
Computational Estimation of the Acidities of Pyrimidines and Related Compounds †

Abstract
:1. Introduction
2. Methods
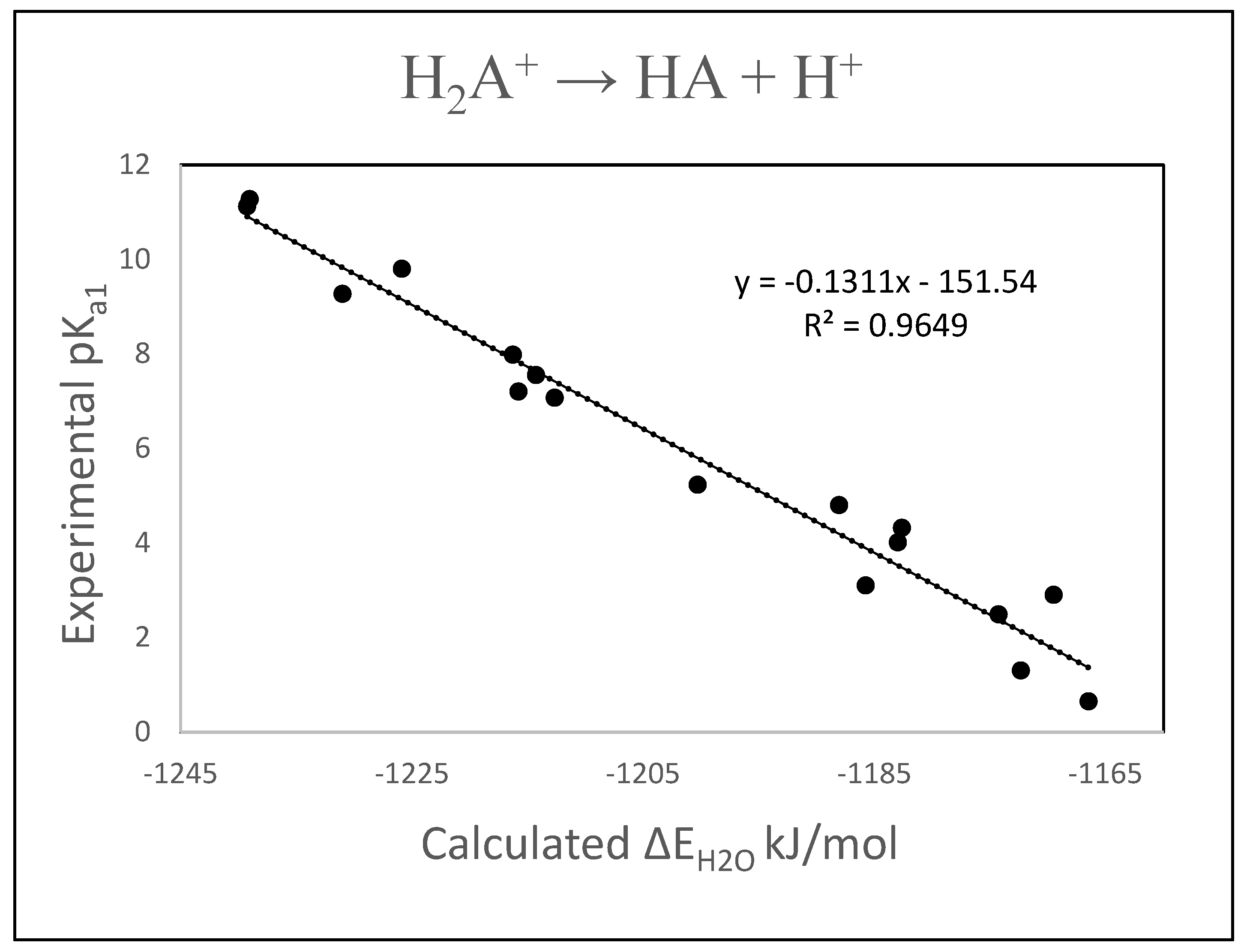
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Shields, G.C.; Seybold, P.G. Computational Approaches for the Prediction of pKa Values; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Ho, J. Predicting pKa in Implicit Solvents: Current Status and Future Directions. Aust. J. Chem. 2014, 67, 1441–1460. [Google Scholar] [CrossRef]
- Pracht, P.; Grimme, S. Efficient Quantum-Chemical Calculations of Acid Dissociation Constants from Free-Energy Relationships. J. Phys. Chem. A 2021, 125, 5681–5692. [Google Scholar] [CrossRef]
- Morency, M.; Néron, S.; Iftimie, R.; Wuest, J.D. Predicting pKa Values of Quinols and Related Aromatic Compounds with Multiple OH Groups. J. Org. Chem. 2021, 86, 14444–14460. [Google Scholar] [CrossRef] [PubMed]
- Haslak, Z.P.; Zareb, S.; Dogan, I.; Aviyente, V.; Monard, G. Using Atomic Charges to Describe the pKa of Carboxylic Acids. J. Chem. Inf. Model. 2021, 61, 2733–2743. [Google Scholar] [CrossRef]
- Thapa, B.; Raghavachari, K. Accurate pKa Evaluations for Complex Bio-Organic Molecules in Aqueous Media. J. Chem. Theory Comput. 2019, 15, 6025–6035. [Google Scholar] [CrossRef]
- Hunt, P.; Hosseini-Gerami, L.; Chrien, T.; Plante, J.; Ponting, D.J.; Segall, M. Predicting pKa Using a Combination of Semi-Empirical Quantum Mechanics and Radial Basis Function Methods. J. Chem. Inf. Model. 2019, 35, 7896–7904. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.R.; de Souza Silva, C.; Custodio, R. On the Accuracy of the Direct Method to Calculate pKa from Electronic Structure Calculations. J. Phys. Chem. A 2021, 125, 65–73. [Google Scholar] [CrossRef]
- Pereira, R.W.; Ramabhadran, R.O. pK-Yay: A Black-Box Method Using Density Functional Theory and Implicit Solvation Models to Compute Aqueous pKa Values of Weak and Strong Acids. J. Phys. Chem. A 2020, 124, 9061–9074. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, Y.; Zhang, L.; Luo, S.; Cheng, J.-P. Holistic Prediction of the pKa in Diverse Solvents Based on a Machine Learning Approach. Angew. Chem. Int. Ed. 2020, 59, 19282–19291. [Google Scholar] [CrossRef]
- Geremia, K.L.; Seybold, P.G. Computational Estimation of the Acidities of Purines and Indoles. J. Mol. Model. 2019, 25, 12. [Google Scholar] [CrossRef]
- Seybold, P.G.; Shields, G.C. Computational estimation of pKa values. WIREs Comput. Mol. Sci. 2015, 5, 290–297. [Google Scholar] [CrossRef]
