness. Cold diethyl ether was added, and the mixture was stirred. Then the white crystals were filtered off. This compound must be stored in the refrigerator. Yield 1.59 g (71%), m.p. 95.5–99 °C (Ref. [43] 115–116 °C). 1H-NMR (CDCl3, 400 MHz): δ 6.33 (br s, 2H), 5.26 (br s, 1H), 4.36 (s, 1H), 2.78 (d, J = 4.9 Hz, 3H), 1.84 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 171.3, 156.0, 86.6, 25.6, 22.3. Anal. Calc. for C5H10N2O: C, 52.61; H, 8.81; N, 24.54. Found: C, 52.58; H, 8.93; N, 23.81%.
3-Amino-N-methyl-3-phenylprop-2-enamide (1f). The title compound was prepared according to the published procedure [44]. N-Methyl-3-oxo-3-phenylpropanamide (4d) (7 g, 40 mmol), ammonium acetate (16.3 g, 200 mmol) and methanol (100 mL) were stirred at 55 °C for 3 days. Then, aqueous ammonia (50 mL) was added. The reaction mixture was extracted with dichloromethane (2 × 50 mL). The organic layer was dried with anhydrous sodium sulphate, and solvent was removed by distillation. Dichloromethane (50 mL) was added to the residue, and the organic layer was again extracted with water (1 × 50 mL). The organic layer was dried with anhydrous sodium sulphate, and solvent was removed by distillation. Recrystallization from ethyl acetate was obtained 3.78 g (55%). White crystals, m.p. 127–130 °C. 1H-NMR (CDCl3, 400 MHz): δ 7.28–7.40 (m, 5H), 6.56 (s, 2H), 5.47 (s, 1H), 4.8 (s, 1H), 2.83 (d, J = 5.2 Hz, 3H); 13C-NMR (CDCl3, 100 MHz): δ 171.1, 157.3, 138.5, 129.7, 128.7, 126.1, 87.5, 25.9. Anal. Calc. for C10H12N2O: C, 68.16; H, 6.86; N, 15.90. Found: C, 68.24; H, 6.98; N, 15.71%.
N-Methyl-3-(methylamino)but-2-enamide (1g). N-Methyl-3-oxobutanamide (4c) (2.65 g, 23 mmol): 33 wt% solution of methylamine in ethanol (8.6 mL, 69 mmol) and montmorillonite K-10 (5.8 g) was boiled ca 60 min. After cooling, dichloromethane (10 mL) was added, and montmorillonite was filtered and washed with dichloromethane (4 × 20 mL). Dichloromethane was distilled off, and the residue solidified in the refrigerator overnight. Then the beige crystals were filtered off. This compound must be stored in the refrigerator. Yield 2.63 g (89%), m.p. 50–54 °C. 1H-NMR (CDCl3, 400 MHz): δ 5.87 (br s, 1H), 5.23 (br s, 1H), 4.33 (s, 1H), 2.85 (d, J = 5.2 Hz, 3H), 2.75 (d, J = 4.9 Hz, 3H), 1.86 (s, 3H); 13C-NMR (CDCl3, 100 MHz): δ 171.7 (br), 158.9 (br), 84.3 (br), 29.2, 25.5 (br), 19.0. HR-MS (MALDI), cald. for C6H13N2O: [M + H]+ 129.1022, found: 129.1023 [M + H]+.
N-Methyl-3-(methylamino)-3-phenylprop-2-enamide (1h). The method according to Štefane and Polanc was used [33]. The solution of 3b (3.38 g, 15 mmol) and 33 wt% methylamine in ethanol (9.3 mL) in propan-1-ol (65 mL) was heated with stirring in an Ace pressure tube at 130–140 °C. After heating for 4 h, the reaction mixture was evaporated under reduced pressure, and the residue was cooled. Slightly yellowish crystals were filtered off. Yield 4 g (96%), m.p. 101–105 °C. 1H-NMR (CDCl3, 400 MHz): δ 8.86 (br s, 1H), 7.34–7.36 (m, 3H), 7.29–7.32 (m, 2H), 5.39 (br s, 1H), 4.45 (s, 1H), 2.79 (d, J = 4.9 Hz, 3H), 2.64 (d, J = 5.3 Hz, 3H); 13C-NMR (CDCl3, 100 MHz): δ 171.3, 162.4, 136.6, 128.7, 128.2, 127.9, 87.9, 31.2, 25.7. For elemental analysis, this compound was recrystallized from the cyclohexane/toluene mixture. Anal. Calc. for C11H14N2O: C, 69.45; H, 7.42; N, 14.73. Found: C, 69.58; H, 7.50; N, 14.70%.
General procedure for the preparation of 4-ethoxy-2,2-difluoro-1,3,2-dioxoborinanes 2a, b. A modified method according to Štefane and Polanc was used [33]. To a solution of ethyl benzoylacetate (156 mmol, 30 g) in dichloromethane (35 mL) 48% BF3–Et2O (312 mmol, 39.6 mL) was added at 23–25 °C. After stirring at 23–25 °C for 24 h, the precipitated material was filtered off. All volatile materials were removed under vacuum from the filtrate. Cooled diethyl ether (100 mL) was added to the residue, and after stirring for 5 min, the crystals were filtered off. Filtrate was evaporated again, and the residue was stirred with a small amount of cooled diethyl ether. The precipitated crystals were filtered off. The total yield of white crystals was 26.95 g (72%). M.p. 105–106 °C (Ref. [45] 108–110 °C). For compound 2a: After stirring at 23–25 °C for 26.5 h, all volatile materials were removed under vacuum, and the yellow oily product was used for the preparation of compound 3a without purification.
2,2-Difluoro-4-methyl-6-(methylamino)-1,3,2-dioxoborinane (3a). The method according to Štefane and Polanc was used [33]. To a stirred solution of 33 wt% methylamine in ethanol (37 mL, 297 mmol) and acetonitrile (245 mL), crude dioxoborinane 2a was added at 23 °C. The reaction mixture was stirred 30 min at 23 °C. The solvents were evaporated, and the residue was separated by column chromatography using dichloromethane as an eluent to give 16.85 g (45%). Light-yellow crystals, m.p. 95–99 °C. 1H-NMR (CDCl3, 400 MHz): δ 7.30 (br s, 1H, maj.), 6.51 (br s, 0,09H, min.), 5.36 (s, 1H, min.), 5.33 (s, 1H, maj.), 3.00 (d, J = 4,8 Hz, 3.26 H, maj. + min.), 2.16 (s, 0,30H, min.), 2.05 (s, 3H, maj.); 13C-NMR (CDCl3, 100 MHz): δ 181.1 (min.), 176.8 (maj.), 169.2 (min.), 168.8 (maj.), 87.8 (maj.), 84.0 (min.), 28.7 (min.), 27.2 (maj.), 23.3 (min.), 22.5 (maj.); 19F NMR (CDCl3, 376 MHz): δ −142.20 (19F–11B, 0.8F, min.), −142.14 (19F–10B, 0.2F, min.), –142.09 (19F–11B, 0.8F, maj.), –142.03 (19F–10B, 0.2F, maj.). For elemental analysis, this compound was recrystallized from ethyl acetate. Anal. Calc. for C5H8BF2NO2: C, 36.86; H, 4.95; N, 8.60. Found: C 37.12; H 5.07; N 8.63%.