- Alongi, K.S.; Shields, G.C. Theoretical Calculations of Acid Dissociation Constants: A Review Article. Ann. Rep. Comput. Chem. 2010, 6, 113–138. [Google Scholar]
- Liptak, M.D.; Gross, K.C.; Seybold, P.G.; Feldgus, S.; Shields, G.C. Absolute pKa determinations for substituted phenols. J. Am. Chem. Soc. 2002, 124, 6421–6427. [Google Scholar] [CrossRef]
- Seybold, P.G. Quantum Chemical QSPR Estimation of the Acidities and Basicities of Organic Compounds. Adv. Quantum Chem. 2012, 64, 83–104. [Google Scholar]
- Seybold, P.G. Quantum Chemical Estimation of the Acidities of Some Inorganic Oxoacids. Mol. Phys. 2015, 113, 232–236. [Google Scholar] [CrossRef]
- Seybold, P.G. Computational Estimation of the Acidities of Some Inorganic Nitrogen Acids. Mol. Phys. 2016, 114, 389–393. [Google Scholar] [CrossRef]
- Baldasare, C.A.; Seybold, P.G. Computational Estimation of the Gas-Phase and Aqueous Acidities of Carbon Acids. J. Phys. Chem. A 2020, 125, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Nicklaus, M.C. Comparison of nine programs predicting pKa values of pharmaceutical substances. J. Chem. Inf. Model. 2009, 49, 2801–2812. [Google Scholar] [CrossRef]
- Dearden, J.C.; Rotureau, P.; Fayet, G. QSPR prediction of physico-chemical properties for REACH. SAR QSAR Environ. Res. 2013, 34, 279–318. [Google Scholar] [CrossRef]
- Gross, K.C.; Seybold, P.G. Substituent Effects on the Physical Properties and pKa of Aniline. Int. J. Quantum Chem. 2000, 80, 1107–1115. [Google Scholar] [CrossRef]
- Gross, K.C.; Seybold, P.G.; Hadad, C.M. Comparison of Different Atomic Charge Schemes for Predicting pKa Variations in Substituted Anilines and Phenols. Int. J. Quantum Chem. 2002, 90, 445–458. [Google Scholar] [CrossRef]
- Gross, K.C.; Hadad, C.M.; Seybold, P.G. Charge Competition in Halogenated Hydrocarbons. Int. J. Quantum Chem. 2011, 112, 219–229. [Google Scholar] [CrossRef]
- Scheiner, S.; Seybold, P.G. Quantum chemical analysis of the energetics of the anti and gauche conformers of ethanol. Struct. Chem. 2009, 20, 43–48. [Google Scholar] [CrossRef]
- Varekova, R.S.; Geidl, S.; Ionescu, C.-M.; Skrehota, O.; Kudera, M.; Sehnal, D.; Bouchal, T.; Abagyan, R.; Huber, H.J.; Koca, J. Predicting pKa Values of Substituted Phenols from Atomic Charges: Comparison of Different Quantum Mechanical Methods and Charge Distribution Schemes. J. Chem. Inf. Model. 2011, 51, 1795–1806. [Google Scholar] [CrossRef]
- Ugur, I.; Marion, A.; Parant, S.; Jensen, J.H.; Monard, G. Rationalization of the pKa Values of Alcohols and Thiols Using Atomic Charge Descriptors and Its Application to the Prediction of Amino Acid pKa’s. J. Chem. Inf. Model. 2014, 54, 2200–2213. [Google Scholar] [CrossRef]
- Mull, H.F.; Turney, J.M.; Douberly, G.E.; Schaefer, H.F., III. Kinetic Stability of Pentazole. J. Phys. Chem. A 2021, 125, 9092–9098. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural Bond Orbital Methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Advanced Chemistry Development, Inc.: Toronto, ON, Canada. Available online: https://www.acdlabs.com/ (accessed on 24 November 2021).
- Katritzky, A.R.; Ramsden, C.A.; Joule, J.A.; Zhdankin, V.V. Handbook of Heterocyclic Chemistry, 3nd ed.; Elsevier: New York, NY, USA, 2010. [Google Scholar]
- Serjeant, E.P.; Dempsey, B. Ionization Constants of Organic Acids in Aqueous Solution; UPAC Chemical Data Series-No. 23; International Union of Pure and Applied Chemistry: Oxford, UK, 1979. [Google Scholar]
- CRC Handbook of Chemistry and Physics, 89th ed.; CRC Press: Boca Raton, FL, USA, 2008.
- Dean, J.A. Lange’s Handbook of Chemistry, 13th ed.; McGraw-Hill: New York, NY, USA, 1985. [Google Scholar]
- Levene, P.A.; Bass, L.W.; Simms, H.S. The ionization of pyrimidines in relation to the structure of pyrimidine nucleosides. J. Biol. Chem. 1926, 70, 229–241. [Google Scholar] [CrossRef]
- Darnall, K.R.; Townsend, L.B.; Robins, R.K. The structure of showdomycin, a novel carbon-linked nucleoside antibiotic related to uridine. Proc. Nat. Acad. Sci. USA 1967, 57, 548–553. [Google Scholar] [CrossRef] [Green Version]
- Privat, E.J.; Sowers, L.C. A proposed mechanism for the mutagenicity of 5-formyluracil. Mutat. Res. 1996, 354, 151–156. [Google Scholar] [CrossRef]
- Handschmacher, R.E.; Welch, A.D. Microbial studies of 6-azauracil, an antagonist of uracil. Cancer Res. 1956, 16, 965–969. [Google Scholar]
- NIST Chemistry WebBook. Available online: http://webbook.nist.gov/chemistry/ (accessed on 13 July 2016).
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Truhlar, D.G. A Universal Approach to Solvation Modeling. Accts. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Hunter, N.E.; Seybold, P.G. Theoretical estimation of the aqueous pKas of thiols. Mol. Phys. 2014, 112, 340–348. [Google Scholar] [CrossRef]
- Wessner, R.A. Theoretical Estimation of pKas of Pyrimidines and Related Heterocycles. Master’s Thesis, Wright State University, Dayton, OH, USA, 2016. [Google Scholar]
- Scheiner, S.; Seybold, P.G. (Utah State University, Logan, Utah). Unpublished work. 2009.