2,2-Difluoro-6-(methylamino)-4-phenyl-1,3,2-dioxoborinane (3b). The method according to Štefane and Polanc was used [33]. To a stirred solution of 33 wt% methylamine in ethanol (18.1 mL, 146 mmol) and acetonitrile (118 mL), dioxoborinane 2b (26.95 g, 112 mmol) was added at room temperature. The reaction mixture was stirred for 30 min at room temperature. The solvents were evaporated, and the residue was stirred in diethyl ether (40 mL). The crystals were filtered off. White crystals, yield 25 g (84%), m.p. 164–168 °C. 1H-NMR (DMSO-d6, 500 MHz): δ 8.07–8.08 (m, 0.21H, min.), 7.87–7.88 (m, 2H, maj.), 7.54–7.62 (m, 3.37H, maj. + min.), 6,42 (s, 0.1H, min.), 6.14 (s, 1H, maj.), 3.05 (s, 0.36H, min.), 2.96 (s, 3H, maj.); 13C-NMR (DMSO-d6, 125 MHz): δ 171.9 (min.), 169.0 (maj.), 168.9 (min.), 168.2 (maj.), 133.0 (min.), 132.9 (maj.), 132.6 (min.), 132.2 (maj.), 129.0 (maj.), 128.9 (min.), 126.6 (min.), 126.5 (maj.), 85.6 (maj.), 81.2 (min.), 28.5 (min.), 26.9 (maj.); 19F-NMR (DMSO-d6, 470 MHz): δ −141.77 (19F–11B, 0.8F, min.), −141.71 (19F–10B, 0.2F, min.), −141.49 (19F–11B, 0.8F, maj.), −141.44 (19F–10B, 0.2F, maj.). For elemental analysis, this compound was recrystallized from ethanol. Anal. Calc. for C10H10BF2NO2: C, 53.38; H, 4.48; N, 6.23. Found: C 53.68; H 4.60; N 6.23%.
3-Oxo-3-phenylprop-2-enamide (4b). This compound was prepared according to the procedure in Ref. [32]. 3-Amino-3-phenylprop-2-enamide (1b) (8 g, 49 mmol) was heated in water (33 mL) for 4 h. After cooling, the solid product was filtered off. Crystals were crystallized from water. Yellowish crystals, yield 4.62 g (57%), m.p. 108–111 °C (Ref. [32] 110–111 °C).
N-Methyl-3-oxobutanamide (4c). This compound was prepared according to the procedure in Ref. [33]. The mixture of 2,2-difluoro-4-methyl-6-(methylamino)-1,3,2-dioxoborinane (3a) (9 g, 55 mmol), sodium acetate (22,7 g, 276 mmol), and ethanol/water (36 mL/36 mL) was refluxed with stirring for 6 h. The solvents were evaporated, and the residue was washed several times with CH2Cl2 (25 mL). Dichloromethane was evaporated, and the residue was purified by vacuum distillation. Light yellow liquid, yield 4.56 g (72%), b.p. 95–99 °C/2–3 mbar (Ref. [46] 96–98 °C/0.13 mbar).
N-Methyl-3-oxo-3-phenylpropanamide (4d). This compound was prepared according to the procedure in Ref. [33]. The mixture of 2,2-difluoro-4-(methylamino)-6-phenyl-1,3,2-dioxoborinane (3b) (11.4 g, 50 mmol), sodium acetate (20.5 g, 250 mmol), and ethanol/water (50 mL/50 mL) was refluxed with stirring for 13 h. The reaction mixture was extracted with dichloromethane (2 × 100 mL). The organic layer was dried with anhydrous sodium sulphate, and the solvent was removed by distillation. White crystals, yield 7.52 g (85%), m.p. 101.5–103.5 °C (Ref. [47] 101–102 °C).
4-Methylbenzenediazonium tetraphenylborate. 4-Methylaniline (0.73 g, 6.84 mmol) was dissolved in boiling aqueous hydrochloric acid (3 mL, 1:1). The solution was cooled to −5 °C and diazotized by gradual addition of a cold solution of sodium nitrite (0.49 g, 7.11 mmol) in water (1.5 mL). The temperature during diazotization was maintained between −5 and 0 °C. The excess of nitrous acid (presence tested by starch iodide paper) was decomposed using the required amount of sulfamic acid. A solution of sodium tetraphenylborate (2.34 g, 6.84 mmol) in water (75 mL) was added at once. The precipitated diazonium salt was filtered and washed with cold ethanol (1 × 30 mL) and diethyl ether (2 × 30 mL). The diazonium salt was prepared immediately prior to use and dried in vacuo at room temperature in a desiccator for about 1 h (CAUTION: Dry diazonium tetraphenylborates can undergo violent decomposition when the crystalline material is ground!). The yield of diazonium salt was 2.81 g (94%), yellow powder.
General Procedure for the Reaction of 1a–h with 4-Methylbenzenediazonium Tetraphenylborate
Method A. A stoichiometric amount of freshly prepared 4-methylbenzenediazonium tetraphenylborate (2.19 g, 5 mmol) was added to a stirred solution of β-enaminoamide 1a–h (5 mmol) in dry dichloromethane (35 mL) at 10 °C. The reaction mixture was stirred at room temperature for 3–6 days. Then it was filtered, and the solvent was removed in vacuo. The crude residue was separated by column chromatography.
Method B. The procedure was the same as for Method A, except remelted sodium acetate (1.23 g, 15 mmol) was finely ground and added to a stirred solution of β-enaminoamide 1a–h and 4-methylbenzenediazonium tetraphenylborate. The reaction mixture was stirred at room temperature for 3–6 days. Then it was filtered, and the solvent was removed in vacuo. The crude residue was separated by column chromatography.
Method C. To a cold (5 °C) solution of β-enaminoamides 1a–h (5 mmol) in dry dichloromethane (15 mL) and dry toluene (25 mL) was added freshly prepared 4-methylbenzenediazonium tetraphenylborate (2.19 g, 5 mmol). The reaction mixture was stirred 30 min at room temperature and then refluxed for 3–5 h. The reaction mixture was cooled to room temperature and the solvents evaporated. The crude residue was separated by column chromatography.
6-(1-Aminoethylidene)-2-(4-methylphenyl)-3,3-diphenyl-4H-1,2,4,3λ4-triazaborine-5-one (5a). After column chromatography (silica gel/CH2Cl2) and boiling in ethanol (5 mL), the title compound was obtained as yellow crystals. Yield 1.16 g (61%, Method A, 72 h), 1.10 g (58%, Method B, 72 h) and 1.16 g (61%, Method C, reflux 2.5 h), m.p. 237–240 °C. 1H-NMR (DMSO-d6, 400 MHz): δ 12.12 (s, 1H), 10.63 (s, 1H), 7.32–7.34 (m, 4H), 7.23–7.25 (m, 2H), 7.17–7.20 (m, 4H), 7.09–7.12 (m, 2H), 6.94–6.96 (m, 2H), 6.80 (s, 1H), 2.54 (s, 3H), 2.20 (s, 3H); 13C-NMR (DMSO-d6, 100 MHz): δ 176.1, 160.7, 150.7 (br s), 146.0, 135.4, 133.4, 128.4, 127.0, 125.4, 123.3, 122.2, 20.5, 18.5. For elemental analysis, compound 5a was recrystallized from toluene. Anal. Calc. for C23H23BN4O: C, 72.27; H, 6.06; N, 14.66. Found: C, 71.99; H, 6.13; N, 14.39%.
6-Amino-4-methyl-5-[4-(methylphenyl)diazenyl)]-2,2-diphenyl-3H-1,3,2λ4-oxazaborine (6a). After column chromatography (silica gel/CH2Cl2) and recrystallization from the ethanol/ethyl acetate mixture, compound 6a was obtained as yellow crystals. Yield 0.08 g (4%, Method A, 72 h), 0.14 g (7%, Method B, 72 h), m.p. 215–218 °C. 1H-NMR (CDCl3, 500 MHz): δ 11.32 (s, 1H), 7.40–7.44 (m, 6H), 7.28–7.31 (m, 4H), 7.22–7.25 (m, 2H), 7.16–7.20 (m, 2H), 6.84 (s, 1H), 6.15 (br d, J = 3 Hz, 1H), 2.54 (s, 3H), 2.36 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 170.3, 162.1, 150.4, 149.1 (br s), 137.8, 132.0, 129.7, 127.6, 126.7, 120.7, 114.6, 21.5, 21.3. Anal. Calc. for C23H23BN4O: C, 72.27; H, 6.06; N, 14.66. Found: C, 72.10; H, 6.21; N, 14.72%.