- Gauch, H.G., Jr. Prediction, Parsimony and Noise. Am. Sci. 1993, 81, 468–478. [Google Scholar]


{kind=link}
{kind=link}
| No. | Compound | Formula | pKa1 | ACD pKa1 | pKa2 | ACD pKa2 |
|---|---|---|---|---|---|---|
| 1 | azauracil | C3H3N3O2 | - | −4.4 ± 0.2 | - | 7.8 ± 0.2 |
| 2 | aziridine | C2H5N | 7.98 [32], 8.05 [33] | 8.1 ± 0.2 | - | - |
| 3 | creatinine | C4H7N3O | 4.8 [34] | 4.8 ± 0.1 | - | - |
| 4 | cytosine | C4H5N3O | 4.32 [30], 4.58 [35], 4.6 [31] | 4.4 ± 0.1 | 13 [33], 12.15 [36] | 12.3 ± 0.1 |
| 5 | flucytosine | C4H3FN2O2 | 3.26 [34] | 2.6 ± 0.1 | - | 10.5 ± 0.1 |
| 6 | imidazole | C3H4N2 | 7.15 [33], 6.99 [34,35] | 7.2 ± 0.6 | 14.44 [33] | 13.9 ± 0.1 |
| 7 | 1-methylimidazole | C4H6N2 | 6.95 [34] | 7.0 ± 0.1 | N/A | - |
| 8 | 4-methylimidazole | C4H6N2 | 7.55 [35] | 7.7 ± 0.6 | - | 14.3 ± 0.1 |
| 9 | isocytosine | C4H5N3O | 4.01 [34] | 3.4 ± 0.5 | 9.42 [36] | 9.6 ± 0.4 |
| 10 | isoxazole | C3H6NO | −2.0 [34] | −2.0 ± 0.5 | N/A | |
| 11 | maleimide | C4H3NO2 | - | −5.7 ± 0.2 | 9.46 [37] | 8.5 ± 0.2 |
| 12 | morpholine | C4H9NO | 8.492 [35] | 9.0 ± 0.2 | N/A | - |
| 13 | piperidine | C5H11N | 11.12 [35] | 10.4 ± 0.1 | - | - |
| 14 | piperazine | C4H10N2 | 9.78 [35], 9.73 [34] | 9.6 ± 0.1 | - | - |
| 15 | 1-methylpiperazine | C5H12N2 | 10.19 [35] | 9.3 ± 0.1 | - | - |
| 16 | oxazole | C3H6NO | 0.8 [34] | 1.0 ± 0.1 | N/A | - |
| 17 | pyrazine | C4H4N2 | 0.65 [34] | 1.2 ± 0.1 | N/A | - |
| 18 | pyrazole | C3H4N2 | 2.61 [35] | 2.8 ± 0.1 | 14.21 [33] | 14.0 ± 0.5 |
| 19 | pyridazine | C4H43N2 | 2.3 [32] | 3.1 ± 0.1 | N/A | - |
| 20 | pyridine | C5H5N | 5.23 [34] | 5.2 ± 0.1 | N/A | - |
| 21 | pyrimidine | C4H4N2 | 1.3 [32] | 1.8 ± 0.1 | N/A | - |
| 22 | pyrrole | C4H5N | −3.8 [34] | −0.3 ± 0.5 | 17.0 [33] | 17.0 ± 0.5 |
| 23 | pyrrolidine | C4H9N | 12.10 [33], 11.31 [34,35] | 10.5 ± 0.1 | - | - |
| 24 | succinimide | C4H5NO2 | - | −4.4 ± 0.2 | 9.62 [34,35], 9.68 [36] | 9.6 ± 0.1 |
| 25 | thymine | C5H6N2O2 | - | −4.1 ± 0.4 | 9.9 [33], 9.79 [35], 9.44 [34] | 9.2 ± 0.1 |
| 26 | uracil | C4H4N2O2 | - | −4.2 ± 0.1 | 9.43 [38], 9.45 [34,36] | 8.9 ± 0.1 |
| 27 | 5-bromouracil | C4H3BrN2O2 | - | - | 7.91 [38] | 6.8 ± 0.1 |
| 28 | 5-chlorouracil | C4H3CIN2O2 | - | - | 7.92 [38] | 6.8 ± 0.1 |
| 29 | fluorouracil | C4H4FN3O | - | - | 8.04 [30], 8.00 [39], 7.93 [38] | 6.7 ± 0.1 |
| 30 | 5-formyluracil | C5H4N2O3 | - | - | 6.84 [38] | 7.3 ± 0.1 |
| 31 | 5-nitrouracil | C4H3N3O4 | - | - | 5.3 [38] | 5.2 ± 0.1 |
| Compound | Exp. ΔrG° a | Calc. ΔrG° b | Calc. ΔE b |
|---|---|---|---|
| pyridine | 1601 | 1605 | 1648 |
| pyrazine | 1605 | 1605 | 1643 |
| pyrimidine | 1577 | 1579 | 1614 |
| pyridazine | 1565 | 1562 | 1601 |
| imidazole | 1433 | 1432 | 1466 |
| succinimide | 1414 | 1401 | 1436 |
| Compound | ΔE kJ/mol | Exp. pKa1 | Calc. pKa1 a | Residual |