6-(Amino(phenyl)methylidene)-2-(4-methylphenyl)-3,3-diphenyl-4H-1,2,4,3λ4-triazaborine-5-one (5b). After column chromatography (silica gel/CH2Cl2) and recrystallization from the ethanol/toluene mixture, the title compound was obtained as yellow crystals. Yield 1.11 g (50%, Method A, 72 h), 0.82 g (37%, Method B, 72 h) and 1.22 g (55%, Method C, reflux 2.5 h), m.p. 133–137 °C. 1H-NMR (CDCl3, 500 MHz,): δ 12.00 (br s, 1H), 8.26 (br s, 1H), 7.42–7.44 (m, 2H), 7.32–7.36 (m, 5H), 7.21–7.27 (m, 6H), 7.20–7.18 (m, 2H), 6.99–7.01 (m, 2H), 6.74–6.76 (m, 2H), 5.18 (br s, 1H), 2.13 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 173.3, 161.9, 149.4 (br s), 145.7, 137.0, 133.6, 133.4, 132.1, 129.8, 128.6, 128.3, 127.4, 126.1, 122.8, 122.7, 21.0. Anal. Calc. for C28H25BN4O: C, 75.69, H, 5.67, N, 12.61. Found: C, 75.78, H, 5.86, N, 12.60%.
5-[(4-Methylphenyl)hydrazono]-2,2,6-triphenyl-1H,3H-1,3,2λ4-diazaborine-4-one (7b). After column chromatography (silica gel/CH2Cl2) and boiling in cyclohexane (4 mL), the title compound was obtained as yellow crystals. Yield 0.22 g (10%, Method A, 72 h), 0.46 g (21%, Method B, 72 h), m.p. 255–260 °C. 1H-NMR (CDCl3, 500 MHz): δ 15.35 (br s, 1H), 8.46 (br s, 1H), 7.53–7.57 (m, 3H), 7.45–7.48 (m, 2H), 7.39–7.41 (m, 4H), 7.28–7.31 (m, 4H), 7.21–7.23 (m, 2H), 7.07–7.09 (m, 2H), 6.96–6.97 (m, 2H), 5.73 (br s, 1H), 2.29 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 170.5, 164.7, 150.0 (br), 139.3, 135.7, 134.2, 132.4, 132.0, 130.2, 129.4, 128.5, 127.8, 126.5, 122.1, 116.1, 21.1. Anal. Calc. for C28H25BN4O: C, 75.69, H, 5.67, N, 12.61. Found: C, 75.89, H, 5.78, N, 14.46%.
6-[1-(Methylamino)ethylidene]-2-(4-methylphenyl)-3,3-diphenyl-4H-1,2,4,3λ4-triazaborine-5-one (5c). After column chromatography (silica gel/CH2Cl2) and recrystallization from toluene, the title compound was obtained as yellow crystals. Yield 0.85 g (43%, Method A, 72 h), 0.95 g (48%, Method B, 72 h) and 1.38 g (69%, Method C, reflux 2.5 h), m.p. 224–227 °C. 1H-NMR (CDCl3, 400 MHz): δ 13.40 (s, 1H), 7.38–7.40 (m, 4H), 7.19–7.23 (m, 6H), 7.12–7.17 (m, 2H), 6.89–6.91 (m, 2H), 5,51 (s, 1H), 3.17 (d, J = 5.3 Hz, 3H), 2.50 (s, 3H), 2.21 (s, 3H); 13C-NMR (CDCl3, 100 MHz): δ 174.9, 162.2, 149.8 (br s), 146.2, 136.4, 133.7, 128.6, 127.2, 126.0, 123.4, 122.8, 31.7, 21.0, 13.8. Anal. Calc. for C24H25BN4O: C, 72.74, H, 6.36, N, 14.14. Found: C, 73.01, H, 6.45, N, 13.95%.
1,6-Dimethyl-5-[(4-methylphenyl)hydrazono]-2,2-diphenyl-3H-1,3,2λ4-diazaborine-4-one (7c). After column chromatography (silica gel/CH2Cl2) and boiling in ethanol (3 mL), the title compound was obtained as yellow crystals. Yield 0.15 g (8%, Method B, 72 h), m.p. 213–216 °C. 1H-NMR (CDCl3, 400 MHz): δ 15.24 (s, 1H), 7.39–7.42 (m, 4H), 7.27–7.31 (m, 4H), 7.21–7.25 (m, 4H), 7.14–7.16 (m, 2H), 5.61 (s, 1H), 3.10 (s, 3H), 2.54 (s, 3H), 2.32 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 171.7, 163.2, 148.5 (br s), 139.7, 134.9, 133.3, 130.2, 127.7, 126.5, 123.8, 115.7, 39.7, 21.1, 15.5. Anal. Calc. for C24H25BN4O: C, 72.74, H, 6.36, N, 14.14. Found: C, 73.04, H, 6.62, N, 13.85%.
6-[(Methylamino)(phenyl)methyliden]-2-(4-methylphenyl)-3,3-diphenyl-4H-1,2,4,3λ4-triazaborine-5-one (5d). After column chromatography (silica gel/CH2Cl2/EtOAc (10:1)) and boiling in ethanol, the title compound was obtained as yellow crystals. Yield 1.5 g (65.5%, Method A, 104 h), 1.8 g (79%, Method B, 72 h), and 1.57 g (68.5%, Method C, reflux 5h), m.p. 184–187 °C. 1H-NMR (CDCl3, 500 MHz): δ 13.22 (br q, J = 5 Hz, 1H), 7.39–7.45 (m, 7H), 7.25–7.26 (m, 2H), 7.18–7.21 (m, 4H), 7.11–7.13 (m, 2H), 6.89–6.92 (m, 2H), 6.65–6.67 (m, 2H), 5.7 (s, 1H), 2.88 (d, J = 5 Hz, 3H), 2.05 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 174.3, 162.0, 149.6 (br s), 145.7, 136.1, 133.5, 130.4, 130.0, 128.5, 128.3, 128.2, 127.2, 125.8, 123.6, 122.2, 33.1, 20.7. For elemental analysis, compound 5d was recrystallized from the cyclohexane/toluene mixture. Anal. Calc. for C24H25BN4O: C, 72.74, H, 6.36, N, 14.14. Found: C, 73.01, H, 6.45, N, 13.95%.
1-Methyl-5-[(4-methylphenyl)hydrazono]-2,2,6-triphenyl-3H-1,3,2λ4-diazaborine-4-one (7d). After column chromatography (silica gel/CH2Cl2/EtOAc (10:1)) and boiling in ethanol for 5 min, the title compound was obtained as yellow crystals. Yield 0.04 g (1.7%, Method A, 104 h), 0.24 g (10.5%, Method B, 72 h), and 0.22 g (9.6%, Method C, reflux 5 h), m.p. 207.5–211 °C. 1H- NMR (CDCl3, 500 MHz): δ 15.07 (s, 1H), 7.49–7.51 (m, 7H), 7.32–7.35 (m, 4H), 7.26–7.27 (m, 4H), 6.98–7.00 (m, 2H), 6.75–6.76 (m, 2H), 5.68 (s, 1H), 2.89 (s, 3H), 2.24 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 172.6, 163.7, 148.6 (br s), 139.5, 134.9, 133.3, 133.1, 130.0, 129.6, 128.5, 127.7, 127.5, 126.6, 124.8, 115.6, 41.1, 21.0. For elemental analysis, compound 7d was recrystallized from toluene. Anal. Calc. for C29H27BN4O: C, 75.99, H, 5.94, N, 12.22. Found: C, 76.20, H, 6.01, N, 12.15%.
4-Methyl-6-(methylamino)-5-[(4-methylphenyl)diazenyl]-2,2-diphenyl-3H-1,3,2λ4-oxazaborine (6e). After column chromatography (silica gel/CH2Cl2 or n-hexane/EtOAc) and boiling in ethanol (4 mL) for 5 min, the title compound was obtained as yellow crystals. Yield 0.54 g (29%, Method A, 72 h), 1.01 g (51%, Method B, 72 h), and 0.63 g (32%, Method C, reflux 5 h), m.p. 173–176 °C. 1H-NMR (CDCl3, 400 MHz): δ 11.95 (br s, 1H), 7.41–7.44 (m, 6H), 7.26–7.29 (m, 4H), 7.20–7.23 (m, 2H), 7.16–7.19 (m, 2H), 6.79 (br s, 1H), 3.11 (d, J = 4.4 Hz, 3H), 2.54 (s, 3H), 2.35 (s, 3H); 13C-NMR (CDCl3, 100 MHz): δ 169.4, 161.6, 150.5, 149.8 (br s), 137.2, 131.7, 129.7, 127.4, 126.4, 120.5, 115.3, 26.3, 21.2 (2 × CH3). For elemental analysis, compound 6e was recrystallized from the cyclohexane/toluene mixture. Anal. Calc. for C24H25BN4O: C, 72.74, H, 6.36, N, 14.14. Found: C, 73.13, H, 6.39, N, 14.45%. HR-MS (MALDI), calc. for C24H26BN4O: [M + H]+ 397.2194, found: 397.2201 [M + H]+; calc. for C24H26BN4ONa: [M + Na]+ 419.2014, found: 419.2022 [M + Na]+; calc. for C24H26BN4OK: [M + K]+ 435.1723, found 435.1753 [M + K]+.
1,4-Dimethyl-2,2-diphenyl-5-[(4-methylphenyl)hydrazono]-1H-1,3,2λ4-diazaborine-4-one (7e). After column chromatography (silica gel/CH2Cl2) and boiling for a few minutes in ethanol (4 mL), the title compound was obtained as yellow crystals. Yield 0.5 g (27%, Method A, 72 h) and 0.31 g (16%, Method C, reflux 5 h), m.p. 148.5–151.5 °C. 1H-NMR (CDCl3, 500 MHz): δ 15.6 (s, 1H), 8.10 (br s, 1H), 7.40–7.41 (m, 4H), 7.30–7.33 (m, 4H), 7.22–7.27 (m, 4H), 7.16–7.18 (m, 2H), 2.68 (s, 3H), 2.48 (s, 3H), 2.35 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 170.4, 163.7, 147.7 (br s), 139.6, 135.1, 133.4, 130.2, 127.7, 126.7, 122.9, 115.8, 30.7, 21.2, 21.1. For elemental analysis, compound 7e was recrystallized from the cyclohexane/toluene mixture. Anal. Calc. for C24H25BN4O: C, 72.74, H, 6.36, N, 14.14. Found: C, 72.49, H, 6.37, N, 14.30%.
5,N-Dimethyl-2-(4-methylphenyl)-3,3-diphenyl-4H-1,2,4,3λ4-triazaborine-6-carboxamide (8e). After column chromatography (silica gel/CH2Cl2 → EtOAc), the title compound was obtained as orange crystals. Yield 0.51 g (26%, Method C, reflux 5 h), m.p. 184–187 °C. 1H-NMR (CDCl3, 500 MHz): δ 7.63 (br s, 1H), 7.31–7.32 (m, 4H), 7.16–7.22 (m, 8H), 7.02 (q, J = 5 Hz, 1H), 6.89–6.91 (m, 2H), 2.83 (s, J = 5 Hz, 3H), 2.53 (s, 3H), 2.21 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 164.9, 158.8, 147.1 (br s), 145.3, 136.5, 133.6, 128.7, 127.5, 126.7, 123.0, 25.9, 23.8, 21.1. For elemental analysis, compound 8e was recrystallized from cyclohexane. Anal. Calc. for C24H25BN4O: C, 72.74, H, 6.36, N, 14.14. Found: C, 72.73, H, 6.37, N, 14.39%.
6-(Methylamino)-5-[(4-methylphenyl)diazenyl]-2,2,4-triphenyl-3H-1,3,2λ4-oxazaborine (6f). After column chromatography (silica gel/CH2Cl2) and recrystallization from the of ethanol/toluene mixture, the title compound was obtained as yellow crystals. Yield 0.18 g (8%, Method A, 96 h), 0.98 g (43%, Method B, 72 h), and 0.48 g (21%, Method C, reflux 2.5 h), m.p. 228–230 °C. 1H-NMR (CDCl3, 500 MHz): δ 12.09 (br s, 1H), 7.61–7.63 (m, 2H), 7.49–7.52 (m, 5H), 7.44–7.47 (m, 2H), 7.28–7.31 (m, 4H), 7.21–7.23 (m, 4H), 7.08–7.10 (m, 2H), 6.84 (s, 1H), 3.20 (d, J = 4.9 Hz, 3H), 2.31 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 168.8, 163.1, 150.3, 149.7 (br s), 137.4, 136.2, 131.8, 131.0, 129.8, 129.7, 128.1, 127.5, 126.5, 120.8, 115.2, 26.7, 21.3. Anal. Calc. for C29H27BN4O: C, 75.99, H, 5.94, N, 12.22. Found: C, 76.28, H, 6.08, N, 12.09%.
3-Methyl-5-[(4-methylphenyl)hydrazono]-2,2,6-triphenyl-1H-1,3,2λ4-diazaborine-4-one (7f). After column chromatography (silica gel/CH2Cl2) and recrystallization from the ethanol/toluene mixture, the title compound was obtained as yellow crystals. Yield 1.13 g (51%, Method A, 96 h), 0.2 g (9%, Method B, 72 h), and 0.3 g (13%, Method C, reflux 2.5 h), m.p. 238–240 °C. 1H NMR (CDCl3, 500 MHz): δ 15.75 (s, 1H), 8.12 (s, 1H), 7.53–7.54 (m, 3H), 7.45–7.49 (m, 6H), 7.31–7.35 (m, 4H), 7.24–7.27 (m, 2H), 7.08–7.09 (m, 2H), 6.99–7.01 (m, 2H), 2.75 (s, 3H), 2.29 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 169.5, 164.4, 148.1 (br), 139.5, 135.3, 134.1, 133.4, 131.9, 130.2, 129.4, 128.5, 127.8, 126.6, 122.2, 115.9, 30.9, 21.1. Anal. Calc. for C29H27BN4O: C, 75.99, H, 5.94, N, 12.22. Found: C, 76.16, H, 6.11, N, 12.49%.
4-Methyl-6-[1-(methylamino)ethyliden]-2-(4-methylphenyl)-3,3-diphenyl-1,2,4,3λ4-triazaborine-5-one (5g). After column chromatography (Method A: silica gel/n-hexan → CH2Cl2/n-hexan (1:4) → CH2Cl2 → EtOAc, Method C: silica gel/n-hexane/EtOAc (15:1) → EtOAc), the title compound was obtained as yellow crystals. Yield 0.53 g (26%, Method A, 72 h), 0.56 g (30%, Method B, 72 h), and 0.99 g (48%, Method C, reflux 4 h), m.p. 193–196 °C. 1H-NMR (CDCl3, 500 MHz): δ 13.88 (br q, J = 4.5 Hz, 1H), 7.45–7.47 (m, 4H), 7.20–7.22 (m, 4H), 7.13–7.15 (m, 2H), 7.08–7.1 (m, 2H), 6.82–6.83 (m, 2H), 3.08 (d, J = 5.4 Hz, 3H), 2.57 (s, 3H), 2.43 (s, 3H), 2.16 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 174.6, 161.1, 147.5 (br), 146.3, 136.0, 134.0, 128.3, 127.1, 125.8, 123.0, 122.8, 31.5, 30.5, 20.9, 13.6. For elemental analysis, compound 5g was recrystallized from the cyclohexane/toluene mixture. Anal. Calc. for C25H27BN4O: C, 73.18, H, 6.63, N, 13.65. Found: C, 73.39, H, 6.73, N, 13.56%.