|---|---|---|---|---|
| azauracil | −1084 | - | <<0 | - |
| aziridine | −1216 | 8.01 | 7.76 | 0.25 |
| creatinine | −1188 | 4.8 | 4.09 | 0.71 |
| cytosine | −1183 | 4.5 | 3.43 | 1.07 |
| flucytosine | −1169 | 3.26 | 1.60 | 1.66 |
| imidazole | −1213 | 7.07 | 7.36 | −0.29 |
| 1-methylimidazole | −1216 | 7.95 | 7.76 | 0.19 |
| 4-methylimidazole | −1215 | 7.55 | 7.63 | −0.08 |
| isocytosine | −1183 | 4.01 | 3.43 | 0.58 |
| isoxazole | −1115 | −2 | <0 | - |
| maleimide | −1035 | - | <<0 | - |
| oxazole | −872 | 0.8 | <<0 | - |
| piperidine | −1239 | 11.12 | 10.77 | 0.35 |
| piperazine | −1226 | 9.76 | 9.07 | 0.69 |
| 1-methylpiperazine | −1231 | 10.19 | 9.72 | 0.47 |
| pyrazine | −1166 | 0.65 | 1.21 | −0.56 |
| pyrazole | −1174 | 2.61 | 2.25 | 0.36 |
| pyridazine | −1186 | 2.3 | 3.83 | −1.53 |
| pyridine | −1200 | 5.23 | 5.66 | −0.43 |
| pyrimidine | −1172 | 1.3 | 1.99 | −0.69 |
| pyrrolidine | −1239 | 11.71 | 10.77 | 0.94 |
| succinimide | −1007 | - | <<0 | - |
| Compound | ΔE kJ/mol | Exp. pKa2 | Calc. pKa2 a | Residuals |
|---|---|---|---|---|
| azauracil | −1179 | - | 6.82 | - |
| aziridine | −1325 | - | 27.41 | - |
| creatinine | −1217 | - | 12.18 | - |
| cytosine | −1216 | 12.57 | 12.04 | 0.53 |
| isocytosine | −1196 | 9.42 | 9.22 | 0.20 |
| flucytosine | −1254 | - | 17.39 | - |
| imidazole | −1216 | 14.4 | 12.04 | 2.36 |
| 4-methylimidazole | −1224 | - | 13.16 | - |
| maleimide | −1196 | 9.5 | 9.22 | 0.28 |
| piperazine | −1313 | - | 25.71 | - |
| 1-methylpiperazine | −1395 | - | 37.28 | - |
| piperidine | −1310 | - | 25.29 | - |
| pyrrole | −1243 | 17 | 15.84 | 1.16 |
| pyrrolidine | −1390 | - | 36.57 | - |
| thymine | −1199 | 9.71 | 9.64 | 0.07 |
| uracil | −1200 | 9.44 | 9.78 | −0.34 |
| 5-bromouracil | −1184 | 7.91 | 7.52 | 0.39 |
| 5-chlorouracil | −1182 | 7.92 | 7.24 | 0.68 |
| fluorouracil | −1193 | 7.99 | 8.79 | −0.80 |
| 5-formyluracil | −1180 | 6.84 | 6.96 | −0.12 |
| 5-nitrouracil | −1159 | 5.3 | 4.00 | 1.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holt, R.A.; Seybold, P.G. Computational Estimation of the Acidities of Pyrimidines and Related Compounds. Molecules 2022, 27, 385. https://doi.org/10.3390/molecules27020385
Holt RA, Seybold PG. Computational Estimation of the Acidities of Pyrimidines and Related Compounds. Molecules. 2022; 27(2):385. https://doi.org/10.3390/molecules27020385
Chicago/Turabian StyleHolt, Rachael A., and Paul G. Seybold. 2022. "Computational Estimation of the Acidities of Pyrimidines and Related Compounds" Molecules 27, no. 2: 385. https://doi.org/10.3390/molecules27020385
APA StyleHolt, R. A., & Seybold, P. G. (2022). Computational Estimation of the Acidities of Pyrimidines and Related Compounds. Molecules, 27(2), 385. https://doi.org/10.3390/molecules27020385