3,4-Dimethyl-6-(methylamino)-5-[(4-methylphenyl)diazenyl]-2,2-diphenyl-1,3,2λ4-oxazaborine (6g). After column chromatography (see 5g) and recrystallization from the ethanol/toluene mixture, the title compound was obtained as yellow crystals. Yield 0.65 g (32%, Method A, 72 h), 0.32 g (17%, Method B, 72 h) and 0.11 g (5.4%, Method C, reflux 4 h), m.p. 215.5–221 °C. 1H-NMR (CDCl3, 500 MHz): δ 12.22 (br s, 1H), 7.41–7.43 (m, 2H), 7.38–7.39 (m, 4H), 7.27–7.30 (m, 4H), 7.22–7.25 (m, 2H), 7.16–7.17 (m, 2H), 2.99 (s, 3H), 2.64 (s, 3H), 2.35 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 170.1, 161.3, 150.7, 148.3 (br), 136.6, 133.1, 129.7, 127.3, 126.5, 120.3, 116.2, 37.6, 26.3, 21.3, 15.2. HR-MS (MALDI), calc. for C25H28BN4O: [M + H]+ 411.2351, found: 411.2358 [M + H]+; calc. for C25H27BN4ONa: [M + Na]+ 433.2170, found: 433.2178 [M + Na]+; calc. for C19H22BN4O: [M + H − Ph]+ 333.1881, found: 333.1886 [M + H − Ph]+.
1,3,6-Trimethyl-5-[(4-methylphenyl)hydrazono]-2,2-diphenyl-1,3,2λ4-diazaborine-4-one (7g). After column chromatography (see 5g) and recrystallization from the ethanol/toluene mixture, the title compound was obtained as yellow crystals. Yield 0.13 g (6.3%, Method C, reflux 4 h), m.p. 263.5–266 °C. 1H-NMR (CDCl3, 500 MHz): δ 15.59 (s, 1H), 7.47–7.48 (m, 4H), 7.31–7.34 (m, 4H), 7.24–7.28 (m, 4H), 7.17–7.19 (m, 2H), 3.01 (s, 3H), 2.56 (s, 3H), 2.53 (s, 3H), 2.35 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 171.2, 163.0, 146.3 (br), 140.0, 134.6, 133.9, 130.2, 127.7, 126.6, 123.8, 115.6, 39.7, 30.5, 21.1, 15.4. Anal. Calc. for C25H27BN4O: C, 73.18, H, 6.63, N, 13.65. Found: C, 73.58, H, 6.64, N, 13.43%.
N,4,5-Trimethyl-2-(4-methylphenyl)-3,3-diphenyl-1,2,4,3λ4-triazaborine-6-carboxamide (8g). After column chromatography (silica gel/n-hexane/EtOAc (15:1) → EtOAc) and boiling in ethanol (2 mL) for 5 min, the title compound was obtained as dark yellow crystals. Yield 0.05 g (2.4%, Method C, reflux 4 h), m.p. 186–189 °C. 1H-NMR (CDCl3, 500 MHz): δ 7.30–7.31 (m, 4H), 7.16–7.22 (m, 6H), 7.08 (br q, J = 4.9 Hz, 1H), 7.00–7.02 (m, 2H), 6.86–6.88 (m, 2H), 3.00 (s, 3H), 2.87 (d, J = 4.9 Hz, 3H), 2.65 (s, 3H); 13C-NMR (CDCl3, 100 MHz): δ 165.2, 159.5, 145.8, 145.6 (br), 135.7, 134.0, 128.9, 128.4, 127.5, 126.6, 123.4, 39.1, 26.1, 21.0, 17.8; 11B-NMR (CDCl3, 128 MHz): δ 0.98. HR-MS (MALDI), calc. for C25H28BN4O: [M + H]+ 411.2356, found: 411.2358 [M + H]+; calc. for C25H27BN4ONa: [M + Na]+ 433.2176, found: 433.2178 [M + Na]+; calc. for C25H27BN4OK: [M + K]+ 449.1915, found: 449.1918 [M + K]+; calc. for C19H22BN4O: [M + H − Ph]+ 333.1887, found: 333.1887 [M + H − Ph]+.
4,7,8-Trimethyl-2-(4-methylphenyl)-3,3,6,6-tetraphenyl-4,6-dihydro-3H-2λ4,3λ4,6λ4,7λ4-[1,3,2]oxazaborinino[6,5-e][1,2,4,3]triazaborinine (9g). After column chromatography (silica gel/n-hexane → CH2Cl2 → EtOAc) and boiling in ethanol (2 mL) for 5 min, the title compound was obtained as yellow crystals. Yield 0.11 g (4%, Method A, 72 h), m.p. 217–218 °C. 1H-NMR (CDCl3, 500 MHz): δ 7.39–7.40 (m, 4H), 7.33–7.35 (m, 4H), 7.26–7.30 (m, 6H), 7.17–7.22 (m, 6H), 7.08–7.10 (m, 2H), 6.82–6.84 (m, 2H), 3.08 (s, 3H), 2.66 (s, 3H), 2.54 (s, 3H), 2.16 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 169.3, 155.8, 146.8 (br), 145.6 (br), 145.5, 136.2, 134.1, 132.9, 128.6, 127.6, 127.5, 126.9, 126.5, 122.9, 120.0, 38.6, 31.9, 21.0, 15.1. HR-MS (MALDI), calc. for C37H37B2N4O: [M + H]+ 575.3148, found: 575.3160 [M + H]+; calc. for C37H36B2N4ONa: [M + Na]+ 597.2967, found: 597.2979 [M + Na]+; calc. for C31H31B2N4O: [M + H − Ph]+ 497.2679, found: 497.2692 [M + H − Ph]+.
4-Methyl-6-[(methylamino)(phenyl)methylidene]-2-(4-methylphenyl)-3,3-diphenyl-1,2,4,3λ4-triazaborine-5-one (5h). After column chromatography (silica gel/CH2Cl2) and boiling in ethanol (5 mL) for 5 min, the title compound was obtained as yellow crystals. Yield 0.35 g (14.8%, Method C, reflux 3 h), m.p. 206–211 °C. 1H-NMR (CDCl3, 500 MHz): δ 13.80 (br q, J = 4.6 Hz, 1H), 7.45–7.49 (m, 7H), 7.29–7.30 (m, 2H), 7.20–7.23 (m, 4H), 7.13–7.15 (m, 2H), 6.80–6.82 (m, 2H), 6.61–6.63 (m, 2H), 2.94 (d, J = 5.4 Hz, 3H), 2.60 (s, 3H), 2.03 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 174.4, 161.1, 147.4 (br), 145.7, 135.8, 133.9, 130.4, 130.2, 128.4, 128.3, 128.1, 127.2, 125.9, 123.3, 122.4, 33.1, 30.5, 20.8. For elemental analysis, compound 5h was recrystallized from ethyl acetate. Anal. Calc. for C30H29BN4O: C, 76.28, H, 6.19, N, 11.86. Found: C, 76.31, H, 6.27, N, 11.68%.
3-Methyl-6-(methylamino)-5-[(4-methylphenyl)diazenyl]-2,2,4-triphenyl-1,3,2λ4-oxazaborine (6h). After flash chromatography (n-hexane → chloroform) and washing with cyclohexane, the title compound was obtained as yellow crystals. Yield 0.53 g (22%, Method A, 96 h), m.p. 187–189.5 °C. 1H-NMR (CDCl3, 500 MHz): δ 11.94 (br s, 1H), 7.42–7.50 (m, 7H), 7.30–7.33 (m, 6H), 7.24–7.27 (m, 2H), 6.94–7.00 (m, 4H), 3.05 (d, J = 4.5 Hz, 3H), 2.76 (s, 3H), 2.25 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 171.4, 162.1, 150.4, 148.1 (br), 136.7, 134.7, 133.1, 129.5, 128.8, 128.2, 128.1, 127.4, 126.6, 120.3, 117.0, 39.4, 26.5, 21.2. HR-MS (MALDI), calc. for C25H28BN4O: [M + H]+ 473.25072, found: 473.25162 [M + H]+; calc. for C25H27BN4ONa: [M + Na]+ 495.2327, found: 495.2335 [M + Na]+; calc. for C25H27BN4OK: [M + K]+ 511.2066, found: 511.2075 [M + K]+.
1,3-Dimethyl-5-[(4-methylphenyl)hydrazono]-2,2,6-triphenyl-1,3,2λ4-diazaborine-4-one (7h). After column chromatography (silica gel/CH2Cl2) and boiling in ethanol (5 mL) for 5 min, the title compound was obtained as yellow crystals. Yield 0.37 g (16%, Method C, reflux 3 h) with m.p. 160.5–163.5 °C. After flash chromatography (n-hexane → chloroform) and boiling in ethanol, the title compound was obtained as yellow crystals. Yield 0.07 g (3%, Method A, 96 h). 1H-NMR (CDCl3, 500 MHz): δ 15.43 (s, 1H), 7.57–7.59 (m, 4H), 7.49–7.50 (m, 3H), 7.35–7.38 (m, 4H), 7.27–7.30 (m, 2H), 7.22–7.24 (m, 2H), 7.00–7.01 (m, 2H), 6.76–6.77 (m, 2H), 2.78 (s, 3H), 2.61 (s, 3H), 2.25 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 172.1, 163.3, 146.2 (br), 139.8, 134.6, 133.9, 133.1, 130.1, 129.5, 128.4, 127.7, 127.5, 126.6, 124.8, 115.4, 41.0, 30.5, 21.0. For elemental analysis, compound 7h was recrystallized from ethyl acetate. Anal. Calc. for C30H29BN4O: C, 76.28, H, 6.19, N, 11.86. Found: C, 76.56, H, 6.24, N, 11.75%.
5,N-Dimethyl-2-(4-methylphenyl)-3,3-diphenyl-1,2,4,3λ4-triazaborine-6-carboxamide (8h). After column chromatography (silica gel/CH2Cl2) and boiling in ethanol (5 mL) for 5 min, the title compound was obtained as orange crystals. Yield 0.51 g (21%, Method C, reflux 3 h), m.p. 207.5–209 °C. 1H-NMR (CDCl3, 500 MHz): δ 7.41–7.48 (m, 3H), 7.34–7.36 (m, 4H), 7.18–7.24 (m, 8H), 7.10–7.12 (m, 2H), 6.96 (br q, J = 5 Hz, 1H), 6.92–6.93 (m, 2H), 2.78 (s, 3H), 2.72 (d, J = 5 Hz, 3H), 2.25 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 163.9, 159.0, 145.8, 145.4 (br s), 136.3, 134.5, 134.1, 129.38, 129.36, 128.7, 128.5, 127.6, 126.7, 126.5, 123.8, 40.6, 26.0, 21.1. For elemental analysis, compound 8h was recrystallized from ethyl acetate. Anal. Calc. for C30H29BN4O: C, 76.28, H, 6.19, N, 11.86. Found: C, 76.24, H, 6.28, N, 11.75%.
4,7-Dimethyl-2-(4-methylphenyl)-3,3,6,6,8-pentaphenyl-4,6-dihydro-3H-2λ4,3λ4,6λ4,7λ4-[1,3,2]oxazaborinino[6,5-e][1,2,4,3]triazaborinine (9h). After column chromatography (silica gel/n-hexane → CH2Cl2 → EtOAc) and recrystallization from cyclohexane (or boiling in acetic acid), the title compound was obtained as yellow crystals. Yield 0.45 g (15%, Method A, 96 h), 0.11 g (3%, Method C, reflux 3 h), m.p. 216–217 °C. 1H-NMR (CDCl3, 500 MHz): δ 7.39–7.40 (m, 4H), 7.45–7.52 (m, 7H), 7.27–7.39 (m, 12H), 7.16–7.23 (m, 6H), 6.77–6.79 (m, 2H), 6.63–6.65 (m, 2H), 2.93 (s, 3H), 2.70 (s, 3H), 2.07 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 170.3, 156.3, 146.9 (br), 145.3 (br), 145.2, 135.9, 134.0, 132.9, 132.4, 129.9, 128.6, 128.4, 128.1, 127.7, 127.6, 127.0, 126.5, 122.2, 120.8, 40.5, 31.8, 20.9. HR-MS (MALDI), calc. for C42H39B2N4O: [M + H]+ 637.3304, found: 5637.3318 [M + H]+; calc. for C42H38B2N4ONa: [M + Na]+ 659.3124, found: 659.3136 [M + Na]+; calc. for C36H33B2N4O: [M + H − Ph]+ 559.2835, found: 559.2851 [M + H − Ph]+.
Rearrangement of oxazaborines 6a, 6e–h. The appropriate oxazaborine 6a, 6e–h was heated at 200–250 °C, and the reaction was monitored by TLC with dichloromethane as the eluent. The residue was subjected to column chromatography on silica gel (CH2Cl2).
5-[(4-Methylphenyl)hydrazono]-2,2-diphenyl-1H-1,3,2λ4-diazaborine-4-one (7a). Oxazaborine 6a (0.99 g, 2.59 mmol) was used. After 6 min at 250 °C, the crude mixture was dissolved in dichloromethane. An amount of 0.42 g was not dissolved in dichloromethane, and according to 1H NMR it was found that it was pure diazaborinone 7a. After column chromatography, the title compound was obtained as yellow crystals (0.39 g). Total yield 0.81 g (82%), m.p. 263.5–266 °C. 1H-NMR (CDCl3, 500 MHz): δ 15.29 (br s, 1H), 8.39 (br s, 1H), 7.349–7.35 (m, 4H), 7.27–7.29 (m, 4H), 7.21–7.24 (m, 4H), 7.17–7.20 (m, 2H), 5.73 (br s, 1H), 2.53 (s, 3H), 2.35 (s, 3H); 13C-NMR (CDCl3, 125 MHz): δ 171.2, 164.0, 149.7 (br), 139.4, 135.6, 132.4, 130.3, 127.8, 126.6, 122.8, 116.0, 21.3, 21.1. Anal. Calc. for C13H23BN4O: C, 72.27 H, 6.06, N, 14.64. Found: C, 72.47, H, 5.99, N, 14.54%.
Diazaborinone 7e. Oxazaborine 6e (0.79 g, 2 mmol) was used: 25 min at 200 °C and 25 min at 250 °C. After column chromatography, the title compound was obtained as yellow crystals. Yield 0.5 g (63%).
Diazaborinone 7f. Oxazaborine 6f (0.92 g, 2 mmol) was used: 70 min at 250 °C. After column chromatography, the title compound was obtained as yellow crystals. Yield 0.45 g (49%).
Diazaborinone 7g. Oxazaborine 6g (0.82 g, 2 mmol) was used: 30 min at 250 °C. After column chromatography, the title compound was obtained as yellow crystals. Yield 0.64 g (78%).
Diazaborinone 7h. Oxazaborine 6h (0.74 g, 0.74 mmol) was used: 90 min at 230 °C. After column chromatography, the title compound was obtained as yellow crystals. Yield 0.2 g (57%).
4. Conclusions
The novel boron containing heterocyclic compounds—triazaborinones 5a–d, 5g, and 5h—were synthesized from the reaction of simple β-enaminoamides and 4-methylbenzenediazonium tetraphenylborate. Other products that were formed during the reaction were oxazaborines 6a, 6e, 6f, and 6g; diazaborinones 7b–h; and triazaborines 8e, 8g, and 8h. Heating (250 °C, 6–70 min) of oxazaborines 6 without solvent resulted in their transformation into diazaborinones 7a–h in 49–82% yields. Compound 7a was not formed by the reaction of enaminoamide 1a and diazonium tetraphenylborate, but it was formed by ring transformation of oxazaborine 6a. Although N-phenylenaminoamides 1 (R3 = Ph) previously used produced diazaborinones only when there was an –NH2 group at position 3 of N-phenylenaminoamide, the enaminoamides 1 (R3 = H or Me) used in this work gave diazaborinones even when there was –NHMe at position 3 of the enaminoamide.
Structural, spectral, and crystallographic properties of the prepared structures were studied. Triazaborinones 5c and 5d and oxazaborine 6a form dimers in the solid state. There are hydrophobic cavities in the structure of triazaborinone 5d in which toluene as a solvent is enclosed. The measured quantum yields of the fluorescence in the solid-state of selected compounds are not very high; for diazaborinone 7a, 14.62% is the best.
Supplementary Materials
The following supporting information can be downloaded at, Scheme S1: Preparation of starting compounds; 1H, 13C, 11B, and 15N NMR spectra of heterocyclic compounds 5–9: Figures S1–S111; UV–Vis spectra: Figures S112–S124; Table S1: UV–Vis parameters of prepared heterocyclic compounds in CH2Cl2; Table S2: UV–Vis parameters of triazaborine 5g; Table S3: UV–Vis parameters of oxazaborine 6g; Table S4: UV–Vis parameters of diazaborinone 7a; Table S5: Crystal Data; Crystallographic data: Figures S125–S136; Fluorescence: Figures S137–S139.
Author Contributions
Conceptualization, M.S. (Markéta Svobodová); methodology, M.S. (Markéta Svobodová); validation, M.S. (Markéta Svobodová) and J.S.; analysis, M.S. (Markéta Svobodová), J.S., V.B., L.M., and B.-H.L.; investigation, M.S. (Markéta Svobodová), L.S., and L.M.; data curation, M.S. (Markéta Svobodová), J.S., V.B., and B.-H.L.; writing—original draft preparation, M.S. (Markéta Svobodová), J.S., M.S. (Miloš Sedlák), and B.-H.L.; writing—review and editing, M.S. (Markéta Svobodová), J.S., M.S. (Miloš Sedlák), and B.-H.L.; visualization, M.S. (Markéta Svobodová) and J.S.; supervision, M.S. (Miloš Sedlák) All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Education, Youth and Sports of the Czech Republic.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented are available in the manuscript Supplementary Materials. The data files in cif format for the crystal structures in this paper can be requested from the Cambridge Structural Database. CCDC 2127610–2127615 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures (accessed on 3 December 2021).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 6–9 are available from the authors.
References
- Mai, A.; Perrone, A.; Nebbioso, A.; Rotili, D.; Valente, S.; Tardugno, M.; Massa, S.; De Bellis, F.; Altucci, L. Novel Uracil-Based 2-Aminoanilide and 2-Aminoanilide-like Derivatives: Histone Deacetylase Inhibition and in-Cell Activities. Bioorg. Med. Chem. Lett. 2008, 18, 2530–2535. [Google Scholar] [CrossRef] [PubMed]
- Zandersons, A.; Lusis, V.; Vigante, B.; Mutsenietse, D.; Dubur, G. Synthesis of Ethoxycarbonyl-1,4- and -1,2-Dihydropyridinecarboxylic Acid Amides. Chem. Heterocycl. Compd. 1991, 27, 1339–1347. [Google Scholar] [CrossRef]
- Cooper, K.; Fray, M.J.; Parry, M.J.; Richardson, K.; Steele, J. 1,4-Dihydropyridines as Antagonists of Platelet Activating Factor. 1. Synthesis and Structure-Activity Relationships of 2-(4-Heterocyclyl)Phenyl Derivatives. J. Med. Chem. 1992, 35, 3115–3129. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.C.; Chiu, G.; Wetzel, J.M.; Marzabadi, M.R.; Nagarathnam, D.; Wang, D.; Fang, J.; Miao, S.W.; Hong, X.; Forray, C.; et al. Identification of a Dihydropyridine as a Potent A1a Adrenoceptor-Selective Antagonist That Inhibits Phenylephrine-Induced Contraction of the Human Prostate. J. Med. Chem. 1998, 41, 2643–2650. [Google Scholar] [CrossRef]
- Sainani, J.B.; Shah, A.C.; Arya, V.P. Synthesis of 1,4-Dihydro-2,6-Dimethyl-4-(Substituted Phenyl)-5-N-Methylaminocarbonyl-Pyridine-3-Carboxylate. Indian J. Chem. Sect. B Org. Chem. Incl. Med. Chem. 1994, 33, 573–575. [Google Scholar]
- Hisaki, M.; Kashima, K.; Sakamoto, Y.; Hojo, M.; Katayama, O.; Hata, H. 3-Aminocarbonyl-1,4-dihydropyridine-5-carboxylic Acid Compounds, and Pharmaceutical Composition Containing the Same. Patent US4874773A, 17 October 1989. [Google Scholar]
- Kato, T.; Noda, M. Studies on Ketene and Its Derivatives. LXXXI. Reaction of β-Aminocrotonamide with α,β-Unsaturated Ketones. Chem. Pharm. Bull. 1976, 24, 1408–1410. [Google Scholar] [CrossRef][Green Version]
- Chiba, T.; Takahashi, T. Studies on Amino Acid Derivatives. IV. Synthesis of 3-Amino-2 (1H)-Pyridone Derivatives Using 4-Ethoxymethylene-2-Phenyl-5-Oxazolone. Chem. Pharm. Bull. 1985, 33, 2731–2734. [Google Scholar] [CrossRef][Green Version]
- Brown, D.J.; Cowden, W.B. Unfused Heterobicycles as Amplifiers of Phleomycin. V. A Range of Pyridinylpyrimidines with Strongly Basic Side Chains. Aust. J. Chem. 1982, 35, 1203–1207. [Google Scholar] [CrossRef]
- Kato, T.; Chiba, T.; Sasaki, M. Reaction of 3-Aminocrotonamide with Nitriles. Heterocycles 1981, 16, 577–580. [Google Scholar] [CrossRef]
- Sato, M.; Ogasawara, H.; Kato, T. Reaction of β-Aminocrotonamide with Dibasic Acid Derivatives. J. Heterocycl. Chem. 1983, 20, 87–91. [Google Scholar] [CrossRef]
- Katagiri, N.; Koshihara, A.; Atsuumi, S.; Kato, T. Reaction of β-Aminocrotonamide with N-Acylated Amino Acid Esters to Give 2-Acylaminoalkyl-6-Methylpyrimidin-4 (3H)-Ones and Their Ring Closure with Polyphosphoric Acid (PPA). Chem. Pharm. Bull. (Tokyo) 1983, 31, 2288–2295. [Google Scholar] [CrossRef]
- Katagiri, N.; Koshihara, A.; Atsuumi, S.; Kato, T. 2-[(Acylamino)Methyl]-6-Methylpyrimidin-4(3H)-Ones. Novel Precursors for the Synthesis of Imidazo[1,5-a]Pyrimidines and Imidazo[4,5-b]Pyridines. J. Org. Chem. 1982, 47, 167–169. [Google Scholar] [CrossRef]
- Bischoff, C.; Schröder, E. Heterocyclenbildung Mit Enaminen. J. Prakt. Chem./Chem.-Ztg. 1992, 334, 711–715. [Google Scholar] [CrossRef]
- Zankowska-Jasinska, W.; Golus, J.; Kamela, Z.; Kolasa, A. Oxalyl Chloride in Furan- and 1H-Pyrrole-2,3-Dione Syntheses. Pol. J. Chem. 1987, 61, 141–148. [Google Scholar]
- Shaw, G.; Sugowdz, G. IsoOxazolones. Part VI. The Hydrogenation of 5-Aminoisooxazoles. A New Synthesis of Pyrimidines. J. Chem. Soc. 1954, 0, 665–668. [Google Scholar] [CrossRef]
- Goerdeler, J.; Horn, H. Über Isothiazole, IV. Substituierte 5-Amino-Isothiazole Und Isothiazolo[5.4-d]Pyrimidine. Chem. Ber. 1963, 96, 1551–1560. [Google Scholar] [CrossRef]
- Baldwin, A.G.; Rivers-Auty, J.; Daniels, M.J.D.; White, C.S.; Schwalbe, C.H.; Schilling, T.; Hammadi, H.; Jaiyong, P.; Spencer, N.G.; England, H.; et al. Boron-Based Inhibitors of the NLRP3 Inflammasome. Cell Chem. Biol. 2017, 24, 1321–1335. [Google Scholar] [CrossRef]
- Brough, D.; Allan, S.M.; Freeman, S.; Baldwin, A.G. Cyclic Diarylboron Derivatives as NLRP3 Inflammasome Inhibitors. WO2017017469A1, 2 February 2017. [Google Scholar]
- Šimůnek, P.; Pešková, M.; Bertolasi, V.; Macháček, V.; Lyčka, A. Synthesis, NMR and X-Ray Characterisation of 6-Substituted 4-Amino-5-Aryldiazenyl-1-Arylpyridazinium Salts. Tetrahedron 2005, 61, 8130–8137. [Google Scholar] [CrossRef]
- Josefík, F.; Svobodová, M.; Bertolasi, V.; Šimůnek, P. A Simple, Enaminone-Based Approach to Some Bicyclic Pyridazinium Tetrafluoroborates. Beilstein J. Org. Chem. 2013, 9, 1463–1471. [Google Scholar] [CrossRef]
- Šimůnek, P.; Pešková, M.; Bertolasi, V.; Lyčka, A.; Macháček, V. Formation of Pyridazinium Salts by Azo Coupling of N-Substituted 3-Amino-1-phenylbut-2-en-1-ones and Diazonium Salts. Eur. J. Org. Chem. 2004, 2004, 5055–5063. [Google Scholar] [CrossRef]
- Šimůnek, P.; Svobodová, M.; Bertolasi, V.; Macháček, V. Facile and Straightforward Method Leading to Substituted 4-Amino-1-Arylpyrazoles. Synthesis 2008, 11, 1761–1766. [Google Scholar] [CrossRef]
- Šimůnek, P.; Svobodová, M.; Macháček, V. Synthesis and Characterization of Some 3-Acyl-4-Amino-1-Aryl-1H-Pyrazoles. J. Heterocycl. Chem. 2009, 46, 650–655. [Google Scholar] [CrossRef]
- Šimůnek, P.; Macháček, V.; Svobodová, M.; Růžička, A. Some New Information on the Formation of Substituted 4-Amino-1-Substituted Phenyl-1H-Pyrazoles from β-Enaminones and Diazonium Tetrafluoroborates. J. Heterocycl. Chem. 2011, 48, 780–786. [Google Scholar] [CrossRef]
- Brož, B.; Padělková, Z.; Bertolasi, V.; Šimůnek, P. A Simple New Hydrazine-Free Synthesis of Methyl 1,4,5-Trisubstituted 1H-Pyrazole-3-carboxylates. Mon. Chem.—Chem. Mon. 2013, 144, 1013–1019. [Google Scholar] [CrossRef]
- Pešková, M.; Šimůnek, P.; Bertolasi, V.; Macháček, V.; Lyčka, A. Novel 5-(4-Substituted-Phenyldiazenyl)-1,3,2λ4-Oxazaborines and Their Rearrangement to 1,2,4,3λ4-Triazaborines. Organometallics 2006, 25, 2025–2030. [Google Scholar] [CrossRef]
- Svobodová, M.; Bárta, J.; Šimůnek, P.; Bertolasi, V.; Macháček, V. Straightforward Access to Oxazaborines, Diazaborinones and Triazaborines by Reactions of β-Enaminoamides with 4-Methylbenzenediazonium Tetraphenylborate. J. Organomet. Chem. 2009, 694, 63–71. [Google Scholar] [CrossRef]
- Josefík, F.; Svobodová, M.; Bertolasi, V.; Šimůnek, P.; Macháček, V.; Almonasy, N.; Černošková, E. A New Bicyclic Oxazaborines with a Bridged Nitrogen Atom, Their Thermic Rearrangement and Fluorescence Properties. J. Organomet. Chem. 2012, 699, 75–81. [Google Scholar] [CrossRef]
- Svobodová, M.; Šimůnek, P.; Macháček, V.; Štruncová, L.; Růžička, A. Four-Coordinate Organoboron Compounds from β-Enaminonitriles and Diazonium Salts. Tetrahedron 2012, 68, 2052–2060. [Google Scholar] [CrossRef]
- Josefík, F.; Mikysek, T.; Svobodová, M.; Šimůnek, P.; Kvapilová, H.; Ludvík, J. New Triazaborine Chromophores: Their Synthesis via Oxazaborines and Electrochemical and DFT Study of Their Fundamental Properties. Organometallics 2014, 33, 4931–4939. [Google Scholar] [CrossRef]
- Giardina, G.A.M.; Sarau, H.M.; Farina, C.; Medhurst, A.D.; Grugni, M.; Raveglia, L.F.; Schmidt, D.B.; Rigolio, R.; Luttmann, M.; Vecchietti, V.; et al. Discovery of a Novel Class of Selective Non-Peptide Antagonists for the Human Neurokinin-3 Receptor. 1. Identification of the 4-Quinolinecarboxamide Framework. J. Med. Chem. 1997, 40, 1794–1807. [Google Scholar] [CrossRef]
- Štefane, B.; Polanc, S. A New Regio- and Chemoselective Approach to β-Keto Amides and β-Enamino Carboxamides via 1,3,2-Dioxaborinanes. Synlett 2004, 4, 698–702. [Google Scholar] [CrossRef]
- Macháček, V.; Bertolasi, V.; Šimůnek, P.; Svobodová, M.; Svoboda, J.; Černošková, E. A Three-Dimensional Channel Supramolecular Architecture Based on 3-Amino-2-(4-dimethylaminophenyldiazenyl)-1-phenylbut-2-en-1-one and Aromatic Guests. Cryst. Growth Des. 2010, 10, 85–91. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Macromol. Crystallogr. Part A 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Blessing, R.H. An Empirical Correction for Absorption Anisotropy. Acta Crystallogr. Sect. A 1995, 51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A New Tool for Crystal Structure Determination and Refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, M. PARST95—An Update to PARST a System of Fortran Routines for Calculating Molecular Structure Parameters from the Results of Crystal Structure Analyses. J. Appl. Crystallogr. 1995, 28, 659. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX Suite for Small-Molecule Single-Crystal Crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Kato, T.; Yamanaka, H.; Shibata, T. Studies on Ketene and Its Derivatives—XIII: Synthesis of β-Aminocrotonamide and the Structure of Its Pyrolysed Product. Tetrahedron 1967, 23, 2965–2971. [Google Scholar] [CrossRef]
- Kato, T.; Yamanaka, H.; Kawamata, J.; Shimomura, H. Studies on Ketene and Its Derivatives. XXVIII. Reaction of β-Aminocrotonamide with Ketene and Diketene. Chem. Pharm. Bull. 1969, 17, 1889–1895. [Google Scholar] [CrossRef][Green Version]
- Hansen, K.B.; Rosner, T.; Kubryk, M.; Dormer, P.G.; Armstrong, J.D. Detection and Elimination of Product Inhibition from the Asymmetric Catalytic Hydrogenation of Enamines. Org. Lett. 2005, 7, 4935–4938. [Google Scholar] [CrossRef] [PubMed]
- Stahl, I. 1,3-Dithienium- Und 1,3-Dithioleniumsalze, III. Synthese Cyclischer Dithioacetale von β-Ketoestern Aus Keten-Silylacetalen. Chem. Ber. 1985, 118, 3159–3165. [Google Scholar] [CrossRef]
- Gelbard, G.; Lin, J.; Roques, N. Reductions with NADH Models. 3. The High Reactivity of Hantzsch Amides. J. Org. Chem. 1992, 57, 1789–1793. [Google Scholar] [CrossRef]
- Sato, T.; Yamamoto, K.; Fukui, K.; Saito, K.; Hayakawa, K.; Yoshiie, S. Metal-Catalysed Organic Photoreactions. Photoreactions of 3,5-Dimethylisoxazole with and without Catalytic Assistance by Copper(II) Salts. J. Chem. Soc. Perkin Trans. 1 1976, 7, 783–787. [Google Scholar] [CrossRef]






















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).